

Abstract
The current study was undertaken to investigate the relative contribution of calcium and myosin binding to thin filament activation. Using the in vitro motility assay, myosin strong binding to the thin filament was controlled by three mechanisms: 1), varying the myosin concentration of the motility surface, and adding either 2), inorganic phosphate (Pi) or 3), adenosine diphosphate (ADP) to the motility solutions. At saturating myosin conditions, Pi had no effect on thin filament motility. However, at subsaturating myosin concentrations, velocity was reduced at maximal and submaximal calcium in the presence of Pi. Adding ADP to the motility buffers reduced thin filament sliding velocity but increased the pCa50 of the thin filament. Thus by limiting or increasing myosin strong binding (with the addition of Pi and ADP, respectively), the calcium concentration at which half maximal activation of the thin filament is achieved can be modulated. In experiments without ADP or Pi, the myosin concentration on the motility surface required to reach maximal velocity inversely correlated with the level of calcium activation. Through this approach, we demonstrate that myosin strong binding is essential for thin filament activation at both maximal and submaximal calcium levels, with the relative contribution of myosin strong binding being greatest at submaximal calcium. Furthermore, under conditions in which myosin strong binding is not rate limiting (i.e., saturating myosin conditions), our data suggest that a modulation of myosin cross-bridge kinetics is likely responsible for the graded response to calcium observed in the in vitro motility assay.
INTRODUCTION
Muscle shortening is the result of myosin cross-bridges cyclically interacting with the thin filament. This process is controlled through thin filament mediated regulation of myosin binding, which is primarily triggered by calcium. However, it is increasingly understood that myosin strong binding is essential for full thin filament activation. A currently accepted model of thin filament regulation (McKillop and Geeves, 1993) dictates that in the absence of calcium, and the resultant enhanced affinity of troponin I (TnI) to actin, the position of tropomyosin on actin essentially blocks the myosin cross-bridge from binding to the thin filament. At increased calcium concentrations, TnI and troponin C demonstrate enhanced affinity to one another (Gordon et al., 2000). TnI is released from actin allowing tropomyosin to shift on the thin filament, partially exposing the myosin binding site on actin. Full thin filament activation and complete exposure of myosin binding sites on the thin filament occurs through myosin strong binding further displacing tropomyosin on actin.
The relative role of calcium and myosin strong binding in thin filament activation is not completely understood. Here we have used the in vitro motility assay to investigate the effect of myosin strong binding on thin filament activation as a function of calcium. Myosin binding and cross-bridge kinetics were controlled through the use of adenosine triphosphate (ATP) hydrolysis products (inorganic phosphate (Pi) and adenosine diphosphate (ADP) and by varying the number of myosin cross-bridges interacting with the thin filament. Our hypothesis is that the contribution of myosin strong binding to thin filament activation is greatest at subsaturating calcium levels.
METHODS
Actin and myosin were isolated from chicken pectoralis muscle by standard techniques (Margossian and Lowey, 1982; Pardee and Spudich, 1982). Troponin and tropomyosin were isolated from beef cardiac muscle as previously described (Potter, 1982; VanBuren et al., 1999). Thin filaments were reconstituted (i.e., actin, troponin, and tropomyosin) and labeled with equimolar rhodamine-phalloidin before use (VanBuren et al., 2002).
The in vitro motility assay has been previously described in detail (VanBuren et al., 1999, 2002). In brief, monomeric myosin (12.5–150 μg/ml) was adhered to a nitrocellulose coated coverslip in loading buffer (300 mM KCl, 25 mM imidazole (pH 7.4), 10 mM dithiothreitol, 5 mM MgCl2, 2 mM EGTA). Nonspecific protein binding to the motility surface was blocked with a bovine serum albumin (0.5 mg/ml) wash in low salt buffer (25 mM KCl, 25 mM imidazole (pH 7.4), 10 mM dithiothreitol, 5 mM MgCl2, 2 mM EGTA). To eliminate noncycling “rigor” cross-bridges on the motility surface, actin without rhodamine-phalloidin labeling (1 μM in low salt buffer) was placed on the myosin-coated surface followed by an ATP wash (1 mM in low salt buffer) to release actin from actively cycling myosin cross-bridges. This was followed by labeled actin or regulated thin filaments (∼10 nM in low salt buffer), and finally motility buffer.
