

Abstract
Femtosecond excitation of the red edge of the chlorophyll a QY transition band in photosystem I (PSI), with light of wavelength ≥700 nm, leads to wide transient (subpicosecond) absorbance changes: positive ΔA between 635 and 665 nm, and four negative ΔA bands at 667, 675, 683, and 695 nm. Here we compare the transient absorbance changes after excitation at 700, 705, and 710 nm at 20 K in several PSI preparations of Chlamydomonas reinhardtii where amino acid ligands of the primary donor, primary acceptor, or connecting chlorophylls have been mutated. Most of these mutations influence the spectrum of the absorbance changes. This supports the view that the chlorophylls of the electron transfer chain as well as the connecting chlorophylls are engaged in the observed absorbance changes. The wide absorption spectrum of the electron transfer chain revealed by the transient measurements may contribute to the high efficiency of energy trapping in photosystem 1. Exciton calculations, based on the recent PSI structure, allow an assignment of the ΔA bands to particular chlorophylls: the bands at 675 and 695 nm to the dimers of primary acceptor and accessory chlorophyll and the band at 683 nm to the connecting chlorophylls. The subpicosecond transient absorption bands decay may reflect rapid charge separation in the PSI reaction center.
INTRODUCTION
Excitonic interaction between chlorophyll molecules is a common phenomenon in photosynthetic molecular systems, both in antenna and in the reaction centers (RC). Strong interactions between closely located chlorophylls lead to a situation where these molecules lose their individual identity and their spectral properties change significantly. A well-known and widely studied example of this phenomenon is the LH2 antenna system from the purple bacterium Rhodopseudomonas acidophila containing two regular rings of bacteriochlorophyll a (BChls a) molecules (McDermott et al., 1995). The nine BChls a constituting one of these rings behave essentially like monomers with an absorption maximum at 800 nm. However, the absorption of the other ring of similar diameter, but containing two times more densely packed molecules, peaks at 850 nm. It was estimated that almost half of this red shift was due to excitonic coupling (see Sundström et al. (1999) for review). A similar but bigger ring of BChls a in the LH1 antenna surrounds the reaction center and has an absorption maximum at 870–880 nm. Again, this red shift is caused in part by excitonic interactions. Thus, interactions between BChls in LH1 and LH2 significantly contribute to the down shift of the excited-state energy to promote excitation energy flow toward the reaction center. Excitonic interactions have also been studied in other antenna systems, like Fenna-Matthews-Olson (FMO) proteins (Freiberg et al., 1997; Melkozernov et al., 1998a; Wendling et al., 2002) and LHCII (Palacios et al., 2002).
In reaction centers, the manifestation of excitonic interaction is the lower excited-state energy of the primary donor relative to neighboring antenna and electron transfer chain (ETC) Chls, and this again promotes excitation energy transfer to the primary donor. This red shift is due to the fact that the primary donor is always a (B)Chl dimer, although the interaction between its components depends on the type of RC, being the strongest in purple bacteria (550–950 cm−1), intermediate in PSI (∼420 cm−1; Byrdin et al., 2002), and weak in PSII (∼140 cm−1; Durrant et al., 1995). In addition to these RC traps, other species with very low-energy absorption bands, called red Chls, were found in antenna systems of LHCI (Jansson, 1994; Melkozernov et al., 1998b, 2000a; Gobets et al., 1994) and PSI (see Gobets and van Grondelle (2001) for review). Red Chls are characterized by an excited-state energy even lower than that of the primary donor and thus compete for trapping of excitation energy. They have been proposed to be dimers or trimers of excitonically coupled Chls (Gobets et al., 1994; Pålsson et al., 1996; Melkozernov et al., 2000b; Frese et al., 2002). Their function is still not very well understood. It has been proposed that they may increase the efficiency of energy trapping (Trissl, 1993; Holzwarth et al., 1993; van Grondelle et al., 1994) or promote photoprotection (Karapetyan et al., 1999). There have also been attempts to consider the ETC Chls of PSII and PSI as excitonically coupled systems (Durrant et al., 1995; Beddard, 1998). It has been pointed out that the observed wide, multiband absorption spectrum likely resulting from excitonic interaction between ETC Chls may increase spectral overlap between different spectral forms of antenna Chls and ETC Chls in PSI and thus increase efficiency of energy transfer to the RC (Gibasiewicz et al., 2002).
