

Abstract
Mammalian synaptic membranes appear to contain high proportions of specific, sn-1-stearoyl-2-docosahexaenoyl- and sn-1-stearoyl-2-arachidonoyl phosphoglycerides, but the structural significance of this is unclear. Here we used a standardized approach to compare the properties of homogeneous monolayers of the corresponding phosphatidylcholines, phosphatidylethanolamines, phosphatidylserines, and phosphatidic acids with those of control monolayers of sn-1-stearoyl-2-oleoyl- and sn-1-palmitoyl-2-oleoyl phosphoglycerides. Major findings were: 1), that the presence of an sn-2-docosahexaenoyl group or an sn-2-arachidonoyl group increases the molecular areas of phosphoglycerides by 3.8 Å2 (7%) relative to the presence of an sn-2-oleoyl group; 2), that the phosphorylcholine headgroup independently increases molecular areas by a larger amount, 7.1 Å2 (13%); and 3), that the dipole moments of species having an arachidonoyl moiety or an oleoyl moiety are 83 mD (19%) higher than those of comparable docosahexaenoic acid-containing phosphoglycerides. These and other results provide new information about the molecular packing properties of polyenoic phosphoglycerides and raise important questions about the role of these phosphoglycerides in synapses.
The phosphoglycerides of mammalian cell membranes consist of many different molecular species that differ in content of polar headgroups, sn-1-acyl-, alkyl-, or alk-1′-enyl chains and sn-2-acyl chains. For example, the phosphatidylcholine (PC) of the human erythrocyte membrane, which together with sphingomyelin and glycolipids comprises the bulk of the outer leaflet of the membrane, consists largely of sn-1-palmitoyl-2-oleoyl- (16:0-18:1-) and sn-1-palmitoyl-2-linoleoyl- (16:0-18:2-) molecular species, whereas the phosphatidylethanolamine (PE), plasmenylethanolamine (PLE), phosphatidylserine (PS), and phosphatidylinositol (PI), which comprise the bulk of the inner leaflet lipids, consist largely of molecular species that contain arachidonic acid (20:4) or some other very long chain polyunsaturated fatty acid (Myher et al., 1989). The functional significance of these differences remains unclear, though induced changes in the relative distribution of molecular species of PC in erythrocyte membranes have been found to adversely affect specific mechanisms of membrane ion transport (Engelmann et al., 1990).
Less is known about the distribution of phosphoglyceride molecular species in other mammalian cell plasma membranes. However, the synaptic plasma membranes of mammalian neurons may represent a different example. Analyses of the fatty acid compositions of phosphoglyceride classes from these membranes have suggested that the PC may consist largely of molecular species that contain palmitic acid (16:0) and oleic acid (18:1), whereas the PE and PS may consist largely of stearic acid (18:0) and either docosahexaenoic acid (22:6) or arachidonic acid (20:4), the PLE may consist largely of comparable molecular species that contain an 18-carbon alk-1′-enyl chain, and a molecular species that contains 18:0 and 20:4 may comprise the bulk of the PI (Breckenridge et al., 1972; Sun and Sun, 1974). Furthermore, molecular species of PE and PS that contain these polyunsaturated acyl moieties appear to be conserved in the brains of many mammals (Yabuuchi and O'Brien, 1968; Crawford et al., 1977; Farkas et al., 2000).
What could be the structural significance of the differences between the fatty acyl chain composition of PC and the fatty acyl chain compositions of PE, PS, PLE, and PI? This question cannot be answered at present because relatively little is known about the physical properties of polyunsaturated phosphoglycerides that contain headgroups other than phosphorylcholine. Molecular species of PC that contain 20:4 or 22:6 have been studied by several groups of investigators (see, for example, Demel et al., 1972; Evans et al., 1987; Huster et al., 1997; Koenig et al., 1997; Mitchell and Litman, 1998; Dumaual et al., 2000), but few studies, (e.g., Shaikh et al., 2001a), have been done of corresponding molecular species of other phosphoglycerides.
As a first step toward understanding the structural roles of 20:4 and 22:6 in the phosphoglycerides of cell membranes, we have compared the packing properties of homogeneous 18:0-20:4 and 18:0-22:6 molecular species (hereafter referred to as SA and SD) of phosphoglycerides with each other and with similar species having the acyl moieties, 16:0-18:1 and 18:0-18:1 (referred to as PO and SO). We compared the lipids in monomolecular films for several reasons. Firstly, the lateral packing density of lipids in fluid bilayers and other three dimensional fluid phases is difficult to determine (Nagle and Tristram-Nagle, 2000), whereas in monolayers lipid packing is user controllable (Brockman, 1999) and packing properties can be compared at surface pressures of 35 mN/m, a value considered to be bilayer relevant (Marsh, 1996; MacDonald, 1996). Secondly, as components of multicomponent bilayer membranes in cells, PEs and other lipids are part of a globally planar structure. However, in pure form the more unsaturated PEs tend to form nonbilayer phases under physiological conditions of temperature, pH and ionic strength (Allen et al., 1990; Shaikh et al., 2001a). By using monolayers, the packing properties of PEs and other nonbilayer forming lipids can be more directly compared to other lipids in a phase of the same, biologically relevant curvature. In addition, monolayers provide quantitative information concerning other membrane properties, such as dipole potential and compressibility, that also are relatively difficult to measure in bilayers (Clarke, 2001).
Using monolayers, we examined the packing properties of four groups of phosphoglyceride classes, namely 1), a group of SA molecular species of PC, PS, PE, PI, and phosphatidic acid (PA); 2), a group of SD molecular species of PC, PS, PE, and PA; 3), a control group of SO molecular species of PC, PS, PE, and PA; and 4), a control group of PO species of PC, PS, PE, and PA. Because the cytosol of normal mammalian cells contains Mg2+ (Raju et al., 1989) the acidic phosphoglycerides, PS and PA, were studied in both the presence and absence of 1.0 mM Mg2+ ions in the subphase. Important features of the experimental approach were that: 1), Steps were taken not only to identify and exclude oxidized contaminants of the polyunsaturated, i.e., SA and SD phosphoglycerides before analysis but also to prevent the oxidation of the SA and SD phosphoglycerides during analysis. 2), An automated film balance was used to examine at least six replicates of each sample during the course of each set of analyses. 3), Samples of POPC were interspersed among these replicates and used as reference standards to facilitate intragroup and intergroup comparisons.
From this relatively rigorous monolayer study, the packing and electrostatic properties of the phosphoglycerides in a planar interface could be directly compared. Intragroup comparisons revealed that the phosphorylcholine headgroup of PC and the sn-2 polyunsaturated acyl chains were apparently independent determinants of packing density for the phosphoglycerides. Intergroup comparisons showed that polyenoic species other than PCs packed closer than did monoenoic PCs. Moreover, they revealed a significant difference in dipole potential between lipids within the same phospholipid class but having 22:6 compared to 20:4 or 18:1 in the sn-2 position. These and other results challenge current concepts concerning the packing properties of polyenoic phosphoglycerides and focus attention on specific properties of 18:0-22:6 phosphoglycerides that could potentially be of physiological relevance.
MATERIALS AND METHODS
Purchased reagents
Butylated hydroxytoluene was from Calbiochem (La Jolla, CA). Hexane (nonspectro 99.9+%) and isopropanol (ultrapure) for monolayer studies were from Burdick and Jackson (Muskegon, MI). For lipid purification, high performance liquid chromatography (HPLC)-grade ammonium acetate, chloroform, hexane, methanol, isopropanol, anhydrous diethyl ether (stabilized with butylated hydroxytoluene), and HClO4 (70%) were from Baker (Phillipsburg, NJ). Phospholipase D (Streptomyces chromofuscus and Streptomyces sp.), L-serine, choline chloride, phosphate standards, molybdate reagent, and Fiske and Subbarow reducer for the Barlett phosphate assay were from Sigma/Sigma-Aldrich (St. Louis, MO). Bovine heart PE, bovine liver PI, SOPC, SOPS, SOPE, SOPA, and POPC, POPS, POPE, and POPA were from Avanti Polar Lipids (Alabaster, AL).
Phosphoglycerides
SA phosphoglycerides
SAPE was isolated from bovine heart PE (Avanti Polar Lipids) by isocratic elution with methanol/10 mM ammonium acetate from a 5 μm particle size, 1 cm × 25 cm, semi-preparative C18 HPLC column (Rainin/Varian, Walnut Creek, CA) essentially as described (Holte et al., 1990); identified by gas chromatographic analysis of its fatty acid content (Lepage and Roy, 1986); and purified from any contaminating material by rechromatography. Nonoxidized SAPE could be distinguished from contaminating, oxidized SAPE by its ratio of absorbance at 206 nm to absorbance at 234 nm of >200 (Holte et al., 1990) and could be separated from oxidized material because it eluted later from the C18 column, at approximately twice the retention time. Some of the purified SAPE was reserved for later use in monolayer experiments (see below), but most of it was converted to the corresponding molecular species of PC, PS, and PA. The PE was converted into SAPC and SAPS by treatment with Streptomyces sp. phospholipase D in the presence of choline [1 M] or serine [3 M] (Juneja et al., 1988; Juneja et al., 1989). SAPE was converted into SAPA by treatment with phospholipase D from Streptomyces chromofuscus, as described (Imamura and Horiuti, 1979).
