

Abstract
M13 major coat protein was derivatized with BODIPY (n-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl iodoacetamide), and its aggregation was studied in 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and DOPC/1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (DOPG) or 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE)/DOPG (model systems of membranes with hydrophobic thickness matching that of the protein) using photophysical methodologies (time-resolved and steady-state self-quenching, absorption, and emission spectra). It was concluded that the protein is essentially monomeric, even in the absence of anionic phospholipids. The protein was also incorporated in pure bilayers of lipids with a strong mismatch with the protein transmembrane length, 1,2-dierucoyl-sn-glycero-3-phosphocholine (DEuPC, longer lipid) and 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine (DMoPC, shorter lipid), and in lipidic mixtures containing DOPC and one of these lipids. The protein was aggregated in the pure vesicles of mismatching lipid but remained essentially monomeric in the mixtures as detected from BODIPY fluorescence emission self-quenching. From fluorescence resonance energy transfer (FRET) measurements (donor-n-(iodoacetyl)aminoethyl-1-sulfonaphthylamine (IAEDANS)-labeled protein; acceptor-BODIPY labeled protein), it was concluded that in the DEuPC/DOPC and DMoPC/DOPC lipid mixtures, domains enriched in the protein and the matching lipid (DOPC) are formed.
INTRODUCTION
M13 coat protein has been shown to exist in many aggregation states, depending on factors like isolation, reconstitution procedure, pH, ionic strength, and amphiphile composition (Hemminga et al., 1993; Stopar et al., 1997). The mechanism of phage assembly in the Escherichia coli membrane is not yet completely understood, but the assembly site has been proposed to be composed of a dynamic protein-lipid network, characterized by absence of preferential association between M13 coat protein and/or lipids (Hemminga et al., 1993), which allows storage of monomeric coat protein at very high local concentrations, as the insertion of the protein in the assembling phage particle is only possible on the monomeric form (Russel, 1991). The type of interactions between lipids and coat protein which allows for formation of this structure is largely unknown and in this study it is intended to obtain information on the influence of lipid bilayer composition on the lateral distribution and oligomerization properties of M13 coat protein in membrane model systems, to better understand the phage particle assembly mechanism.
It has been proposed that self-association behavior of transmembrane proteins while incorporated in lipid membranes is influenced by hydrophobic matching conditions on the protein-lipid interface (Mouritsen and Bloom, 1984; Killian, 1998). The monomeric protein is expected to be stable under perfect matching conditions with the phospholipids surrounding it. In case of hydrophobic mismatch at the protein-lipid interface, it is possible that the boundary lipids reorganize themselves, to lower the tension created by exposure of hydrophobic acyl chains or amino-acid residues, which can be achieved by ordering/disordering of the phospholipids (Nezil and Bloom, 1992). Moreover, if the hydrophobic mismatch is too high for correction with small adjustments of bilayer hydrophobic thickness, this might result in protein aggregation to obtain minimization of the protein-lipid contacts (Ren et al., 1999; Lewis and Engelman, 1983, Mobashery et al., 1997; Mall et al., 2001). Other responses to hydrophobic mismatch might be transmembrane helix tilt (for long transmembrane hydrophobic protein domains), change in conformation or decrease of bilayer partitioning of the protein, transition of phospholipids to nonlamellar phases, and molecular sorting of proteins and lipids in the presence of a binary lipid system (for reviews see Killian, 1998; Dumas et al., 1999).
Some studies have already related the oligomerization state of proteins to the hydrophobic mismatch extent in the bilayer. Aggregation was shown to depend on hydrophobic mismatch for bacteriorhodopsin at extreme hydrophobic mismatch conditions (Lewis and Engelman, 1983) and gramicidin (Mobashery et al., 1997). Mall and co-workers (Mall et al., 2001) concluded that, for a synthetic peptide, the free energy of dimerization increased linearly with each additional carbon of the phospholipid fatty acyl chain, with a slope of 0.5 kJ mol−1. Apparently, this effect is much more significant upon negative hydrophobic thickness (thicker hydrophobic section for the bilayer than for the protein; Ren et al., 1999; Lewis and Engelman, 1983; Mall et al., 2001). Meijer et al. (2001) found by electron spin resonance that the M13 major coat protein incorporated in di(22:1)PC (1,2-dierucoylphosphatidylcholine) aggregated or existed in several orientations/conformations, whereas in di(14:1)PC (1,2-dimyristoleoylphosphatidylcholine) no indication was found toward aggregation.
In addition, for proteins incorporated in binary phospholipid mixtures, strong selectivity to one lipid component, and phase separation or lipid sorting effects (depending on their miscibility), was predicted to occur as a result of hydrophobic mismatch (Sperotto and Mouritsen, 1993). This phenomenon has also been observed experimentally in a mixture of di(12:0)PC (dilauroylphosphatidylcholine)/di(18:0)PC(distearoylphosphatidylcholine), in which bacteriorhodopsin was shown to preferentially associate with the short chain lipid in the gel-gel and gel-fluid coexistence regions (Dumas et al., 1997). Although the preferential association of bacteriorhodopsin with short chain lipid in the gel-fluid coexistence region could be understood by an eventual preference for the disordered phase, these results were rationalized as being a consequence of lipid-protein hydrophobic matching interactions. A similar effect was observed for E. coli lactose permease (Lehtonen and Kinnunen, 1997), and for gramicidin (Fahsel et al., 2002). In the case of bacteriorhodopsin, Dumas and co-workers (Dumas et al., 1997), using a theoretical model, concluded that the protein was preferentially associated with the longer chain lipid on the mixed fluid lipid phase. In this mixture no macroscopic phase separation was occurring, but only preference of protein association with the phospholipid which allowed for more favorable energetic interactions, resulting in bacteriorhodopsin being surrounded by di(18:0)PC. This phenomenon is also denominated by wetting, and may extend to multiple layers of phospholipids (Gil et al., 1998). The interfacial stress, which is likely to occur between the wetting phase and the lipid matrix, can lead to sharing of these microdomains by many proteins, to minimize the interface area. As a result, formation of protein-enriched domains would occur in the bilayer. This process has been proposed as a mechanism for protein aggregation inducement (Gil et al., 1998).
Some studies have also dealt with transmembrane protein/peptide interaction selectivity toward anionic phospholipids, based on electrostatic interactions of the lipid headgroup and basic residues on the protein (Horvàth et al., 1995a,b). A glucosyltransferase from Acholeplasma laidlawii was found to exhibit preference for localization on PG domains formed in a PC matrix (Karlsson et al., 1996).
The purpose of this work is to study the influence of the bilayer composition on the aggregation state of the M13 coat protein. In addition, the protein was also incorporated in binary lipidic systems, where there is strong hydrophobic mismatch of the protein with one of the components. The possibility of protein segregation to lipidic domains in this situation was also investigated.
For these purposes, several fluorescence methodologies (fluorescence self-quenching, absorption and emission spectra, and energy transfer) were applied, using the protein derivatized with n-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl iodoacetamide (BODIPY FL C1-IA) or n-(iodoacetyl)aminoethyl-1-sulfonaphthylamine (IAEDANS), as described in more detail below.
Through the use of the self-quenching of the BODIPY fluorescence, it is expected to obtain information about the aggregation/oligomerization state of protein. With the same objective, the presence of BODIPY ground-state dimers is investigated. These techniques allow us to check for molecular contacts between BODIPY groups labeled on the transmembrane section of the mutant protein.
To solve the question of whether the presence of M13 major coat protein is capable of inducing lipid domain formation through electrostatic interactions or hydrophobic mismatch, FRET measurements are carried out on different lipid mixtures with M13 major coat protein incorporated. As the C-terminal of the M13 major coat protein is heavily basic, it is intended to know if the presence of protein would lead to formation of domains enriched in anionic phospholipids and protein. Additionally, by using mixtures of phospholipids with different acyl-chain lengths, formation of protein-enriched domains due to preferential binding to hydrophobically matching lipids is checked.