The availability of a high-resolution PSI structure (Fromme et al., 1996, 2001; Schubert et al, 1997; Jordan et al., 2001) allows for a detailed theoretical and experimental study of excitonic interactions (Stewart et al., 1999; Melkozernov et al., 2000b, 2001; Byrdin et al., 2002; Gibasiewicz et al., 2002). Studies on cyanobacterial PSI have focused on red Chls and low-energy states were ascribed to antenna dimers located close to the RC (Melkozernov et al., 2000b,c). On the other hand, theoretical calculations based on the latest PSI structure show that there are many Chl dimers in different locations, some distant from the RC (Byrdin et al., 2002). Low-temperature femtosecond transient absorption studies of Synechocystis sp. 6803 with selective excitation at ≥700 nm revealed coupling between long-wavelength ΔA bands and two resonant bands: the more prominent one at 683 nm and a poorly resolved one at 675 nm. These two bands were originally assigned to the upper excitonic states of two Chl dimers located in the vicinity of the RC (Melkozernov et al., 2000b). Later, it was suggested that the two resonant bands originate from a borrowing of the oscillator strength from the excited-state absorption of Chl dimers to the ground-state absorption of nearby Chl monomers (Melkozernov et al., 2001; see also Valkunas et al. (1999)). Similar transient ΔA bands were reported for PSI from C. reinhardtii strain CC2696 after excitation at 700 nm and 10 K. However, these two bands were accompanied by two other negative bands at 667 nm and 695 nm and four positive bands between 635 and 661 nm (Gibasiewicz et al., 2002). This wide structure was interpreted in terms of excitonic coupling between the six ETC Chls: the negative bands were ascribed to photobleaching of the transitions to four different one-exciton states, and the positive bands were ascribed to excited state absorption from the lowest one-exciton state to four different two-exciton states. The experimentally measured energy levels of the one-exciton states fitted very well to those theoretically calculated by Beddard (1998). However, these calculations were not based on the 2.5-Å structure of PSI (Jordan et al., 2001), and the directions of QY dipole moments of Chl molecules were obtained by fitting to experimental data (White et al., 1996; Mathis et al., 1988).
In this work, transient absorbance changes at 20 K, after 150-fs excitation pulses at ≥700 nm, were measured for PSI from five different mutants of C. reinhardtii with altered ligands to the primary donor P, primary electron acceptor A0, and the two connecting Chls, and compared to results obtained for wild-type (WT) PSI. The connecting Chls are in the vicinity of A0 Chls and were proposed to be excitation energy mediators between the rest of antenna and RC (Jordan et al., 2001). All mutants, except for the primary donor mutant, display alterations in the pattern of excitonic coupling, thus demonstrating the engagement of A0 Chls and connecting Chls in excitonic coupling. Based on the precise structure of PSI, resolved so far only for a cyanobacterium S. elongatus (Jordan et al., 2001), and using simplified exciton theory, one-exciton states were calculated and compared to the experimental results for WT and mutant PSI. Using the PSI structure from different organism seems to be justified as this structure, especially of the core electron transfer chlorophylls, is believed to be conserved in all PSI reaction centers (Webber and Bingham, 1998; Nechushtai et al., 1996; Golbeck, 1994).
MATERIALS AND METHODS
Samples preparation
PSI particles were prepared as described (Gibasiewicz et al., 2001; Krabben et al., 2000). The control PSI particles were prepared from two different strains of C. reinhardtii, CC2696 and KRC51-3A(154-1A), both of which lack PSII and have reduced light-harvesting complexes; they gave similar results. Three A0 mutants were obtained in strain CC2696 by replacing PsaB–Met-664, which coordinates the A0 Chl in branch B, with histidine (MH(B664)), serine (MS(B664)), or leucine (ML(B664)).