In each case the resulting SAPC, SAPS, or SAPA was separately recovered from the ether phase and several ether washes of the aqueous phase of the reaction mixture. The ether phase and ether washes were combined and evaporated almost to dryness under argon, redissolved in chloroform/methanol, and partitioned by the method of Bligh and Dyer (Bligh and Dyer, 1959). The phosphoglyceride-containing chloroform phase was concentrated under a stream of argon, and traces of water were removed by adding a small amount of benzene. In most cases, the SAPC, SAPS, and SAPA showed less than 1% contamination by either the starting SAPE or oxidized phosphoglycerides. This analysis was performed by semi-quantitative comparisons of eluted peak areas from analytical, 5-μm particle size, 0.4 cm × 25 cm, HPLC silica columns (Rainin/Varian), using a modification of the normal phase solvent system described by Patton, et al. (Patton et al., 1982). Specifically, we used a solvent system that contained hexane/isopropanol/10 mM ammonium acetate/ethanol/glacial acetic acid (367:490:62:100:0.6) instead of one that contained phosphate buffer to avoid phosphate precipitation and filtration problems. For samples containing SAPS, we found that increasing the concentration of acetic acid to 1.2 parts gave a pH 4.5 solvent that improved the sharpness and resolution of the SAPS peak.
Further purification of SAPA preparations was not required. However, SAPC and SAPS preparations were usually contaminated with 5%–10% SAPA byproduct, as determined by thin layer chromatography with acidic systems (Skipski et al., 1964; Rouser et al., 1969; Rouser et al., 1970) on plastic-backed silica gel H thin layer chromatography plates (EM Reagents; Gibbstown, NJ). The SAPA could be removed by chromatography using the normal phase solvent system modified from Patton (Patton et al., 1982) on semi-preparative, 5-μm particle size, 1 cm × 25 cm, silica HPLC columns. Samples containing the purified phosphoglycerides were then concentrated under argon or using a Speed-Vac (Savant, Holbrook, NY); extracted immediately by the method of Bligh and Dyer to remove traces of ammonium acetate; dried under argon in the presence of benzene; immediately redissolved in chloroform that contained 0.05% BHT; and stored at −20°C under argon in Pyrex tubes capped with Mininert valves (Aldrich, Milwaukee, WI or Pierce, Rockford, IL).
SAPI was isolated from bovine liver PI by chromatography on a C18 HPLC column. SAPI eluted too rapidly with the methanol-ammonium acetate solvent system of (Holte et al., 1990). However, a modified isocratic solvent system containing 10 mM ammonium acetate in methanol/acetonitrile/water (90:7.5:2.5), similar to one described by Patton et al. (1982), gave optimum resolution of the PI mixtures. Sample workup and storage were as described above for the other SA phosphoglycerides except that it was necessary to redissolve and store the SAPI in chloroform/methanol/water (75/25/2) (Gonzalez-Sastre and Folch-Pi, 1968) to maintain complete solubility.
SD phosphoglycerides
Livers harvested from hatchery-grown Atlantic salmon were used by Avanti Polar Lipids for a large custom preparation of PE. We later isolated SDPE from this preparation using the same HPLC methods described above for the purification of SAPE. Some of the SDPE was used to synthesize and purify SDPC, SDPS, and SDPA by methods that were similar to those described above for the corresponding SA molecular species.
SO and PO phosphoglycerides
A sample of each synthetic phosphoglyceride molecular species from Avanti Polar Lipids was dried with nitrogen, redissolved in benzene and brought again to dryness to help remove residual water before being dissolved in its appropriate solvent (see below).
Preparation of samples for shipment from Seattle to Austin
The phosphorus content of individual stored solutions of SA and SD phosphoglycerides was measured using the method of Bartlett (Bartlett, 1959). Then an appropriate aliquot of each sample was dried under argon and a stock solution containing 1.75 mM phosphoglyceride phosphorus was made by dilution in the appropriate solvent. For most lipids the solvent was hexane/ethanol (95:5, v/v) but for PS molecular species, hexane/isopropanol/water [70:30:2.5, v/v] was used (Ali et al., 1994). The concentration of butylated hydroxytoluene in the solution was adjusted to comprise 0.5 mol % of the total phosphoglyceride and the concentration of phosphoglyceride phosphorus in the stock solution was determined. Then 0.25 ml aliquots of the final stock solution were transferred under argon into six to eight, prewashed, preweighed, threaded, 0.3 ml autosampler vials (National Scientific, Lawrenceville, GA), and the vials were tightly closed with caps that had Teflon-silicon rubber-Teflon septa (National Scientific). The loaded vials were reweighed several times over a 10–20 min period to check for seal leakage and shipped overnight to Austin on gel packs frozen at −20°C.
Upon receipt in Austin, the vials were equilibrated to room temperature and reweighed, and samples showing evidence of leakage were discarded. Generally, for each compound or condition being tested, at least six vials were selected for monolayer characterization. One of these samples was used to redetermine the phosphate concentration and this value was used for setting up monolayer experiments and for subsequent analysis.
Monolayer experiments
Surface pressure-molecular area-surface potential isotherms were determined at 24°C using an automated Langmuir-type film balance (Brockman et al., 1980; Brockman et al., 1984) housed in a room supplied with HEPA- and charcoal-filtered air. The surface pressure sensor was calibrated using lipids of known equilibrium spreading pressure (Momsen et al., 1990). As a consequence of the irregular geometry at the sensor end of the monolayer trough, the reported trough working area was empirically calibrated with respect to added lipid. This was accomplished by measuring surface pressure-molecular area isotherms for samples with different relative numbers of molecules of the same lipid (Smaby and Brockman, 1985). Plotting the reported surface area versus number of molecules spread at each of several arbitrarily chosen surface pressures gave a series of straight lines with a common intercept on the abscissa. From this information a software offset was adjusted, if necessary, to achieve proportionality of reported monolayer area with molecules spread.
Water for buffer preparation was purified by reverse osmosis, charcoal filtration, deionization, ion exchange, and UV irradiation in a system (Millipore, Bedford, MA) designed to lower total organics to <5 ppb. The subphase buffer used for monolayer characterization contained 5.0 mM HEPES Free Acid (Ultrol grade, Calbiochem), 5.0 mM HEPES Sodium Salt (Ultrol grade, Calbiochem), 0.1 M NaCl (ACS grade, Mallinckrodt, Paris, KY), 0.1 mM EGTA (Sigma) and 1.5 mM sodium azide (Sigma) ± 1.0 mM MgCl2 (Sigma). The buffer was prepared as a 36.6-fold concentrate. To remove possible surface-activity impurities, 200-ml aliquots were sparged with argon in a 250-ml graduated cylinder while the surface was continuously aspirated. When the volume had been reduced to 150 ml, the cleaned buffer concentrate was diluted with water to 5.5 l in a glass jug and, if necessary, adjusted to pH 7.4. The buffer was kept under an argon atmosphere until it was pumped into the trough of the film balance through Teflon tubing via a pump having only ceramic working parts. The surface of the aqueous phase in the trough was overfilled and repeatedly cleaned by automated surface compression and aspiration. After cleaning was complete, the subphase liquid level was set using an aspirator. This insured reproducibility in liquid level of ±14 μm, thereby minimizing the effects of subphase volume on effective trough area through curvature at the trough edges (Brockman et al., 1984). The absence of surface-active contaminants in the subphase was checked periodically using dipole potential measurements as described (Smaby and Brockman, 1991a). The trough was maintained in an atmosphere of humidified argon that had been passed through a series of seven filters selected to remove possible organic and particulate contaminants (Li et al., 2001). Samples (51.67 μl) were applied from 250-μl vials onto the buffer surface using an autosampler as described (Brockman et al., 1984) and, after a waiting period of 4 min to allow for solvent evaporation, the monolayer was compressed at ≤4 Å2/min·molecule.
Before the first experimental sample and after each two samples a surface pressure-molecular area isotherm of a reference standard, POPC, was measured to monitor instrument performance. POPC was used because of its ready availability and its stability during storage. Deviations from normal balance performance due to, for example, solvent impurities could generally be detected as a change in POPC isotherm shape and/or a gross change in isotherm molecular area relative to reference isotherms. If detected, the problem was corrected and the affected isotherms were excluded from analysis. Acceptable POPC isotherms were highly reproducible for any given day's experiments, but varied slightly for experiments done several months or years apart. Over the course of the entire study the 39 sets of POPC curves that were determined showed an average molecular area at 35 mN/m of 60.5 ± 0.9 Å2.
DATA ANALYSIS
Data obtained using the automated film balance were analyzed using Filmfit (Creative Tension, Austin, MN), a program optimized for analyzing surface pressure-surface potential-molecular area isotherms. Using this software phase transitions (including monolayer collapse) were identified, compressibilities were calculated, and pressure-potential-area isotherm shapes in the liquid-expanded state were analyzed using equations of state as recently summarized (Dahim et al., 2002).
To correct for film balance variations over time, the set of POPC surface pressure-molecular area isotherms obtained on each day during characterization of each lipid preparation were averaged. A subset of data containing 41 pressure-molecular area values was generated from the average isotherm at 1 mN/m intervals over the range of 2–42 mN/m. This was compared by linear regression to a data set comprised of the average of all POPC areas at each surface pressure. In almost all cases the linear fits to the data were essentially perfect (R2 > .9999). The slopes and intercepts of the POPC calibration lines were then used to normalize the molecular area values for each sample isotherm to the common reference. In most cases, this procedure reduced the variability of isotherms measured in separate experiments with a phosphoglyceride species separated widely in date. Importantly, this procedure also normalized our measurements of phosphoglycerides of different headgroup and acyl chain composition to a common reference. It should be noted, however, that this normalization does not correct for small differences in the chemical determination of the concentration of POPC in different stock solutions over time.