The final pCa motility buffer was varied as a function of free calcium and a function of free Pi or ADP, as follows, using the public domain software of Brooks and Storey (1992). The final motility buffer contained 2 mM ATP, 0.6% methylcellulose, 25 mM imidazole, 2 mM free MgCl2, 5 mM EGTA, 10 mM dithiothreitol, an oxygen scavenger system (glucose oxidase 0.1 mg/ml, catalase 0.0018 mg/ml, and glucose 2.3 mg/ml), and KCl. Potassium chloride in the motility buffer was adjusted to fix the final ionic strength of the buffer. In experiments containing no excess Pi or ADP, the ionic strength of the motility buffers was 51 mM. In the solutions containing Pi, the ionic strength of the solution was fixed at 120 mM. Free calcium (pCa 10–4) and Pi (0–30 mM) were varied in the motility buffers. In the buffers containing ADP, the ionic strength was fixed at 100 mM, with free calcium (pCa 10–4) and ADP (0–5 mM) both being varied. The ADP experiments were performed with 1 mM ATP. Thus experiments were conducted in which the motility buffers (pCa 10–4) contained either 1), no Pi or ADP, 2), Pi (0, 8, 16, and 30 mM), or 3), ADP (0, 0.5, 2, and 5 mM). In addition, the myosin concentration in the loading buffer was also varied (12.5–150 μg/ml). Previous experiments have demonstrated a linear correlation of the myosin surface density and the concentration of myosin in the loading buffer up to saturating surface conditions (VanBuren et al., 1999, 2002). All experiments were conducted at 30°C.
The velocity (VanBuren et al., 2002) and length (VanBuren et al., 1999) of the thin filaments were determined as previously described. Typically >250 thin filament velocities were averaged to determine a single data point. Mean values for each pCa point were used to fit the pCa:velocity data to the Hill equation. This fit provides the parameters: Vmax (maximal velocity), pCa50, and the Hill coefficient with associated standard errors (SE). Statistical significance was determined from the parameters of the fit with analysis of variance (ANOVA). Specifically, two-way ANOVA was applied to the parameters of the fit for the Pi and ADP experiments (as represented in Tables 2–5) with experimental variables being myosin concentration and Pi or ADP, respectively. For the experiments represented in Fig. 2 and Table 1, a one-way ANOVA was performed. All values are expressed as mean ± SE.
TABLE 2.
Maximal velocity of regulated thin filaments as a function of Pi
| Myosin concentration
|
||||
|---|---|---|---|---|
| Pi | 150 μg/ml | 100 μg/ml | 50 μg/ml | 25 μg/ml |
| 0 | 5.32 ± 0.66 | 5.67 ± 0.75 | 5.26 ± 0.68 | 4.33 ± 0.46 |
| 8 | 5.03 ± 0.97 | 5.32 ± 0.22 | 4.99 ± 0.23 | 3.93 ± 0.41 |
| 16 | 5.05 ± 0.72 | 5.20 ± 0.64 | 4.41 ± 0.16 | 3.71 ± 0.19 |
| 30 | 4.65 ± 0.56 | 4.36 ± 0.15 | 4.04 ± 0.22 | 2.70 ± 0.14 |
Maximal thin filament velocity (μm/s) as determined from the fit of the Hill equation as a function of Pi concentration (mM) in the motility buffer and myosin concentration in the loading buffer.
TABLE 5.
The effect of ADP on regulated thin filament half maximal activation
| Myosin concentration
|
|||
|---|---|---|---|
| ADP | 100 μg/ml | 50 μg/ml | 25 μg/ml |
| 0 | 6.36 ± 0.02 | 6.38 ± 0.02 | 6.45 ± 0.05 |
| 0.5 | 6.47 ± 0.02 | 6.46 ± 0.01 | 6.43 ± 0.03 |
| 2 | 6.46 ± 0.72 | 6.60 ± 0.03 | 6.67 ± 0.07 |
| 5 | 6.78 ± 0.79 | 6.67 ± 0.04 | 6.56 ± 0.13 |
The negative log of the calcium concentration at which the thin filament is half maximally activated (i.e., pCa50 ± SE) as a function of ADP concentration (mM) in the motility buffer and myosin concentration in the loading buffer.
FIGURE 2.

(A) Velocity: pCa relations of regulated thin filaments as a function of myosin concentration in the loading buffer. Decreasing the myosin density on the motility surface resulted in a decrease in maximal and submaximal velocities. (B) Myosin activation of the thin filament as determined by thin filament velocity at varying calcium concentrations. These data are a replotting of the data depicted in Table 1. At lower calcium concentrations, greater amounts of myosin are required to achieve maximal velocity.
TABLE 1.
Effect of surface myosin concentration on maximal and half maximal regulated thin filament function
| Actin* | Regulated thin filaments† | ||
|---|---|---|---|
| Myosin concentration‡ | Velocity§ | Vmax¶ | pCa50‖ |
| 100 | 4.57 ± 0.37 | 5.71 ± 0.36 | 6.42 ± 0.04 |
| 50 | 4.51 ± 0.35 | 5.93 ± 0.23 | 6.27 ± 0.03 |
| 37.5 | 4.38 ± 0.07 | 5.41 ± 0.20 | 6.10 ± 0.02 |
| 25 | 4.01 ± 0.11 | 4.11 ± 0.32 | 6.09 ± 0.08 |
| 18.75 | 3.95 ± 0.34 | 2.74 ± 0.18 | 6.00 ± 0.58 |
Actin velocities are mean values ± SE.