In reconstituted systems we can have two different orientations for the M13 coat protein in the bilayer (with the N-terminus sticking to the inside or the outside of the lipid vesicle), and in this way, interactions between proteins might involve parallel or antiparallel protein orientations. For this reason, the mutants chosen for the present work were T36C and A35C, because for the coat protein in vesicles of DOPC, the Thr36 and Ala35 residues are located near the center of the bilayer, as was shown by AEDANS wavelength of maximum emission (Spruijt et al., 2000). This increases the possibility of self-quenching due to the formation of complexes or from collisions between fluorophores, and allowed us to ignore situations with complex oligomers containing proteins with parallel and antiparallel orientations as well as to simplify the energy transfer analysis to the two-dimensional case, as, for both orientations, both residues should be located at approximately the same position.
MATERIALS AND METHODS
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (DOPG), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dierucoyl-sn-glycero-3-phosphocholine (DEuPC) and 1,2-dimyristoleoyl-sn-glycero-3-phosphocholine (DMoPC), were obtained from Avanti Polar Lipids (Birmingham, AL). N-(iodoacetyl)aminoethyl-1-sulfonaphthylamine (1,5-IAEDANS) and N-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-yl)methyl) iodoacetamide (BODIPY FL C1-IA) were obtained from Molecular Probes (Eugene, OR). Fine chemicals were obtained from Merck (Darmstadt, Germany). All materials were used without further purification.
Coat protein isolation and labeling
The wild-type M13 major coat protein and the T36C mutant were grown, purified from the phage (Spruijt et al., 1996) and the A35C mutant was obtained from transformed cells of E. coli B21 (DE3) (Spruijt et al., 2000). Both mutants were labeled with BODIPY and IAEDANS as described previously (Spruijt et al., 1996). The remaining impurities were extracted using size exclusion chromatography and the protein was eluted with 50 mM sodium cholate, 150 mM NaCl, and 10 mM Tris-HCl pH 8.
Coat protein reconstitution in lipid vesicles
The labeled protein mutant was reconstituted in the DOPC/DOPG (80/20 mol/mol), DOPC, DOPE/DOPG (70/30 mol/mol), DMoPC/DOPC (60/40 mol/mol), DEuPC/DOPC (60/40 mol/mol), DMoPC, and DEuPC vesicles using the cholate-dialysis method (Spruijt et al., 1989). The phospholipid vesicles were produced as follows: the chloroform from solutions containing the desired phospholipid amount was evaporated under a stream of dry N2 and last traces removed by a further evaporation under vacuum. The lipids were then solubilized in 50 mM sodium cholate buffer (150 mM NaCl, 10 mM Tris-HCl, and 1 mM EDTA) at pH 8 by brief sonication (Branson 250 cell disruptor) until a clear opalescent solution was obtained, and then mixed with the wild-type and labeled protein. Samples had a phospholipid concentration of 1 mM and the lipid-to-protein ratio (L/P) was always kept at 50, with addition of wild-type protein when necessary, except for the studies with pure DMoPC and DEuPC bilayers in which the L/P was always <50 (due to a smaller labeling efficiency obtained in the preparation of A35C mutant) and no wild-type protein was added. Dialysis was carried at room temperature and in the dark (to prevent IAEDANS degradation), with a 100-fold excess buffer containing 150 mM NaCl, 10 mM Tris-HCl, and 1 mM EDTA at pH 8. The buffer was replaced 5× every 12 h.
Fluorescence spectroscopy
Absorption spectroscopy was carried out with a Jasco V-560 spectrophotometer (Tokyo, Japan). The absorption of the samples was kept <0.1 at the wavelength used for excitation.
CD spectroscopy was performed on a Jasco J-720 spectropolarimeter with a 450 W Xe lamp (Easton, MD).
Steady-state fluorescence measurements were carried out with an SLM-Aminco 8100 Series 2 spectrofluorimeter (Rochester, NY; with double excitation and emission monochromators, MC400) in a right-angle geometry. The light source was a 450-W Xe arc lamp and for reference a Rhodamine B quantum counter solution was used. Correction of emission spectra was performed using the correction software of the apparatus. 5 × 5 mm quartz cuvettes were used. All measurements were performed at room temperature.
In the fluorescence quenching measurements, the BODIPY emission spectra were recorded with an excitation wavelength of 460 nm and a bandwidth of 4 nm for both excitation and emission.
AEDANS-labeled protein quantum yield was determined using quinine sulfate (φ = 0.55) (Eaton, 1988) as reference.
The excitation wavelength for the energy transfer measurements was 340 nm and the emission spectra were recorded with an excitation and emission bandwidth of 4 nm.
Fluorescence decay measurements of IAEDANS were carried out with a time-correlated single-photon counting system, which is described elsewhere (Loura et al., 2000). Measurements were performed at room temperature. Excitation and emission wavelengths were 340 and 440 nm, respectively. The timescales used were between 12 and 67 ps/ch, depending on the amount of BODIPY-labeled protein present in the sample. Data analysis was carried out using a nonlinear, least-squares iterative convolution method based on the Marquardt algorithm (Marquardt, 1963). The goodness of the fit was judged from the experimental χ2-value, weighted residuals, and autocorrelation plot.
In all cases, the probes florescence decay were complex and described by a sum of exponentials,
 |
(1) |
where ai are the normalized amplitudes.
The average lifetimes are defined by
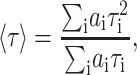 |
(2) |
and the amplitude average or lifetime-weighted quantum yield (Lakowicz, 1999), is obtained as
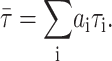 |
(3) |
THEORETICAL BACKGROUND
Fluorescence self-quenching
Collisional quenching is responsible for a decrease in both the quantum yield and lifetime of a fluorophore, whereas static quenching only affects the fluorescence intensity due to the dark (“nonfluorescent”) character of the self-quenching complex. The effects of the collisional contribution of self-quenching on the fluorescence lifetime are described by the Stern-Volmer equation,
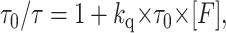 |
(4) |
where τ0 is the lifetime of the fluorophore in the absence of quenching, τ is the lifetime of the fluorophore in presence of quenching, kq is the bimolecular diffusion rate constant, and [F] is the concentration of the fluorophore.
The diffusion coefficient of the fluorophore (D) can be calculated from kq using the Smoluchowski equation (Lakowicz, 1999), taking into account transient effects (Umberger and Lamer, 1945),
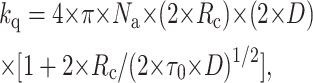 |
(5) |
where Na is the Avogadro number and Rc is the collisional radius. Diffusion in membranes is here considered to take place in an isotropic three-dimensional medium. If the membrane were strictly bidimensional, different boundary conditions for the Smoluchowski formalism should be applied (Razi-Naqvi, 1974). The best approach to the specific situation of probe diffusion in a membrane is the one used by Owen (1975), in which the finite bilayer width is considered (cylindrical geometry). Owen introduced the parameter τs, which defines the crossover from the spherical (three-dimensional) to the cylindrical geometry, its value being τs ≅ 42 ns when considering the bilayer and the peptide. This value is much longer than the longest fluorescence lifetime of the probes in the peptide (6.1 ns), and longer than the experimental time-window (28 ns = 28 ps/channel × 1000 channels) so the three-dimensional framework approximation is essentially correct. Almgren (1991), in a comparative study of quenching in restricted dimensionality, also states that deviations from the three-dimensional occur only for very long fluorescence lifetimes.
The static quenching component can be described through a sphere of action, which accounts for statistical contact pairs formed at the moment of excitation. These contact pairs are nonfluorescent, although they do not form a complex, i.e., the interaction energy is <k × T. The combined contributions of the collisional and the sphere-of-action effects on the fluorescence intensity are given by Loura et al. (2000) as
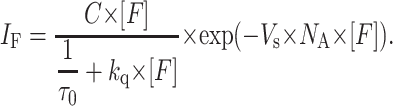 |
(6) |
Here, IF is the fluorescence intensity, C is a constant, and Vs is the sphere-of-action volume. The sphere-of-action radius is obtained by
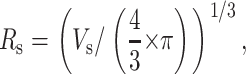 |
(7) |
and for a collisional quenching mechanism it should be close to the sum of the Van der Waals radii.