Each surface pressure-molecular area isotherm, after being normalized as described above, was analyzed for the existence of monolayer and collapse phase transitions by the use of second and third derivatives of the data as previously described (Brockman et al., 1980), and transition pressures from replicate isotherms were averaged. This procedure also identified the data point at which the second derivative changed sign and this provided one limit to define the surface pressure-surface potential-molecular area data set for further analysis. The other limit was arbitrarily taken as the molecular area at a surface pressure of 1.0 mN/m. This subset of data from the liquid-expanded state was fitted to an osmotic-based equation of state for liquid-expanded monolayers (Feng et al., 1994),
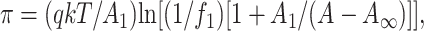 |
(1) |
where π is the surface pressure and A is the molecular area. The cross sectional area of a water molecule, A1, is taken as 9.65 Å2, k is the Boltzmann constant and T is the absolute temperature. The fitting parameter, A∞, is the lipid molecular area extrapolated to infinite pressure. In the context of the model, it is essentially the cross sectional area of the totally dehydrated lipid molecule in the plane of the interface. The fitting parameters f1 and q describe isotherm shape and reflect the activity coefficient of interfacial water. The fits of the data were excellent and allowed theoretical isotherms to be generated over any range of surface pressures. Parameters from fitting replicate isotherms were averaged. Evaluation of Eq. 1, solved for A, using the fitting parameters obtained for each isotherm at the collapse surface pressure provided the reported collapse area. For those species that exhibited a transition from the liquid expanded to a more condensed monolayer state at surface pressures below collapse, collapse areas were estimated by extrapolating the fitted liquid-expanded portion of the isotherm to the collapse surface pressure of the corresponding PC species (see Results). The molecular area and modulus of compression of each film, Cs−1 = A(dπ/dA), were determined at 35 mN/m and values from replicate isotherms were averaged. For each lipid that was not in the liquid-expanded state at 35 mN/m, molecular area and modulus of compression were estimated using the calculated value of A and dπ/dA determined from the fitting parameters, Eq. 1 and its derivative with respect to A.
The measured electrostatic potential across the monolayer during compression, ΔV, was analyzed over the same range of areas as the surface pressure using the equation of state,
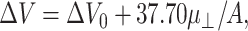 |
(2) |
where ΔV is the measured potential at A relative to a lipid-free interface, μ⊥ is the dipole moment of the monolayer perpendicular to the plane of the interface and ΔV0 is the area-independent component of the measured potential (Brockman, 1994). It should be noted that for lipids with a net charge, these parameters contain contributions from molecular charges and subphase counter ions as well as from molecular dipoles.
RESULTS
Surface pressure-molecular area isotherms
Fig. 1, A–D, shows a representative surface pressure-molecular area isotherm for each of the 17 lipid species (acidic phosphoglyceride isotherms were determined in the presence of 1.0 mM Mg2+). The open symbols in each trace show a subset of the ∼2000 experimental points from each isotherm. As molecular area is decreased, a liftoff from the baseline into the liquid-expanded monolayer state is observed that is typical of biological lipids having an unsaturated acyl chain. For 12 of the 17 isotherms (Fig. 1, A–D) the curves are monotonic until the monolayers collapse from the liquid-expanded state at an essentially constant surface pressure of 45.7 ± 1.1 mN/m (Table 1). Within error, this constant value agrees with the average value of ∼48 mN/m determined for a variety of phosphoglycerides by a novel pipette technique (Lee et al., 2001). The exceptions are the monoenoic phosphoglycerides, POPA, POPE, SOPA, SOPE, and SOPS, each of which shows one or more transitions to more condensed monolayer state(s) before collapse at a pressure above that of those species that collapse from the liquid-expanded state. The surface pressures at which seven of the acidic species collapsed directly from the liquid expanded state without Mg2+ (Fig. 2, A and B) were essentially the same as in its presence at 46.2 + 1.1 mN/m (Table 2). For those species that exhibited a monolayer phase transition before collapse in the absence of Mg2+, the expanded to condensed transition pressures were higher by 10–12 mN/m than for the same species in the presence of Mg2+. This increase presumably reflects lateral charge repulsion in the absence of the divalent cation and its size is roughly the same as that observed when the sn-1 chain was changed from stearate to palmitate in the presence of Mg2+ (Fig. 1, A–D).
FIGURE 1.

Surface pressure-molecular area isotherms of phosphoglycerides at the argon-buffer interface. (Panel A) PO series; (B) SO series; (C) SA series; (D) SD series. In all panels; PA (□), PE (▵), PC (○), PS (▿), PI (◊). Solid lines show range over which data were fit to Eq. 1; dotted lines show extrapolations of fitted curves to the collapse pressure of PC for species that did not collapse directly from the liquid-expanded state.
TABLE 1.
Analysis of the liquid-expanded phase of monolayers in the presence of magnesium ion
| Lipid | n | πc, mN/m | Ac, Å2 | A35, Å2 | A∞, Å2 | f1 | q | Cs,35−1, mN/m | ΔV0, mV | μ⊥, mD |
|---|---|---|---|---|---|---|---|---|---|---|
| POPA | 6 | n/a | [49.5] | [52.1] | 42.4 ± 0.4 | 1.261 ± 0.003 | 1.801 ± 0.046 | [204] | −23 ± 3 | 550 ± 6 |
| POPS | 6 | 45.1 ± 0.3 | 51.3 ± 0.3 | 54.2 ± .3 | 40.3 ± 0.2 | 1.176 ± 0.002 | 2.259 ± 0.023 | 140 ± 1 | −14 ± 1 | 527 ± 4 |
| POPC | 6 | 47.8 ± 0.3 | 56.2 ± 0.1 | 60.6 ± .2 | 41.9 ± 0.3 | 1.157 ± 0.001 | 3.023 ± 0.068 | 134 ± 1 | 114 ± 1 | 516 ± 3 |
| POPE | 4 | n/a | [50.6] | 53.7 ± .7 | 41.3 ± 0.7 | 1.206 ± 0.006 | 2.138 ± 0.052 | 148 ± 1 | 75 ± 4 | 540 ± 4 |
| SOPA | 5 | n/a | [50.5] | 52.3 ± .2 | 46.0 ± 0.2 | 1.294 ± 0.001 | 1.224 ± 0.007 | [262] | −29 ± 1 | 567 ± 1 |
| SOPS | 8 | n/a | [48.4] | 51.3 ± .3 | 38.7 ± 0.3 | 1.178 ± 0.001 | 2.063 ± 0.018 | 119 ± 4 | −11 ± 1 | 461 ± 3 |
| SOPC | 16 | 46.4 ± 1.3 | 53.9 ± 0.7 | 58.0 ± .7 | 39.3 ± 2.2 | 1.155 ± 0.004 | 3.082 ± 0.125 | 128 ± 5 | 112 ± 5 | 494 ± 13 |
| SOPE | 6 | n/a | [50.6 ] | 53.3 ± .2 | 42.7 ± 0.1 | 1.201 ± 0.002 | 1.785 ± 0.020 | [180] | 80 ± 4 | 521 ± 9 |
| SAPA | 6 | 44.1 ± 0.2 | 52.4 ± 0.2 | 54.6 ± .2 | 43.5 ± 0.2 | 1.219 ± 0.001 | 1.931 ± 0.018 | 165 ± 1 | 19 ± 5 | 543 ± 6 |
| SAPS | 23 | 45.6 ± 0.4 | 53.2 ± 2.5 | 57.1 ± 2.6 | 38.1 ± 2.7 | 1.148 ± 0.005 | 3.013 ± 0.307 | 123 ± 6 | 15 ± 17 | 492 ± 38 |
| SAPC | 16 | 45.3 ± 0.1 | 59.2 ± 0.5 | 63.6 ± .6 | 40.3 ± 0.5 | 1.135 ± 0.002 | 3.725 ± 0.073 | 124 ± 1 | 121 ± 2 | 528 ± 7 |
| SAPE | 12 | 46.3 ± 0.3 | 52.8 ± 0.4 | 56.4 ± .4 | 40.5 ± 0.8 | 1.167 ± 0.003 | 2.682 ± 0.337 | 138 ± 4 | 86 ± 6 | 505 ± 15 |
| SAPI | 6 | 46.5 ± 0.1 | 51.3 ± 0.6 | 55.2 ± .7 | 37.5 ± 0.4 | 1.155 ± 0.002 | 2.821 ± 0.041 | 131 ± 1 | 4 ± 1 | 450 ± 6 |
| SDPA | 6 | 44.2 ± 0.2 | 54.0 ± 0.1 | 56.4 ± .1 | 43.4 ± 0.1 | 1.192 ± 0.001 | 2.191 ± 0.012 | 138 ± 1 | 21 ± 1 | 501 ± 2 |
| SDPS | 7 | 45.4 ± 0.2 | 52.1 ± 0.2 | 56.0 ± .2 | 37.6 ± 0.2 | 1.138 ± 0.002 | 2.816 ± 0.040 | 119 ± 1 | 25 ± 2 | 387 ± 4 |
| SDPC | 13 | 44.9 ± 0.1 | 59.5 ± 1.4 | 64.0 ± 1.4 | 39.7 ± 1.5 | 1.125 ± 0.002 | 3.831 ± 0.100 | 116 ± 4 | 153 ± 7 | 431 ± 21 |
| SDPE | 6 | 44.8 ± 0.1 | 53.8 ± 0.2 | 57.4 ± .2 | 39.6 ± 0.2 | 1.151 ± 0.001 | 2.795 ± 0.035 | 126 ± 1 | 117 ± 2 | 431 ± 6 |
FIGURE 2.

