

Abstract
A reaction probability is required to calculate the rate constant of a diffusion-dominated reaction. Due to the complicated geometry and potentially high dimension of the reaction probability problem, it is usually solved by a Brownian dynamics simulation, also known as a random walk or path integral method, instead of solving the equivalent partial differential equation by a discretization method. Building on earlier work, this article completes the development of a robust importance sampling algorithm for Brownian dynamics—i.e., biased Brownian dynamics with weight control—to overcome the high energy and entropy barriers in biomolecular association reactions. The biased Brownian dynamics steers sampling by a bias force, and the weight control algorithm controls sampling by a target weight. This algorithm is optimal if the bias force and the target weight are constructed from the solution of the reaction probability problem. In reality, an approximate reaction probability has to be used to construct the bias force and the target weight. Thus, the performance of the algorithm depends on the quality of the approximation. Given here is a method to calculate a good approximation, which is based on the selection of a reaction coordinate and the variational formulation of the reaction probability problem. The numerically approximated reaction probability is shown by computer experiments to give a factor-of-two speedup over the use of a purely heuristic approximation. Also, the fully developed method is compared to unbiased Brownian dynamics. The tests for human superoxide dismutase, Escherichia coli superoxide dismutase, and antisweetener antibody NC6.8, show speedups of 17, 35, and 39, respectively. The test for reactions between two model proteins with orientations shows speedups of 2578 for one set of configurations and 3341 for another set of configurations.
INTRODUCTION
This article considers an elliptic partial differential equation for reaction probability in high dimensions and constructs path integral, or random walk, methods for solving it. The methods are oriented toward the calculation of rate constants for diffusion-limited reactions. The challenge of the random walk method is variance reduction. An importance sampling method for random walk methods, biased Brownian dynamics, is proposed in an earlier article (Zou et al., 2000). That method makes use of weighted averages, but unfortunately the possibility of large weights sometimes cancels the benefit of importance sampling. To avoid this problem, a method of weight control has been developed (Zou, 2002) and submitted to SIAM J. Sci. Comput. for publication (G. Zou and R. D. Skeel, “Robust Variance Reduction for Random Walk Methods,” http://bionum.cs.uiuc.edu/p.html). The main contribution of this present work is to give an effective and systematic technique to approximate the reaction probability, from which the bias force in biased Brownian dynamics and the target weight in weight control are constructed. We demonstrate the effectiveness of the algorithm with four numerical tests.
The application considered here is the rate constant computation for diffusion-limited reactions, for which the encounter of the reactants is the time-limiting stage of the reaction. Biochemical reactions such as enzyme-substrate and antibody-antigen association fit well in this category. Brownian dynamics (BD) simulation is widely used in this area (Antosiewicz et al., 1995; Brune and Kim, 1994; Getzoff et al., 1992; Guddat et al., 1994; Kozack et al., 1995; Northrup et al., 1993; Wade et al., 1994). Northrup, Allison, and McCammon (NAM) developed a method for rate constant calculation for diffusion-limited reactions in 1984. The NAM method connects the reaction rate with a reaction probability, which is the solution of the Smoluchowski equation, an elliptic partial differential equation (PDE), in a finite domain (Zhou, 1990). Solving the PDE by a discretization method is difficult due to the possible high dimension and the complicated geometry of configuration space. Instead, the random walk method is used, which involves the calculation of the expectation of an exit value for a stochastic differential equation (SDE). A BD simulation code for biomolecules, University of Houston Brownian Dynamics (UHBD), that calculates biomolecular rates of association with the NAM method, is distributed by McCammon and co-workers (Davis et al., 1991). Our rate constant computation is based mainly on the NAM method and UHBD.
Standard BD samples only a few reacted trajectories, especially for problems with high dimensions or with high energy barriers. Thus it gives a large variance for the random exit value. To reduce the variance, an enhanced sampling method—weighted ensemble Brownian dynamics (WEBD)—was developed by Huber and Kim (1996). WEBD maintains an ensemble of particles (in configuration space) and associates a probability weight with each particle. All particles carry out Brownian motion without influencing each other. Periodically, WEBD splits and merges particles according to their position and weight. This procedure enables better sampling in regions inaccessible to standard BD. Thus, particles are sampled more frequently near reaction regions and the variance is reduced. WEBD was originally proposed for use with the flux-over-population method (Hänggi et al., 1990, p. 258), but it has also been combined with the NAM method (Rojnuckarin et al., 2000).
However, WEBD is a fairly complicated algorithm, and biased BD (Zou et al., 2000) is offered as a simpler, and delightfully parallel, alternative. Biased BD does importance sampling by 1), introducing a bias force in addition to the existing force; and 2), associating a weight with the particle. The expectation of the weighted exit value—the product of the exit value and the weight at the exit time—gives an unbiased estimate of the reaction probability of standard BD. The variance of the weighted exit value is related to the bias force. It turns out there is an optimal bias force that makes the variance of the weighted exit value 0, but in practice a rough approximation must be used.
One difficulty that biased BD faces is that the particle weight can grow without bound due to the imperfect choice of the bias force and the approximation in the numerical integration. The large weight fluctuations result in a large variance of the weighted exit value. Under the optimal bias force, the particle has an ideal weight, which is not trajectory-dependent, but a function only of the current configuration. This is the basis of a weight control algorithm (Zou, 2002), which uses a target weight and a tolerance range to force the particle weight into a range. The weight control algorithm operates as follows. At the beginning of each time step, the particle weight is checked. If the weight exceeds the upper limit, the particle splits into two, each with one-half the weight, and they are simulated independently. The final result is the sum of the two weighted exit values. Conversely, if the weight falls below the lower limit, the weight is doubled with 50% probability and zeroed out (and the trajectory abandoned) otherwise. Thus, the expectation of the exit value is unchanged.
In practice, an approximate reaction probability function is used to construct the bias force and the target weight. Hence, the quality of the approximation determines the performance of the algorithm. Given in the section “Construction of bias force” of this article is a method to calculate a good approximation, based on the selection of a reaction coordinate and the variational formulation of the reaction probability problem.
The benefit of using a partly numerical rather than a purely heuristic estimate of reaction probability is investigated for a model of Escherichia coli superoxide dismutase (SOD) 1eso. The speedup is 1.8 without weight control and 2.0 with it. Also, the fully developed method is compared to unbiased Brownian dynamics for this and two other systems. A Homo sapiens SOD 1spd test shows a speedup of 17, the SOD 1eso test shows a speedup of 35, and an antisweetener antibody NC6.8 test shows a speedup of 39.
Results are also obtained for a more difficult test problem of a reaction between two model proteins with orientations. The speedup due to a partly numerical estimate of reaction probability is 2.3 without weight control and 1.3 with it. The speedup for the fully developed method over unbiased Brownian dynamics is a factor of 2578 for one set of configurations and a factor of 3341 for another set.