In case that a complex is formed, the model to describe static quenching effects should take into account its equilibrium constant. For a monomer/dimer equilibrium of only one molecular species, the fluorescence intensity is given by
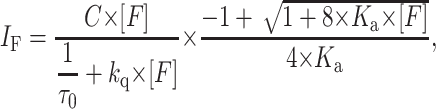 |
(8) |
where Ka is the oligomerization constant.
However, for our system, in which there are two different protein species (labeled and unlabeled, where the unlabeled class includes both nonlabeled mutant and wild-type protein), there will be several combinations of protein species within an aggregate. For a dimer, there will be three different combinations available—labeled protein/labeled protein, labeled protein/unlabeled protein, and unlabeled protein/unlabeled protein—but only formation of the first one induces self-quenching of BODIPY. A complexation model describing fluorescence static quenching in our system will have to account for this fact. From the knowledge of the concentration of each species, the fraction of labeled protein participating in oligomers (dimers/trimers) containing more than one BODIPY labeled protein (and as a result nonfluorescent), can be obtained for a given Ka. The resulting set of nonlinear equations was solved (for a given total protein concentration, labeling efficiency, labeled protein concentration, aggregation number, and Ka) using Maple V (Waterloo, Ontario).
Fluorescence resonance energy transfer
The energy transfer between fluorophores can be used to characterize the lateral distribution of labeled coat protein mutant in the bilayer. The degree of fluorescence emission quenching of the donor caused by the presence of acceptors is used to calculate the experimental energy transfer efficiency (E):
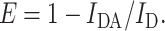 |
(9) |
Here, IDA is the donor fluorescence emission intensity in the presence of acceptor, and ID is the donor fluorescence emission intensity in the absence of acceptor.
Wolber and Hudson (1979) obtained the analytical solution for energy transfer efficiencies in a random distribution of acceptors in a bidimensional space,
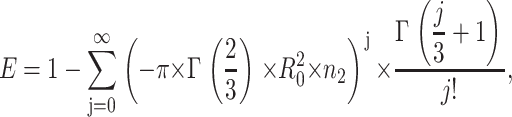 |
(10) |
where R0 is the Förster radius, Γ is the complete gamma function, and n2 is the acceptor numerical density (number of acceptors per unit area).
The Förster radius is given by
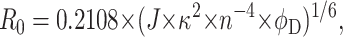 |
(11) |
where J is the spectral overlap integral, κ2 is the orientation factor, n is the refractive index of the medium, and φD is the donor quantum yield. J is calculated from
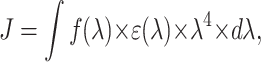 |
(12) |
where f(λ) is the normalized emission spectra of the donor and ɛ(λ) is the absorption spectra of the acceptor. If the λ-units in Eq. 12 are nm, the calculated R0 in Eq. 11 has Å-units (Berberan-Santos and Prieto, 1987).
RESULTS
The M13 coat protein is known to form large irreversible aggregates under specific conditions. These aggregates are not found in vivo, and therefore are regarded as an artifact (Hemminga et al., 1993). Due to their large percentage of β-sheet conformation, they can be detected by CD spectroscopy. Both the wild-type and labeled mutant protein conformation were checked by CD spectroscopy in all lipid systems used, and all spectra obtained were typical for α-helices (data not shown), indicating the absence of significant β-sheet protein conformation.
The fluorescence decay of BODIPY in the labeled mutant proteins incorporated in DOPC, DMoPC/DOPC, DEuPC/DOPC, DMoPC, and DEuPC bilayers was described by two components τ1 = 6.23 ns (a1 = 0.9) and τ2 = 3.27 ns, which leads to an average lifetime of 〈τ0〉 = 6.1 ns (Eq. 2), as measured in samples with [BD-M13 coat protein]eff <10−3 M (BODIPY-labeled protein effective membrane concentration). For the determination of protein effective concentration in the membranes, the lipid molar volumes were calculated from the reported lipid areas (72 Å2) and membrane thicknesses (Tristam-Nagle et al., 1998; Lewis and Engelman, 1983).
Considering a random protein distribution in the bilayers, and fitting Eq. 4 to the experimental average lifetimes, linear Stern-Volmer plots are obtained (Fig. 1) and kq values are recovered (Table 1).
FIGURE 1.

Transient state Stern-Volmer plots, describing BODIPY fluorescence self-quenching at different labeled protein concentrations. The lines are fits of Eq. 4 to the data. (A) Labeled protein incorporated in DOPC (▴); DMoPC/DOPC (60/40 mol/mol) (○); and DEuPC/DOPC (60/40 mol/mol) (•). (B) Labeled protein incorporated in DMoPC (▪); DEuPC (▵); and Stern-Volmer fit to DOPC data (see A) (- - -). The BODIPY-labeled mutant used in the DMoPC and DEuPC lipid systems was A35C, and for the other three lipid compositions the mutant used was T36C.
TABLE 1.
| Systems | kq/(109 M−1 s−1) | D/(10−7 cm2 s−1) | Rs (Å) |
|---|---|---|---|
| T36C in DOPC | 2.3 | 0.7 | 14 |
| A35C in DOPC | 1.7 | 0.4 | 14 |
| A35C in DMoPC | 2.6 | 0.8 | 23 |
| A35C in DEuPC | 5.0 | 2.6 | 27 |
| T36C in DMoPC/DOPC | 6.6 | 4.2 | 14 |
| T36C in DEuPC/DOPC | 20 | 22 | 14 |
Bimolecular diffusion rate constants (kq), diffusion coefficients (D), and apparent sphere-of-action radii (Rs) recovered from BODIPY fluorescence emission self-quenching from BODIPY-labeled T36C and A35C mutants. Values of Rs > 14 Å are evidence of molecular aggregation (see text for details).
The fluorescence steady-state quenching profiles for BD-M13 coat protein in DOPC, DMoPC/DOPC, DEuPC/DOPC, DMoPC, and DEuPC are presented in Fig. 2. This data can be fitted to Eq. 6 using the already-obtained kq-values to retrieve the fluorescence quenching contribution of the sphere-of-action effect (Table 1).
FIGURE 2.

Fluorescence steady-state data for BODIPY fluorescence self-quenching at different labeled protein concentrations. (A) Protein incorporated in DOPC (▴); DMoPC/DOPC (60/40 mol/mol) (○); and DEuPC/DOPC (60/40 mol/mol) (•). Eq. 6 is fitted to the data on the basis of dynamical quenching and a sphere-of-action quenching model (14.4 Å of radius) (—) for the protein in all lipid systems. (B) Protein incorporated in DOPC (▴); DMoPC (○); and DEuPC (□). Eq. 6 fit of data from DOPC bilayers using a sphere-of-action radii of 14 Å (–), from DEuPC bilayers with a sphere-of-action radii of 27 Å (- - -), and from DMoPC bilayers using a sphere-of-action of 23 Å (–). These higher values are evidence of aggregation. See text for details.
Many studies on protein and peptide aggregation make use of fluorophore spectral changes induced by ground or excited state dimer formation (Otoda et al., 1993; Liu et al., 1998; Melnyk et al., 2002; Shigematsu et al., 2002). Recently, under restricted geometry, BODIPY was shown to form ground-state dimers in two different conformations (D| and D‖), with distinct spectroscopic properties from the monomer. The D| BODIPY dimer (parallel transition dipoles) is characterized by a strong excitation peak at 477 nm and a null quantum yield. The D‖ dimer (planar transition dipoles) exhibits an absorption peak at 570 nm and a broad fluorescence emission band centered at 630 nm (Bergström et al., 2001). In our BODIPY-derivatized protein none of the spectral properties assigned to the D| and D‖ dimers are observed, and both the excitation and fluorescence emission spectra obtained for the samples with higher concentration of BODIPY-labeled protein were that expected for BODIPY monomers and identical in all lipid systems (Fig. 3).