Surface pressure-molecular area isotherms of acidic phosphoglycerides at the argon-buffer interface without Mg2+. (Panel A) PA series; (B) PS series with SAPI (◊). Acyl compositions for the PA and PS series; PO (○), SO (□), SA (▵) and SD (▿). Lines are as in Fig. 1.
TABLE 2.
Analysis of the liquid-expanded phase of monolayers in the absence of magnesium ion
| Lipid | n | πc, mN/m | Ac, Å2 | A35, Å2 | A∞, Å2 | f1 | q | Cs,35−1, mN/m | ΔV0, mV | μ⊥, mD |
|---|---|---|---|---|---|---|---|---|---|---|
| POPA | 8 | n/a | [51.1] | 54.7 ± 0.6 | 40.7 ± 0.2 | 1.140 ± 0.003 | 2.246 ± 0.077 | 137 ± 3 | −45 ± 4 | 531 ± 3 |
| POPS | 5 | 46.8 ± 0.2 | 52.7 ± 0.3 | 56.8 ± 0.4 | 39.3 ± 0.2 | 1.153 ± 0.002 | 2.769 ± 0.061 | 130 ± 1 | −13 ± 3 | 480 ± 5 |
| SOPA | 4 | n/a | [51.6] | 54.8 ± 0.3 | 42.3 ± 0.2 | 1.160 ± 0.000 | 1.941 ± 0.009 | [158] | −40 ± 1 | 534 ± 2 |
| SOPS | 8 | 48.1 ± 0.3 | 48.0 ± 0.4 | 52.3 ± 0.3 | 35.3 ± 0.4 | 1.158 ± 0.002 | 2.696 ± 0.054 | 123 ± 1 | −9 ± 2 | 403 ± 4 |
| SAPA | 6 | 45.1 ± 0.6 | 52.7 ± 0.3 | 55.8 ± 0.4 | 41.7 ± 0.2 | 1.159 ± 0.005 | 2.158 ± 0.052 | 140 ± 3 | −8 ± 2 | 489 ± 8 |
| SAPS | 12 | 45.8 ± 0.2 | 57.1 ± 1.9 | 61.8 ± 1.9 | 38.9 ± 2.6 | 1.121 ± 0.001 | 3.455 ± 0.139 | 117 ± 6 | 18.1 ±10 | 442 ± 45 |
| SAPI | 4 | 45.9 ± 0.1 | 51.5 ± 0.2 | 55.7 ± 0.3 | 35.3 ± 0.2 | 1.141 ± 0.001 | 3.222 ± 0.030 | 120 ± 1 | −1 ± 1 | 379 ± 2 |
| SDPA | 6 | 45.4 ± 0.1 | 55.7 ± 0.3 | 59.7 ± .2 | 41.8 ± 0.5 | 1.124 ± 0.000 | 2.613 ± 0.064 | 121 ± 1 | 12 ± 2 | 424 ± 5 |
| SDPS | 8 | 44.9 ± 0.2 | 51.0 ± 0.3 | 55.1 ± .2 | 34.1 ± 0.3 | 1.131 ± 0.001 | 3.217 ± 0.036 | 109 ± 1 | 14 ± 2 | 315 ± 2 |
Because we are primarily interested in comparing the properties of the monounsaturated lipids to those of the more polyunsaturated species that all remain in the chain melted state, only the expanded phase of each of the isotherms shown in Figs. 1 and 2 was further characterized. At lower surface pressures in the liquid-expanded state the isotherms for each fatty acyl composition, i.e., each panel in Fig. 1, show a consistent pattern. In each set the PC is the largest, the PA is the smallest and the others are nearly identical. For comparison, isotherms for the acidic species were also measured in the absence of Mg2+ (Fig. 2, A and B). For six of the nine acidic species the isotherms (Fig. 2, A and B) were more expanded at low pressures in the absence of Mg2+, as might be expected on the basis of charge repulsion. However, in side-by-side comparisons (not shown) isotherms for SOPS, SDPS, and SAPI ± Mg2+ from Figs. 1 and 2 were nearly identical. As controls we also measured isotherms for the zwitterionic species, POPC, SOPE and SAPE, in the absence of Mg2+ (data not shown). The isotherms for these lipids were unaffected by the divalent ion.
The solid line through each set of data points in Figs. 1 and 2 shows the fitted curve for the liquid-expanded portion of each phosphoglyceride isotherm, determined using Eq. 1 as described in the experimental section. For those lipids that did not collapse from the liquid-expanded state, the fitted curve at lower pressures was extrapolated (dotted lines) to the molecular area at which it reached the collapse pressure of the corresponding PC molecular species. This surface pressure was chosen because all PCs collapsed from the liquid-expanded state and all lipids that collapsed from the liquid-expanded state exhibited essentially the same collapse pressure (45.7 ± 1.1 mN/m). The average values of fitting parameters for all lipids studied are given in Table 1 and Table 2. For those lipids that did not collapse from the liquid-expanded state and for which parameters could not be directly determined, values obtained from extrapolated isotherms are shown in brackets in the tables. Inspection of the fitted/extrapolated isotherms (Fig. 1, A–D, solid and dotted lines) shows an obvious pattern. Namely, at higher, physiologically relevant surface pressures around 35 mN/m the isotherms of each set of chain-matched phosphoglycerides, with the exception of the PCs, converge into a narrow range of molecular areas. Excluding PCs, the averages of the molecular areas of the PO, SO, SA, and SD series at 35 mN/m (Table 1) are 53.3 ± 1.1, 52.3 ± 1.0, 55.8 ± 1.1, and 56.6 ± 0.7 Å2. Likewise, the collapse areas of those species that collapse from the liquid-expanded state, like the molecular areas measured at 35 mN/m, are similar if PCs are excluded (Table 1). Across all series, the PCs were larger than other species with the same acyl chains by an average of 7.1 ± 1.2 Å2, an increase of 13 ± 1%. These data suggest that, with the exception of the PCs, the molecular areas under biologically relevant conditions are determined solely by the area of the fatty acyl chains of the lipids or, at the least, that the headgroup contribution is constant. The moduli of compression of the SD lipids at 35 mN/m are consistently smaller than those of the SA lipids by 3%–20% and, for a given fatty acyl composition, the moduli generally fall in the order PA > PE > PS ≈ PC (Table 1).
For each phosphoglyceride class except PI, for which only the SA species was studied, the polyunsaturated SA and SD species are very similar in molecular area at 35 mN/m (Table 1). These areas are, however, larger than their monoenoic PO and SO counterparts by an average 3.8 Å2, an increase of 7.0%. Thus, at higher, physiologically relevant surface pressures the presence of a PC headgroup has approximately twice the effect on molecular area than does polyunsaturation in the acyl moiety at the sn-2 position of glycerol. Moreover, the area contributions of the PC headgroup and the glycerol ester chains appear additive. Another consequence of the combined effects of chains and headgroups is that at surface pressures >10mN/m the molecular areas of the sn-2 polyunsaturated SA and SD species, other than PCs, are actually smaller than those of the monounsaturated species, POPC and SOPC.
For all 17 lipids studied in the presence of Mg2+, comparison of the fitting parameter A∞, i.e., molecular area (A) extrapolated to infinite surface pressure, gives a range of values between 38 Å2 and 45 Å2 with no clear differences between sn-2 polyunsaturated and monounsaturated species. A plot of the fitting parameters f1 vs. q from these isotherms (Fig. 3) shows what appears to be a continuous relationship that can be described by a second order polynomial (line). A similar continuous relationship was previously observed in the absence of Mg2+ for lipids with less variation in acyl chains (Smaby and Brockman, 1991b) and was later explained by Feng et. al. who showed that both parameters were related to the activity of interfacial water (Feng et al., 1994). If the parameter values for the PA and PS acidic species studied without Mg2+ are added to the graph (not shown) the PS data fall along the solid line but the PA data are displaced downward. Thus, Mg2+ appears to be a more direct determinant of PA surface behavior.
FIGURE 3.

Relationship of fitting parameters f1 and q. Solid line is a second order polynomial fit. PO series (□), SO series (○), SA series (▵), SD series (▿).
Potential-concentration isotherms
Fig. 4, A–D (+Mg2+) and Fig. 5, A and B (−Mg2+) present the experimental values of the change in electrostatic potential measured across the interface during monolayer compression of the lipids shown in Figs. 1 and 2. Following the usual convention, the data are plotted against the surface concentration of lipid, i.e., the reciprocal of molecular area. For those isotherms for which the data were collected at sufficiently low lipid concentrations, it can be seen that the potential rises abruptly in the gaseous-liquid expanded coexistence region (over which the surface pressure is essentially zero). It becomes linear with respect to the two-dimensional lipid concentration in the range corresponding to the liquid-expanded region of the pressure-area isotherms and becomes approximately constant following monolayer collapse. The solid lines (Figs. 4, A–D, and 5, A and B) show the range of data fitted to Eq. 2. The range of lipid concentrations fitted corresponds to the range of molecular areas used for analysis of the corresponding surface pressure isotherms (Figs. 1, A–D, and 2, A and B, solid lines). From the linear behavior exhibited, the parameters, ΔV0 and μ⊥, were determined from the intercept and slope of each line and are given in Tables 1 and 2.
FIGURE 4.