Regulated thin filament values represent the parameters of the fit to the Hill equation ± SE.
Myosin concentration (μg/ml) in the loading buffer.
Velocity (μm/s).
Vmax, maximal velocity (μm/s).
pCa50, negative log of the calcium concentration at which half maximal thin filament velocity is achieved.
RESULTS
The velocity of regulated thin filaments as a function of free calcium has been previously reported by our laboratory (VanBuren et al., 2002). By reconstituting actin filaments with troponin and tropomyosin, complete calcium regulation of motility is achieved (Fig. 1). Calcium-regulated motility can be analyzed in terms of: 1), the velocity of thin filaments (Fig. 1 A); 2), the percent of filaments moving in a smooth and continuous fashion (Fig. 1 B; defined as those filaments in which the standard deviation of velocity is less than one-half of the mean of velocity (Homsher et al., 1992)); or 3), the percent of filaments that are mobile (Fig. 1 B; defined as all filaments moving at velocities >0.7 μm/s). We have represented the regulated thin filament motility data in this manuscript using the pCa:velocity method (Fig. 1 A).
FIGURE 1.
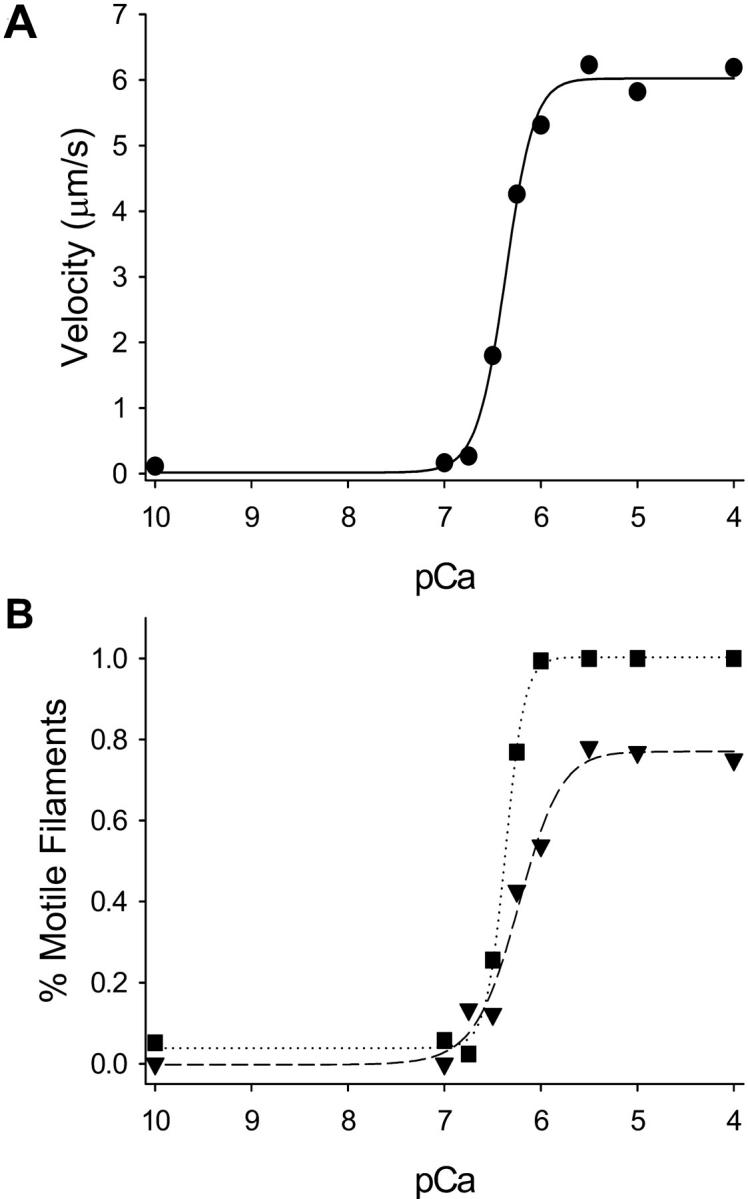
Calcium regulation in the motility assay using fully regulated thin filaments under standard conditions (i.e., no excess Pi or ADP) and 100 μg/ml myosin. A represents the pCa:velocity relation fit to the Hill equation with a maximal velocity of 6.02 ± 0.22 μm/s and a pCa50 of 6.37 ± 0.02. B depicts the percent of filaments that are motile (squares, i.e., any filament moving >0.7 μm/s) and the percent of filaments moving smoothly (triangles down, see Results) as a function of calcium concentration. At maximal calcium activation, 96 ± 2% of all filaments are motile and 77 ± 6% of all filaments are moving smoothly. Using these methods of analysis, the pCa50 is 6.37 ± 0.01 for the all motile filament group and 6.24 ± 0.06 for the smoothly moving filament group.