Discussion
The partly numerical estimate of the reaction probability gives a worthwhile speedup and may serve as a safeguard against a poor heuristic estimate. The overall method is shown by numerical experiments to be a great success and as fast as WEBD. The relative simplicity of biased BD makes implementation easier and facilitates analysis and improvement of the method. In any case, these dramatic reductions in the cost of calculating rate constants make it practical to employ more detailed models of the biomolecular system and thus to close the gap between experiment and computation.
RATE CONSTANTS OF DIFFUSION-LIMITED REACTIONS
Equations of motion
Consider two types of molecules, enzyme and substrate, diffusing in solvent. Assume dilute concentrations, and, therefore, consider the movement between a pair of molecules. Model the enzyme by a set of atoms that moves like a rigid body, and neglect its rotational movement. Each atom has a partial charge and van der Waals parameter. Model the substrate by a set of spherical subunits with partial charges and van der Waals parameters and with subunits connected by constraints or bonded forces. Neglect the rotational movement of each subunit. Electrostatic and excluded volume forces act between subunits of the substrate, and between the substrate and the enzyme.
Let r1, r2, …, and rN be the coordinates of the subunits of the substrate relative to the geometry center of all atoms of the enzyme. These coordinates describe a configuration of the system, represented by a column vector R of dimension 3N. Specify a set of reacted configurations Ωrc such that if the particle diffuses to the reaction surface ∂Ωrc, a reaction happens and motion terminates. Assume, as in UHBD, that reaction happens if, and only if, a certain number of distance criteria are satisfied, say, m out of n distances are less than a distance ξrc.
Define a potential energy function U(R) and a 3N × 3N symmetric positive definite diffusion tensor D(R). The movement of the substrate relative to the enzyme is described by a stochastic differential equation
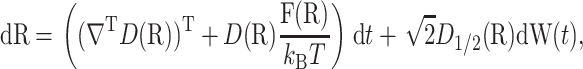 |
(1) |
where D1/2 satisfies D1/2D1/2T = D, ∇ is the column vector of 3N partial derivative operators, F(R) = −∇U(R) are the forces, W(t) is a 3N-dimensional canonical Wiener process, kB is the Boltzmann constant, and T is the temperature. A canonical Wiener process W(t) has a Gaussian distribution with mean zero and covariance EWi(s)Wj(t) = min {s, t}δij. A typical choice for the tensor D(R) is the Rotne-Prager tensor, whose diagonal blocks are
 |
(2) |
and whose off-diagonal blocks are
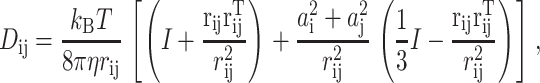 |
(3) |
where η is the viscosity of the solvent, ai is the radius of the ith subunit, amol is the radius of the enzyme, and I is the 3 × 3 identity matrix.
Rate constant calculation
Select a center rc for the substrate. In UHBD, rc is the geometry center of all subunits. The rate constant k is approximated by the formula (Northrup et al., 1984)
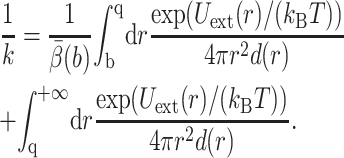 |
(4) |
where b < q are two values for |rc|, d(|rc|) is the diffusion coefficient of the center rc, and 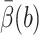 is defined shortly. Assume that the 3N-dimensional set defined by |rc| ≤ b contains Ωrc, and let Ωq be the 3N-dimensional set defined by |rc| ≤ q. For a high-dimensional problem, UHBD computes d(|rc|) during the BD simulation by collecting the mobility information of the center rc. The term Uext(|rc|) is usually computed by treating the enzyme as a Debye-Hückel sphere with its net charge at the center and treating the substrate as a point charge.
is defined shortly. Assume that the 3N-dimensional set defined by |rc| ≤ b contains Ωrc, and let Ωq be the 3N-dimensional set defined by |rc| ≤ q. For a high-dimensional problem, UHBD computes d(|rc|) during the BD simulation by collecting the mobility information of the center rc. The term Uext(|rc|) is usually computed by treating the enzyme as a Debye-Hückel sphere with its net charge at the center and treating the substrate as a point charge.
The value 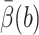 is defined in terms of a reaction probability β(R), which satisfies the PDE and boundary condition (Zhou, 1990),
is defined in terms of a reaction probability β(R), which satisfies the PDE and boundary condition (Zhou, 1990),
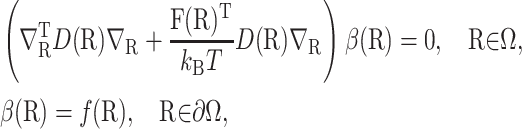 |
(5) |
where ∇R acts on all that follows it, the domain Ω = Ωq\Ωrc, and the function f(R) is defined to be 0 on the escape surface ∂Ωq and 1 on the reaction surface ∂Ωrc.
In terms of SDEs, the reaction probability β(R) is (see Appendix) the path integral
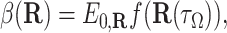 |
where R(t) is a trajectory of the Brownian particle governed by Eq. 1 with initial condition R(0) = R, τΩ is the first exit time from domain Ω, and E0,R is the expectation with respect to the probability law for the random trajectory R(t) starting at R(0) = R.
The average reaction probability  on the b-surface for high-dimensional problems is taken as the average of β(R) with respect to some distribution pb(R) on the b-surface, in which the center rc is uniformly distributed on the |rc| = b spherical surface and conformations are Boltzmann-distributed.
on the b-surface for high-dimensional problems is taken as the average of β(R) with respect to some distribution pb(R) on the b-surface, in which the center rc is uniformly distributed on the |rc| = b spherical surface and conformations are Boltzmann-distributed.
In practice, Eq. 1 is approximated by the scheme
 |
(6) |
where Δt is the time step and Zn+1 is a vector of 3N independent standard Gaussian random numbers (of mean 0 and variance 1).
BIASED BROWNIAN DYNAMICS WITH WEIGHT CONTROL
The expectation of f(R(τΩ)) can be estimated with smaller relative statistical error if reactions are more numerous. This motivates the addition of a bias force Fb(R):
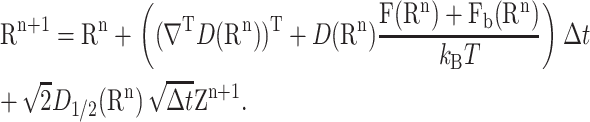 |
(7) |
The bias thus introduced is compensated for perfectly if the exit value is multiplied by the appropriate weight: the weight associated with Rn is expressed as exp un where u0 = 0 and
 |
(8) |
In the continuum limit Δt → 0, E0,R exp u(τΩ)f(R(τΩ)) = β(R), where u(t) satisfies the Δt → 0 limit of Eq. 8 (Milstein, 1988).
The purpose of the bias force is to reduce the variance of the estimate. Remarkably, in the continuum limit Δt → 0, there is an optimal bias force,
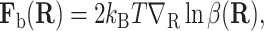 |
(9) |
that reduces the variance to zero (Milstein, 1988). In practice, the bias force is constructed from an estimate of β(R).