The primary donor mutant was obtained from the strain CC2696 by replacing PsaB–His-656, which coordinates the B-side Chl of the primary donor, with serine (HS(B656)). The mutant affecting the connecting Chls (HL(A730)/HL(B714)) was obtained by transforming a strain lacking psaA and psaB with plasmids containing mutant genes in which the two histidines that coordinate the connecting Chls (PsaA–His-730 and PsaB–His-714) were converted to leucines (Redding et al., 1998). This strain was then crossed to a mt− nac2 cbn1 strain to obtain nac2 cbn1 progeny that had the two mutations in PSI, as well as the lack of PSII and reduced antenna (Boudreaux et al., 2001). The KRC51-3A(154-1A) strain has WT PSI, lacks PSII, has reduced antenna, and served as the appropriate control strain.
No systematic differences between steady-state absorption spectra in the QY region (maximum at ∼675 nm) of all these preparations were observed, except for the (HL(A730)/HL(B714)) mutant for which the red slope of the main band was steeper and the amount of absorption around 700 nm was ∼30% lower than in the other preparations.
Femtosecond transient absorption spectroscopy
The setup and measurements were described earlier (Gibasiewicz et al., 2001, 2002; Freiberg et al., 1998). The samples mixed with glycerol (1:2 v/v) and with addition of 40 mM sodium ascorbate and 20 μM phenazine methosulfate were placed between two plexiglass plates separated with a rubber ring. The final OD at ∼675 nm (steady-state absorption maximum of PSI from C. reinhardtii) in this ∼1-mm thick cuvette was 1–1.2. The measurements were performed in a helium cryostat (APD) at ∼20 K. Excitation energies were kept on the low level of <1 photon per RC, to avoid annihilation effects. Selective excitation of different populations of antenna Chls was obtained by applying 5-nm wide (fwhm) ∼150-fs pulses centered at 700, 705, and 710 nm. The transient spectra were measured between 600 and 740 nm with a step size of 54 fs from 1 ps before to 2 ps after the excitation pulse. The spectral resolution of the setup was 1 nm and the spectra were averaged over 2-nm intervals. Thus, the number of data points (spectral channels times (×) delay times) taken to one global analysis was typically 70 × 55 = 3850. Decay-associated spectra (DAS) were calculated from global fitting, accounting for deconvolution of the recorded signals with the instrument response function using locally written software (ASUFIT, www.public.asu.edu/laserweb/asufit/asufit.html). The instrument response function was modeled by a Gaussian function with a width of ∼0.3 ps. The time versus wavelength absorbance change surfaces were corrected for the spectral dispersion of the probe beam.
Exciton calculations
Interaction energies between Chls (Vij) were calculated from the formula for dipole–dipole interaction (van Amerongen et al., 2000; Freiberg et al., 1999):
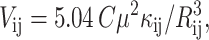 |
(1) |
where κij is the orientation factor:
 |
(2) |
μ2 is the dipole strength of the isolated Chl a molecule, C is a constant dependent on the dielectric constant of the medium (van Amerongen et al., 2000), Rij is the distance between the centers of the planes of interacting Chls, μi and μj are the unit vectors associated with the transition moment of the ith and jth Chls, respectively, and Rij is the unit vector along the line joining centers of the ith and jth Chls. The values of μi, μj, Rij, and Rij were calculated based on the atomic coordinates for PSI (PDB file 1JBO), assuming that the QY transition moment runs through nitrogens NB and ND. The value of Cμ2 = 19 debye2 was taken from Byrdin et al. (2002) and Shipman (1977). If the distances and transition dipole moments are given in nanometers and debye units, respectively, Vij is read in wavenumbers.