To determine the effect of myosin strong binding on regulated thin filament activation, the myosin concentration on the motility surface was varied from 12.5 to 100 μg/ml. For actin filaments, varying myosin concentration on the motility surface had no effect on velocity when myosin concentrations were ≥18.5 μg/ml in the loading buffer (Table 1; p = NS). Regulated thin filament velocity is faster than actin velocity at saturating myosin and maximal calcium (Table 1) as has been previously reported (Homsher et al., 1996). However, at subsaturating myosin conditions, regulated thin filament velocity is reduced as a function of the myosin surface concentration at maximal calcium activation (Fig. 2 A, Table 1; p = 0.002). Furthermore, reducing the myosin concentration on the motility surface resulted in an increase in the amount of calcium required to achieve half maximal activation, i.e., a decrease in the pCa50 (Fig. 2 A, Table 1; p = 0.016). There was no significant change in the Hill coefficient in these experiments. These results demonstrate an effect of myosin strong binding on unloaded shortening, and are consistent with force experiments in muscle fibers (Fitzsimons et al., 2001).
To better illustrate the effect of myosin strong binding on thin filament activation, the pCa: velocity data are replotted using myosin concentration on the x axis (Fig. 2 B), with a sigmoidal increase in regulated thin filament velocity as a function of myosin concentration being demonstrated. Higher surface myosin concentrations are required at lower calcium concentrations to half maximally activate the thin filament when compared to higher calcium concentrations. Thus we show that myosin strong binding is required for thin filament activation at all calcium concentrations, with the relative contribution of myosin strong binding to thin filament activation being greatest at submaximal calcium. This constitutes to our knowledge the first direct evidence of the relative role of myosin strong binding on regulated thin filament motility at maximal and submaximal calcium activation.
To further probe the effect of myosin strong binding on thin filament activation, myosin strong binding was limited with the addition of Pi to the motility solutions. With unregulated actin filaments, inorganic phosphate (0, 8,16, and 30 mM) in the motility buffers had no effect on velocity irrespective of the myosin concentration in the myosin loading buffer (p = NS, Fig. 3). In experiments in which regulated thin filaments were used, baseline maximal and half maximal pCa:velocity relations differed slightly for the three experimental approaches (myosin concentration var-ied alone, Pi, and ADP), reflecting the different solution conditions of these experiments. The addition of Pi to the motility buffers had no significant effect on regulated thin filament pCa:velocity relations at saturating myosin conditions (myosin concentration 150 μg/ml) as measured by either maximal calcium-activated velocity or half maximal velocity (Fig. 4, Tables 2 and 3). Similar to our results at high myosin concentrations, no effect of Pi on shortening was observed in muscle fiber experiments at high calcium concentrations (Metzger, 1996). However, a concentration-dependent effect of Pi was observed at reduced myosin concentrations (i.e., <150 μg/ml), with both maximal velocity (Fig. 4, Table 2) and pCa50 being reduced as a function of the Pi concentration (Fig. 4, Table 3). Two-way ANOVA of the parameters of the fits indicates the significant effect of myosin concentration and Pi at maximal calcium activation at all myosin concentrations (p < 0.001 and p = 0.001, respectively) and at half maximal activation for myosin concentrations ≤100 μg/ml (p < 0.002 and p = 0.020, respectively). In addition, Pi appeared to increase the steepness of the activation transition (Hill coefficient). However, in some of these experiments, the transition occurred very rapidly, making it difficult to apply meaningful cooperativity values. Although a reduction in half maximal activation and the increase in the Hill coefficient with added Pi are observed in muscle fiber force experiments (Millar and Homsher, 1990), this is the first study to demonstrate an effect of Pi on half maximal activation for unloaded shortening. The inhibitory effect of reducing the surface myosin density and increasing the Pi concentration in the motility buffers was additive, with the maximal velocity at 25 μg/ml myosin and 30 mM Pi being reduced by 50% when compared to conditions with saturating myosin surface conditions and no Pi. Furthermore, as these effects were not observed for actin filaments, it indicates that this is a unique effect of myosin strong binding on regulated thin filament function.
FIGURE 3.

The velocity of unregulated actin filaments as a function of myosin surface concentration and Pi in the motility buffers. No effect of Pi on actin filament motility was observed at myosin loading concentrations of 150 μg/ml (circles), 100 μg/ml (squares), 50 μg/ml (triangles up), and 25 μg/ml (triangles down). Pi was added to the motility buffers at concentrations of 0 mM, 8 mM, 16 mM, and 30 mM.
FIGURE 4.

