

Abstract
μ-Conotoxins (μ-CTX) are peptides that inhibit Na+ flux by blocking the Na+ channel pore. Toxin residue arginine 13 is critical for both high affinity binding and for complete block of the single channel current, prompting the simple conventional view that residue 13 (R13) leads toxin docking by entering the channel along the pore axis. To date, the strongest interactions identified are between μ-CTX and domain II (DII) or DIII pore residues of the rat skeletal muscle (Nav1.4) Na+ channels, but little data is available for the role of the DI P-loop in μ-CTX binding due to the lack of critical determinants identified in this domain. Despite being an essential determinant of isoform-specific tetrodotoxin sensitivity, the DI-Y401C variant had little effect on μ-CTX block. Here we report that the charge-changing substitution Y401K dramatically reduced the μ-CTX affinity (∼300-fold). Using mutant cycle analysis, we demonstrate that K401 couples strongly to R13 (ΔΔG > 3.0 kcal/mol) but not R1, K11, or R14 (≪1 kcal/mol). Unlike K401, however, a significant coupling was detected between toxin residue 14 and DI-E403K (ΔΔG = 1.4 kcal/mol for the E403K-Q14D pair). This appears to underlie the ability of DI-E403K channels to discriminate between the GIIIA and GIIIB isoforms of μ-CTX (p < 0.05), whereas Y401K, DII-E758Q, and DIII-D1241K do not. We also identify five additional, novel toxin-channel interactions (>0.75 kcal/mol) in DII (E758-K16, D762-R13, D762-K16, E765-R13, E765-K16). Considered together, these new interactions suggest that the R13 side chain and the bulk of the bound toxin μ-CTX molecule may be significantly tilted with respect to pore axis.
INTRODUCTION
μ-Conotoxins (μ-CTX) are sodium channel pore blockers (Catterall, 1988) produced by the sea snail Conus geographus (Nakamura et al., 1983; Cruz et al., 1985; Olivera et al., 1990) that consist of 22 amino acids with three internal disulfide bonds imparting extreme backbone rigidity (Graham et al., 1977; Nakamura et al., 1983; Cruz et al., 1985; Moczydlowski et al., 1986b; Yanagawa et al., 1986; Catterall, 1988; Hidaka et al., 1990; Olivera et al., 1990; Lancelin et al., 1991; Sato et al., 1991; Wakamatsu et al., 1992; Hill et al., 1996). Their well defined 3D structures (Lancelin et al., 1991; Sato et al., 1991; Wakamatsu et al., 1992; Hill et al., 1996) render them useful structural probes of the Na+ channel pore vestibule. The best studied forms of μ-CTXs are GIIIA and GIIIB, whose amino acid sequences are highly homologous and have essentially identical 3D backbone structures. Six of the seven positively charged residues of GIIIA are crucial for its activity (Sato et al., 1991; Becker et al., 1992; Chahine et al., 1995). Unlike tetrodotoxin (TTX) and saxitoxin (STX), whose receptor in the channel consists of only a few key residues as a result of their smaller sizes (∼300 mol wt versus ∼2600 mol wt of μ-CTX) (Noda et al., 1989; Terlau et al., 1991; Backx et al., 1992; Heinemann et al., 1992; Satin et al., 1992; Lipkind and Fozzard, 1994; Perez-Garcia et al., 1996; Penzotti et al., 1998, 2001), high-affinity binding of μ-CTX results from the summed effects of numerous weaker toxin-channel interactions (Becker et al., 1992; Chen et al., 1992; Stephan et al., 1994; Li et al., 1997, 1999, 2000, 2001a,b, 2002a). Although this makes the search for μ-CTX-Na+ channel interactions substantially more difficult, studies with μ-CTX can potentially provide more structural information over a much broader channel surface.
As a result of Na+ channel pore asymmetry (Chiamvimonvat et al., 1996; Perez-Garcia et al., 1996), μ-CTX is likely to bind Na+ channels in only one energetically favorable orientation (Li et al., 1997, 1999, 2000, 2001a,b, 2002a; Chang et al., 1998; Dudley et al., 2000). A detailed examination of partially blocking μ-CTX derivatives was consistent with a single preferred docking orientation (Hui et al., 2002). Earlier studies show that the domain II (DII) P-loop residue E758 in rat skeletal muscle (μ1, Nav1.4) Na+ channels interacts strongly with R13 of μ-CTX (Chang et al., 1998), that DII P-S6 residues D762 and E765 interact with Q14 and R19 of μ-CTX, and that DIII-D1241 interacts with K16. (Li et al., 2001a,b) This pattern of toxin-channel interactions and the known structure of μ-CTX supported the idea that the four channel domains are arranged in a clockwise configuration (Dudley et al., 2000; Li et al., 2001a). However, numerous possible interactions between other channel and toxin residues have not yet been evaluated. For instance, relatively little is known about the extent to which the DI P-loop also participates in toxin binding. Although the isoform-specific substitution Y401C (tyrosine in the skeletal muscle Nav1.4 but cysteine in the cardiac Nav1.5) is sufficient to explain the vast differences in TTX and Cd2+ block between the heart and skeletal muscle Na+ channels, it had virtually no effect on μ-CTX block (Backx et al., 1992; Heinemann et al., 1992; Satin et al., 1992; Chahine et al., 1995; Li et al., 1997). To date, the largest reduction of μ-CTX block reported for DI was the >10-fold decrease observed with the mutations Y401A and E403Q (vs. >100-fold reduction of E758C, D762C, and E765C from DII) (Li et al., 1997, 2000; Chang et al., 1998; Dudley et al., 2000). Due to the paucity of information available, further studies of DI promise to provide structural insights into the Na+ channel pore structure. Successful identification of strong μ-CTX interactions with DI would allow its spatial relationships with other domains to be constrained.
To look for novel toxin-channel contact points, we studied the molecular couplings between the cationic μ-CTX GIIIA sites R1, K11, R13, Q14 (R14 in GIIIB; cf. Li et al., 2001b), K16, and R19 and defined channel pore residues. μ-CTX GIIIA, GIIIB, and GIIIA-based derivatives were systematically applied to wild type (WT) and various Nav1.4 channel constructs, followed by mutant cycle analysis to assess the individual interaction (or coupling) energies between these toxins and channels. Our results reveal novel μ-CTX interactions with residues from DI (and also DII), thereby enabling us to restrain the toxin docking orientation. The structural implications for the pore from these data are discussed in the context of the known 3D structure of the toxin and current pore models.