Surface potential-concentration isotherms of phosphoglycerides at the argon-buffer interface (acidic phosphoglycerides with 1.0 mM Mg2+). Panel (A) PO series; (B) SO series; (C) SA series; (D) SD series. In all panels; PA (□), PE (▵), PC (○), PS (▿), PI (◊). Solid lines show fits to Eq. 2 over the range of data shown by the solid lines in Fig. 1.
FIGURE 5.

Surface potential-concentration isotherms of acidic phosphoglycerides at the argon-buffer interface without Mg2+. (Panel A) PA series; (B) PS series with SAPI (◊). Acyl compositions for the PA and PS series; PO (○), SO (□), SA (▵), and SD (▿). Lines are as in Fig. 4.
The parameter, ΔV0, is an area-independent potential proposed to arise from an epitaxial ordering of water molecules by the lipid headgroups, relative to the air-buffer interface against which the potentials are referenced (Brockman, 1994). In Fig. 6 the values of ΔV0 for all SO, SA, and SD species, except SAPI, are plotted against those of the PO series. Not surprisingly, the values for the SO series form a line with a slope of 1.0 and an intercept of approximately zero. This shows that substituting stearate for palmitate in the sn-1 position of glycerol has a negligible effect on the extents to which the various headgroups structure interfacial water. Within error, the same slope is observed for the SD series but the line is displaced upward by 41 mV. This shows that having 22:6 in the sn-2 position has a significant and consistent effect on water structure, as probed by this parameter. The values for the SA series are linear but with a slope of only 0.77. Essentially, for the acidic species, PA and PS, ΔV0 values for the SA species are offset like those of the SD series whereas for the zwitterionic species, PE and PC, the values are not. The removal of Mg2+ had little effect on ΔV0 values of the acidic species (Table 2), except for SAPA which was between the SO and SA lines (data not shown), consistent with the ambivalent behavior of the SA series.
FIGURE 6.

Variation of the area-independent component of the dipole potential (ΔV0) with acyl composition. Values for SO, SA, and SD species, except SAPI, are plotted against those of the PO series. SO series (□), SA series (○), SD series (▵). The solid lines show the linear fit for each set of points.
Another heretofore unrecognized difference in electrostatic behavior related to polyunsaturation in the sn-2 position is seen with dipole moments. Fig. 7 shows that, compared to their range of 400–500 mD, the dipole moments of each of the PO, SO, SA, and SD series vary little with phosphoglyceride headgroup. This small variation is indicated by the generally upward slope of each set of data in the figure moving from left to right. For all the SD species the average is 438 ± 48 mD. However, for the PO, SO, and SA series combined, the values average 521 ± 29 mD, an increase of 83 mD or 19%. This same trend is preserved in the absence of Mg2+ (not shown) for which the dipole moments of the acidic SD and other species (Table 2) differ by an average of 96 mD. A second trend related to Mg2+ is that it raises the dipole moments of the acidic species by an average of 52 ± 22 mD, irrespective of the fatty acid in the sn-2 position.
FIGURE 7.

Effect of acyl structure on phosphoglyceride dipole moment. PO series (□), SO series (○), SA series (▵), and SD series (▿).
DISCUSSION
This study was undertaken to provide insight as to how the polyenoic structures of arachidonic and docosahexaenoic acids in the sn-2 position of phosphoglyceride species affect their packing in synaptic membranes. Emphasis was on making standardized comparisons at a membrane relevant surface pressure, 35 mN/m, and Mg2+ was included in the aqueous phase at an approximately physiological level. The results do not always agree with previously published monolayer properties of phosphoglycerides in monolayers. For example, the molecular area of SDPC at ∼30–35 mN/m has been reported to be 115 Å2 (Stillwell et al., 1993), 68 Å2 (Ghosh et al., 1973), 64 Å2 (Zerouga et al., 1995), and 51 Å2 (Dumaual et al., 2000) with collapse surface pressures that range from 34 to 41 mN/m. Indeed, before adding 0.5 mol % butylated hydroxytoluene to samples of polyunsaturated lipids, we observed much greater scatter in their molecular areas as compared to the interspersed POPC controls as well as a progressive increase in molecular areas for one particular preparation of SDPE of 15% over a period of three days. Given the potential for oxidation of polyunsaturated lipids, differences in experimental conditions and the number of compounds studied, we have not attempted a comprehensive comparison with earlier data. Rather, we focus on patterns within our data and how these relate to prior observations and the properties of polyunsaturated lipids in membranes.
The collapse pressure of liquid-expanded phospholipid monolayers observed in this study and by a pipette technique (Lee et al., 2001) is ∼47 mN/m. Based on the equivalence of the surface tension of ∼25 mN/m (72–47 mN/m) at monolayer collapse and the surface tension of a hydrocarbon-air interface, Lee et al. proposed that monolayer collapse occurs when the surface tension of the monolayer reaches that of the hydrocarbon-air interface, i.e., that collapse depends only on the acyl chains of the lipids. Thus, there is no reason, a priori, to equate monolayer properties at collapse with those of phospholipids in unstressed bilayers. Rather, a surface pressure of 35 mN/m was chosen for comparison because prior work suggested the relevance of lipid packing densities at this surface pressure to those in large, unstressed bilayers (Marsh, 1996; MacDonald, 1996).Considering differences in conditions, the monolayer areas at 35 mN/m of SOPC (58.0 ± 0.7 Å2) and SDPC (64.0 ± 1.4 Å2) agree reasonably well with reported bilayer values of 61.4 Å2 (Koenig et al., 1997) and 65 Å2 (Feller et al., 2002), respectively. Note also that the difference between these bilayer values is consistent with that observed in monolayers as a consequence of having a polyunsaturated versus a monounsaturated chain at sn-2.
Another physical property that further supports the validity of the monolayer-bilayer comparison at 35 mN/m is elasticity. It was recently reported for a group of lipids with varying degrees of unsaturation in the sn-2 chain that the stretching modulus was relatively independent of precise acyl structure with a mean value of 243 ± 24 mN/m (Rawicz et al., 2000). For all 14 lipids characterized in the present study that had a compressibility modulus directly determinable from the isotherm (Table 1) the average value, doubled for comparison with bilayers, is in good agreement at 264 ± 26 mN/m. Consistent with this agreement is the observation that small quantities of hydrocarbon do not affect the bilayer stretching modulus (D. Needham, personal communication), suggesting the absence of interdigitation effects in controlling bilayer elasticity. Thus, the results of this study strongly support the use of the monolayer-derived data at 35 mN/m as a predictor of the packing and physical behavior of phospholipids in bilayer membranes.
Foremost, the results of this study show; a), that sn-2 polyunsaturation (SA and SD species) increases the molecular areas of phosphoglycerides by 3.8 Å2 (7%) relative to their sn-2 monoenoic counterparts (SO and PO species), b), that the phosphorylcholine headgroup independently increases molecular areas by a larger amount, 7.1 Å2 (13%), than does sn-2 polyunsaturation, and c), that species with an arachidonoyl or oleoyl moiety at sn-2 have dipole moments 83 mD (19%) higher than those of comparable docosahexaenoyl phosphoglycerides. The implications of these observations will be addressed below.
Polyunsaturation and phosphoglyceride packing
Some years ago Applegate and Glomset simulated the structures of selected polyunsaturated hydrocarbons, diacylglycerols and diacylglycerol arrays (Applegate and Glomset, 1986, 1991a,b). They concluded that: 1), The rotational energy thresholds of single carbon-carbon bonds that are adjacent to cis double bonds in unsaturated hydrocarbons are lower than those of single carbon-carbon bonds in saturated hydrocarbons. 2), As a result of this, a hexaenoic hydrocarbon model of docosahexaenoic acid can adopt multiple, highly bent conformations or alternate, angle iron-shaped or helical conformations that have straight molecular axes. 3), Single molecules of sn-1-stearoyl-2-docosahexaenoyl diacylglycerol or sn-1-stearoyl-2-arachidonoyl diacylglycerol having angle-iron shaped or helical polyunsaturated chains can adopt low energy conformations that have straight molecular axes. 4), Groups of such diacylglycerol molecules having angle-iron shaped polyunsaturated chains can exhibit close packing as a consequence of back-to-back, intermolecular contacts involving these chains (Applegate and Glomset, 1986, 1991a,b). Though the simulations were done without reference to potential effects of polar headgroups, water of hydration, or applied thermal energy, the molecular areas obtained for the model diacylglycerols, ∼40 Å2, are in good agreement with the average A∞ for sn-2 polyunsaturated phosphoglycerides, 40.0 ± 2.2 Å2 (Table 1), obtained here by mathematically extrapolating surface pressure-area isotherms to infinite surface pressure, i.e., to the dehydrated state (Feng et al., 1994). This raises the possibility that corresponding natural phosphoglycerides may be able to pack closely together in monolayers and bilayers if their headgroups do not interfere.
Previous evidence related to this possibility has largely come from studies of PCs. In part this is because polyunsaturated PCs, particularly dipolyunsaturated PCs, are important components of retinal rod outer segment membranes (Miljanich et al., 1979), whereas species of pure polyunsaturated PEs form hexagonal phases under physiologically relevant conditions of pH and temperature (see, for example, Shaikh et al., 2001a). Comparisons of phase transition temperatures of hydrated PCs as a function of the extent and position of acyl unsaturation have supported the notion that polyunsaturation can have an ordering effect relative to monounsaturation (Huang, 2001; Ichimori et al., 1999; Holte et al., 1995) as have the results of recent simulations of PCs containing sn-1-saturated and sn-2-polyunsaturated fatty acyl chains (Huber et al., 2002; Saiz and Klein, 2001; Huang, 2001). Ordered segments of individual polyene chains are present and relatively long lived (Saiz and Klein, 2001), though not all double bonds in a particular chain participate in ordered structures, leading overall to a relatively high degree of chain disorder (Huber et al., 2002). These results are consistent with NMR results for polyunsaturated PCs (Feller et al., 2002; Huber et al., 2002).