Representative graphs of the effect of Pi on regulated thin filament motility at myosin loading concentrations of 150 μg/ml and 25 μg/ml (see Tables 2 and 3). Pi was added to the motility buffers at concentrations of 8 mM (squares, dotted regression), 16 mM (triangles up, dashed regression), and 30mM (triangles down, dash-dot regression) in comparison to control experiments with no added Pi (circles, solid regression). The effect of increasing Pi and decreasing myosin concentration was additive at both maximal and submaximal calcium concentrations (see Results).
TABLE 3.
The effect of Pi on regulated thin filament half maximal activation
| Myosin concentration
|
||||
|---|---|---|---|---|
| Pi | 150 μg/ml | 100 μg/ml | 50 μg/ml | 25 μg/ml |
| 0 | 6.37 ± 0.05 | 6.39 ± 0.08 | 6.31 ± 0.09 | 6.21 ± 0.05 |
| 8 | 6.41 ± 0.01 | 6.36 ± 0.01 | 6.28 ± 0.02 | 6.19 ± 0.06 |
| 16 | 6.37 ± 0.07 | 6.28 ± 0.05 | 6.27 ± 0.02 | 6.08 ± 0.05 |
| 30 | 6.42 ± 0.05 | 6.22 ± 0.02 | 6.20 ± 1.86 | 6.14 ± 0.15 |
The negative log of the calcium concentration at which the thin filament is half maximally activated (i.e., pCa50 ± SE) as a function of Pi concentration (mM) in the motility buffer and myosin concentration in the loading buffer.
Myosin strong binding to the thin filament was increased with the addition of ADP to the motility buffers (Baker et al., 2002). A concentration-dependent reduction of actin filament velocity was observed with the addition of ADP (data not shown) similar to that reported by Warshaw et al. (1991) using smooth muscle myosin. Two-way ANOVA was applied to the parameters of the Hill equation fit represented in Tables 4 and 5. Using regulated thin filaments, maximal calcium-activated velocity was reduced as a function of the ADP concentration (Fig. 5, Table 4; p < 0.001). In contrast, increasing myosin strong binding to the thin filament with the addition of ADP resulted in activation of the thin filament at lower calcium concentrations as reflected by an increase in the pCa50 as a function of ADP (Fig. 5, Table 5; p = 0.041). There was no effect of ADP on the Hill coefficient. In summary, we demonstrate that half maximal activation of the thin filament (pCa50) can be shifted through the modulation of myosin strong binding with Pi or ADP.
TABLE 4.
Maximal velocity of regulated thin filaments as a function of ADP
| Myosin concentration
|
|||
|---|---|---|---|
| ADP | 100 μg/ml | 50 μg/ml | 25 μg/ml |
| 0 | 5.32 ± 0.17 | 5.15 ± 0.21 | 2.99 ± 0.19 |
| 0.5 | 4.01 ± 0.21 | 3.43 ± 0.14 | 2.63 ± 0.17 |
| 2 | 2.20 ± 0.29 | 1.29 ± 0.10 | 1.17 ± 0.17 |
| 5 | 0.96 ± 0.32 | 1.10 ± 0.08 | 0.74 ± 0.16 |
Maximal thin filament velocity (μm/s) as determined from the fit of the Hill equation as a function of ADP concentration (mM) in the motility buffer and myosin concentration in the loading buffer.
FIGURE 5.

Representative graphs of the effect of ADP on regulated thin filament motility at myosin loading concentrations of 100 μg/ml and 25 μg/ml (see Tables 4 and 5). ADP was added to the motility buffers at concentrations of 0.5 mM (squares, dotted regression), 2 mM (triangles up, dashed regression), and 5 mM (triangles down, dash-dot regression) in comparison to control experiments with no added ADP (circles, solid regression). ADP significantly decreased maximal velocity but increased the pCa50 of the thin filament in a concentration-dependent manner (see Results).
DISCUSSION
The ability of myosin strong binding to activate the thin filament was first demonstrated using limiting ATP concentrations (Bremel et al., 1973) and has been subsequently demonstrated using strong binding myosin analogs (Swartz and Moss, 1992). However, factors affecting unloaded shortening in a calcium-regulated system are not completely understood. Specifically, it has been proposed that the calcium-dependent increase in unloaded shortening is the result of cross-bridge recruitment (Homsher et al., 1996, 2000), whereas others have postulated a modulation in the kinetics of the cross-bridge (Swartz and Moss, 2001). In this study, myosin strong binding to regulated thin filaments was controlled by three perturbations: 1), varying the amount of ADP in the motility buffers, 2), varying the amount of Pi in the motility buffers, and 3), varying the myosin concentration on the motility surface. Through this approach, we demonstrate that myosin strong binding is essential for thin filament activation at both maximal and submaximal Ca2+ levels. However, under conditions in which myosin strong binding is not rate limiting (i.e., high myosin concentrations), other factors must be responsible for the graded motile response of the thin filament to increased calcium.