In the continuum limit Δt → 0, the use of the optimal bias force produces an ideal weight
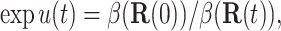 |
(10) |
that does not depend on the history of the trajectory. An estimate 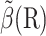 of β(R) can be used to define a target weight and an acceptable range of weights, and this can be used to control the weight as described in the Introduction. The numerical tests reported here use the range u*(Rn) − 2 ≤ un ≤ u*(Rn) + 1.
of β(R) can be used to define a target weight and an acceptable range of weights, and this can be used to control the weight as described in the Introduction. The numerical tests reported here use the range u*(Rn) − 2 ≤ un ≤ u*(Rn) + 1.
The actual calculation is that of an average 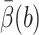 on the b-surface, not that of a value β(R0) at a single point. A better target weight is given by
on the b-surface, not that of a value β(R0) at a single point. A better target weight is given by
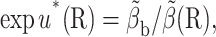 |
(11) |
where 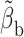 approximates
approximates 
CONSTRUCTION OF BIAS FORCE
Biased BD with weight control requires a bias force and a target weight, which can be constructed from an approximate solution 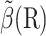 using Eqs. 9 and 10. In particular, the bias force is
using Eqs. 9 and 10. In particular, the bias force is
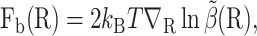 |
(12) |
which is computed by one-sided finite differences applied to 
The method proposed here for computing 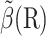 uses a reaction coordinate ξ = ξ(R) that is easy to evaluate for every configuration R and that correlates well with β, in particular, 1), there is a value ξrc for which the set of configurations with ξ(R) < ξrc is exactly Ωrc; and 2), there is a value ξq for which the set of configurations with ξ(R) ≤ ξq encloses Ωq and equals Ωq approximately. See Fig. 1 for an illustration. The reaction probability β(R) is approximated by
uses a reaction coordinate ξ = ξ(R) that is easy to evaluate for every configuration R and that correlates well with β, in particular, 1), there is a value ξrc for which the set of configurations with ξ(R) < ξrc is exactly Ωrc; and 2), there is a value ξq for which the set of configurations with ξ(R) ≤ ξq encloses Ωq and equals Ωq approximately. See Fig. 1 for an illustration. The reaction probability β(R) is approximated by  where
where  is to be determined.
is to be determined.
FIGURE 1.

Reaction coordinate.
The choice of reaction coordinate is discussed first. Then a two-point boundary value problem is obtained for 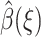 by minimizing a functional. Finally, a Monte Carlo method is described for computing a density function ρ(ξ), which is required to compute
by minimizing a functional. Finally, a Monte Carlo method is described for computing a density function ρ(ξ), which is required to compute 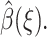
A single reaction coordinate ξ = ξ(R) is theoretically sufficient for representing β(R). The closer the constant ξ hyper-surfaces are to the constant β hyper-surfaces, the better is the choice of reaction coordinate. The reaction coordinate should attempt to measure closeness to reaction in the same way that β does, particularly near the binding site, which is most important for sampling. That is also why ξ(R) = ξrc is required to be the exact boundary of Ωrc.
As stated earlier, the reaction condition in the test problems is that m out of n distances be less than the reaction distance ξrc. Hence, the reaction coordinate ξ(R) is defined as the mth smallest of n distances.
The value ξq is defined as the maximum of ξ(R) on the q-surface. The ξ(R) = ξq surface is not the same as the q-surface, but is close to it. It is enough that 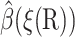 satisfies the boundary condition on the q-surface only approximately, because this does not affect
satisfies the boundary condition on the q-surface only approximately, because this does not affect 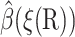 much for the more interesting configurations with small reaction coordinates.
much for the more interesting configurations with small reaction coordinates.
To determine 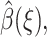 we use the variational formulation of Eq. 5, namely, that β(R) minimize the functional
we use the variational formulation of Eq. 5, namely, that β(R) minimize the functional
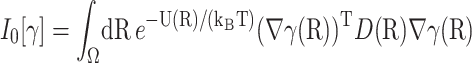 |
for all functions γ(R) satisfying the same boundary condition as β(R), which is 0 at the q-surface ∂Ωq, and 1 at the reaction site ∂Ωrc. We seek a function 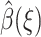 that minimizes the much more restricted and slightly different functional
that minimizes the much more restricted and slightly different functional
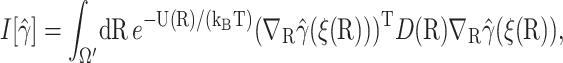 |
(13) |
subject to the boundary condition  and
and 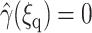 where Ω′ is the set of configurations with ξrc ≤ ξ(R) ≤ ξq. Noting that
where Ω′ is the set of configurations with ξrc ≤ ξ(R) ≤ ξq. Noting that 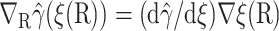 and inserting
and inserting  into the right-hand side of Eq. 13, where δ(x) is the one-dimensional Dirac delta function, we obtain
into the right-hand side of Eq. 13, where δ(x) is the one-dimensional Dirac delta function, we obtain
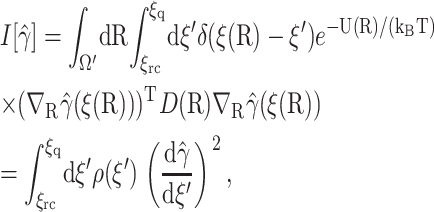 |
(14) |
where the density function is
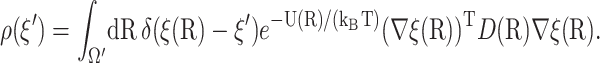 |
(15) |
To minimize the functional in Eq. 14, 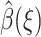 must satisfy
must satisfy 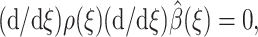 with boundary condition
with boundary condition 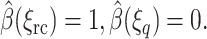 The solution is
The solution is
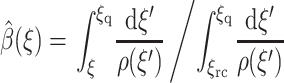 |
(16) |
To compute the density function ρ(ξ′) defined in Eq. 15 numerically, let ξrc = ξ0 < … < ξn = ξq be a partition of the reaction coordinate and use the piecewise constant approximation
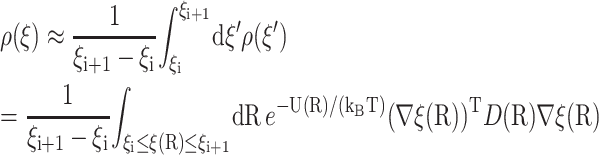 |
(17) |
for ξi ≤ ξ ≤ ξi+1. The above integral is computed using Monte Carlo trials. Note that a uniform scaling for ρ(ξ) does not affect the solution  only the relative magnitude for different ξ matters. Uniformly distributed random configurations R are generated, and for every such configuration, the reaction coordinate ξ(R) and the integrand in Eq. 17 are evaluated. A sum of integrand values is accumulated for each range of reaction coordinates. A rough piecewise constant function ρ(ξ) is obtained through millions of such Monte Carlo trials and is stored as a set of (ln ξ, ln ρ(ξ)) pairs.
only the relative magnitude for different ξ matters. Uniformly distributed random configurations R are generated, and for every such configuration, the reaction coordinate ξ(R) and the integrand in Eq. 17 are evaluated. A sum of integrand values is accumulated for each range of reaction coordinates. A rough piecewise constant function ρ(ξ) is obtained through millions of such Monte Carlo trials and is stored as a set of (ln ξ, ln ρ(ξ)) pairs.