However, synaptic membranes appear to contain little or no SDPC or SAPC, as mentioned previously, and data from the present study suggests that in a planar environment PC should be regarded as the exception rather than the model for SD and SA phosphoglycerides. This is because the molecular areas of SDPC and SAPC, at a pressure of 35 mN/m, were significantly larger than those of corresponding non-PC phosphoglycerides (Fig. 1, Table 1). The difference between the molecular areas of the polyunsaturated PC and non-PC phosphoglycerides probably depended in part on the area expanding effect of the choline headgroup because the addition of 1–3 methyl groups to PE has been noted earlier to progressively increase molecular area in monolayers (Evans et al., 1980). Beyond this, our data revealed additive effects of polyunsaturated acyl chains and phosphorylcholine headgroups on the molecular areas of the SD- and SAPCs, which suggest that the polyunsaturated fatty acyl chains compete laterally for interfacial area with the hydrated choline methyl groups. Supporting evidence for polyunsaturated chain-choline interactions comes from NMR measurements (Everts and Davis, 2000) and one simulation of SDPC that has shown “remarkable overlap between water distribution and the double bond region of the lipid chains” (Saiz and Klein, 2001). Additional support comes from the dipole potential data discussed below under “Potential-concentration isotherms”.
The fact that the molecular areas of all non-PC SD and SA phosphoglycerides determined in our study were considerably smaller than those of the corresponding PCs is consistent with the smaller sizes of the headgroups of PE, PS, and PA as well as with the effect of Mg2+ on PA, that we have demonstrated. The fact that the molecular area of SAPI also was smaller than that of the corresponding PC implies that, at a pressure of 35 mN/m, its relatively large headgroup may have had a perpendicular orientation with respect to the monolayer surface (Bradshaw et al., 1999) and so, had little effect on the molecular area of that phosphoglyceride. Moreover, a recently obtained surface pressure-area isotherm of sn-1-stearoyl-2-arachidonoylglycerol (M.M. Momsen and H.L. Brockman, unpublished), when fitted below its collapse pressure of ∼32 mN/m and mathematically extrapolated to higher pressures as described for some lipids in this study, has a molecular area at 35 mN/m of 56.6 Å2, a value essentially equal to the average of 55.8 ± 1.1 Å2 reported above for non-PC SA phosphoglycerides. We suggest, therefore, that the acyl chains, not the headgroups, determine molecular area of the non-PCs. In addition, the conformations of the docosahexaenoyl and arachidonoyl chains in the non-PC phosphoglycerides may have been more ordered than they were in the PCs. In support of this, unpublished dynamic simulations of homogeneous arrays of hydrated molecules of SDPC and SDPE that initially contained angle iron-shaped docosahexaenoyl chains (K. R. Applegate and J. A. Glomset) showed that the conformations of these chains remained relatively ordered in SDPE during the simulations (the effective chain lengths decreased by only 10%), but became more disordered in SDPC. If the polyenoic chains of synaptic membrane non-PC phosphoglycerides also have relatively ordered conformations, this might promote close packing of the phosphoglycerides and reduce the nonspecific permeability of the membranes to Ca+2 ions. Moreover, the inherent flexibility of the polyenoic chains (see above) might facilitate fast membrane fusion reactions during the secretion of neutrotransmitters.
It should be noted, however, that the molecular areas of the non-PC SD and SA phosphoglycerides were larger than those of the corresponding SO and PO phosphoglycerides at a pressure of 35 mN/m (Fig. 1, Table 1). Furthermore, the monolayers of polyenoic non-PC phosphoglycerides remained fluid until they collapsed, whereas the monolayers of corresponding, non-PC SO and PO phosphoglycerides showed a tendency to form pressure-dependent condensed phases (Fig. 1). These differences may have been caused not by sn-2 chain disorder, but by the relative inability of the angle-iron- or helical-shaped docosahexaenoyl or arachidonoyl chains to form attractive intra- and intermolecular contacts, as implied by the simulation studies of the corresponding diacylglycerols (Applegate and Glomset, 1986, 1991a,b). Similar simulation studies of static arrays of SO diacylglycerol (Applegate and Glomset, 1991b) failed to predict effective packing for this species, perhaps because the simulations were not dynamic and did not accurately predict attractive intra- and intermolecular interactions involving saturated segments of the monounsaturated chains.
One consequence of changes in headgroup or chain areas of lipids is to alter the tendency of the lipids to adopt nonbilayer phases. This is reflected in the critical packing parameter, f = v/al (Israelachvili et al., 1980) which can be related to spontaneous membrane curvature (Hui and Sen, 1989). This factor reflects differences in headgroup and chain geometry because it is calculated from the volume of the acyl chains, v, the effective length of the acyl chains perpendicular to the interface, l, and the effective area of the lipid headgroup in the interfacial plane, a. As originally postulated, the values of f over which the spontaneous curvature results in a bilayer state is approximately 0.5–1, a twofold range. Attempts to calculate f are complicated by the lack of precise definitions of effective chain volume, length, and headgroup area (Hui and Sen, 1989; Sen and Hui, 1988). Despite these limitations, the effect of the introduction of polyunsaturation at sn-2 can be crudely estimated from the molecular area measurements at 35 mN/m. At this pressure, the areas of the SA and SD species were the same, on average, but larger than those of the SO species by about 4.8% for any phospholipid class. Since the quantity v/l in the Israelachvili equation is the effective area of the acyl chains, the effect of polyunsaturation at sn-2 on f and, hence, on spontaneous curvature should be small. This can be contrasted to the effect of changing the phospholipid headgroup from PE to PC which will change the term a in the equation by 54–106% depending on the extent to which headgroup hydration contributes to its effective area (Hui and Sen, 1989; Sen and Hui, 1988).
Potential-concentration isotherms
The dipole potential between the aqueous phase and the center of membranes is an important determinant of lipid lateral organization in membranes (McConnell, 1991), ion transport regulation (Clarke, 2001) and the behavior of proteins in membranes (Porschke, 1997; McIntosh et al., 2002). However, it is difficult to measure in bilayers (Clarke, 2001) and, hence, little data with respect to membranes is available. However, in monolayers the potential across the monolayer is relatively simple to measure (Brockman, 1994; Clarke, 2001). Its value across monolayers can be divided into molecular area-independent (ΔV0) and area-dependent contributions and it is from the area-dependent portion that the dipole moment, μ⊥, which corresponds to that obtained from bilayer data, is calculated (Brockman, 1994). The value of μ⊥ is determined primarily by the glycerol ester portion of the phosphoglyceride molecule and associated water (Brockman, 1994), as confirmed in this study for the PO, SO, and SA series (Fig. 7) which averaged 521 ± 29 mD across all phosphoglyceride species, including PCs. A surprising observation was that the average μ⊥ for the SD series was 438 ± 48 mD, a difference that was unaffected by the presence of Mg2+. This suggests some unique property of the docosahexaenoyl chain that is not shared with the arachidonoyl chain. Given the origins of dipole moments in lipids, the double bond close to the aqueous phase at position 4 of the docosahexaenoyl chain is a likely possibility. Because of its proximity to the interface, this double bond may assume a unique conformation or interact in a unique way with water as compared with the double bond at position 5 of the arachidonoyl chain. Supporting this idea is the 41 mV offset of ΔV0 values for the SD series of phosphoglycerides (Fig. 6) and the dependence of ΔV0 on the headgroup and its interactions with water.
This difference between the dipole moments of SD and other lipids translates into a change in electrical field strength across the interfacial region of the monolayer of approximately 4 × 107 V m−1. For comparison, a typical transmembrane potential of 40 mV generates a total field across the membrane of ∼1 × 107 V m−1 (Clarke, 2001). It is, therefore, likely that the unique electrostatic properties of docosahexaenoyl lipids, whatever their origin, will have consequences for ion transport, protein conformation and other events that are regulated by such local fields.
Future directions
The results of the present study provide new information about the packing and electrostatic behavior of non-PC SD and SA phosphoglycerides that may relate to the biological roles that these phosphoglycerides play in mammalian synaptic plasma membranes. But the study represents only a first step toward an examination of these roles, as mentioned previously, and additional information is clearly needed. For example, synaptic membranes appear to contain not only SD and SA phosphoglycerides, but also corresponding PLEs (Breckenridge et al., 1972; Sun and Sun, 1974). Therefore, it will be important to examine the behavior of these polyunsaturated PLEs. Because the PLEs lack sn-1-chain carbonyl groups, monolayers containing them might pack even more compactly than do monolayers of diacyl phosphoglycerides, as suggested by an earlier study of species monounsaturated at sn-2 (Smaby et al., 1983). On the other hand, docosahexaenoic acid- or arachidonic acid-containing PLE might have special properties that depend on the combined presence of an alk-1′-enyl double bond in the sn-1-chain and proximally located cis double bonds in the sn-2-chain. Note that both types of double bonds are adjacent to single carbon-carbon bonds, that the latter occupy opposed positions in energy-minimized models of docosahexaenoic-containing diradylglycerol molecules (K. R. Applegate and J. G. Glomset, unpublished results) and that the single, carbon-carbon bonds would be predicted to have low energy thresholds for rotation.