Limiting the weak to strong cross-bridge isomerization with Pi has a profoundly negative effect on force (Cooke and Pate, 1985; Millar and Homsher, 1990). In contrast, no effect of phosphate on unloaded shortening has been demonstrated in either the motility assay with unregulated actin (Warshaw et al., 1991) or intact muscle fibers at maximal calcium activation (Metzger, 1996; Cooke and Pate, 1985). Consistent with these prior studies, no discernable effect of Pi on regulated thin filament velocity at high myosin concentrations was observed in this study. However, at lower myosin concentrations, we delineate a Pi concentration-dependent reduction in regulated thin filament velocity at both maximal and submaximal calcium concentrations, thus demonstrating an effect of myosin strong binding on activation of the regulated thin filament in the motility assay.
Although the rate-limiting step for unloaded shortening in muscle is generally felt to be cross-bridge detachment (Huxley, 1957; Cooke and Pate, 1985), at subsaturating myosin concentrations other factors may affect thin filament motility. Understanding that the principal mechanical effect of excess Pi is to impede the weak to strong cross-bridge isomerization (Gordon et al., 2001), the reduction in velocity in the presence of Pi is likely the result of a reduction in the rate of cross-bridges transitioning to a strongly bound state and thus deactivates the thin filament by limiting strong binding. Therefore, by limiting the number of cross-bridges in a strongly bound state (by adjusting myosin and Pi concentrations) the regulated thin filament is less activated (even in the setting of maximal calcium). This effect of Pi and myosin concentration on velocity is specific to regulated thin filaments as this was not observed for actin alone. These data are consistent with previous fiber studies that indicate that myosin strong binding in addition to calcium is essential for maximal thin filament activation (Swartz et al., 1996) and extends these observations to unloaded shortening. Based on the fact that greater myosin concentrations are required to activate the thin filament at submaximal calcium levels, the relative role of myosin strong binding activation is greatest at submaximal calcium and is confirmatory of experiments in muscle fibers (Swartz and Moss, 2001).
Could the effect of Pi be accounted for by the fact that thin filaments are moving under loaded conditions in the motility assay? The fact that no reduction in velocity is observed with actin filaments in the presence of high Pi concentrations even at low myosin concentrations argues that there is no significant internal loading of the thin filament induced by the conditions of the motility assay itself. Thus the velocity reduction at low myosin concentrations and in the presence of Pi can be accounted for by several potential contributing mechanisms: 1), an increased number of cross-bridges in a weakly bound “preforce” state, causing an internal loading of the thin filament, thus slowing velocity; 2), a reduced number of cross-bridges interacting with the thin filament such that the number of cross-bridges interacting with the thin filament is rate limiting; and 3), an activation-dependent modulation in cross-bridge kinetics such that with limited myosin strong binding, and hence reduced thin filament activation, the kinetic rate transitions of the myosin cross-bridge are slowed and rate limiting for velocity. Weakly bound cross-bridges have been shown to create a load at lower ionic strength conditions in the motility assay (Warshaw et al., 1990). However, as these Pi experiments were conducted at 120 mM, it is unlikely that an internal loading by weakly bound cross-bridges is contributing to the reduced thin filament velocity seen with the addition of Pi (Gordon et al., 1997). As is discussed below, both cross-bridge number and cross-bridge kinetics likely contribute to thin filament activation.
To further test the role of myosin strong binding in thin filament activation, ADP was added to the motility assay. ADP is known to reduce thin filament sliding velocity in a concentration-dependent fashion (Yamashita et al., 1994) by reducing the rate of myosin cross-bridge detachment (Siemankowski et al., 1985). Furthermore, the addition of ADP increases the relative number of myosin cross-bridges in the strongly bound state (Pate and Cooke, 1989) by increasing the attachment time of the individual myosin cross-bridge (Baker et al., 2002). When the effect of ADP was assessed as a function of calcium activation, we demonstrate that the addition of ADP increased the pCa50 for the thin filament in a concentration-dependent fashion. Thus by increasing the relative number of cross-bridges in the strongly bound state with the addition of ADP, two primary effects are observed: a reduction in velocity (due to an internal loading of the thin filament by strongly bound cross-bridges) and an increase in the half maximal activation of the thin filament. These results are consistent with the results for Pi and indicate that half maximal calcium activation can be shifted by modulating the number of cross-bridges strongly bound to the thin filament.