A single Monte Carlo simulation usually does not give enough samples in the region with small ξ-values because of its small volume. Therefore, in practice, several Monte Carlo simulations with nested ranges are performed. As shown in the figures for the test problems, each Monte Carlo simulation gives a set of (ln ξ, ln ρ(ξ)) pairs that is “good” in a certain range of ξ. The sets of (ln ξ, ln ρ(ξ)) pairs from different Monte Carlo simulations are aligned to each other to form a merged density that is defined in the full range [ξrc, ξq], which is further smoothed to obtain a smoothed density.
The smoothing for (ln ξ, ln ρ(ξ)) is done by least-squares fitting ln ρ(ξ) to piecewise linear functions of ln ξ to obtain a set of (ln ξ, ln ρ(ξ), d ln ρ(ξ)/d ln ξ) triples, which are then used with Hermite interpolation to obtain a smoothed function. The reason to use linear functions to fit (ln ξ, ln ρ(ξ)) is that there is an approximate power relation between ξ and ρ(ξ), i.e., ρ(ξ) ∝ ξα, for small and for large ξ.
Alignment of two sets of (ln ξ, ln ρ(ξ)) pairs is done by computing an offset between the two sets of data. An offset is computed as the following: select a range of ξ such that both sets are “good” in this range. Smooth both sets in the range. The average difference between the smoothed data in this range is then used as the offset.
RATE CONSTANTS FOR SOD AND NC6.8
Implementation details and cost measurement
The simulations reported here are performed by a C program written by the first author, which uses methods from UHBD. Bonded forces are implemented as constraints and excluded volume forces as hard sphere bumping. The hard spheres representing the enzyme have radii equal to their van der Waals radii. When an integration step results in penetration, one simply retries the step with another random vector. Intermolecular electrostatic forces use a test charge approximation, where the force on the subunit is the product of its charge and the gradient of an electrostatic potential for the enzyme only. The potential for the enzyme's point charges in surrounding ionized solvent is modeled by the Poisson-Boltzmann equation and precomputed by UHBD on a three-dimensional grid.
The computational cost is measured by tmethod, the CPU hours needed to make the relative error 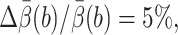 where
where 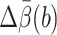 is the 95% confidence interval of the estimate
is the 95% confidence interval of the estimate  Let N′trials be the required number of trajectories to make
Let N′trials be the required number of trajectories to make 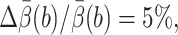 tstep be the CPU seconds per integration step of the SDE,
tstep be the CPU seconds per integration step of the SDE, 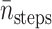 be the average number of integration steps per trajectory,
be the average number of integration steps per trajectory, 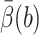 be our best estimate of the average reaction probability on the b-surface (obtained as a weighted average of the available estimates with weights proportional to the reciprocals of the variances), and VarB be the estimated variance of the exit value B = exp u(τΩ)f(R(τΩ)) whose expectation
be our best estimate of the average reaction probability on the b-surface (obtained as a weighted average of the available estimates with weights proportional to the reciprocals of the variances), and VarB be the estimated variance of the exit value B = exp u(τΩ)f(R(τΩ)) whose expectation  Then
Then
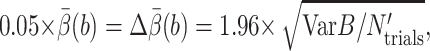 |
so
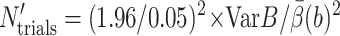 |
(for standard BD, 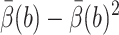 is used instead of VarB), and
is used instead of VarB), and
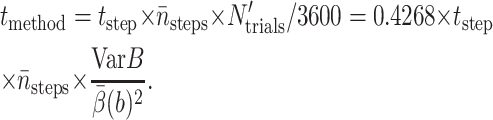 |
(18) |
To get tmethod for a 10% relative error, one simply divides it by 4. All tests in this and the next section were run on Linux clusters of dual-processor Pentium III 1-GHz machines.
The relative error for  is a good measure of the relative error of the rate constant k, because the second term on the right-hand side of Eq. 4 is usually much smaller than the first.
is a good measure of the relative error of the rate constant k, because the second term on the right-hand side of Eq. 4 is usually much smaller than the first.
SOD tests
The enzyme CuZn SOD converts toxic O2− ions to oxygen and hydrogen peroxide. SOD is extremely reactive with a rate constant close to that obtained in the diffusion limit. SOD has been extensively studied by BD simulations (e.g., Sines et al., 1990; Getzoff et al., 1992). The experimental rate constant of SOD of bovine Bos taurus and shark Prionace glauca is reported to be 3.92 × 109 M−1s−1 (Polticelli et al., 1994) compared to a calculated value of 5 × 109 M−1s−1 (Rojnuckarin et al., 2000). Two types of SOD are examined here, H. sapiens SOD 1spd and E. coli SOD 1eso. The computed rate constant of SOD 1spd is ∼1.4 × 109 M−1s−1. The computed rate constant of SOD 1eso is one order of magnitude less.
Coordinate data for SOD are taken from the Protein Data Bank (http://www.rcsb.org/pdb) and the partial charge and van der Waals parameter tables are taken from the standard CHARMM force field (Brooks et al., 1983) incorporated into UHBD. The substrate O2− is modeled by a subunit with a hard-sphere radius 1.5 Å and a charge of −e. Reaction is said to happen when O2− is within 7 Å of the copper atom of SOD. The diffusion tensor D is diagonal as in Eq. 2 where T = 300 K, water viscosity η = 0.89 g m−1s−1, the O2− hydrodynamic radius a1 = 2.05 Å, and that of SOD is amol = 25 Å. The simulations use b = 80 Å and q = 400 Å.
Fig. 2 shows the Monte Carlo simulation results for ρ(ξ) for SOD 1spd. In the figure, each of the three original densities is valid for a certain range of ξ-values. They are aligned to each other by shifting vertically to form a merged density that is valid in the full range. This merged density is then smoothed to form a smoothed density in the full range by the process given in the preceding section.
FIGURE 2.

Monte Carlo simulations for SOD 1spd give three original densities for differing ξ-ranges. These are merged and smoothed to determine ρ(ξ).