MATERIALS AND METHODS
Site-directed mutagenesis and heterologous expression
The gene encoding for the μ1 sodium channel α-subunit (Trimmer et al., 1989) was cloned into the pGFP-IRES vector with an internal ribosomal entry site separating it from the GFP reporter gene (Yamagishi et al., 1997). Mutagenesis was performed in pGFP-IRES using polymerase chain reaction (PCR) with overlapping mutagenic primers. All mutations were made in duplicate and confirmed by sequencing the region where mutagenesis was performed. Na+ channel constructs were transfected into tsA-201 cells, which constitutively express t-antigen to enhance the level of channel expression, using Lipofectamine Plus (Gibco-BRL, Gaithersburg, MD) according to the manufacturer's protocol. Briefly, plasmid DNA encoding the WT or mutant α-subunit (1 μg/60-mm dish) was added to the cells with lipofectamine, followed by incubation at 37°C in a humidified atmosphere of 95% O2-5% CO2 for 48–72 h before electrical recordings. Transfected cells were identified by epifluorescence microscopy.
Synthesis of point-mutated μ-CTX GIIIA
μ-CTX (GIIIA) derivatives were synthesized as previously described (Sato et al., 1991; Becker et al., 1992; Chang et al., 1998). Briefly, peptides were synthesized using 9-fluorenylmethoxycarbonyl chemistry and were HPLC-purified. Peptide composition was verified by quantitative amino acid analysis and/or mass spectroscopy. For some derivatives, one-dimensional proton NMR spectra of the synthesized toxin were compared to those of the native toxin to ensure proper folding.
Electrophysiology and data analysis
Electrophysiological recordings were performed using the whole-cell patch clamp technique (Hamill et al., 1981) at room temperature. Pipet electrodes had final tip resistances of 1–3 MΩ. The bath solution contained (in mM) 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH adjusted to 7.4 with NaOH. Designated concentrations of toxin were added to the bath as indicated. The internal pipet recording solution contained (in mM) 35 NaCl, 105 CsF, 1 MgCl2, 10 HEPES, 1 EGTA, pH adjusted to 7.2 with CsOH. Only cells expressing peak sodium currents within the range of 2–5 nA were chosen for experiments in order to ensure proper voltage control and current resolution. Series resistance was typically compensated at 50–60%.
Continuous superfusion of toxin was maintained during experimental recordings. The flow rate was maintained at 10–20 ml/min (bath volume was 150 μl). Washout began only after the peak currents had reached a steady-state level. Equilibrium half-blocking concentrations (IC50) for toxins were determined from the following binding isotherm by assuming a Hill coefficient of 1 known for the 1:1 stoichiometry of μ-CTX binding to Na+ channels (Dudley et al., 1995; Li et al., 1997):
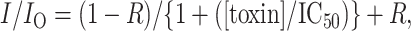 |
where IC50 is the half-blocking concentration, R is the fractional residual current or subconductance when all channels are bound by toxin, IO and I are the peak currents measured from a step depolarization to −10 mV from a holding potential of −100 mV before and after application of the toxin, respectively. The value of R for each of the toxin derivatives was determined by single-channel recordings of block of bilayer-incorporated WT Nav1.4 channels. R was set as 0.31 for R13A and 0 for other derivatives used in this study (French and Horn, 1997; Chang et al., 1998; Hui et al., 2002), and, for each toxin derivative, R was assumed to be identical for all channel constructs studied.
Kinetic analysis of μ-CTX block (for determining the toxin association (kon) and dissociation (koff) rate constants, and the kinetically derived KD) (Li et al., 2000) and mutant cycle analysis (for estimating coupling energies) were performed as described previously (Serrano et al., 1990; Hidalgo and MacKinnon, 1995; Schreiber and Fersht, 1995; Chang et al., 1998; Dudley et al., 2000; Li et al., 2001a,b, 2002a). In general, a coupling energy with an absolute magnitude of 0.75 kcal/mol is considered significant in this study.
All data are reported as mean ± SE. Statistical significance was determined using a paired student's t-test at the 5% level.
RESULTS
Effects of alanine mutations on μ-conotoxin block of WT Nav1.4 Na+ channels
We initially examined the half-blocking concentrations (IC50) for block of WT Nav1.4 Na+ channels by WT GIIIA and alanine-substituted GIIIA-based toxin derivatives. Fig. 1 summarizes these results. Neutralization of the positively charged toxin residues R1, K11, R13, K16, and R19 by alanine significantly (p < 0.05) altered μ-CTX IC50 compared to WT GIIIA (30.7 ± 6.2 nM, n = 6). R13A (19.7 ± 4.7 μM, n = 8) displayed the largest reduction in blocking affinity (note that this derivative shows a residual current at saturating concentrations and in single channel studies—see Materials and Methods), followed by R19A (6.1 ± 1.7 μM, n = 4), K16A (2.5 ± 0.4 μM, n = 6), R1A (532.7 ± 74.5 nM, n = 9), and K11A (319.4 ± 45.3 nM, n = 9). To characterize further the mechanisms underlying these changes in equilibrium IC50, we studied also the effects of alanine substitutions on the kinetics of toxin block. The time course of onset (and offset) of toxin block was fitted with a single-exponential function and the resulting time constants were used to derive the corresponding toxin association and dissociation rate constants (i.e., kon and koff) and kinetic equilibrium constants (KD). These kinetic parameters are summarized in Fig. 2. K11A almost exclusively affected the association rate (sixfold decrease). R13A displayed the largest effects on koff (14-fold increase; cf. Fig. 2 A), whereas R19A had the largest impact on kon (50-fold decrease).
FIGURE 1.

Effects of alanine mutations on μ-CTX-GIIIA block of WT μ1 Na+ channels. (Top) Representative Na+ currents through WT μ1 sodium channels in the absence and presence of μ-CTX mutants R1A, K11A, R13A, K16A, and R19A as indicated. Currents were elicited by depolarization to -10 mV from a holding potential of -100 mV. Control peak currents were normalized to 1.0. (Bottom) The dose-response relationship for the blockade of WT channels by the same μ-CTX mutants shown in the top panel. The mutation R13A had the largest effect on toxin block followed by R19A, K16A, R1A, and K11A (in this order). Data presented are mean ± SE from 4 to 10 individual determinations. GIIIA, R1A, and R19A from Li et al., 2001a are shown for comparison. Normalized peak Na+ currents at −10 mV were plotted as a function of the extracellular toxin concentrations. Data points were fitted with a single-site binding isotherm to estimate the IC50s of μ1 channels for block by different toxin derivatives (see Materials and Methods).
FIGURE 2.

Kinetic analysis of block by mutant μ-CTX derivatives. (A) Left Panel: Time courses of onset and offset of R13A (30 μM) block of WT μ1 channels during toxin wash in (solid squares) and wash out (open circles). Normalized peak sodium currents elicited by depolarization to −10 mV from a holding potential of −100 mV were plotted versus time. Data were fitted with a mono-exponential function to estimate τon and τoff for toxin binding and unbinding respectively. The equations used for deriving kon and koff from τon and τoff have been previously described (Li et al., 2000). Right Panel: Representative records of μ1 currents elicited during wash in (top) and wash out (bottom) of R13A (30 μM). Sweeps (15 msec duration) shown were separated by 6-s intervals for clarity. (B) Logarithmic plot of the dissociation rate constants (koff) versus the reciprocal of the association rate constants (kon). The horizontal and vertical dotted lines respectively represent the levels of 1/kon and koff for the WT channels. All mutant toxins studied (except K11A) had substantial effects on both kon and koff (see text for details). The largest effects on kon and koff were found with R19A and R13A, respectively.