FIGURE 3.

Corrected excitation spectra of IAEDANS-labeled protein (—), and of BD-M13 coat protein (- - -). Corrected emission spectra of IAEDANS-labeled protein (–), and of BD-M13 coat protein (- - -).
The donor-acceptor pair chosen for the energy transfer study shows a wide overlap of donor (AEDANS) fluorescence emission and acceptor (BODIPY) absorption (Fig. 3). Energy transfer studies were performed for the labeled protein incorporated in DOPC, DOPC/DOPG (80/20 mol/mol), DOPE/DOPG (70/30 mol/mol), DEuPC/DOPC (60/40 mol/mol), and DMoPC/DOPC (60/40 mol/mol). It was intended to study the influence of electrostatic interactions, hydrophobic mismatch, and the presence of nonlamellar lipids in the aggregational and compartmentalization properties of the M13 coat protein. Due to the nonlamellar character of phosphatidylethanolamines, it was necessary to include a fraction of lamellar lipids (PG) in the lipid mixture for bilayer stabilization.
The fluorescence emission spectra of the T36C mutant labeled with IAEDANS and reconstituted in these lipid systems were identical in the DOPC, DOPC/DOPG, DOPE/DOPG, DEuPC/DOPC, and DMoPC/DOPC lipid systems (results not shown), their wavelength of maximum fluorescence emission (477 nm) being characteristic of a very apolar environment (Hudson and Weber, 1973) and very similar to the one obtained by Spruijt and co-workers for the same mutant (478 nm; Spruijt et al., 2000). The wavelengths of maximum emission for the IAEDANS-labeled protein in DMoPC and DEuPC were slightly different, 478 nm and 475 nm, respectively.
Donor fluorescence intensities (ID and IDA in Eq. 9) obtained by steady-state measurements and by integrated donor decays were identical. The IAEDANS-labeled protein quantum yield determined by us was φ = 0.64. Using Eqs. 11 and 12, assuming κ2 = 2/3 (the isotropic dynamic limit) and n = 1.4 (Davenport et al., 1985), we obtain R0 = 48.8 Å for this FRET pair. The value κ2 = 2/3 was considered because for fluorophores in the center of a fluid bilayer, the rotational freedom should be sufficiently high to randomize orientations. This is supported by the reasonably low steady-state anisotropy values obtained for the IAEDANS and BODIPY probes labeled on the T36C M13 coat protein mutant (〈r〉AEDANS = 0.14, 〈r〉BODIPY = 0.23; for a detailed discussion, see Loura et al., 1996).
The results for BD-M13 coat protein in DOPC, DOPC/DOPG, DOPE/DOPG, DMoPC/DOPC, and DEuPC/DOPC bilayers are presented in Fig. 4.
FIGURE 4.

(A) Donor (AEDANS) fluorescence quenching by energy transfer acceptor (BODIPY). Experimental data for DOPC (▴); DOPC/DOPG (80/20 mol/mol) (•); and DOPE/DOPG bilayers (70/30 mol/mol) (□). Theoretical expectation (Eq. 10) for energy transfer in a random distribution of acceptors (—). Energy transfer simulation for a total co-localization of M13 coat protein in 20% (- - -), and 30% (— - —) of the surface area available. (B) Donor (AEDANS) fluorescence quenching by energy transfer acceptor (BODIPY). Theoretical expectation for energy transfer in a random distribution of acceptors (—). Energy transfer simulation for a segregation of M13 coat protein (mcp) to 60% of the surface area available (- - -). Experimental data for DEuPC/DOPC (60/40 mol/mol) (•); experimental data for DMoPC/DOPC (60/40 mol/mol) (○). IDA and ID were obtained by integration of donor decays.
DISCUSSION
For membrane proteins incorporated in lipid bilayers, the net tendency for protein aggregation should be weaker under good lipid-protein hydrophobic matching conditions (Mouritsen and Bloom, 1984). As the hydrophobic α-helix of the M13 coat protein is composed by 20 amino-acid residues, its length is ∼30 Å. The lipids used in this work, with the exception of DMoPC (21 Å) and DEuPC (35 Å), form bilayers with almost the same hydrophobic thickness (28 Å) (Lewis and Engelman, 1983). Therefore, the oligomerization properties of the M13 major coat protein in DMoPC and DEuPC should differ from those in the hydrophobically matching phospholipid DOPC.
The conditions of hydrophobic mismatch considered in this study do not seem to be able to induce any protein conformational change, as checked by CD spectroscopy in both lipid systems (no spectral change found). The absence of β-sheet conformation for the M13 coat protein implies no irreversible aggregation in the bilayer, and consequently any self-association observed should be due to reversible interactions between the α-helices.
The monitoring of IAEDANS maximum fluorescence emission (λmax) of the labeled mutant also rules out any change in conformation or exclusion from the bilayer of the M13 coat protein when incorporated in hydrophobically mismatching phospholipid, for the IAEDANS λmax in these samples (475–478 nm) are almost identical to that observed with DOPC and the other hydrophobic matching phospholipids, and are typical for the fluorophore located near the center of the bilayer (Spruijt et al., 2000).
Although BODIPY fluorescence decay is essentially monoexponential, the complex decay we obtained (dominated by a component of 6.3 ns) is also reported for derivatized proteins (Karolin et al., 1994).
The BODIPY fluorescence emission self-quenching studies were performed with two different mutants. For the lipid mixtures T36C was used, and the A35C mutant was employed in the studies with pure mismatching lipid. As a control, the experiments were performed for both mutants in pure DOPC bilayers and the results were identical (Table 1).
The bimolecular quenching constants calculated from the self-quenching results for BD-M13 coat protein incorporated in DOPC, DMoPC/DOPC (60/40 mol/mol) DEuPC/DOPC (60/40 mol/mol), DEuPC, and DMoPC allow the estimation of the labeled protein molecular diffusion coefficient through Eq. 5. Considering for BODIPY a collisional radius of 6 Å, we obtain for DBD-M13 coat protein in DOPC bilayers a value of 7.0 × 10−8 cm2 s−1, which is the same order of magnitude of the values of D for the M13 coat protein incorporated in fluid bilayers reported in the literature (Smith et al., 1979, 1980). However, for the lipid mixtures in which the predominant lipid does not hydrophobically match with the hydrophobic core of the M13 coat protein, the values obtained for D are unreasonably high. For the DMoPC/DOPC bilayers, a value of 4.2 × 10−7 cm2 s−1 is obtained, whereas in DEuPC/DOPC it is even higher (2.2 × 10−6 cm2 s−1). If DBD-M13 coat protein in pure vesicles of DOPC is considered to report a random distribution in the bilayer, the values in these mixtures are likely to be reporting protein segregation effects in the bilayer. We believe that this is caused by the hydrophobic mismatch constraints the protein finds when incorporated in bilayers that have too-long, or too-short phospholipids in their composition, probably leading to formation of localized areas with increased content of DOPC and protein. In this way, the effective apparent concentration in Eq. 4, should be higher than the one assumed on the basis of a random distribution, leading to an overestimation of kq, and so of D. However, the increase in local protein concentration arising from this effect would still be insufficient to explain the one order-of-magnitude increase of D from pure DOPC bilayers to the studied DEuPC/DOPC mixture (see below for further discussion).
This rationalization is supported by D values obtained from BODIPY labeled protein in the pure mismatching lipid DMoPC and DEuPC (Table 1). These values are smaller than the ones obtained from the mixtures, and the diffusion coefficient in pure DMoPC is almost identical to the value in pure DOPC. For pure vesicles of DEuPC, DBD-M13 coat protein is larger than in DOPC, but it is still much smaller than the value obtained from the DEuPC/DOPC mixture. The results from dynamical self-quenching indicate therefore that although in pure vesicles of DEuPC there are already more collisions between BODIPY groups than what could be expected from a random distribution of labeled protein in the bilayer (probably due to aggregation), when DOPC is added the probability of collision greatly increases, and this can in part be explained in terms of protein segregation to DOPC-enriched microdomains. In DMoPC/DOPC the effect is similar, but the bimolecular quenching constant is smaller than in DEuPC/DOPC.