The 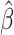 function is computed from the smoothed ρ(ξ) with Eq. 16. The bias potential
function is computed from the smoothed ρ(ξ) with Eq. 16. The bias potential  as a function of reaction coordinate ξ is shown in Fig. 3. The turning point of the curve is at ∼ξ = 20 Å where Ub(ξ) = 5kBT. The magnitude of the bias potential indicates the strength of the free energy barrier needed to be overcome for the substrate to get to the reaction site of the enzyme. Compared to results for the tests that follow, the value 5kBT is relatively low. The corresponding reaction probability and rate constant are also large compared to those in other tests. Near ξ = ξq, the bias potential prevents the particle from reaching the ξ = ξq surface.
as a function of reaction coordinate ξ is shown in Fig. 3. The turning point of the curve is at ∼ξ = 20 Å where Ub(ξ) = 5kBT. The magnitude of the bias potential indicates the strength of the free energy barrier needed to be overcome for the substrate to get to the reaction site of the enzyme. Compared to results for the tests that follow, the value 5kBT is relatively low. The corresponding reaction probability and rate constant are also large compared to those in other tests. Near ξ = ξq, the bias potential prevents the particle from reaching the ξ = ξq surface.
FIGURE 3.

The bias potential for SPD 1spd with escape distance q = 400 Å.
Table 1 shows the rate constants of SOD 1spd and computing costs for biased BD and standard BD. In addition to the quantities defined earlier, the tabulated value Ntrials is the actual number of trajectories, EBest is the estimate produced for 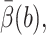 and CPU is the actual CPU time. The CPU cost per integration step of biased BD is 2.7× that of the standard BD. The number of integration steps per trajectory of biased BD is 43% of that of the standard BD due to the weight control algorithm. The variance of biased BD is only 5% of that of the standard BD. The overall speedup of biased BD relative to standard BD is 17. It is interesting to compare this speedup with the sevenfold speedup obtained for biased BD without weight control several years ago (Zou et al., 2000).
and CPU is the actual CPU time. The CPU cost per integration step of biased BD is 2.7× that of the standard BD. The number of integration steps per trajectory of biased BD is 43% of that of the standard BD due to the weight control algorithm. The variance of biased BD is only 5% of that of the standard BD. The overall speedup of biased BD relative to standard BD is 17. It is interesting to compare this speedup with the sevenfold speedup obtained for biased BD without weight control several years ago (Zou et al., 2000).
TABLE 1.
Results for unbiased and full-featured biased Brownian dynamics
| Unbiased BD
|
Biased BD
|
||||
|---|---|---|---|---|---|
| System | SOD 1spd | SOD 1eso | NC6.8 | SOD 1spd | NC6.8 |
| tstep(s) | 2.55 × 10−6 | 2.58 × 10−6 | 7.86 × 10−6 | 6.83 × 10−6 | 1.65 × 10−5 |
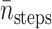 |
1.94 × 105 | 1.96 × 105 | 1.29 × 105 | 8.32 × 104 | 7.58 × 104 |
| Ntrials | 200,000 | 200,000 | 160,000 | 200,000 | 80,000 |
| CPU(h) | 27.5 | 28.2 | 45.1 | 31.6 | 27.8 |
| EBest | 1.49 × 10−2 | 1.73 × 10−3 | 6.50 × 10−4 | 1.45 × 10−2 | 6.82 × 10−4 |
| VarB | 1.47 × 10−2 | 1.73 × 10−3 | 6.50 × 10−4 | 7.21 × 10−4 | 1.40 × 10−5 |
| k(M−1s−1) | 1.46 × 109 | 1.71 × 108 | 5.85 × 107 | 1.42 × 109 | 6.13 × 107 |
| Δk(M−1s−1) | 5.19 × 107 | 1.79 × 107 | 1.12 × 107 | 1.15 × 107 | 2.33 × 106 |
| tmethod(h) | 14.4 | 123.3 | 635.5 | 0.8 | 16.1 |
E. coli SOD 1eso has a lower attractive force to steer the O2− ions to its binding site, which results in a higher energy barrier for association and a lower rate constant. Its bias potential has a graph that is qualitatively similar to that in Fig. 3. The turning point of the curve is at ∼ξ = 20 Å where Ub(ξ) = 7kBT. This is larger than that of SOD 1spd and the rate constant is smaller as a result. The effect of using partly numerical instead of heuristic estimates of the reaction probability is shown for SOD 1eso in Table 2. The heuristic choice is 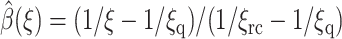 where ξrc = 7 Å and ξq = 405.26545 Å (Zou, 2002). The best result in this table is 35× that of unbiased BD given in Table 1.
where ξrc = 7 Å and ξq = 405.26545 Å (Zou, 2002). The best result in this table is 35× that of unbiased BD given in Table 1.
TABLE 2.
Results for SOD 1eso with heuristic and partly numerical estimates of reaction probability
| Heuristic
|
Partly numerical
|
|||
|---|---|---|---|---|
| wt ctl? | No | Yes | No | Yes |
| tstep(s) | 5.50 × 10−6 | 6.91 × 10−6 | 5.59 × 10−6 | 6.83 × 10−6 |
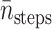 |
6.14 × 105 | 1.01 × 105 | 1.57 × 105 | 8.78 × 104 |
| Ntrials | 200,000 | 200,000 | 200,000 | 200,000 |
| CPU(h) | 187.8 | 38.9 | 48.7 | 33.3 |
| EB | 1.74 × 10−3 | 1.74 × 10−3 | 1.74 × 10−3 | 1.75 × 10−3 |
| VarB | 6.86 × 10−5 | 7.20 × 10−5 | 1.46 × 10−4 | 4.16 × 10−5 |
| k(M−1s−1) | 1.71 × 108 | 1.71 × 108 | 1.72 × 108 | 1.72 × 108 |
| Δk(M−1s−1) | 3.57 × 106 | 3.66 × 106 | 5.21 × 106 | 2.78 × 106 |
| tmethod(h) | 32.9 | 7.1 | 18.0 | 3.5 |
NC6.8 test
This section considers the antibody-antigen binding reaction between the antisweetener antibody NC6.8 (Protein Data Bank entry 2cgr) and the sweet-tasting ligand n-(p-cyanophenyl)-n′-(diphenylmethyl)-guanidinium acetic acid, formula C23H20N4O2). The antibody is the enzyme and the ligand is the substrate. The NC6.8 binding reaction was previously studied for the WEBD (Rojnuckarin et al., 2000). Here we follow the same setup and test for biased BD.
The NC6.8 antibody and the sweet-tasting ligand structure files are from the Protein Data Bank. In the simulation, only the Fv fragment of NC6.8 is used. The setup is similar to that of SOD except the following: the sweet-tasting ligand is modeled as a two-subunit dumbbell with a distance constraint of 4.374 Å between subunit centers. The negative subunit, centered at the carbonyl carbon (C19), has a radius of 1.5 Å and a charge of −e. The positive subunit, centered at the most cationic guanidinium nitrogen (N16), has a radius of 2.0 Å and a charge of +e. The reaction condition is that the positive subunit is within 5.0 Å of Glu H:162 OE2 (H:50), and the negative subunit is within 5.0 Å of Arg H:169 NH1 (H:57). The diffusion tensor D is a Rotne-Prager tensor as in Eqs. 2 and 3 where the hydrodynamic radius of the positive subunit a1 = 2.0 Å, that of the negative subunit a2 = 1.5 Å, and that of the enzyme amol = 25 Å. The values b = 80 Å and q = 300 Å are used.