DI-Y401K is coupled to R13A, and DI-E403K to Q14D
Although the Y-to-C isoform variant between the skeletal muscle and cardiac Na+ channels located in the DI pore loop had no effect on μ-CTX binding (Chahine et al., 1995; Li et al., 1997), we reasoned that residue 401 might influence μ-CTX block if its charge was altered. Indeed, we have used the same charge-conversion approach to study the proximity between the neutral μ-CTX residue A22 and the Na+ channel pore (Li et al., 2002a). Therefore, we studied the effect of substituting the neutral native tyrosine by a positively charged amino acid by creating the charge-change mutation DI-Y401K. Consistent with our hypothesis, unlike Y401C, Y401K dramatically reduced μ-CTX block by ∼300-fold (IC50 for GIIIA = 8.4 ± 1.1 μM, n = 4; Fig. 3). Mutant cycle analysis further showed that Y401K was strongly coupled to R13A (Ω = 288.9, ΔΔG = +3.4 ± 0.1 kcal/mol) but not to R1A, K11A, or Q14R of μ-CTX (Figs. 3, B and C).
FIGURE 3.

Interactions of Y401K with R1A, K11A, R13A, and Q14R. (A) Representative Na+ currents through Y401K channels recorded in the absence and presence of WT μ-CTX GIIIA and its derivatives K11A, R13A, and Q14R as indicated. (B) Bar graphs summarizing the half-blocking concentrations (IC50) for block of Y401K channels by WT, R1A, K11A, R13A, and Q14R μ-CTX. (C) ΔΔGs for the interactions of Y401K with the same toxins as in parts A and B. Y401K was strongly coupled to R13A but not to R1A, K11A, and Q14R.
E403, which constitutes part of the outer charge ring, is located at a more superficial location than Y401 (cadmium block electrical distance δ ∼ 0.13 of E403 vs. ∼0.24 of Y401) (Backx et al., 1992; Chiamvimonvat et al., 1996). In contrast to the lack of coupling between toxin residue 14 and Y401K (Ω = 1.6, ΔΔG = +0.3 ± 0.1 kcal/mol for the Y401K-Q14R pair; cf. Fig. 3 C), a significant positive interaction was detected between E403K channels and the peptide derivative Q14D (Ω = 9.1, ΔΔG = +1.4 ± 0.1 kcal/mol; Fig. 4). As anticipated from this result, E403K channels also displayed differential affinities to the GIIIA and GIIIB forms of μ-CTX (p < 0.05), which have a glutamine and an arginine, respectively, at toxin position 14 (Li et al., 2001b). This discriminatory ability of E403K channels was site-specific. None of the DI-Y401K, DII-E758Q, and DIII-D1241K channels could distinguish between the two toxin forms (Fig. 4 C), suggesting that these channel sites do not interact with R14.
FIGURE 4.

DI-E403K interacts with Q14D, and can discriminate between GIIIA and GIIIB. (A) Representative Na+ currents through DI-E403K channels recorded in the absence and presence of WT μ-CTX GIIIA and the derivative Q14D as indicated. (B) Bar graphs summarizing the half-blocking concentrations (IC50) for block of WT μ1 and E403K channels by GIIIA and Q14D (left panel) and ΔΔGs for the interactions of E403K with Q14D (right panel). However, there is no significant coupling between DI-Y401K, which is separated by only one amino acid from residue 403, and Q14R (from Fig. 3 C; shown here for comparison). (C) Conotoxin GIIIA and GIIIB sensitivities of WT, Y401K, E403K, E758Q, and D1241K channels. As anticipated from the positive coupling between E403K and Q14D, E403K channels discriminated between GIIIA and GIIIB (p < 0.05) in a manner similar to the DII P-S6 lysine-substituted channels D762K and E765K. However, none of the following channels: WT, Y401K, E758Q, and D1241K, distinguished between the two toxin forms (p > 0.05).
E758 interacts with R13 and K16
To provide a clearer picture of the channel outer vestibule structure, additional toxin-channel interaction pairs are needed. The DII pore residue E758 has been shown to be critical for μ-CTX block (Dudley et al., 1995; Li et al., 1997; Chang et al., 1998) and interacts strongly with toxin residue R13 (Chang et al., 1998). However, this anionic channel residue is likely to interact also with other cationic toxin residues. We next explored the interactions between E758 and the toxin sites R1, K11, R13, K16, and R19 using mutant cycle analysis (Fig. 5). First, we studied the block of E758Q and E758C channels by WT μ-CTX GIIIA and its derivatives R1A, K11A, R13A, K16A, and R19A. The half-blocking concentrations (IC50) are summarized in Fig. 5 B; those for R19A block of both E758Q and E758C channels, however, could not be determined because as much as 3 μM of this derivative did not result in measurable current blockade. From the IC50s, the corresponding ΔΔG were calculated (Fig. 5 C). The largest interaction energies were between E758Q and R13A, followed by K16A. Couplings with R1A and K11A were not significant. For R19A, interaction energy could not be precisely determined because of the lack of sensitivity of E758Q channels to block by R19A. However, an interaction energy comparable to those observed for the E758Q-R13A and E758Q-K16A pairs would predict a current blockade of ∼10–40% by 3 μM R19A assuming a Hill coefficient of 1 (Dudley et al., 1995; Li et al., 1997), which clearly was not observed. Taken together, our observations are consistent with a lack of positive coupling between E758 and R19. The same pattern of couplings was observed with E758C channels (i.e., R13A > K16A ≫ R19A; Fig. 5).
FIGURE 5.

Interactions of E758 with R1, K11, R13, K16, and R19. (A) Representative Na+ currents through E758Q (top panels) and E758C (bottom panels) channels recorded in the absence and presence of WT, R13A, K16A, and R19A GIIIA μ-CTX as indicated. (B) Bar graphs summarizing the IC50s of E758Q (open bars) and E758C (solid bars) channels for block by WT μ-CTX GIIIA and the derivatives R1A, K11A, R13A, K16A, and R19A. The IC50 for block of both E758Q and E758C channels by R19A could not be determined; n.a. denotes not available. (C) Interaction (coupling) energies (ΔΔG) of E758Q (open bars) and E758C (solid bars) channels with R1A, K11A, R13A, and K16A as estimated from the experimental IC50s in (B). Both E758Q and E758C interact most strongly with R13A followed by K16A but there is a relatively weak coupling of E758Q with R1A and K11A.