Information is also needed concerning the properties of monolayers that contain mixtures of non-PC SD and SA phosphoglycerides, corresponding PLEs, PO molecular species of PC or PA, and stearoyl sphingomyelin. The possibility that some of these mixtures may show evidence of acyl-chain-dependent lateral phase separation warrants attention because differences in acyl-chain structure and addition of cholesterol have recently been shown to promote similar phenomena in the case of other lipid mixtures (Shaikh et al., 2001b). Moreover, acyl-chain-dependent phase separation involving polyunsaturated phosphoglycerides, monounsaturated phosphoglycerides, and sphingomyelin might conceivably promote the formation of functionally important domains in synaptic membranes. The domains could potentially cause interfacial proteins to bind selectively to the membrane through headgroup-dependent effects or effects that depend on special properties of the polyunsaturated phosphoglycerides. Note that the bending modulus of unsaturated fatty acyl chains in phosphoglycerides has been found to be markedly altered by the presence of polyunsaturation versus monounsaturation (Rawicz et al., 2000) and that a role for dipole potential in regulating protein function has been demonstrated (Cladera et al., 1999).
Finally, studies of isolated synaptic membranes that play a direct role in synaptic signaling are needed to determine the precise composition and distribution of lipids in these membranes. The membranes of neurosecretory vesicles might be excellent models for study because they can be isolated, apparently intact, from homogenates of synaptosomes by methods of size exclusion and immunoadsorption. Techniques of lipid mass spectroscopy could potentially be used to obtain precise information about their content of lipid molecular species, and lipid asymmetry between the leaflets of the vesicle membrane lipid bilayer could be analyzed. Since detailed information about the protein content of the vesicles is already available, biophysical studies designed to correlate polyunsaturated phosphoglyceride content and asymmetry with protein binding might eventually become possible.
Acknowledgments
The authors appreciate helpful comments from Dr. David Needham concerning the interpretation and significance of isotherm data.
This work was supported by the Howard Hughes Medical Institute (J.A.G.), United States Public Health Service (USPHS) Grant HL49180 (H.L.B.), and the Hormel Foundation (H.L.B.). Support for Dr. Applegate was provided by USPHS Grant RR00166 to the Regional Primate Research Center at the University of Washington.
K. R. Applegate is deceased.
References
- Ali, S., J. M. Smaby, H. L. Brockman, and R. E. Brown. 1994. Cholesterol's interfacial interaction with galactosylceramides. Biochemistry. 33:2900–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, T. M., K. Hong, and D. Papahadjopoulos. 1990. Membrane contact, fusion, and hexagonal (HII) transitions in phosphatidylethanolamine liposomes. Biochemistry. 29:2976–2985. [DOI] [PubMed] [Google Scholar]
- Applegate, K. R., and J. A. Glomset. 1986. Computer-based modeling of the conformation and packing properties of docosahexaenoic acid. J. Lipid Res. 27:658–680. [PubMed] [Google Scholar]
- Applegate, K. R., and J. A. Glomset. 1991a. Effect of acyl chain unsaturation on the conformation of model diacylglycerols: a computer modeling study. J. Lipid Res. 32:1635–1644. [PubMed] [Google Scholar]
- Applegate, K. R., and J. A. Glomset. 1991b. Effect of acyl chain unsaturation on the packing of model diacylglycerols in simulated monolayers. J. Lipid Res. 32:1645–1655. [PubMed] [Google Scholar]
- Bartlett, G. R. 1959. Phosphorus assay in column chromatography. J. Biol. Chem. 234:466–469. [PubMed] [Google Scholar]
- Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:912–917. [DOI] [PubMed] [Google Scholar]
- Bradshaw, J. P., R. J. Bushby, C. C. D. Giles, and M. R. Saunders. 1999. Orientation of the headgroup of phosphatidylinositol in a model biomembrane as determined by neutron diffraction. Biochemistry. 38:8393–8401. [DOI] [PubMed] [Google Scholar]
- Breckenridge, W. C., G. Gombos, and I. G. Morgan. 1972. The lipid composition of adult rat brain synaptosomal plasma membranes. Biochim. Biophys. Acta. 266:695–707. [DOI] [PubMed] [Google Scholar]
- Brockman, H. L. 1994. Dipole potential of lipid membranes. Chem. Phys. Lipids. 73:57–79. [DOI] [PubMed] [Google Scholar]
- Brockman, H. L. 1999. Lipid Monolayers: why use half a membrane to characterize protein-membrane interactions? Curr. Opin. Struct. Biol. 9:438–443. [DOI] [PubMed] [Google Scholar]
- Brockman, H. L., C. M. Jones, C. J. Schwebke, J. M. Smaby, and D. E. Jarvis. 1980. Application of a microcomputer controlled film balance system to collection and analysis of data from mixed monolayers. J. Colloid Interface Sci. 78:502–512. [Google Scholar]
- Brockman, H. L., J. M. Smaby, and D. E. Jarvis. 1984. Automation of surface cleaning and sample addition for surface balances. J. Phys. [E] Sci. Instrum. 17:351–353. [Google Scholar]
- Cladera, J., I. Martin, J.-M. Ruysschaert, and P. O'Shea. 1999. Characterization of the sequence of interactions of the fusion domain of the simian immunodeficiency virus with membranes. Role of the membrane dipole potential. J. Biol. Chem. 274:29951–29959. [DOI] [PubMed] [Google Scholar]
- Clarke, R. J. 2001. The dipole potential of phospholipid membranes and methods for its detection. Adv. Colloid Interface Sci. 89:263–281. [DOI] [PubMed] [Google Scholar]
- Crawford, M. A., A. G. Hassam, G. Williams, and W. Whitehouse. 1977. Fetal accumulation of long-chain polyunsaturated fatty acids. Adv. Exp. Med. Biol. 83:135–143. [DOI] [PubMed] [Google Scholar]
- Dahim, M., N. K. Mizuno, X.-M. Li, W. E. Momsen, M. M. Momsen, and H. L. Brockman. 2002. Physical and photophysical characterization of a BODIPY phosphatidylcholine as a membrane probe. Biophys. J. 83:1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demel, R. A., W. S. M. Guerts van Kessel, and L. L. M. van Deenen. 1972. The properties of polyunsaturated lecithins in monolayers and liposomes and the interactions of these lecithins with cholesterol. Biochim. Biophys. Acta. 266:26–40. [DOI] [PubMed] [Google Scholar]
- Dumaual, A. C., L. J. Jenski, and W. Stillwell. 2000. Liquid crystalline/gel state phase separation in docosahexaenoic acid-containing bilayers and monolayers. Biochim. Biophys. Acta. 1463:395–406. [DOI] [PubMed] [Google Scholar]
- Engelmann, B., J. A. F. op den Kamp, and B. Roelofsen. 1990. Replacement of molecular species of phosphatidylcholine: influence on erythrocyte Na transport. Am. J. Physiol. 258:C682–C691. [DOI] [PubMed] [Google Scholar]
- Evans, R. W., M. A. Williams, and J. Tinoco. 1980. Surface viscosities of phospholipids alone and with cholesterol in monolayers at the air-water interface. Lipids. 15:524–533. [Google Scholar]
- Evans, R. W., M. A. Williams, and J. Tinoco. 1987. Surface areas of 1-palmitoyl phosphatidylcholines and their interactions with cholesterol. Biochem. J. 245:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everts, S., and J. H. Davis. 2000. 1H and 13C NMR of multilamellar dispersions of polyunsaturated (22:6) phospholipids. Biophys. J. 79:885–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas, T., K. Kitajka, E. Fodor, I. Csengeri, E. Lahdes, Y. K. Yeo, Z. Krasznai, and J. E. Halver. 2000. Docosahexaenoic acid-containing phospholipid molecular species in brains of vertebrates. Proc. Natl. Acad. Sci. USA. 97:6362–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, S. E., K. Gawrisch, and A. D. MacKerell, Jr. 2002. Polyunsaturated fatty acids in lipid bilayers: intrinsic and environmental contributions to their unique physical properties. J. Am. Chem. Soc. 124:318–326. [DOI] [PubMed] [Google Scholar]
- Feng, S., H. L. Brockman, and R. C. MacDonald. 1994. On osmotic-type equations of state for liquid-expanded monolayers of lipids at the air-water interface. Langmuir. 10:3188–3194. [Google Scholar]
- Ghosh, D., M. A. Williams, and J. Tinoco. 1973. The influence of lecithin structure on their monolayer behavior and interactions with cholesterol. Biochim. Biophys. Acta. 291:351–362. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Sastre, F., and J. Folch-Pi. 1968. Thin-layer chromatography of the phosphoinositides. J. Lipid Res. 9:532–533. [PubMed] [Google Scholar]
- Holte, L. L., S. A. Peter, T. M. Sinnwell, and K. Gawrisch. 1995. 2H nuclear magnetic resonance order parameter profiles suggest a change of molecular shape for phosphatidylcholines containing a polyunsaturated acyl chain. Biophys. J. 68:2396–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holte, L. L., F. J. G. M. van Kuijk, and E. A. Dratz. 1990. Preparative high-performance liquid chromatography purification of polyunsaturated phospholipids and characterization using ultraviolet derivative spectroscopy. Anal. Biochem. 188:136–141. [DOI] [PubMed] [Google Scholar]