The bias potential Ub(ξ) for NC6.8 is again qualitatively similar to that in Fig. 3. The turning point of the curve is at ∼ξ = 20 Å where Ub(ξ) = 8kBT, which is larger than for both SOD tests and results in a smaller rate constant. Table 1 shows a 39× speedup of biased BD over standard BD for the NC6.8 test. A speedup of 8 is obtained from the WEBD/NAM method of Rojnuckarin et al. (2000).
ARTIFICIAL TEST PROBLEM
Problem description
The model problem described here is proposed in a 1992 article by Northrup and Erickson. It is later used by Huber and Kim in 1996 to show the speedup of the WEBD algorithm. Although this problem is outside the class of problems defined previously, the modification is straightforward.
In solvent are two types of proteins, each modeled as a sphere of radius 18 Å. For the purpose of defining a reaction condition, imagine that 17 Å × 17 Å squares are attached at their centers tangentially to each sphere as shown in Fig. 4. The set of reacted configurations Ωrc consists of those configurations with a position and orientation such that at least three of the four vertex pairs, A1A2, B1B2, C1C2, and D1D2, between the two squares, are within 2 Å.
FIGURE 4.

Model protein diagram.
For i = 1, 2, let ri be the center of protein i, Qi be an orthogonal matrix describing the orientation of protein i, and ai = 18 Å be the radius of protein i. Although the orthogonal matrix Qi contains nine entries, it has only 3 degrees of freedom because of the constraints QiTQi = I. Any point fixed in the body frame of protein i can be converted to lab frame coordinates with rlab = Qirbody + ri.
The tangent space for the three-dimensional manifold QiTQi = I at a particular point Qi has as a basis 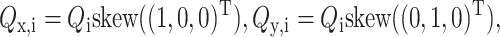 and
and 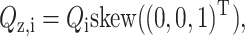 where skew is the mapping
where skew is the mapping
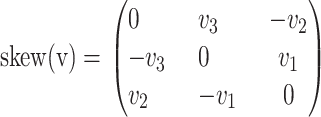 |
from a vector to a skew matrix. The basis is orthogonal with respect to the double-dot inner product (A:C = tr(ATC)) for matrices. Hence, the motion of two proteins is described by their translational and rotational diffusion in the liquid,
 |
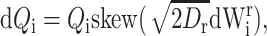 |
(19) |
for i = 1, 2, where Dt and Dr are translational and rotational diffusion coefficients given by the Stokes-Einstein relations Dt = kBT/6πηai = 1.36 × 10−10 m2 s−1; Dr = kBT/8πηai3 = 3.16 × 107 s−1 for T = 298 K; η = 0.89g m−1s−1; and where W1, W2, W1r, and W2r are independent canonical three-dimensional Wiener processes. There is no interaction between the proteins except hard sphere bumping.
Consider the relative position rc = r1 − r2. We have
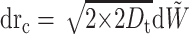 |
(20) |
where  is a canonical Wiener process. The b-surface and q-surface are the set of configurations with |rc| = b and |rc| = q, respectively. The distribution of starting points on the b-surface is uniform for the center rc and uniform with respect to the orientation of protein 1.
is a canonical Wiener process. The b-surface and q-surface are the set of configurations with |rc| = b and |rc| = q, respectively. The distribution of starting points on the b-surface is uniform for the center rc and uniform with respect to the orientation of protein 1.
We compute the rate constant k with Eq. 4, where Uext(r) = 0 and d(r) = 2Dt, although rigorous justification is not given here.
Numerical integration and bias force
For the rc coordinates, there is no difference from previous sections for the numerical integration, bias force evaluation, and weight computation. However, these are all slightly different for the Qi coordinates.
Directly applying a numerical integration method to Eq. 19 for Qi usually does not preserve the orthogonality constraints of Qi. To preserve the constraints, we update Qi by multiplying it by orthogonal matrices. Let
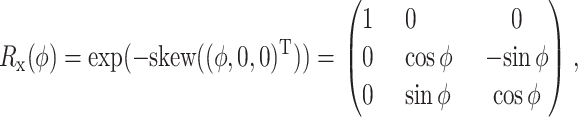 |
in which Rx(φ) is the matrix that rotates an angle φ about the x-axis. Similarly define Ry and Rz. For a finite time step Δt, let  be the Wiener increment Wir(t + Δt) − Wir(t), and let
be the Wiener increment Wir(t + Δt) − Wir(t), and let
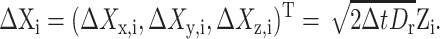 |
Then to integrate Qi numerically, use
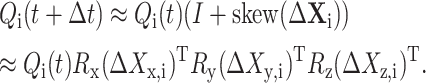 |
Let
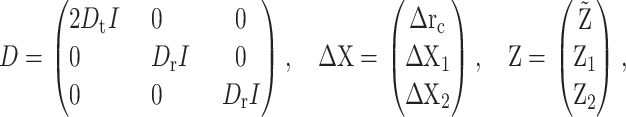 |
where Δrc is the increment of rc during the time period [t, t + Δt] and  is the increment of
is the increment of  The unbiased BD numerical scheme is thus
The unbiased BD numerical scheme is thus
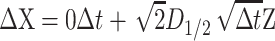 |
where D1/2D1/2T = D.
Similar to Eqs. 7 and 8 for biased BD, here we add a bias vector to the stochastic increment and associate a weight exp(u) with the system. Let 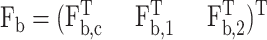 where Fb,c, Fb,1, and Fb,2 are the bias forces for rc, Q1, and Q2, respectively. The unbiased numerical scheme changes to the biased scheme
where Fb,c, Fb,1, and Fb,2 are the bias forces for rc, Q1, and Q2, respectively. The unbiased numerical scheme changes to the biased scheme
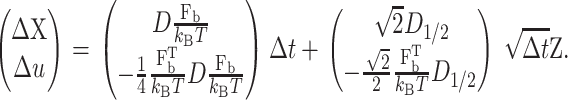 |
The bias force Fb/(kBT) is computed numerically from Eq. 12. For example, to compute the derivative of the approximate reaction probability  in the direction of Qx,i, use
in the direction of Qx,i, use 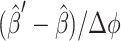 where
where 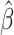 is the approximate solution for the current configuration and
is the approximate solution for the current configuration and 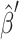 is the approximate solution for the configuration obtained by replacing Qi by QiRx(Δφ)T for some small angle Δφ.
is the approximate solution for the configuration obtained by replacing Qi by QiRx(Δφ)T for some small angle Δφ.