DII P-S6 residues D762 and E765 interact with R13, K16, and R19
We previously reported that the DII P-S6 channel residues D762 and E765 interact significantly with Q14 (or R14 of GIIIB) and R19 but not R1 (Li et al., 2001a). Here, we have further studied the interactions between these DII P-S6 channel sites and the μ-CTX residues K11, R13, and K16. The IC50 values for block and the estimated ΔΔG for interactions of D762K and E765K channels with the derivatives K11A, R13A, and K16A are summarized in Fig. 6. Our previous results with R1A and R19A are also shown in Fig. 6 B for comparison (Li et al., 2001a). These results show that D762 interacts moderately with R13, K16, and R19 but not significantly with R1 and K11. Further inspection reveals an interesting pattern: the interaction energy (coupling constant) of D762 was greatest with R13 and decreased progressively toward K16 and R19. Similar to D762, coupling energies of E765K indicate that this channel residue interacts strongly with R13, K16, and R19 but weakly with R1 and K11. However, the interactions of E765 with R13, K16, and R19 were of similar magnitude, implying that this channel residue may be roughly equidistant from these toxin sites.
FIGURE 6.

Interactions of D762K and E765K with R1A, K11A, R13A, K16A, and R19A. (A) Bar graphs summarizing the half-blocking concentrations (IC50) for block of WT, D762K, and E765K channels by WT μ-CTX and the point-mutated derivatives K11A, R13A, and K16A. (B) Estimated ΔΔGs of D762K and E765K with K11A, R13A, and K16A. Our previous results with R1A and R19A (Li et al., 2001a) are also shown for comparison. Both D762 and E765 interact most strongly with R13, K16, and R19 but relatively weakly with R1 and K11. The interaction of D762K was strongest with R13 and decreased progressively toward K16 and R19; those of E765 with R13, K16, and R19 were of similar magnitude.
Interactions with DIII-D1241
We have previously demonstrated that the DIII P-loop residue D1241 interacts most strongly with K16, followed by K11, but not with R1 (Li et al., 2001a). Here we studied the block of D1241K channels (IC50 for GIIIA = 8.4 ± 2.8 μM, n = 5) by the toxin derivatives R13A and R19A. Unfortunately, even very high concentrations of these toxins (30 μM of R13A and 10 μM of R19A) did not result in any blockade of Na+ flux through D1241K channels, rendering the assessment of the IC50s impossible. However, similar to D1241C channels (Li et al., 2001a), significant interactions were detected when K16A (IC50 = 0.9 ± 0.1 μM, n = 3; ΔΔG = 4.0 ± 0.2 kcal/mol) and K11A (IC50 = 7.6 ± 2.3 μM, n = 4; ΔΔG = 1.5 ± 0.2 kcal/mol) were applied to D1241K channels. D1241K did not display any significant interaction with R1A (data not shown). Note that the rank order of potency of these interactions (i.e., K16A > K11A ≫ R1A) was identical to that observed with D1241C channels (Li et al., 2001a).
DISCUSSION
Important cationic residues on μ-CTX
Our experiments with the μ-CTX derivatives R1A, K11A, R13A, K16A, and R19A are consistent with previous studies using either glutamine or alanine neutralization of the same toxin residues (Sato et al., 1991; Becker et al., 1992). Despite the different assays and mutations used in these studies (rat diaphragm bioassay of Sato et al. and bilayer-incorporated single Na+ channels of Becker et al.), the order of reduction in blocking affinity reported was identical to ours (i.e., R13 > R19 > K16 > R1K11). However, our results do differ in some details compared with those reported by Dudley et al. (2000) for the same derivatives studied in heterologously expressed Na+ channels in Xenopus oocytes. These investigators reported that K16A has an ∼sixfold higher affinity than R1A (versus our ∼fivefold lower affinity); K16A in turn was only 20 times (versus ∼100 times here) less potent than WT GIIIA. Their reported affinity for Q14D was also ∼10 times higher than our previously published results (Li et al., 2001b). Some of these discrepancies may arise from differences in the expression systems employed (oocyte versus mammalian cells) and/or simply the ionic conditions (Li et al., 2003b) used in these experiments (e.g., 96 mM Na+ of Dudley et al. versus 140 mM Na+ here). Irrespective of the sources of differences, these studies show that all of the above mentioned cationic sites are important for the normal biological activity of μ-CTX.
Kinetic analysis of block further suggests that among these charges, K11 and R19 are important for guiding the toxin to the pore receptor because of their predominant effects on the toxin association rate (see Fig. 2, and Becker et al., 1992). In contrast, R1A, R13A, and K16A had significant effects on both kon and koff, implying that these residues interact with the channel both before and after the formation of the high-energy intermediate (or activated) complex. Since the side chains of these cationic residues protrude from different faces of the toxin, these observations further support the notion that toxin interactions involve multiple channel residues.
Novel insights into the DI pore region
Although TTX/STX and μ-CTX show competitive binding to the Na+ channel pore, (Moczydlowski et al., 1986a; French et al., 1996) the isoform-specific variant residue at position 401 in DI (Y401C) responsible for the distinct TTX/STX block affinities of skeletal muscle and cardiac channels (Backx et al., 1992; Satin et al., 1992) has little effect on μ-CTX binding (Chahine et al., 1995; Li et al., 1997). These observations led to the hypothesis that the TTX/STX and μ-CTX sites overlap but are not identical (see also Becker et al., 1992). However, here we report that when the native tyrosine in the skeletal muscle channel was replaced by lysine (i.e., Y401K), μ-CTX block was drastically reduced (by ∼300-fold). It seems that Y401 (or C401), located ∼20% across the transmembrane voltage from the outside (Backx et al., 1992), does not directly participate in μ-CTX binding, despite being a critical determinant for TTX/STX block, because the latter toxins are much more compact in size (∼300 mol wt versus ∼2600 mol wt of μ-CTX) and can therefore reach this deeper pore region. However, the substituted lysine of Y401K channels would be capable of interacting with μ-CTX via electrostatic interactions at a distance. This hypothesis is consistent with our observation that Y401K interacts exclusively with R13, which is thought to enter most deeply into the pore to inhibit Na+ currents (Chang et al., 1998; Hui et al., 2002). In contrast, the other toxin residues tested (i.e., R1, K11, and Q(R)14) are predicted to be more superficial in the toxin-bound channel complex (see below). Based on similar reasoning, D400, which forms part of the selectivity filter and is located at an even deeper location than Y401 (Chiamvimonvat et al., 1996), should affect toxin block in a similar manner. Indeed, this premise is also consistent with the minor effects of D400C and D400A mutations on μ-CTX block (Li et al., 1997; Chang et al., 1998). Although we attribute the effects of Y401K on toxin block and its coupling to R13 primarily to electrostatic interactions, it should be noted that a steric role of K401 cannot be excluded. Analysis of the action of partially blocking toxin derivatives suggests that R13 participates in significant steric and electrostatic interactions with ions within the pore (Hui et al., 2002). Indeed, the greater length and flexibility of the K401 side chain, compared to tyrosine, could also lead to the new interactions. Further experiments involving various substitutions of residue 401 and R13 homologs of different lengths, sizes, and charges (Nakamura et al., 2001) might provide additional insights into the role of the inner pore in μ-CTX block.