In Fig. 2, the obtained steady-state quenching profiles are presented together with the theoretical expectation for a sphere-of-action quenching model (Eq. 6). For the BODIPY-labeled protein in the DOPC-containing lipid systems (DOPC, DMoPC/DOPC, and DEuPC/DOPC) the results are well described using a sphere-of-action radius of 14 Å. For the pure mismatching lipids it is necessary to use larger radii to describe the results using Eq. 6.
In Fig. 5, a simulation was included for a small degree of protein aggregation in DOPC, DMoPC, and DEuPC bilayers. In these simulations it was considered that, due to the small degree of self-association considered, there was no change in M13 coat protein distribution and dynamics, and that in an oligomer, the fluorescence intensity of a BODIPY group is reduced to zero by the presence of another BODIPY group in the same aggregate. For DOPC bilayers, the prediction using a low fraction of aggregation (25% for dimerization and 10% for trimerization) clearly overestimates the extent of aggregation at the high labeled protein concentration, the range where this methodology is more sensitive. In agreement, from Fig. 2, it is clear that the data for the three DOPC-containing lipid systems are rationalized on the basis of dynamic quenching and a sphere of action, without need for assumption of aggregation. The recovered radius (Rs = 14 Å) is close to the sum of the Van der Waals radii. These results indicate that BD-M13 coat protein in the studied DOPC-containing bilayers does not oligomerize. This conclusion is further supported by the absence of BODIPY dimers in our samples, which would be revealed in the absorption/emission spectra.
FIGURE 5.
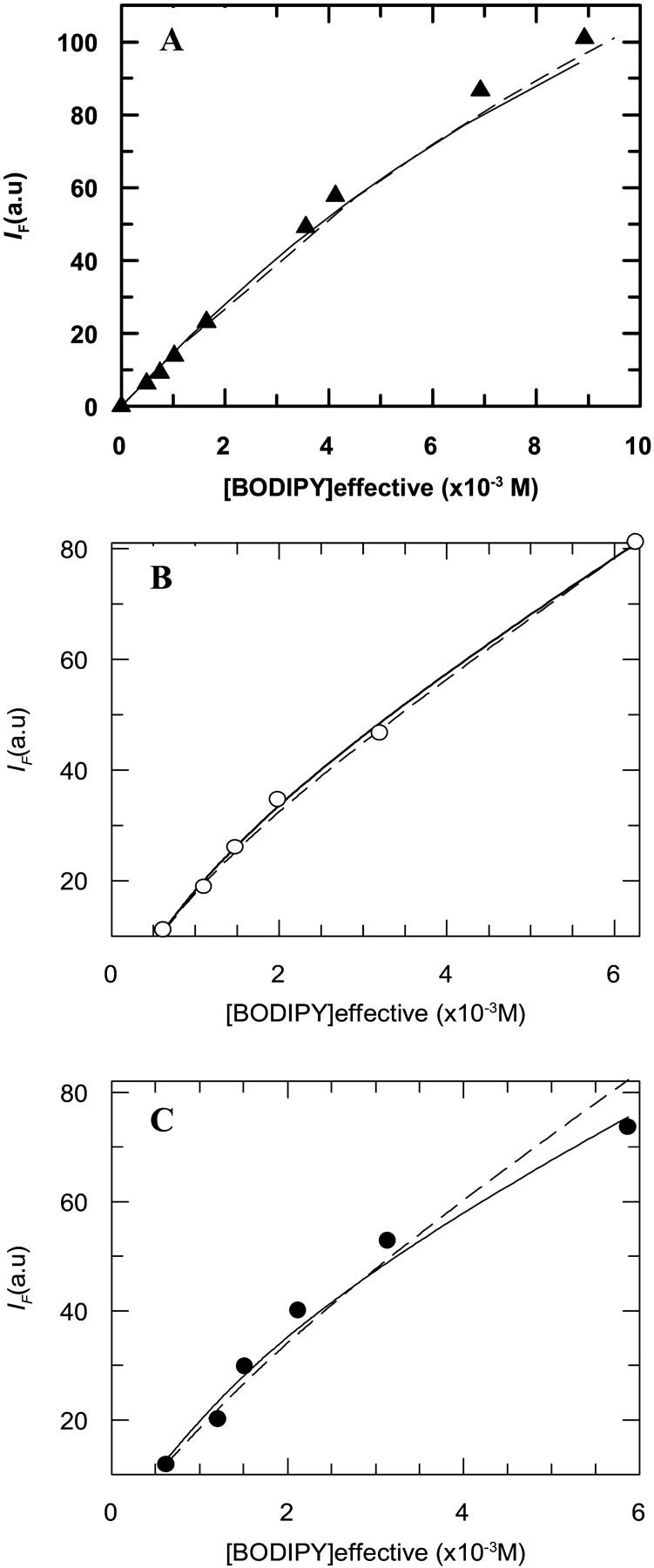
Fluorescence steady-state data for BODIPY fluorescence self-quenching at different labeled protein concentrations. (A) Protein incorporated in DOPC (▴) and simulations considering protein oligomerization including static quenching: due to trimerization of the protein with a Ka = 1000 (≈ 10% total mcp oligomerization) (—); due to dimerization of the protein with a Ka = 10 (≈ 25% total mcp oligomerization) (- - -). (B) Protein incorporated in DMoPC (○) and simulations for dimerization of protein with a Ka = 20 (≈ 13% total mcp oligomerization for the most concentrated data point) (- - -) and trimerization of the protein with a Ka = 1300 (≈ 13% total mcp oligomerization for the most concentrated data point) (—). (C) Protein incorporated in DEuPC (•) and simulations for dimerization of protein with a Ka = 30 (≈ 17% total mcp oligomerization for the most concentrated data point) (- - -) and trimerization of the protein with a Ka = 7500 (≈ 25% total mcp oligomerization for the most concentrated data point) (—). See text for details on the simulations.
It could be possible that in a labeled protein oligomer, there is no contact between the BODIPY groups, and therefore the formation of oligomers would not necessarily induce fluorescence self-quenching. Some mutational studies actually include the Thr36 residue in a lipid-interactive face of the transmembrane segment of the M13 coat protein. The M13 coat protein amino acids in this lipid-interactive face would only have contact with the phospholipid acyl chains inside the bilayer, and would be excluded from the transmembrane domain face involved in protein-protein interactions, where the amino acids responsible for the affinity of identical transmembrane segments are located (Deber et al., 1993; Webster and Haigh, 1998). That positioning of the mutated Cys36 residue would make the contact between BODIPY groups from labeled proteins participating in an oligomer unlikely, and therefore exclude the formation of “dark” (nonfluorescent) dimers. Localization of the BODIPY group on the lipid-interactive face of the transmembrane section of the protein can be seen as an advantage, inasmuch as the mutation of an amino-acid residue to cysteine after the labeling with fluorophore, at the protein-protein interactive face, could cause dramatic changes in the oligomerization behavior of the M13 coat protein. The T36C mutant labeled with BODIPY could be seen, for that reason, as a more ideal model for wild-type protein aggregation studies than the M13 coat protein mutant A35C, in which the mutated amino-acid residue is located in the protein-interactive face of the hydrophobic core domain. However, as can be concluded from the results obtained for both mutants labeled with BODIPY in DOPC (Table 1), the positioning of the BODIPY group is not critical. Probably the fluorophore has sufficient mobility to extend from the surface of the transmembrane helix and probe a large extent of the area surrounding the protein.
The BODIPY self-quenching results obtained in the present study clearly indicate that there is no protein aggregation in presence of hydrophobic matching phospholipids for all hydrophobic headgroup compositions used, and points to a large monomer stability under those conditions, which had already been suggested in other studies (Stopar et al., 1997; Spruijt et al., 1989; Sanders et al., 1991).