Reaction coordinates and Monte Carlo calculation
Uniformly distributed random configurations are generated by randomly placing protein 1 at a distance r with probability density ∝ r2, and rotating protein 1 with Euler angles φ, θ, ψ, where φ is the angle of self-rotation along the z-axis, θ is the angle of nutation along the x-axis, and ψ is the angle of precession along the z-axis. The positive z-axis goes through the center of the square on the protein. Angles φ and ψ are uniformly distributed in [0, 2π] and cos θ is uniformly distributed in [−1, 1].
Uniformly distributed random configurations will not give enough samples for small ξ, for example, for ξ < 10 Å. Importance sampling with restricted distance and restricted nutation are used. The distance is restricted by a small maximum distance rmax. The cosine of the nutation angle θ is restricted to a uniform distribution in [(cos θ)min, 1], for example, (cos θ)min = 0.8 or (cos θ)min = 0.95. Note that restricted Monte Carlo simulation gives correct relative ρ(ξ) only for those ξ that are small enough that they cannot be generated with nutation angle satisfying cos θ < (cos θ)min or distance r > rmax. The restricted and unrestricted Monte Carlo simulation results are aligned at some common range of values of ξ which are valid for and have enough samples from each simulation.
In Fig. 5, each original density is valid for a certain region. These are aligned by vertical shifts and then smoothed to form a density for the full range.
FIGURE 5.

Monte Carlo simulations for the two-spheres problem give five original densities for differing ranges of distance and nutation angle. These are merged and smoothed to determine ρ(ξ). This curve is compared with straight lines corresponding to ρ(ξ) ∝ ξ2 and ρ(ξ) ∝ ξ5.
The bias potential Ub(ξ) is shown in Fig. 6. When ξ is small, the bias force needs to overcome the strong entropy barrier caused by both translational and orientational restrictions. The relative movement of the spheres is like a particle moving in six-dimensional space with the reaction site being a radii 2 Å spherical surface in six-dimensional space. When ξ gets larger, the restriction from orientation vanishes, and the movement is similar to a three-dimensional free diffusion.
FIGURE 6.

The bias potential of the two-spheres problem with escape distance q = 360 Å.
Estimated cost of standard Brownian dynamics
We did not perform extensive numerical tests with the standard BD algorithm because of the time-consuming nature of these tests and because a good theoretical estimate is possible.
Recall Eq. 18 for the cost of an algorithm: it needs CPU seconds per integration step tstep, the average number of integration steps per trajectory 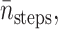 VarB, and
VarB, and 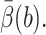 The value of tstep is computed to be 4.49 × 10−6s for this test problem from computer timings. The value of
The value of tstep is computed to be 4.49 × 10−6s for this test problem from computer timings. The value of 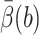 is obtained from the biased BD result. Because the exit values of standard BD are either 1 or 0 depending on whether or not they react, EB2 = EB and VarB = EB2 − (EB)2 = EB − (EB)2 ≈ EB, which is obtained from the biased BD result for
is obtained from the biased BD result. Because the exit values of standard BD are either 1 or 0 depending on whether or not they react, EB2 = EB and VarB = EB2 − (EB)2 = EB − (EB)2 ≈ EB, which is obtained from the biased BD result for 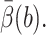 Similar to the method used in Huber and Kim (1996), the average number of integration steps of one trajectory
Similar to the method used in Huber and Kim (1996), the average number of integration steps of one trajectory 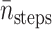 is computed by
is computed by
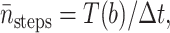 |
where Δt = 2.38 ps is the maximum time step in the numerical integration scheme for Eq. 19, and T(r) is the mean first passage time (MFPT) for protein 1 starting at distance r away from protein 2 with exit boundary at q and reflecting boundary at a = a1 + a2 = 36 Å, the minimum-allowed distance between two proteins. Since the probability of reaction is so low, we neglect the possibility of exit at the reaction site. Thus, the MFPT is assumed to depend only on the distance between two proteins and not on their orientations. It can be shown (see Appendix) that T(r) satisfies the equation
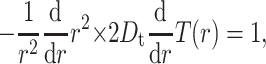 |
with boundary conditions (d/dr)T(r)|r=a = 0 and T(r)|r=q = 0. The solution for the MFPT is
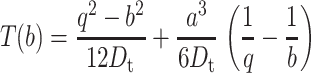 |
Results for the model problem
The effect of using partly numerical instead of heuristic estimates of the reaction probability is shown in Table 3. For a heuristic density function we use Eq. 16 where d ln ρ(ξ)/d ln ξ = a(ξ) and a(ξ) is a step function that takes the value 5 for 2 < ξ < 30, the value 4 for 30 < ξ < 40, the value 3 for 40 < ξ < 50, and the value 2 for 50 < ξ < 600. Comparison to unbiased BD is given in Table 4. Column 5 of each table is an independent computation. The starred entries for unbiased BD denote the theoretical estimates. The per-step cost for biased BD is about twice as expensive as that of standard BD. Biased BD gains in both the average number of steps per trajectory and the variance. Overall, biased BD gives a factor of 2578 speedup with b = 45 Å and q = 360 Å and a factor of 3341 speedup with b = 80 Å and q = 360 Å.
TABLE 3.
Results for model protein with heuristic and partly numerical estimates of reaction probability
| Heuristic
|
Partly numerical
|
|||
|---|---|---|---|---|
| wt ctl? | No | Yes | No | Yes |
| tstep(s) | 7.69 × 10−6 | 8.85 × 10−6 | 7.68 × 10−6 | 8.83 × 10−6 |
 |
1.94 × 105 | 1.48 × 105 | 1.37 × 105 | 1.33 × 105 |
| Ntrials | 200,000 | 200,000 | 200,000 | 200,000 |
| CPU(h) | 82.8 | 73.0 | 58.4 | 65.3 |
| EB | 4.95 × 10−6 | 8.66 × 10−6 | 5.48 × 10−6 | 8.52 × 10−6 |
| VarB | 6.41 × 10−8 | 4.06 × 10−9 | 4.92 × 10−8 | 3.40 × 10−9 |
| k(M−1s−1) | 1.05 × 105 | 1.83 × 105 | 1.16 × 105 | 1.80 × 105 |
| Δk(M−1s−1) | 2.35 × 104 | 5.91 × 103 | 2.06 × 104 | 5.41 × 103 |
| tmethod(h) | 1667.3 | 30.4 | 735.3 | 23.5 |
TABLE 4.