The energies of the N-exciton states of N-interacting Chls characterized by the site energies ɛ were calculated as eigenvalues of the Hamiltonian matrix:
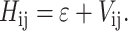 |
(3) |
The squared elements of eigenvectors of this matrix, Dij, describe distribution of particular exciton states f over particular excitonically coupled Chls. Oscillator strengths D(f) of particular exciton states f were calculated from the expression (van Amerongen et al., 2000):
 |
(4) |
where ρi,j(f) = cf,icf,j, ρi,j(f) is the so-called density matrix of state f, cf,i, and cf,j are the elements of eigenvectors associated with the state f, and Mi,j = μi · μj is the dipole strength matrix.
RESULTS
Absorbance changes recorded 0.27–0.32 ps after excitation of WT and mutant PSI at 700 nm are shown in Fig. 1, A–C. For the WT PSI, results from two different preparations are shown to illustrate the reproducibility of the four negative bands (peaking at 667, 675, 683–685, and 695 nm) and four positive bands (peaking at 635, 645, 653, and 661 nm; Fig. 1 A). The temporal evolution of the transient absorption spectra between the earliest delay times and 100 ps have already been published for WT PSI (Gibasiewicz et al., 2002). Similarly as for WT PSI, absorbance changes for three different preparations of the HL(A730)/HL(B714) mutant are shown (Fig. 1 B). In this mutant, the band at 683 nm is missing or significantly reduced. The four bands making up the positive structure is less well resolved compared to WT. Otherwise, the ΔA signal is similar to that of WT with a similar relative contribution of negative bands at 667, 677, and 697 nm. The 2-nm red shift of the 677- and 697-nm bands relative to the corresponding bands in the WT may be not significant, considering the 2-nm spectral resolution of the data. Absorbance changes for the three different A0 mutants are similar to each other but significantly different from the WT and the HL(A730)/HL(B714) mutant (Fig. 1 C). The amplitude of the 683-nm band drops twofold relative to the band at 695 (697) nm compared to WT. The amplitudes of the 667 and 675-nm bands are also smaller than those in WT. Again, the positive part of ΔA signal is less well resolved than in WT.
FIGURE 1.

Normalized absorbance difference spectra (ΔA; A–C) and decay-associated spectra found in 2-ps time windows (DAS; D–F) for WT (A and D) and mutant (B, C, E, F) PSI from C. reinhardtii after ∼150-fs excitation at 700 nm. The ΔA spectra were recorded at Δt = 0.27–0.32 ps after excitation and their absolute magnitude was ∼2 m OD. (A) ΔA for two different preparations of WT PSI. (B) ΔA for three different preparations of HL(A730)/HL(B714) connecting Chls mutant. (C) ΔA for three different A0 mutants. (D) DAS for one of the WT preparations. (E) Averaged DAS calculated for three different preparations of HL(A730)/HL(B714) mutant. (F) DAS of three different A0 mutants. ND: nondecaying components.
The difference spectra recorded at pump-probe delay times of up to 2 ps were globally analyzed and two components were found for all preparations: a 140–190 fs decay associated spectrum (DAS) and a component nondecaying (ND) on the 2-ps timescale (Fig. 1, D–F). The subpicosecond components were resolved at the limit of the temporal resolution of the experimental setup. The kinetics of the absorbance changes in WT PSI at eight different wavelengths are shown in Fig. 2 along with global fits resulting in a 140-fs component and a nondecaying component. The subpiscosecond DAS is preparation dependent and its shape is similar to the shape of the corresponding ΔA spectrum for each preparation (Fig. 1, A–C). In particular, the maximum positions are exactly the same. These similarities demonstrate that the absorbance changes initially observed live only for a few hundreds of femtoseconds after excitation. On the other hand, the ND component, with a maximum at 697 nm and a much smaller band at ∼680 nm, is roughly the same for all mutants and for WT. The amplitude of the ND component could be used as an independent normalization factor for a direct comparison of the subpicosecond DAS amplitudes (Fig. 1, D–F). In particular, the amplitudes of the 675- and 683–685-nm bands in A0 mutants DAS are again shown to be about half the amplitude of the corresponding bands in the WT DAS (Fig. 1 D versus Fig. 1 F). The relations between the amplitudes of the particular bands in transient spectra and DAS, for WT and mutants, after normalization at 695/697 nm are shown in Fig. 3.