Simultaneous interactions of Q(R)14 with DI and DII
Recently, we showed that μ-CTX residue 14 can interact strongly with the critical DII P-S6 channel determinants D762 and E765 in the toxin-bound state, and that the Q-to-R substitution between the GIIIA and GIIIB forms of μ-CTX at this toxin position is responsible for their differential binding to D762K and E765K channels (Li et al., 2001a,b). Interestingly, DI-E403K (but not DI-Y401K) is also coupled to toxin residue 14. Consistent with these results, E403K channels, but not Y401K, E758Q, E758C, D1241C, and D1241K, discriminate between GIIIA and GIIIB (Li et al., 2001b). These observations suggest that the ability of E403K channels to distinguish between GIIIA and GIIIB is site-specific, despite the fact that residues 401 and 403 are separated by only one amino acid. Taken together, our results suggest that Q(R)14 faces both DI and DII simultaneously in the toxin-bound state, thereby providing novel insight into how μ-CTX interacts with a channel domain (DI), for which limited information was previously available.
Toward a more refined toxin-channel docking model
The new interactions described in this paper, along with other published observations, contribute significantly to the essential endeavor of building a precise molecular model of μ-CTX docking in the Na+ channel vestibule. However, significant refinements in our understanding are still needed. Included among the issues, which eventually must be resolved, are the following.
First, most of the residue pairs that show significant couplings have been identified on the basis of charge-changing mutations. This approach has the advantage of being likely to reveal, or generate, more interactions than more conservative mutations, and thus give a broader collection of clues to the underlying structure. However, based on only a limited subset of interacting pairs, it is still difficult to draw definitive conclusions about the precise side-chain orientations or locations since electrostatic interactions are often relatively long range and, in principle, multidirectional. For instance, both R13 and K16, the two toxin residues with the strongest known interactions with the channel, both have been reported to have interaction energies of ≥1 kcal/mol with one residue or another in each of the four channel domains. Although it is intuitively reasonable to assign a side-chain orientation on the basis of either the strongest coupling or a resultant vector (Dudley et al., 2000; Li et al., 2001a,b, 2002a), when this has been done, the discordance among different studies has been strikingly dependent on the particular mutations studied.
Second, given the range over which electrostatic interactions operate, it is not surprising that a single residue might interact with more than one domain in a well defined toxin-channel complex. An empirical “length constant” of ∼7Å has been determined, within the channel vestibule, to quantify the spatial decay of electrostatic interactions of charged groups on bound μ-CTX with ions entering the channel vestibule (Hui et al., 2002). While the effective range of electrostatic interactions so defined is significant, it is also short enough that, in any given conformation, charges on opposite extremes of the toxin would have substantial areas of the channel vestibule in which their interactions do not significantly overlap. Multidomain interactions of a single toxin residue, need not, however, rely exclusively on the intrinsic effective range of electrostatic interactions. An alternative possibility is that there are significant interactions at spatially distinct points in the reaction coordinate as the toxin binds and dissociates, and these might reflect orientations other than that of the stable, bound complex. Further discussion of this issue in the context of μ-CTX and TTX/STX binding may be found in Chang et al. (1998) and Penzotti et al. (1998).
Third, all interaction energies estimated at >2 kcal/mol (R13-Y401, R13-E758, K16-E758, K16-D1241) have been produced by charge-changing mutations. The only case where a pair of charge-conserving mutations has provided a coupling energy as large as ∼1 kcal/mol is for the coupling between hydroxyproline 17 and M1240 (Dudley et al., 2000), making this pair a strong candidate for a close-range interaction.
Fourth, although the NMR-determined structure of μ-CTX provides the current benchmark for structural modeling of the channel vestibule, the flexibility of the crucial arginine and lysine side chains make it impossible, at this stage, to know how close the side chain positions in the solution structure of the toxin are to those in the toxin-channel complex. This does not invalidate the use of the toxins as a template for pore structure, but rather, it places some practical limits on the precision with which spatial positions may be determined based on interactions of side chain pairs.
Finally, the relative “electrical positions” of the DII residues E758, D762, and E765 (Chiamvimonvat et al., 1996; Li et al., 1999) suggest that these residues occupy progressively deeper positions in the pore. Such data must be understood in the context of a structural pore model.
The critical task that we face at this point in the investigation is how to resolve these issues. Collectively, comparison of coupling energies derived from large sets of similar mutation pairs, like those employed in the present study, should help to minimize, if not completely eliminate, uncertainties in attempts to define the structural basis of toxin-channel interactions. We believe that it is important to study individual pairs of interacting toxin and channel sites by multiple substitutions. The resulting data set should allow development of a comprehensive, self-consistent view of the pore vestibule structure and toxin docking. Such a pattern appears to be emerging from the growing set of data now available.
Some major interactions are summarized in a static, pictorial form in Fig. 7. This shows μ-CTX tilted significantly from the more conventional hypothesis in which the R13 side chain enters the channel parallel to the pore axis (see Fig. 7 D). The particular orientation shown was obtained by rotating toxin structure in three dimensions with respect to the four channel “domains,” attempting to place as many of the toxin residues as possible in positions consistent with the experimentally-determined coupling energies (see Table 1) as well as other functional data. There are a number of weak couplings (ΔΔG < 1kcal/mol) that were not taken into consideration here, even though they appear, on statistical grounds, to be experimentally reproducible. Nonetheless, the end result is a toxin orientation that is generally consistent with a large body of diverse experimental measurements (Dudley et al., 1995; Li et al., 1997, 2000, 2001a,b, 2002a, 2003a; Chang et al., 1998; Dudley et al., 2000; Hui et al., 2002). At the very least, we suggest that the hypothetical orientation and location of the critical R13 side chain should continue to face scrutiny in future experiments.
FIGURE 7.

A cartoon representation of some of the interactions between μ-CTX and the sodium channel vestibule. Panel A shows the toxin is viewed here from the extracellular side of the channel. To show the positions of K16 and R19, the toxin has been decapitated by a clipping plane parallel with the plane of the membrane (see panel C). The unclipped view from the top is given in panel B. Coordinates of μ-CTX GIIIA (1TCG) were obtained from the Protein Data Bank. The toxin orientation shown is consistent with most of the accumulated published data on toxin-channel interactions. Coupling energies for interactions with different domains indicated here are all ≥1 kcal/mol; those ≥2 kcal/mol are indicated by bold, underlined type on the figure. Key requirements restricting the docking are that R13 interacts with all domains, Q(R)14 interacts with DI and DII, and K16 interacts with DII and DIII. Hydroxyproline17 (hidden under the K16 side chain in panels A and B, but visible as the brown residue in panel C) interacts with DIII. See text for further details. In this orientation, R1 would face generally toward DI and DIV, but the tilt brings R1 closer to DII, consistent with an earlier report by Dudley et al. (2000)—see Table 1. K11, which interacts with DIII, is more toward DIV than is K16, based on the NMR-determined solution structure of the toxin. When carrying a charge, residue-401 can electrostatically sense R13, although the native tyrosine residue does not directly participate in μ-CTX binding. Panel C shows a side view of the bound μ-CTX molecule from DIII. The pore axis (red arrow) is assumed to be normal to the plane of the membrane, and the approximate position of the clipping plane used in panel A is shown by the black arrow. It is apparent that the R13 side-chain axis is tilted relative to that of the pore; its precise tilt angle, as well as the exact positioning of the toxin relative to the pore axis, however, will have to be determined by additional studies. In panel D, for comparison, the toxin is shown in side view with the R13 side chain parallel to the pore axis. This was the starting hypothesis in the studies of Dudley et al. (2000) and Li et al. (2001a). In neither of those studies, however, were there sufficient constraints to exclude the possibility of some tilt between the axes of the pore and of the R13 side chain, as depicted in the rather dramatic example in panels A–C.