Model protein results for unbiased and full-featured biased Brownian dynamics
| Unbiased BD | Biased BD | ||||
|---|---|---|---|---|---|
| b(Å) | 45.0 | 80.0 | 45.0 | 80.0 | 140.0 |
| q(Å) | 360.0 | 360.0 | 360.0 | 360.0 | 1000.0 |
| tstep(s) | 4.49 × 10−6 | 4.49 × 10−6 | 8.73 × 10−6 | 8.68 × 10−6 | 8.59 × 10−6 |
 |
3.24 × 105* | 3.15 × 105* | 6.65 × 104 | 1.22 × 105 | 7.39 × 105 |
| Ntrials | 120,000 | 120,000 | 40,000 | ||
| CPU(h) | 19.4 | 35.3 | 70.6 | ||
| EBest | 1.72 × 10−5 | 8.56 × 10−6 | 5.42 × 10−6 | ||
| VarB | 1.67 × 10−8 | 3.28 × 10−9 | 1.33 × 10−9 | ||
| k(M−1s−1) | 1.82 × 105 | 1.81 × 105 | 1.81 × 105 | ||
| Δk(M−1s−1) | 7.74 × 103 | 6.85 × 103 | 1.20 × 104 | ||
| tmethod(h) | 36089.5* | 70487.1* | 14.0 | 20.2 | 122.5 |
Acknowledgments
The assistance of A. Rojnuckarin and D. Livesay in the setup for SOD and NC6.8 is gratefully acknowledged.
This material is based upon work supported by the National Science Foundation under grants 9974555 and 020442. It was also supported by a Computational Science and Engineering Fellowship from the University of Illinois.
APPENDIX: FEYNMAN-KAC FORMULA FOR ELLIPTIC PARTIAL DIFFERENTIAL EQUATIONS
Let
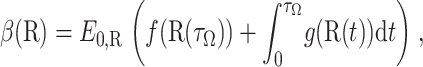 |
(21) |
where R(t) satisfies
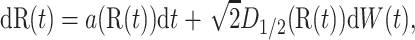 |
with initial condition R(0) = R and where τΩ is the first exit time from domain Ω. The expectation β(R) is the solution of the elliptic boundary value problem
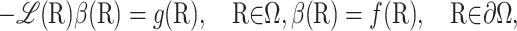 |
(22) |
with ℒ defined by
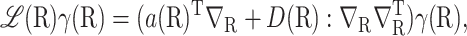 |
where A:C means tr(ATC) for two matrices A and C of the same dimensions, D = D1/2D1/2T, and ∇R∇RT operates only on γ(R).
To apply this to Eq. 5, note that ∇RTD(R)∇R = (∇RTD(R))∇R + D(R):∇R∇RT.
To apply this to the calculation of the MFPT in last section, use Eq. 20 as the SDE, and let f ≡ 0, g ≡ 1 in Eq. 21. Then β(R) is the MFPT, and β(R) satisfies Eq. 22.
References
- Antosiewicz, J., M. K. Gilson, I. H. Lee, and J. A. McCammon. 1995. Acetylcholinesterase: diffusional encounter rate constants for dumbbell models of ligand. Biophys. J. 68:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Brune, D., and S. Kim. 1994. Hydrodynamic steering effects in protein association. Proc. Natl. Acad. Sci. USA. 91:2930–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, M. E., J. D. Madura, B. A. Luty, and J. A. McCammon. 1991. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian dynamics program. Comp. Phys. Comm. 62:187–197. [Google Scholar]
- Getzoff, E. D., C. L. Fisher, H. E. Page, M. S. Viezzoli, L. Banci, and R. A. Hallewell. 1992. Faster superoxide dismutase mutants designed by enhancing electrostatic steering. Nature. 358:347–351. [DOI] [PubMed] [Google Scholar]
- Guddat, L. W., L. Shanand, J. M. Anchin, D. S. Linthicum, and A. B. Edmundson. 1994. Local and transmitted conformational changes on complexation of an anti-sweetener Fab. J. Mol. Biol. 236:247–274. [DOI] [PubMed] [Google Scholar]
- Hänggi, P., P. Talkner, and M. Borkovec. 1990. Reaction-rate theory: fifty years after Kramers. Rev. Mod. Phys. 62:251–342. [Google Scholar]
- Huber, G. A., and S. Kim. 1996. Weighted-ensemble Brownian dynamics simulations for protein association reactions. Biophys. J. 70:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozack, R. E., M. J. D'Mello, and S. Subramaniam. 1995. Computer modeling of electrostatic steering and orientational effects in antibody-antigen association. Biophys. J. 68:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein, G. N. 1988. The Numerical Integration of Stochastic Differential Equations. Urals University Press, Sverdlovsk, Russia. English ed., 1995, Kluwer Academic Publishers, Dordrecht, The Netherlands.
- Northrup, S. H., S. A. Allison, and J. A. McCammon. 1984. Brownian dynamics simulation of diffusion-influenced bimolecular reactions. J. Chem. Phys. 80:1517–1524. [Google Scholar]
- Northrup, S. S., K. A. Thomasson, C. M. Miller, P. D. Barker, L. D. Eltis, J. G. Guillemette, S. C. Inglis, and A. G. Mark. 1993. Effects of charged amino acid mutations on the bimolecular kinetics of reduction of yeast iso-1-ferricytochrome c by bovine ferrocytochrome b5. Biochemistry. 32:6613–6623. [DOI] [PubMed] [Google Scholar]
- Polticelli, F., M. Falconi, P. O'Neill, R. Petruzelli, A. Galtieri, A. Lania, L. Calabrese, G. Rotillio, and A. Desideri. 1994. Molecular modeling and electrostatic potential calculations on chemically modified Cu, Zn superoxide dismutases from Bos taurus and shark Prionace glauca: role of Lys134 in electrostatically steering the substrate to the active site. Arch. Biochem. Biophys. 312:22–30. [DOI] [PubMed] [Google Scholar]
- Rojnuckarin, A., D. R. Livesay, and S. Subramaniam. 2000. Bimolecular reaction simulation using weighted ensemble Brownian dynamics and the University of Houston Brownian dynamics program. Biophys. J. 79:686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sines, J. J., S. A. Allison, and J. A. McCammon. 1990. Point charge distributions and electrostatic steering in enzyme/substrate encounter: Brownian dynamics of modified copper/zinc superoxide dismutases. Biochemistry. 29:9403–9412. [DOI] [PubMed] [Google Scholar]
- Wade, R. C., B. A. Luty, E. Demchuk, J. D. Madura, M. E. Davis, J. M. Briggs, and J. A. McCammon. 1994. Simulation of enzyme-substrate encounter with gated active sites. Struct. Biol. 1:65–69. [DOI] [PubMed] [Google Scholar]
- Zhou, H.-X. 1990. On the calculation of diffusive reaction rates using Brownian dynamics simulations. J. Phys. Chem. 92:3092–3095. [Google Scholar]
- Zou, G. 2002. Robust Biased Brownian Dynamics for Rate Constant Calculation. Ph. D. thesis, University of Illinois at Urbana-Champaign. Also Department of Computer Science Report No. 2294, University of Illinois at Urbana-Champaign, August 2002. Available online at http://www.cs.uiuc.edu/research/reports.html.
- Zou, G., R. D. Skeel, and S. Subramaniam. 2000. Biased Brownian dynamics for rate constant calculation. Biophys. J. 79:638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]