Still, the results from self-quenching on pure bilayers of hydrophobic mismatching lipids (DEuPC and DMoPC) point to some aggregation, as the sphere-of-action radii recovered from the data fit to Eq. 6 were very unrealistic (27 and 23 Å for DEuPC and DMoPC, respectively). Simulations for BODIPY emission self-quenching due to aggregation are compared with the experimental data in these lipid systems in Fig. 5. DMoPC data could be reasonably described using a Ka = 20 for dimerization and a Ka = 1300 for trimerization (13% of aggregated protein at the protein concentration of the most concentrated data point in both simulations).
For the data obtained in DEuPC, the degree of aggregation is higher than in DMoPC (recovered sphere-of-action radius on fit to Eq. 6 was larger), and the fitting of the aggregation models to the data points proved more difficult. Probably the M13 major coat protein in DEuPC bilayers forms somewhat larger aggregates than in DMoPC and that could be an explanation for the increased BODIPY dynamical self-quenching observed in the longer lipid. This result is in agreement with the observations of Meijer et al. (2001), who, from electron spin resonance studies, reported that the protein appeared to exist in several orientations/conformations or in an aggregated form while incorporated in DEuPC bilayers. The larger extent of coat protein aggregation observed in the longer lipid bilayers can be explained by the fact that negative hydrophobic mismatch is considered to be energetically less favorable than positive mismatch (Killian, 1998; Mall et al., 2001).
As mentioned above, for the DEuPC/DOPC and DMoPC/DOPC lipid mixtures used in the present study, no change in conformation or orientation was found (CD spectroscopy/AEDANS fluorescence emission spectra), and even for the protein in DEuPC/DOPC (60/40 mol/mol) no aggregation was detected (BODIPY self-quenching). These results point to stabilization of the protein by the hydrophobic matching phospholipid (DOPC), which was probably achieved by protein segregation to domains enriched in that phospholipid, partly explaining the high M13 coat protein apparent molecular diffusion coefficients obtained for the protein incorporated in DEuPC/DOPC and DMoPC/DOPC bilayers. Although an increase in BODIPY emission dynamical self-quenching was already visible for pure DEuPC bilayers (probably due to the large dimensions of the aggregates, which will have a similar effect as the segregation of protein to microdomains on inducing co-localization of protein in the bilayer), the bimolecular diffusion rate constant value in DEuPC/DOPC bilayers is much higher, and still, this can only be explained by segregation to DOPC-enriched microdomains.
Being that M13 coat protein preferably locates in DOPC-enriched domains, an interesting question is whether these domains are induced by the protein or exist even in the absence of protein (and the latter merely distributes differently among the pre-existing DOPC-rich and DOPC-poor regions). In this regard we obtained preliminary results of 1,6-diphenylhexatriene fluorescence anisotropy upon varying compositions for both DMoPC/DOPC and DEuPC/DOPC mixtures in the absence of protein (data not shown), pointing to the existence of “phase coexistence regions” containing the compositions studied in this work. This experiment is not informative regarding the extent of phase separation, but it is compatible with the existence of small lipid clusters enriched in one of the components, because fluorescence anisotropy only senses the immediate vicinity of the fluorophore. Regarding the DEuPC/DOPC mixtures, these measurements imply that for 60:40 (mol/mol) DEuPC/DOPC, DEuPC-rich domains should coexist with DOPC-rich domains, and the latter should account for approximately one-third of the mixture (not shown). From this result, we could expect at most a threefold increase in protein local concentration for this mixture, and an identical factor for the increase of the recovered kq value relative to that in DOPC. As seen in Table 1, this factor is significantly higher. This suggests that whereas protein segregation into DOPC-rich domains should be occurring, it is not the sole cause for the increase in the apparent diffusion coefficient in 60:40 (mol/mol) DEuPC/DOPC. In this regard the other contributing factors are not clear. It should be pointed out, however, that the estimate presented above for the fraction of DOPC-rich domains was made from experiments performed in the absence of protein, whereas the quenching results refer to L/P = 50.
The centered position of BODIPY in the bilayer allows for a simplification of the energy transfer analysis for a two-dimensional situation, as described by Eq. 10, i.e., there is no need to consider bilayer FRET geometry (Loura et al., 2001). Simulations of energy transfer for random distribution of acceptors using Eq. 10 can therefore be compared with our experimental results (Fig. 4). The energy transfer efficiencies obtained for BD-M13 coat protein in the DMoPC/DOPC and DEuPC/DOPC bilayers support the other results discussed above for these mixtures, as they can only be explained by protein segregation in the bilayer or severe aggregation (Fig. 4 B). However, as discussed above, the data obtained by fluorescence emission self-quenching indicate that segregation into DOPC-enriched domains (rather than aggregation) is the major phenomenon in these lipid mixtures.
Although the results can be reasonably explained on the basis of protein segregation to 60% of the total bilayer area (Fig. 5 B), this rationalization should be considered an oversimplification, and is presented as an illustration. Indeed, the measured efficiencies are only reporting the average BD-M13 coat protein surface density that each IAEDANS-labeled protein is sensing. Probably there will be M13 coat protein interacting with the hydrophobically mismatching phospholipids, but the majority of the proteins will be preferentially surrounded by DOPC, and microdomains enriched in DOPC and M13 coat protein should be formed.
The lipid mixtures used in this work for hydrophobic matching studies (DMoPC/DOPC and DEuPC/DOPC) are thought to be considerably closer to ideality than the ones used in the studies of Dumas and co-workers (gel/fluid coexistence; Dumas et al., 1997) and Lehtonen and Kinunnen (natural and pyrene-derivatized lipids; Lehtonen and Kununnen, 1997). However, protein segregation was observed in our study for both DMoPC/DOPC and DEuPC/DOPC, whereas in DOPC random distribution of protein in the bilayer was confirmed. Also interestingly, the degree of segregation appears to be similar for both mixtures (Fig. 5 B), which was not expected due to the observed larger aggregation degree of coat protein in DEuPC, and therefore to an apparent larger packing difficulty with the longer lipid.
The formation of local structure within the thermodynamic fluid phase for a mixture of two PCs with a 4-carbon difference in acyl-chain lengths (DMPC/DSPC) was theoretically predicted by Mouritsen and Jørgensen (1994). The microdomains size in the Monte Carlo configurations obtained by these authors appears to be very small (10–20 molecules at most). However, significant alterations in FRET efficiency as measured by Lehtonen et al. (1996) in mixtures of unsaturated PCs, and indeed our study of FRET between IAEDANS-labeled protein and BODIPY-labeled protein, require that the domain size should be of the order of magnitude of R0, that is ≅5 nm. In our system, this large domain size might be a consequence of protein-induced phase separation.
In this work we also studied whether similar heterogeneities could be induced by the presence of positively charged M13 coat protein in bilayers composed of mixtures of anionic and neutral phospholipids. Due to the basic character of M13 coat protein C-terminal, it is reasonable to consider the possibility of anionic phospholipid-enriched domain induction by M13 coat protein incorporation in the bilayer. The formation of these domains could actually help explain some of the mechanisms involved in the creation of the phage assembly site.
Assuming that the hypothetical domains were composed by all the protein and DOPG content in the sample, and that the protein would be randomly distributed inside them, we can, using Eq. 10, obtain theoretical curves describing the energy transfer within these domains. These plots are compared with the experimental data points for M13 coat protein incorporated in DOPC/DOPG (80/20 mol/mol) and DOPE/DOPG (70:30 mol/mol) in Fig. 5 A.
It is concluded that the segregation of M13 coat protein to a PG-rich phase in the mixed systems, induced by electrostatic interactions between the positively charged protein and the negatively charged phospholipid, is ruled out on the basis of the obtained data, for this process would lead locally to greater surface densities of acceptor, and therefore, a very significant increase of energy transfer efficiencies should be expected in these conditions.
In addition to being the predominant phospholipids of the E. coli inner membrane (Woolford et al., 1974; Burnell et al., 1980), nonlamellar phospholipids (such as DOPE) are known to interact distinctly from lamellar lipids with proteins, and in some cases to influence their conformation and activity (Hunter et al., 1999; Ahn and Kim, 1998). Our results suggest that these phospholipids have apparently no effect on the lateral distribution (Fig. 4 A) of the protein, which is kept random at the total protein concentration used.