FIGURE 2.

Global analysis fits to the kinetics of absorbance changes at eight wavelengths for which transient ΔA spectrum of C. reinhardtii WT PSI, recorded 0.27–0.32 ps after excitation, has minima or maxima (see Fig. 1 A).
FIGURE 3.

Direct comparison of the ΔA (A and B) and DAS shapes (C, D) for PSI from WT and HL(A730)/HL(B714) mutant (A and C) and WT and A0 mutants (B and D) of C. reinhardtii. The ΔA spectra and DAS are redrawn from Fig. 1 (except for the ΔA spectrum of the HL(A730)/HL(B714) mutant in panel A, for which the averaged spectrum was calculated over the three spectra from Fig. 1 B). The spectra being compared were normalized to the same peak amplitudes at 695/697 nm.
Excitation at wavelengths longer than 700 nm gave absorbance changes of poorer quality due to the smaller signal-to-noise ratio. Comparison of the ΔA traces recorded at 0.32 ps and 2 ps after excitation at 700, 705, and 710 nm in WT PSI and MH(B664) mutant PSI are shown in Fig. 4. In the case of WT PSI (Fig. 4, A–D), independent of excitation wavelength, the transient bands at 673–675 nm and at 683–685 nm are always present and the band at 683–685 nm dominates. At longer excitation wavelengths the contribution of the band at 695 nm decreases. Apparently, most of the amplitude of the 695-nm band comes from photobleaching of a species independent from that or those giving rise to the bands peaking at 675 and 683 nm. Excitation at longer wavelengths does not photobleach this independent species. The positive absorbance changes seem to expand toward the red at longer excitation wavelengths and thus the negative band at 667 nm can no longer be resolved after longer wavelength excitation. The ΔA signals recorded 0.32 ps after excitation at 705 and 710 nm have additional PB/SE bands peaking at 703 and 707–715 nm, respectively. However, increased noise and scattering in this spectral region make the amplitudes and shapes of these bands uncertain. The spectrum recorded 2 ps after excitation is noticeably dependent upon excitation wavelength and the main bands peak at 695, 703, and 707 nm, for excitation at 700, 705, and 710 nm, respectively. It is not clear if the molecules photobleached at these wavelengths are coupled with those giving rise to 675–683-nm bands or if they are independently excited. Similar absorbance change measurements as a function of excitation wavelength were performed for PSI from the MH(B664) mutant that affects A0 (Fig. 4, E–H), the HL(A730)/HL(B714) mutant affecting the connecting Chls (Fig. 4, I–L), and the HS(B656) mutant affecting the primary pair (data not shown). In all these cases, the observed transient spectra are characterized by excitation wavelength dependence very similar to that of WT PSI, with an exception for the HL(A730)/HL(B714) mutant, for which the band at 683 nm was significantly decreased independent of excitation wavelength. Fig. 4, E–H, is shown primarily to demonstrate that the band at 695 nm at Δt = 0.32 ps is also present at excitation wavelengths of 705 nm and 710 nm, although its amplitude is much smaller compared to that seen with 700-nm excitation. Chemical oxidation of the primary donor to P+ does not change significantly the initial absorption changes in PSI either from WT (Fig. 5) or the HS(B656) mutant (data not shown).
FIGURE 4.

Comparison of absorbance changes spectra recorded 0.32 ps and 2 ps after excitation of C. reinhardtii WT PSI (A–D), MH(B664) mutant PSI (E–H), and HL(A730)/HL(B714) mutant PSI (I–L) at 700 (A, E, I), 705 (B, F, J), and 710 nm (C, G, K). (D, H, L) Direct comparison of the shapes of the spectra recorded 0.32 ps after excitation at the three wavelengths.
FIGURE 5.

Absorbance changes recorded 0.32 ps after 700-nm excitation of WT PSI from C. reinhardtii under neutral and oxidizing (primary donor, P, chemically oxidized to P+) conditions at 20 K.