- Huang, C. 2001. Mixed-chain phospholipids: structures and chain-melting behavior. Lipids. 36:1077–1097. [DOI] [PubMed] [Google Scholar]
- Huber, T., K. Rajamoorthi, V. F. Kurze, K. Beyer, and M. F. Brown. 2002. Structure of docosahexaenoic acid-containing phospholipid bilayers as studied by 2H NMR and molecular dynamics simulations. J. Am. Chem. Soc. 124:298–309. [DOI] [PubMed] [Google Scholar]
- Hui, S.-W., and A. Sen. 1989. Effects of lipid packing on polymorphic phase behavior and membrane properties. Proc. Natl. Acad. Sci. USA. 86:5825–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huster, D., A. J. Jin, K. Arnold, and K. Gawrisch. 1997. Water permeability of polyunsaturated lipid membranes measured by 17O NMR. Biophys. J. 73:855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimori, H., T. Hata, H. Matsuki, and S. Kaneshina. 1999. Effect of unsaturated acyl chains on the thermotropic and barotropic phase transitions of phospholipid bilayer membranes. Chem. Phys. Lipids. 100:151–164. [Google Scholar]
- Imamura, S., and Y. Horiuti. 1979. Purification of Streptomyces chromofuscus phospholipase D by hydrophobic affinity chromatography on palmitoyl cellulose. J. Biochem. 85:79–95. [DOI] [PubMed] [Google Scholar]
- Israelachvili, J. N., S. Marcelja, and R. G. Horn. 1980. Physical principles of membrane organization. Q. Rev. Biophys. 13:121–200. [DOI] [PubMed] [Google Scholar]
- Juneja, L. R., T. Kazuoka, N. Goto, T. Yamane, and S. Shimizu. 1989. Conversion of phosphatidylcholine to phosphatidylserine by various phospholipase D in the presence of L- or D-serine. Biochim. Biophys. Acta. 1003:277–283. [Google Scholar]
- Juneja, L. R., T. Kazuoka, T. Yamane, and S. Shimizu. 1988. Kinetic evaluation of conversion of phosphatidylcholine to phosphatidylethanolamine by phospholipase D from different sources. Biochim. Biophys. Acta. 960:334–341. [DOI] [PubMed] [Google Scholar]
- Koenig, B. W., H. H. Strey, and K. Gawrisch. 1997. Membrane lateral compressibility determined by NMR and x-ray diffraction: effect of acyl chain polyunsaturation. Biophys. J. 73:1954–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S., D. H. Kim, and D. Needham. 2001. Equilibrium and dynamic interfacial tension measurements at microscopic interfaces using a micropipet technique. 2. Dynamics of phospholipid monolayer formation and equilibrium tensions at the water-air interface. Langmuir. 17:5544–5550. [Google Scholar]
- Lepage, G., and C. C. Roy. 1986. Direct transesterification of all classes of lipids in a one-step reaction. J. Lipid Res. 27:114–120. [PubMed] [Google Scholar]
- Li, X.-M., M. M. Momsen, J. M. Smaby, H. L. Brockman, and R. E. Brown. 2001. Cholesterol decreases the interfacial elasticity and detergent solubility of sphingomyelins. Biochemistry. 40:5954–5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald, R. C. 1996. The relationship and interactions between lipid bilayers vesicles and lipid monolayers at the air/water interface. In Vesicles. M. Rosoff, editor. Marcel Dekker, New York. 3–48.
- Marsh, D. 1996. Lateral pressure in membranes. Biochim. Biophys. Acta. 1286:183–223. [DOI] [PubMed] [Google Scholar]
- McConnell, H. M. 1991. Structures and transitions in lipid monolayers at the air-water interface. Annu. Rev. Phys. Chem. 42:171–195. [Google Scholar]
- McIntosh, T. J., A. Vidal, and S. A. Simon. 2002. The energetics of peptide-lipid interactions: modulation by interfacial dipoles and cholesterol. Curr. Top. Membr. 52:309–338. [Google Scholar]
- Miljanich, G. P., L. A. Sklar, D. L. White, and E. A. Dratz. 1979. Disaturated and dipolyunsaturated phospholipids in the bovine retinal rod outer segment disk membrane. Biochim. Biophys. Acta. 552:294–306. [DOI] [PubMed] [Google Scholar]
- Mitchell, D. C., and B. J. Litman. 1998. Molecular order and dynamics in bilayers consisting of highly polyunsaturated phospholipids. Biophys. J. 74:896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momsen, W. E., J. M. Smaby, and H. L. Brockman. 1990. The suitability of nichrome for measurement of gas-liquid interfacial tension by the Wilhelmy method. J. Colloid Interface Sci. 135:547–552. [Google Scholar]
- Myher, J. J., A. Kuksis, and S. Pind. 1989. Molecular species of glycerophospholipids and sphingomyelins of human erythrocytes: improved method of analysis. Lipids. 24:396–407. [DOI] [PubMed] [Google Scholar]
- Nagle, J. F., and S. Tristram-Nagle. 2000. Lipid bilayer structure. Curr. Opin. Struct. Biol. 10:474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton, G. M., J. M. Fasulo, and S. J. Robins. 1982. Separation of phospholipids and individual molecular species of phospholipids by high-performance liquid chromatography. J. Lipid Res. 23:190–196. [PubMed] [Google Scholar]
- Porschke, D. 1997. Macrodipoles. Unusual electric properties of biological macromolecules. Biophys. Chem. 66:241–257. [DOI] [PubMed] [Google Scholar]
- Raju, B., E. Murphy, L. A. Levy, R. D. Hall, and R. E. London. 1989. A fluorescent indicator for measuring cytosolic free magnesium. Am. J. Physiol. 256:C540–C548. [DOI] [PubMed] [Google Scholar]
- Rawicz, W., K. C. Olbrich, T. McIntosh, D. Needham, and E. Evans. 2000. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 79:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouser, G., S. Fleischer, and A. Yamamoto. 1970. Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids. 5:494–496. [DOI] [PubMed] [Google Scholar]
- Rouser, G., G. Kritchevsky, A. Yamamoto, G. Simon, C. Galli, and A. J. Bauman. 1969. Diethylaminoethyl and triethylaminoethyl cellulose column chromatographic procedures for phospholipids, glycolipids, and pigments. Methods Enzymol. 14:272–313. [Google Scholar]
- Saiz, L., and M. L. Klein. 2001. Structural properties of a highly polyunsaturated lipid bilayer from molecular dynamics simulations. Biophys. J. 81:204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, A., and S.-W. Hui. 1988. Direct measurement of headgroup hydration of polar lipids in inverted micelles. Chem. Phys. Lipids. 49:179–184. [DOI] [PubMed] [Google Scholar]
- Shaikh, S. R., M. R. Brzustowicz, W. Stillwell, and S. R. Wassall. 2001a. Formation of inverted hexagonal phase in SDPE as observed by solid-state 31P NMR. Biochem. Biophys. Res. Commun. 286:758–763. [DOI] [PubMed] [Google Scholar]
- Shaikh, S. R., A. C. Dumaual, L. J. Jenski, and W. Stillwell. 2001b. Lipid phase separation in phospholipid bilayers and monolayers modeling the plasma membrane. Biochim. Biophys. Acta. 1512:317–328. [DOI] [PubMed] [Google Scholar]
- Skipski, V. P., R. F. Peterson, and M. Barclay. 1964. Quantitative analysis of phospholipids by thin-layer chromatography. Biochem. J. 90:374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaby, J. M., and H. L. Brockman. 1985. Miscibility, chain packing, and hydration of 1-palmitoyl-2-oleoyl phosphatidylcholines and other lipids in surface phases. Biophys. J. 48:701–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaby, J. M., and H. L. Brockman. 1991a. A simple method for estimating surfactant impurities in solvents and subphases used for monolayer studies. Chem. Phys. Lipids. 58:249–252. [DOI] [PubMed] [Google Scholar]
- Smaby, J. M., and H. L. Brockman. 1991b. An evaluation of models for surface pressure-area behavior of liquid-expanded monolayers. Langmuir. 7:1031–1034. [Google Scholar]
- Smaby, J. M., A. Hermetter, P. C. Schmid, F. Paltauf, and H. L. Brockman. 1983. Packing of ether and ester phospholipids in monolayers. Evidence for hydrogen bonded water at the sn-1 acyl group of phosphatidylcholines. Biochemistry. 22:5808–5813. [Google Scholar]
- Stillwell, W., S. R. Wassall, A. C. Dumaual, W. D. Ehringer, C. W. Browning, and L. J. Jenski. 1993. Use of merocyanine (MC540) in quantifying lipid domains and packing in phospholipid vesicles and tumor cells. Biochim. Biophys. Acta. 1146:136–144. [DOI] [PubMed] [Google Scholar]
- Sun, G. Y., and A. Y. Sun. 1974. Synaptosomal plasma membranes: acyl group composition of phosphoglycerides and (Na+ plus K+)-ATPase activity during fatty acid deficiency. J. Neurochem. 22:15–18. [DOI] [PubMed] [Google Scholar]
- Yabuuchi, H., and J. S. O'Brien. 1968. Positional distribution of fatty acids in glycerophosphatides of bovine gray matter. J. Lipid Res. 9:65–67. [PubMed] [Google Scholar]
- Zerouga, M., L. J. Jenski, and W. Stillwell. 1995. Comparison of phosphatidylcholines containing one or two docosahexaenoic acyl chains on properties of phospholipid monolayers and bilayers. Biochim. Biophys. Acta. 1236:266–272. [DOI] [PubMed] [Google Scholar]