Higher protein concentrations than L/P 50 were not tested due to the risk of inducing nonlamellar phases, but it is likely that at smaller L/P ratios than those used in this work attractive interactions between monomers of M13 coat protein could take place, especially as we have two protein orientations in these reconstituted systems (parallel/antiparallel), and the repulsive electrostatic forces between the heavily basic C-terminal of the protein would be eliminated. In vivo, as there is only one orientation, the achievement of high protein concentrations in the monomeric state at the assembly site would be more favorable (Hemminga et al., 1993). The presence of anionic phospholipids, as shown, is not essential for monomer stabilization, and the increase in phosphatidylglycerol and cardiolipin production during virus infection (Chamberlain et al., 1978) should only play a stabilizing role at the very high M13 coat protein concentrations that are expected to exist at the phage assembly site.
CONCLUSIONS
From this study, it is concluded that the M13 coat protein monomeric state is highly stable when incorporated in bilayers containing hydrophobic matching phospholipids. The lack of anionic phospholipids has no effect on the protein oligomerization properties at the protein concentrations used in this study. When the protein is incorporated in pure vesicles of mismatching lipid there is evidence for protein aggregation but for mixtures of lipids containing both hydrophobically matching (DOPC) and mismatching (DEuPC or DMoPC) phospholipids, the protein probably segregates to domains enriched in DOPC, which can explain the stability of the monomeric species of the protein in these lipid systems. This segregation effect is only observed when hydrophobic mismatching phospholipids are present, suggesting that the hydrophobic matching conditions on the protein-lipid interface are more important than electrostatic interactions between the M13 coat protein and the phospholipids, for the protein lateral distribution on the bilayer.
Acknowledgments
The authors thank Dr. A. Fedorov for assistance in the time-resolved measurements.
F.F. acknowledges financial support from Fundação para a Ciência e Tecnologia (FCT), project POCTI/36458/QUI/2000 and COST Action D:22; L.M.S.L. and M.P. acknowledge financial support from FCT, projects POCTI/36458/QUI/2000 and POCTI/36389/FCB/2000.
References
- Ahn, T., and H. Kim. 1998. Effects of non-lamellar-prone lipids on the ATPase activity of SecA bound to model membranes. J. Biol. Chem. 273:21692–21698. [DOI] [PubMed] [Google Scholar]
- Almgren, M. 1991. Kinetics of excited states processes in micellar media. In Kinetics and Catalysis in Microheterogeneous Systems. M. Gratzel, and K. Kalyanasundaram, editors. Marcel Dekker, New York. 63–113.
- Berberan-Santos, M. N., and M. J. E. Prieto. 1987. Energy transfer in spherical geometry. Application to micelles. J. Chem. Soc. Faraday Trans. 283:1391–1409. [Google Scholar]
- Bergström, F., I. Mikhalyov, P. Hägglöf, R. Wortmann, T. Ny, and L. B.-Å. Johansson. 2001. Dimers of dipyrrometheneboron difluoride (BODIPY) with light spectroscopic applications in chemistry and biology. J. Am. Chem. Soc. 124:196–204. [DOI] [PubMed] [Google Scholar]
- Burnell, E., L. Van Alphen, A. Verkleij, and B. De Kruijff. 1980. 31P Nuclear magnetic resonance and freeze-fracture electron microscopy studies on Escherichia coli. I. Cytoplasmic membrane and total phospholipids. Biochim. Biophys. Acta. 597:492–501. [DOI] [PubMed] [Google Scholar]
- Chamberlain, B. K., Y. Nozaki, C. Tanford, and R. E. Webster. 1978. Association of the major coat protein of FD bacteriophage with phospholipid vesicles. Biochim. Biophys. Acta. 510:18–37. [DOI] [PubMed] [Google Scholar]
- Davenport, L., R. E. Dale, R. H. Bisby, and R. B. Cundall. 1985. Transverse location of the fluorescent probe 1,6-diphenyl-1,3,5-hexatriene in model lipid bilayer membrane systems by resonance energy transfer. Biochemistry. 24:4097–4108. [DOI] [PubMed] [Google Scholar]
- Deber, C. M., A. R. Khan, Z. Li, C. Joensson, and M. Glibowicka. 1993. Val → Ala mutations selectively alter helix-helix packing in the transmembrane segment of phage M13 coat protein. Proc. Natl. Acad. Sci. USA. 90:11648–11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas, F., M. M. Sperotto, M.-C. Lebrun, J.-F. Tocanne, and O. G. Mouritsen. 1997. Molecular sorting of lipids by bacteriorhodopsin in dilauroylphosphatidylcholine/distearoylphosphatidylcholine lipid bilayers. Biophys. J. 73:1940–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas, F., M. C. Lebrun, and J.-F. Tocanne. 1999. Is the protein/lipid hydrophobic matching principle relevant to membrane organization and functions? FEBS Lett. 458:271–277. [DOI] [PubMed] [Google Scholar]
- Eaton, D. F. 1988. Reference materials for fluorescence measurement. Pure Appl. Chem. 60:1107–1114. [Google Scholar]
- Fahsel, S., E. M. Pospiech, M. Zein, T. L. Hazlet, E. Gratton, and R. Winter. 2002. Modulation of concentration fluctuations in phase-separated lipid membranes by polypeptide insertion. Biophys. J. 83:334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil, T., J. H. Ipsen, O. G. Mouritsen, M. C. Sabra, M. M. Sperotto, and M. J. Zuckermann. 1998. Theoretical analysis of protein organization in lipid bilayers. Biochim. Biophys. Acta. 1376:245–266. [DOI] [PubMed] [Google Scholar]
- Hemminga, M. A., J. C. Sanders, C. J. A. M. Wolfs, and R. B. Spruijt. 1993. Lipid-protein interactions involved in bacteriophage M13 infection. In Protein-Lipid Interactions, New Comprehensive Biochemistry, Vol. 25. A. Watts, editor. Elsevier, Amsterdam. 191–212.
- Horvàth, L. I., T. Heimburg, P. Kovachev, J. B. Findlay, K. Hideg, and D. Marsh. 1995a. Integration of a K+ channel-associated peptide in a lipid bilayer: conformation, lipid-protein interactions, and rotational diffusion. Biochemistry. 34:3893–3898. [DOI] [PubMed] [Google Scholar]
- Horvàth, L. I., P. F. Knowles, P. Kovachev, J. B. Findlay, and D. Marsh. 1995b. A single-residue deletion alters the lipid selectivity of a K+ channel-associated peptide in the β-conformation; spin-label electron spin resonance studies. Biophys. J. 73:2588–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, E. N., and G. Weber. 1973. Synthesis and characterization of two fluorescent sulfhydryl reagents. Biochemistry. 12:4154–4161. [DOI] [PubMed] [Google Scholar]
- Hunter, G. W., S. Negash, and T. C. Squier. 1999. Phosphatidylethanolamine modulates Ca-ATPase function and dynamics. Biochemistry. 38:1356–1364. [DOI] [PubMed] [Google Scholar]
- Karlsson, O. P., M. Rytomaa, A. Dahqvist, P. K. Kinnunnen, and A. Wieslander. 1996. Correlation between bilayer dynamics and activity of the diglucosyldiacylglycerol synthase from Acholeplasma laidlawii membranes. Biochemistry. 35:10094–10102. [DOI] [PubMed] [Google Scholar]
- Karolin, J., J. B.-Å. Johansson, L. Strandberg, and T. Ny. 1994. Fluorescence and absorption spectroscopic properties of dipyrrometheneboron difluoride (BODIPY) derivatives in lipids, lipid membranes, and proteins. J. Am. Chem. Soc. 116:7801–7806. [Google Scholar]
- Killian, J. A. 1998. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1376:401–416. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy, 2nd Ed. Kluwer Academic/Plenum Press, New York. Chap. 8.