TABLE 1.
Summary of μ-CTX interactions with Nav1.4 Na+ channels identified by mutant cycle analysis to date
| DI
|
DII
|
DIII
|
|||||
|---|---|---|---|---|---|---|---|
| Toxin/channel | K401 | E403 | E758 | T759 | D762 | E765 | D1241 |
| R1 | – | n.s. | – | +‡ | –† | –† | –† |
| K11 | – | n.s. | – | – | – | ±† | |
| R13 | +++ | n.s. | +++* | ++ | + | n.d. | |
| Q(R)14 | – | ± | – | ++† | ++† | – | |
| K16 | – | n.s. | ++ | + | + | +++† | |
| R19 | – | n.s. | – | +† | +† | – | |
The number of plus signs indicates the relative magnitude of coupling. –, no significant interaction; n.s., not studied; n.d., not determined. +, 0.75 < ΔΔG < 1.25 kcal/mol; ++, 1.25 < ΔΔG < 3 kcal/mol; +++, ΔΔG ≥ 3 kcal/mol. Unless otherwise indicated, entries are generated from this report.
Data summarized from Chang et al. (1998).
Data summarized from Li et al. (2000).
Data summarized from Dudley et al. (2000).
A key idea, illustrated in the figure, is that off-axis positions for toxin residues 14 and 16 might allow the observed strong two-domain interactions of each of these residues (14 with DI and DII, and 16 with DII and DIII). The orientation of the Q14 side chain almost certainly differs from that of the substituted R14, which reveals the pair-wise interaction, as it does in the experimentally determined structures of GIIIA and GIIIB (structures 1tcg and 1GIB, respectively, in the Protein Data Bank.). This may provide a clearer rationale for the approximately equal interaction energies found in this study between toxin residue 14 and DI and DII. As noted above, the toxin structure appears to accommodate both a short-range coupling by Hyp17 and a strong interaction of K16 with adjacent residues in DIII. The cartoon in Fig. 7 suggests that there may be a significant tilt between an axis normal to the membrane plane and the direction of the R13 side chain. Such a tilt would fit quite naturally with the present data, and with a generally conical vestibule structure. In addition, the rather flat cross section of the toxin, with at least residues 13, 14, 16, 17, and 19 distributed close to one edge contributing to the interacting surface, might easily fit into a groove between different channel segments or domains, but at this point such a suggestion would be speculative.
Other data, with which Fig. 7 A is generally consistent, include: 1), interactions of particular toxin residues with specific channel domains, which led to the conclusion that channel domains are packed in a clockwise array when viewed from the outside (Dudley et al., 2000; Li et al., 2001a); 2), a “double-dip” conformation of the DII P-S6, which places D762 and E765 relatively deep within the pore (Li et al., 1999), in accord with the rather radical position of R19 in Fig. 7, A and C; and 3), a detailed analysis of the contributions to block of single channels by μ-CTX (Hui et al., 2002), which suggested the exposure of R13 and K16 near the narrowest part of the conducting pathway, while other toxin residues are more distant and/or partially hidden behind the bulk of the toxin.
We note that the detailed studies just cited, of both toxin-channel couplings and partial single-channel block, are consistent with the toxin binding in a single orientation despite various substitutions of individual residues that contribute to binding. As has been clear since the early studies of Sato et al. (1991) and Becker et al. (1992), the strong binding of the native toxin seems to result from a molecular Velcro-like surface interaction made up of many bonds. Thus, disruption of one or two individual attachments may weaken the overall binding without substantially changing the orientation of the bound toxin. The implicit assumption of a single binding orientation is commonly applied in attempts to deduce receptor structure from the known structure of a peptide ligand.
Future studies
Although the identified interacting pairs of charged toxin and channel residues are likely to interact with each other electrostatically, the nature of their interactions needs to be further defined and quantified on a residue-to-residue basis. Mutant cycle analysis identifies the total change in free energy (coupling energy, ΔΔG) but cannot reveal whether a given ΔΔG results from the introduction of new attractions (or repulsions), cancellation of preexisting ones, or combinations of both. It is not possible, for instance, to determine how much of the total ΔΔG is electrostatic and how much results from steric or other factors. It is desirable to utilize multiple approaches with different assumptions to define the pattern of toxin-channel interactions. Future experiments using electrostatic compliance analysis, which involves the substitutions of a chosen interaction pair by different charges in at least nine different permutations (i.e., −1/−1, −1/0, −1/+1, 0/−1, 0/0, 0/+1, +1/−1, +1/0, +1/+1), will allow isolation of electrostatic interactions and estimation of the distances between charges (Stocker and Miller, 1994), thereby providing a more precise calibration of the Na+ channel pore. Interacting pairs identified in this paper are prime candidates for such experiments (Li et al., 2002b).
Comparison with a previous model
Using μ-CTX GIIIA as a probe, Dudley and colleagues (Dudley et al., 2000) proposed that the four sodium channel domains are arranged in a clockwise configuration. Based on a different set of mutations from those used in our studies, these investigators reported that Q14, Hyp17, and K16 of the toxin interacted most strongly with residues of DII, DIII, and DIV, respectively. In contrast to our present working hypothesis, the toxin and side chain orientations, which they proposed, placed R1 close to DII representing an ∼90° rotation of the toxin around the pore axis. The differences can be explained by the choice of channel mutants studied, and the lack of awareness, at the time of their study, of the strong coupling between K16 and D1241. With the larger data set that is now available, some reorientation seems appropriate.
CONCLUSION
In summary, we conclude that the anionic channel residues E758, D762, E765 from DII interact significantly with the μ-CTX pseudohelical binding domain consisting of R13, Q(R)14, K16, Hyp17, and R19. D1241 from DIII interacts with K16 and K11. DI-E403K channels are able to discriminate between GIIIA and GIIIB by interacting with toxin residue 14. The residue at position 401 does not normally participate in μ-CTX binding but can electrostatically sense R13 when carrying a charge. These findings allow us to propose a refined toxin-channel docking model and also provide further validation of the clockwise arrangement of the four sodium channel domains.