At lower myosin concentrations, an effect of myosin strong binding on thin filament activation is demonstrated by modulating myosin strong binding to the thin filament. In contrast, at higher myosin concentrations, limiting myosin binding with Pi had no effect on thin filament motility at both maximal and submaximal calcium activation. What factors then are responsible for the graded velocity response to calcium at saturating myosin conditions? The cooperative unit size for thin filament activation is ∼10–12 actin monomers in a fully regulated system (Geeves and Lehrer, 1994). Thus the reduced velocities observed at high myosin concentrations and low calcium concentrations could be the result of two mechanisms: 1), only a few cooperative units are activated by calcium (at low calcium concentrations) such that velocity is rate limited by cross-bridge number with no apparent effect on cross-bridge kinetics; or 2), the kinetic rate transitions of the myosin cross-bridge are directly modulated as a function of calcium activation. If the first set of conditions (i.e., reduced cross-bridge number with no change in kinetics) is correct, then, similar to the concepts presented by Spudich and colleagues (Uyeda et al., 1990), longer thin filaments would have a greater probability of interacting with cross-bridges than shorter thin filaments. In essence, if the number of activated cooperative units is so low that the number of cross-bridges interacting with the thin filament is the primary determinant for velocity, then longer filaments, having a greater number of activated units in series, would move at greater velocities. Alternatively, if calcium activation affects the kinetics of the individual cross-bridge, then no length dependence of velocity should be observed.
To specifically address this question, the velocity of individual thin filaments as a function of their length was plotted at pCa 6.25 and pCa 4 (Fig. 6). Although on average the thin filaments move more slowly at pCa 6.25 when compared to pCa 4, no length-dependent effect on velocity was observed. This suggests that the reduced thin filament velocities at high myosin concentrations but low calcium may be the result of a calcium-dependent modulation of cross-bridge kinetics. In addition, the fact that adding Pi to the motility buffers (and reducing myosin binding) had no effect on velocity at submaximal calcium concentrations with myosin concentrations of 150 μg/ml indicates that: 1), the number of cross-bridges interacting with the thin filament is not rate limiting for velocity under these conditions, and 2), the reduced velocity is not the result of an internal load on the thin filament due to a relative balance of strongly bound and weakly bound cross-bridges, as adding Pi to the motility buffers would affect both of these scenarios.
FIGURE 6.

The velocity of individual thin filaments as a function of thin filament length at pCa 6.25 (circles) and pCa 4 (triangles down). The mean of velocity for pCa 6.25 and pCa 4 are 2.9 ± 0.1 μm/s and 5.8 ± 0.2 μm/s, respectively. These data demonstrate no correlation between thin filament length and velocity at either pCa 6.25 or pCa 4 (r2 = 0.002 and 0.02, respectively).
The fact that thin filaments at low calcium move at slower velocities independent of filament length indicates that the reduction in velocity is probably the result of a calcium-dependent modulation of rate-limiting steps in the cross-bridge cycle for unloaded shortening. ADP release is likely the rate-limiting step for thin filament motility at maximal calcium activation (Siemankowski et al., 1985). In addition, the rate of ADP release by regulated actomyosin is 15-fold greater in the presence of calcium (Rosenfeld and Taylor, 1987), and suggests that the regulatory proteins are capable of modulating myosin ADP release as a function of calcium. Alternatively, other kinetic rate transitions may be altered as a function of calcium activation and be rate limiting for velocity. Additional experiments (in the laser light trap, for example) tracking single myosin molecular kinetics as a function of calcium would further probe the mechanisms involved in the regulation of myosin cross-bridge kinetics as a function of calcium activation.
Acknowledgments
This work was supported by the National Institutes of Health, grants HL65586 and HL59408.
References
- Baker, J. E., C. Brosseau, P. B. Joel, and D. M. Warshaw. 2002. The biochemical kinetics underlying actin movement generated by one and many skeletal muscle myosin molecules. Biophys. J. 82:2134–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremel, R. D., J. M. Murray, and A. Weber. 1973. Manifestations of cooperative behavior in the regulated actin filament during actin activated ATP hydrolysis in the presence of calcium. Cold Spring Harb. Symp. Quant. Biol. 37:267–275. [Google Scholar]
- Brooks, S. P., and K. B. Storey. 1992. Bound and determined: a computer program for making buffers of defined ion concentrations. Anal. Biochem. 201:119–126. [DOI] [PubMed] [Google Scholar]