- Lewis, B. A., and D. M. Engelman. 1983. Bacteriorhodopsin remains dispersed in fluid phospholipid bilayers over a wide range of bilayer thickness. J. Mol. Biol. 166:203–210. [DOI] [PubMed] [Google Scholar]
- Lehtonen, J. Y. A., J. M. Holopainen, and P. K. J. Kinnunen. 1996. Evidence for the formation of microdomains in liquid crystalline large unilamellar vesicles caused by hydrophobic mismatch of the constituent phospholipids. Biophys. J. 70:1753–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen, J. Y. A., and P. K. J. Kinnunen. 1997. Evidence for phospholipid microdomain formation in liquid crystalline liposomes reconstituted with Escherichia coli lactose permease. Biophys. J. 72:1247–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, W., Y. Chen, H. Watrob, S. G. Bartlett, L. Jen-Jacobson, and M. D. Barkley. 1998. N-termini of EcoRI restriction endonuclease dimers are in close proximity on the protein surface. Biochemistry. 37:15457–15465. [DOI] [PubMed] [Google Scholar]
- Loura, L. M. S., A. Fedorov, and M. Prieto. 1996. Resonance energy transfer in a model system of membranes: application to gel and liquid crystalline phases. Biophys. J. 71:1823–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loura, L. M. S., A. Fedorov, and M. Prieto. 2000. Membrane probe distribution heterogeneity: a resonance energy transfer study. J. Phys. Chem. B. 104:6920–6931. [Google Scholar]
- Loura, L. M. S., R. F. M. de Almeida, and M. Prieto. 2001. Detection and characterization of membrane microheterogeneity by resonance energy transfer. J. Fluoresc. 11:197–209. [Google Scholar]
- Mall, S., R. Broadbridge, R. P. Sharma, J. M. East, and A. G. Lee. 2001. Self-association of model transmembrane helix is modulated by lipid structure. Biochemistry. 40:12379–12386. [DOI] [PubMed] [Google Scholar]
- Marquardt, D. W. 1963. An algorithm for least-squares estimation of non-linear parameters. J. Soc. Ind. Appl. Math. 11:431–441. [Google Scholar]
- Meijer, A. B., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 2001. Membrane-anchoring interactions of M13 major coat protein. Biochemistry. 40:8815–8820. [DOI] [PubMed] [Google Scholar]
- Melnyk, R., A. Partridge, and C. Deber. 2002. Transmembrane domain mediated self-assembly of major coat protein subunits from FF bacteriophage. J. Mol. Biol. 315:63–72. [DOI] [PubMed] [Google Scholar]
- Mobashery, N., C. Nielsen, and O. S. Andersen. 1997. The conformational preference of gramicidin channels is a function of lipid bilayer thickness. FEBS Lett. 412:15–20. [DOI] [PubMed] [Google Scholar]
- Mouritsen, O. G., and M. Bloom. 1984. Mattress model of lipid-protein interactions in membranes. Biophys. J. 46:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouritsen, O. G., and K. Jørgensen. 1994. Dynamical order and disorder in lipid bilayers. Chem. Phys. Lipids. 73:3–25. [DOI] [PubMed] [Google Scholar]
- Nezil, F. A., and M. Bloom. 1992. Combined influence of cholesterol and synthetic amphiphilic peptides upon bilayer thickness in model membranes. Biophys. J. 61:1176–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otoda, K., S. Kimura, and Y. Imanishi. 1993. Orientation and aggregation of hydrophobic helical peptides in phospholipid bilayer membrane. Biochim. Biophys. Acta. 1150:1–8. [DOI] [PubMed] [Google Scholar]
- Owen, C. S. 1975. Two-dimensional diffusion theory: cylindrical diffusion model applied to fluorescence quenching. J. Chem. Phys. 62:3204–3207. [Google Scholar]
- Razi-Naqvi, K. 1974. Diffusion-controlled reactions in two-dimensional fluids: discussion of measurements of lateral diffusion of lipids in biological membranes. Chem. Phys. Lett. 28:280–284. [Google Scholar]
- Ren, J., S. Lew, J. Wang, and E. London. 1999. Control of the transmembrane orientation and interhelical interactions within membranes by hydrophobic helix length. Biochemistry. 38:5905–5912. [DOI] [PubMed] [Google Scholar]
- Russel, M. 1991. Filamentous phage assembly. Mol. Microbiol. 5:1607–1613. [DOI] [PubMed] [Google Scholar]
- Sanders, J. C., N. A. J. van Nuland, O. Edholm, and M. Hemminga. 1991. Conformational and aggregation of M13 coat protein studied by molecular dynamics. Biophys. Chem. 41:193–202. [DOI] [PubMed] [Google Scholar]
- Shigematsu, D., M. Matsutani, T. Furuya, T. Kiyota, S. Lee, G. Sugihara, and S. Yamashita. 2002. Roles of peptide-peptide charge interaction and lipid phase separation in helix-helix association in lipid bilayer. Biochim. Biophys. Acta. 1564:271–280. [DOI] [PubMed] [Google Scholar]
- Smith, L. M., B. A. Smith, and H. M. McConnell. 1979. Lateral diffusion of M-13 coat protein in model membranes. Biochemistry. 18:2256–2259. [DOI] [PubMed] [Google Scholar]
- Smith, L. M., J. L. R. Rubenstein, J. W. Parce, and H. M. McConnel. 1980. Lateral diffusion of M-13 coat protein in mixtures of phosphatidylcholine and cholesterol. Biochemistry. 19:5907–5911. [DOI] [PubMed] [Google Scholar]
- Sperotto, M. M., and O. G. Mouritsen. 1993. Lipid enrichment and selectivity of integral membrane proteins in two-component lipid bilayers. Eur. Biophys. J. 22:323–328. [DOI] [PubMed] [Google Scholar]
- Spruijt, R. B., C. J. A. M. Wolfs, and M. A. Hemminga. 1989. Aggregation related conformational change of the membrane-associated coat protein of bacteriophage M13. Biochemistry. 28:9158–9165. [DOI] [PubMed] [Google Scholar]
- Spruijt, R. B., C. J. A. M. Wolfs, J. W. G. Verver, and M. A. Hemminga. 1996. Accessibility and environment probing using cysteine residues introduced along the putative transmembrane domain of the major coat protein of bacteriophage M13. Biochemistry. 35:10383–10391. [DOI] [PubMed] [Google Scholar]
- Spruijt, R. B., A. B. Meijer, C. J. A. M. Wolfs, and M. A. Hemminga. 2000. Localization and rearrangement modulation of the N-terminal arm of the membrane-bound major coat protein of the bacteriophage M13. Biochim. Biophys. Acta. 1509:311–323. [DOI] [PubMed] [Google Scholar]
- Stopar, D., R. B. Spruijt, C. J. A. M. Wolfs, and M. A. Hemminga. 1997. In situ aggregational state of M13 bacteriophage major coat protein in sodium cholate and lipid bilayers. Biochemistry. 36:12268–12275. [DOI] [PubMed] [Google Scholar]
- Tristam-Nagle, S., H. I. Petrache, and J. F. Nagle. 1998. Structure and interactions of fully hydrated dioleoylphosphatidylcholine bilayers. Biophys. J. 75:917–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umberger, J. Q., and V. K. Lamer. 1945. The kinetics of diffusion-controlled molecular and ionic reactions in solution as determined by measurements of the quenching of fluorescence. J. Am. Chem. Soc. 67:1099–1109. [Google Scholar]
- Webster, R. E., and N. G. Haigh. 1998. The major coat protein of filamentous bacteriophage F1 specifically pairs in the bacterial cytoplasmic membrane. J. Mol. Biol. 279:19–29. [DOI] [PubMed] [Google Scholar]
- Wolber, P. K., and B. S. Hudson. 1979. An analytical solution to the Förster energy transfer problem in two dimensions. Biophys. J. 28:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford, J. L., Jr., J. S. Cashman, and R. E. Webster. 1974. F1 Coat protein synthesis and altered phospholipid metabolism in F1-infected Escherichia coli. Virology. 58:544–560. [DOI] [PubMed] [Google Scholar]