Acknowledgments
We thank Dr. Denis McMasters, University of Calgary, Faculty of Medicine, Core DNA, and Protein Services Facility, for synthesizing several μ-CTX derivatives.
This work was supported by the National Institutes of Health (R01 HL-52768) and a Research Career Development Award from the Cardiac Arrhythmias Research Foundation (to R.A.L.). I.L.E. was supported by a fellowship award from Universidad Nacional de la Plata, Argentina, during the tenure of this work. K.S. was supported by a grant-in-aid from the Ministry of Education, Science, Sports, Culture and Technology of Japan. R.J.F. is a Heritage Medical Scientist of the Alberta Heritage Foundation for Medical Research and receives operating support from the Canadian Institutes of Health Research and the Heart and Stroke Foundation of Alberta, NWT and Nunavut. During the preparation of the manuscript, R.J.F. was supported by a fellowship from the Max Planck Institute for Experimental Medicine, Göttingen, Germany. R.A.L. is the recipient of a Young Investigator Award from the North American Society of Pacing and Electrophysiology.
References
- Backx, P. H., D. T. Yue, J. H. Lawrence, E. Marban, and G. F. Tomaselli. 1992. Molecular localization of an ion-binding site within the pore of mammalian sodium channels. Science. 257:248–251. [DOI] [PubMed] [Google Scholar]
- Becker, S., E. Prusak-Sochaczewski, G. Zamponi, A. G. Beck-Sickinger, R. D. Gordon, and R. J. French. 1992. Action of derivatives of mu-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry. 31:8229–8238. [DOI] [PubMed] [Google Scholar]
- Catterall, W. A. 1988. Structure and function of voltage-sensitive ion channels. Science. 242:50–61. [DOI] [PubMed] [Google Scholar]
- Chahine, M., L. Q. Chen, N. Fotouhi, R. Walsky, D. Fry, V. Santarelli, R. Horn, and R. G. Kallen. 1995. Characterizing the mu-conotoxin binding site on voltage-sensitive sodium channels with toxin analogs and channel mutations. Receptors Channels. 3:161–174. [PubMed] [Google Scholar]
- Chang, N. S., R. J. French, G. M. Lipkind, H. A. Fozzard, and S. Dudley, Jr. 1998. Predominant interactions between mu-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry. 37:4407–4419. [DOI] [PubMed] [Google Scholar]
- Chen, L. Q., M. Chahine, R. G. Kallen, R. L. Barchi, and R. Horn. 1992. Chimeric study of sodium channels from rat skeletal and cardiac muscle. FEBS Lett. 309:253–257. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat, N., M. T. Perez-Garcia, R. Ranjan, E. Marban, and G. F. Tomaselli. 1996. Depth asymmetries of the pore-lining segments of the Na+ channel revealed by cysteine mutagenesis. Neuron. 16:1037–1047. [DOI] [PubMed] [Google Scholar]
- Cruz, L. J., W. R. Gray, B. M. Olivera, R. D. Zeikus, L. Kerr, D. Yoshikami, and E. Moczydlowski. 1985. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 260:9280–9288. [PubMed] [Google Scholar]
- Dudley, S. C., Jr., N. Chang, J. Hall, G. Lipkind, H. A. Fozzard, and R. J. French. 2000. mu-conotoxin GIIIA interactions with the voltage-gated Na(+) channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 116:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley, S. C., Jr., H. Todt, G. Lipkind, and H. A. Fozzard. 1995. A mu-conotoxin-insensitive Na+ channel mutant: possible localization of a binding site at the outer vestibule. Biophys. J. 69:1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French, R. J., and R. Horn. 1997. Shifts of macroscopic current activation in partially blocked sodium channels. Interaction between the voltage sensor and a mu-conotoxin. In From Ion Channels to Cell-to-Cell Conversations. R. Latorre and .J. C. Sáez, editors. Plenum Press, New York. 67–89.
- French, R. J., E. Prusak-Sochaczewski, G. W. Zamponi, S. Becker, A. S. Kularatna, and R. Horn. 1996. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron. 16:407–413. [DOI] [PubMed] [Google Scholar]
- Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–72. [DOI] [PubMed] [Google Scholar]
- Hamill, O. P., A. Marty, E. Neher, B. Sakmann, and F. J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Heinemann, S. H., H. Terlau, and K. Imoto. 1992. Molecular basis for pharmacological differences between brain and cardiac sodium channels. Pflugers Arch. 422:90–92. [DOI] [PubMed] [Google Scholar]
- Hidaka, Y., K. Sato, H. Nakamura, J. Kobayashi, Y. Ohizumi, and Y. Shimonishi. 1990. Disulfide pairings in geographutoxin I, a peptide neurotoxin from Conus geographus. FEBS Lett. 264:29–32. [DOI] [PubMed] [Google Scholar]
- Hidalgo, P., and R. MacKinnon. 1995. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 268:307–310. [DOI] [PubMed] [Google Scholar]
- Hill, J. M., P. F. Alewood, and D. J. Craik. 1996. Three-dimensional solution structure of mu-conotoxin GIIIB, a specific blocker of skeletal muscle sodium channels. Biochemistry. 35:8824–8835. [DOI] [PubMed] [Google Scholar]
- Hui, K., G. Lipkind, H. A. Fozzard, and R. J. French. 2002. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by mu-conotoxin. J. Gen. Physiol. 119:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancelin, J. M., D. Kohda, S. Tate, Y. Yanagawa, T. Abe, M. Satake, and F. Inagaki. 1991. Tertiary structure of conotoxin GIIIA in aqueous solution. Biochemistry. 30:6908–6916. [DOI] [PubMed] [Google Scholar]
- Li, R. A., I. I. Ennis, R. J. French, S. C. Dudley, Jr., G. F. Tomaselli, and E. Marban. 2001a. Clockwise domain arrangement of the sodium channel revealed by mu-conotoxin (GIIIA) docking orientation. J. Biol. Chem. 276:11072–11077. [DOI] [PubMed] [Google Scholar]
- Li, R. A., I. I. Ennis, G. F. Tomaselli, and E. Marban. 2003a. Molecular basis of isoform-specific μ-conotoxin block of cardiac, skeletal muscle, and brain sodium channels. J. Biol. Chem. 278:8717–8724. [DOI] [PubMed] [Google Scholar]
- Li, R. A., I. L. Ennis, G. F. Tomaselli, R. J. French, and E. Marban. 2001b. Latent specificity of molecular recognition in sodium channels engineered to discriminate between two “indistinguishable” mu-conotoxins. Biochemistry. 40:6002–6008. [DOI] [PubMed] [Google Scholar]
- Li, R. A., I. L. Ennis, P. Velez, G. F. Tomaselli, and E. Marban. 2000. Novel structural determinants of mu-conotoxin (GIIIB) block in rat skeletal muscle (mu1) Na+ channels. J. Biol. Chem. 275:27551–27558. [DOI] [PubMed] [Google Scholar]