- Cooke, R., and E. Pate. 1985. The effects of ADP and phosphate on the contraction of muscle fibers. Biophys. J. 48:789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimons, D. P., J. R. Patel, and R. L. Moss. 2001. Cross-bridge interaction kinetics in rat myocardium are accelerated by strong binding of myosin to the thin filament. J. Physiol. 530:263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeves, M. A., and S. S. Lehrer. 1994. Dynamics of the muscle thin filament regulatory switch: the size of the cooperative unit. Biophys. J. 67:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A. M., E. Homsher, and M. Regnier. 2000. Regulation of contraction in striated muscle. Physiol. Rev. 80:853–924. [DOI] [PubMed] [Google Scholar]
- Gordon, A. M., M. A. LaMadrid, Y. Chen, Z. Luo, and P. B. Chase. 1997. Calcium regulation of skeletal muscle thin filament motility in vitro. Biophys. J. 72:1295–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A. M., M. Regnier, and E. Homsher. 2001. Skeletal and cardiac muscle contractile activation: tropomyosin “rocks and rolls”. News Physiol Sci. 16:49–55. [PubMed] [Google Scholar]
- Homsher, E., B. Kim, A. Bobkova, and L. S. Tobacman. 1996. Calcium regulation of thin filament movement in an in vitro motility assay. Biophys. J. 70:1881–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsher, E., D. M. Lee, C. Morris, D. Pavlov, and L. S. Tobacman. 2000. Regulation of force and unloaded sliding speed in single thin filaments: effects of regulatory proteins and calcium. J. Physiol. (Lond.). 524:233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homsher, E., F. Wang, and J. R. Sellers. 1992. Factors affecting movement of F-actin filaments propelled by skeletal muscle heavy meromyosin. Am. J. Physiol. 262:C714–C723. [DOI] [PubMed] [Google Scholar]
- Huxley, A. F. 1957. Muscle structure and theories of contraction. Prog. Biophys. & Biophys. Chem. 7:255–317. [PubMed] [Google Scholar]
- Margossian, S. S., and S. Lowey. 1982. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 85:55–71. [DOI] [PubMed] [Google Scholar]
- McKillop, D. F., and M. A. Geeves. 1993. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 65:693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger, J. M. 1996. Effects of phosphate and ADP on shortening velocity during maximal and submaximal calcium activation of the thin filament in skeletal muscle fibers. Biophys. J. 70:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar, N. C., and E. Homsher. 1990. The effect of phosphate and calcium on force generation in glycerinated rabbit skeletal muscle fibers. A steady-state and transient kinetic study. J. Biol. Chem. 265:20234–20240. [PubMed] [Google Scholar]
- Pardee, J. D., and J. A. Spudich. 1982. Purification of muscle actin. Methods Enzymol. 85:164–181. [DOI] [PubMed] [Google Scholar]
- Pate, E., and R. Cooke. 1989. A model of crossbridge action: the effects of ATP, ADP and Pi. J. Muscle Res. Cell Motil. 10:181–196. [DOI] [PubMed] [Google Scholar]
- Potter, J. D. 1982. Preparation of troponin and its subunits. Methods Enzymol. 85:241–263. [DOI] [PubMed] [Google Scholar]
- Rosenfeld, S. S., and E. W. Taylor. 1987. The dissociation of 1-N6-ethenoadenosine diphosphate from regulated actomyosin subfragment 1. J. Biol. Chem. 262:9994–9999. [PubMed] [Google Scholar]
- Siemankowski, R. F., M. O. Wiseman, and H. D. White. 1985. ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle. Proc. Natl. Acad. Sci. USA. 82:658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz, D. R., and R. L. Moss. 1992. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J. Biol. Chem. 267:20497–20506. [PubMed] [Google Scholar]
- Swartz, D. R., and R. L. Moss. 2001. Strong binding of myosin increases shortening velocity of rabbit skinned skeletal muscle fibres at low levels of Ca(2+). J. Physiol. 533:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz, D. R., R. L. Moss, and M. L. Greaser. 1996. Calcium alone does not fully activate the thin filament for S1 binding to rigor myofibrils. Biophys. J. 71:1891–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda, T. Q., S. J. Kron, and J. A. Spudich. 1990. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. J. Mol. Biol. 214:699–710. [DOI] [PubMed] [Google Scholar]
- VanBuren, P., S. L. Alix, J. A. Gorga, K. J. Begin, M. M. LeWinter, and N. R. Alpert. 2002. Cardiac troponin T isoforms demonstrate similar effects on mechanical performance in a regulated contractile system. Am. J. Physiol. Heart Circ. Physiol. 282:H1665–H1671. [DOI] [PubMed] [Google Scholar]
- VanBuren, P., K. A. Palmiter, and D. M. Warshaw. 1999. Tropomyosin directly modulates actomyosin mechanical performance at the level of a single actin filament. Proc. Natl. Acad. Sci. USA. 96:12488–12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshaw, D. M., J. M. Desrosiers, S. S. Work, and K. M. Trybus. 1990. Smooth muscle myosin cross-bridge interactions modulate actin filament sliding velocity in vitro. J. Cell Biol. 111:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshaw, D. M., J. M. Desrosiers, S. S. Work, and K. M. Trybus. 1991. Effects of MgATP, MgADP, and Pi on actin movement by smooth muscle myosin. J. Biol. Chem. 266:24339–24343. [PubMed] [Google Scholar]
- Yamashita, H., M. Sata, S. Sugiura, S. Momomura, T. Serizawa, and M. Iizuka. 1994. ADP inhibits the sliding velocity of fluorescent actin filaments on cardiac and skeletal myosins. Circ. Res. 74:1027–1033. [DOI] [PubMed] [Google Scholar]