- Li, R. A., K. Hui, R. J. French, C. Henrikson, K. Sato, G. F. Tomaselli, and E. Marban. 2003b. Dependence of μ-conotoxin block of sodium channels on ionic strength but not the permeant [Na+]: Implications into the distinctive mechanistic actions of Na and K channel pore-blocking toxins and their molecular targets. J. Biol. Chem. In press. [DOI] [PubMed]
- Li, R. A., K. Sato, K. Kodama, T. Kohno, T. Xue, G. F. Tomaselli, and E. Marban. 2002a. Charge conversion enables quantification of the proximity between a normally-neutral μ-conotoxin (GIIIA) site and the Na+ channel pore. FEBS Lett. 511:159–164. [DOI] [PubMed] [Google Scholar]
- Li, R. A., K. Sato, K. Kodama, T. Xue, R. J. French, G. F. Tomaselli, and E. Marban. 2002b. Molecular Calibration of the Na channel pore by electrostatic compliance analysis. Biophys. J. 82:88a. [Google Scholar]
- Li, R. A., R. G. Tsushima, R. G. Kallen, and P. H. Backx. 1997. Pore residues critical for mu-CTX binding to rat skeletal muscle Na+ channels revealed by cysteine mutagenesis. Biophys. J. 73:1874–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R. A., P. Velez, N. Chiamvimonvat, G. F. Tomaselli, and E. Marban. 1999. Charged residues between the selectivity filter and S6 segments contribute to the permeation phenotype of the sodium channel. J. Gen. Physiol. 115:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind, G. M., and H. A. Fozzard. 1994. A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophys. J. 66:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moczydlowski, E., B. M. Olivera, W. R. Gray, and G. R. Strichartz. 1986a. Discrimination of muscle and neuronal Na-channel subtypes by binding competition between [3H]saxitoxin and mu-conotoxins. Proc. Natl. Acad. Sci. USA. 83:5321–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moczydlowski, E., A. Uehara, X. Guo, and J. Heiny. 1986b. Isochannels and blocking modes of voltage-dependent sodium channels. Ann. N. Y. Acad. Sci. 479:269–292. [DOI] [PubMed] [Google Scholar]
- Nakamura, H., J. Kobayashi, Y. Ohizumi, and Y. Hirata. 1983. Isolation and amino acid compositions of geographutoxin I and II from the marine snail Conus geographus. Experientia. 39:590–591. [DOI] [PubMed] [Google Scholar]
- Nakamura, M., Y. Niwa, Y. Ishida, T. Kohno, K. Sato, Y. Oba, and H. Nakamura. 2001. Modification of Arg-13 of mu-conotoxin GIIIA with piperidinyl-Arg analogs and their relation to the inhibition of sodium channels. FEBS Lett. 503:107–110. [DOI] [PubMed] [Google Scholar]
- Noda, M., H. Suzuki, S. Numa, and W. Stühmer. 1989. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 259:213–216. [DOI] [PubMed] [Google Scholar]
- Olivera, B. M., J. Rivier, C. Clark, C. A. Ramilo, G. P. Corpuz, F. C. Abogadie, E. E. Mena, S. R. Woodward, D. R. Hillyard, and L. J. Cruz. 1990. Diversity of Conus neuropeptides. Science. 249:257–263. [DOI] [PubMed] [Google Scholar]
- Penzotti, J. L., H. A. Fozzard, G. M. Lipkind, and S. C. Dudley, Jr. 1998. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys. J. 75:2647–2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzotti, J. L., G. Lipkind, H. A. Fozzard, and S. C. Dudley, Jr. 2001. Specific neosaxitoxin interactions with the Na+ channel outer vestibule determined by mutant cycle analysis. Biophys. J. 80:698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia, M. T., N. Chiamvimonvat, E. Marban, and G. F. Tomaselli. 1996. Structure of the sodium channel pore revealed by serial cysteine mutagenesis. Proc. Natl. Acad. Sci. USA. 93:300–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin, J., J. W. Kyle, M. Chen, P. Bell, L. L. Cribbs, H. A. Fozzard, and R. B. Rogart. 1992. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 256:1202–1205. [DOI] [PubMed] [Google Scholar]
- Sato, K., Y. Ishida, K. Wakamatsu, R. Kato, H. Honda, Y. Ohizumi, H. Nakamura, M. Ohya, J. M. Lancelin, D. Kohda, 1991. Active site of mu-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J. Biol. Chem. 266:16989–16991. [PubMed] [Google Scholar]
- Schreiber, G., and A. R. Fersht. 1995. Energetics of protein-protein interactions: analysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 248:478–486. [DOI] [PubMed] [Google Scholar]
- Serrano, L., A. Horovitz, B. Avron, M. Bycroft, and A. R. Fersht. 1990. Estimating the contribution of engineered surface electrostatic interactions to protein stability by using double-mutant cycles. Biochemistry. 29:9343–9352. [DOI] [PubMed] [Google Scholar]
- Stephan, M. M., J. F. Potts, and W. S. Agnew. 1994. The microI skeletal muscle sodium channel: mutation E403Q eliminates sensitivity to tetrodotoxin but not to mu-conotoxins GIIIA and GIIIB. J. Membr. Biol. 137:1–8. [DOI] [PubMed] [Google Scholar]
- Stocker, M., and C. Miller. 1994. Electrostatic distance geometry in a K+ channel vestibule. Proc. Natl. Acad. Sci. USA. 91:9509–9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau, H., S. H. Heinemann, W. Stuhmer, M. Pusch, F. Conti, K. Imoto, and S. Numa. 1991. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 293:93–96. [DOI] [PubMed] [Google Scholar]
- Trimmer, J. S., S. S. Cooperman, S. A. Tomiko, J. Y. Zhou, S. M. Crean, M. B. Kallen, R. G. Sheng, R. L. Barchi, F. J. Sigworth, R. H. Goodman, W. S. Agnew, and G. Mandel. 1989. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 3:33–49. [DOI] [PubMed] [Google Scholar]
- Wakamatsu, K., D. Kohda, H. Hatanaka, J. M. Lancelin, Y. Ishida, M. Oya, H. Nakamura, F. Inagaki, and K. Sato. 1992. Structure-activity relationships of mu-conotoxin GIIIA: structure determination of active and inactive sodium channel blocker peptides by NMR and simulated annealing calculations. Biochemistry. 31:12577–12584. [DOI] [PubMed] [Google Scholar]
- Yamagishi, T., M. Janecki, E. Marban, and G. Tomaselli. 1997. Topology of the P segments in the sodium channel pore revealed by cysteine mutagenesis. Biophys. J. 73:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagawa, Y., T. Abe, and M. Satake. 1986. Blockade of [3H]lysine-tetrodotoxin binding to sodium channel proteins by conotoxin GIII. Neurosci. Lett. 64:7–12. [DOI] [PubMed] [Google Scholar]