DISCUSSION
Exciton calculations
Excitonic coupling in RCs, involving not only the primary donor components, but also the accessory and primary acceptor Chls, has been proposed, based partly on theoretical calculations both for PSII (Durrant et al., 1995) and PSI (Beddard, 1998). Resonance Raman scattering data (Stewart et al., 1999) and femtosecond transient hole burning (Gibasiewicz et al., 2001, 2002) have provided experimental support for the hypothesis of excitonic coupling between the ETC Chls in PSI. The early transient absorption spectra at 10 K (a few hundred femtoseconds) that followed excitation at 700 nm showed a structured shape composed of several bands between 630 and 700 nm (Gibasiewicz et al., 2002). The positions of the four negative bands, peaking between 667 and 695 nm, were successfully fitted to the theoretical calculations of Beddard (1998), which assumed strong interactions between the six ETC Chls. However, the values of the interaction energies between the six molecules were calculated by Beddard based on the earlier 4-Å structure of PSI (Fromme et al., 1996; Schubert et al., 1997) and are significantly different from those calculated using the 2.5-Å resolution structure that now precisely identifies the orientation of the QY transition dipole moments of the Chls (Jordan et al., 2001; Fromme et al., 2001; Byrdin et al., 2002).
Based on the 2.5-Å structure of PSI, we repeated the calculations of interaction energies between the six ETC Chls (Table 1) and based on simple exciton theory (Van Amerongen et al., 2000; Freiberg et al., 1999) we calculated the six one-exciton levels. Analysis of the interaction energies (Fig. 6) leads to the finding that the six ETC Chls may be considered as a set of three dimers: the primary pair (with interaction energy between two Chls of 426 cm−1) and two accessory Chl-A0 (Ac-A0) pairs (with interaction energies of 194 cm−1 and 160 cm−1, respectively), although the dimers are also connected to each other relatively strongly (−86 cm−1, −91 cm−1). The three-dimer structure, together with the twofold symmetry of the ETC Chls, results in a degeneracy of the one-exciton states: instead of six states, there are only four. Two of these states are mainly associated with the upper and lower exciton states of the primary donor. These two states are characterized by the highest and the lowest energy because the site energy splitting is the biggest, due to the strongest, among six ETC Chls, interaction between the primary pair components. The two remaining states could be ascribed essentially to the upper and lower exciton states of both Ac-A0 pairs. These calculations predict a splitting between the lower and upper states in all three dimers that is too big to explain the small difference found in the experimentally measured band positions between 675 and 683 nm. Therefore, to fit the theoretical predictions to the experimentally measured band positions, one needs to modify the calculations. The most straightforward way to produce accordance between theoretical and experimental results is to include the connecting Chls, which are the closest neighbors of the ETC Chls, in the exciton calculations. Because the interaction energies between them and their closest neighbors from the ETC, the A0 Chls, are relatively weak (31 cm−1, 65 cm−1; Table 1, Fig. 6), both connecting Chls keep, to large extent, their monomeric character and again, due to the twofold symmetry, introduce only one additional exciton level. Thus, accounting for the connecting Chls results in a total of five well-separated one-exciton levels shown in Fig. 7 (actually, there are eight levels, but due to the symmetry some of them are close to each other and may not be resolved in the experiment). In the case of WT PSI, only small variations in the site energies, ranging from 682.5 to 688 nm (Table 1), were made to reproduce the positions of four experimentally measured negative ΔA bands (Fig. 1 A). In the experimental data, the lack of the lowest theoretically predicted band at ∼14,100 cm−1 (Table 2, Fig. 7) will be discussed below.
TABLE 1.
Interaction energies between eight central chlorophyll molecules shown in Fig. 6
| Interaction energies [cm−1]
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Chl a | Site energies [cm−1] (λ [nm]) | PA | PB | AcB | AcA | A0A | A0B | cChlA | cChlB |
| PA | 14,536 (688) | – | 426 {12} | −91 | −25 | 14 | 7 | −1 | 3 |
| PB | 14,536 (688) | – | −24 | −86 | 6 | 13 | 3 | 5 | |
| AcB | 14,592 (685) | – | 28 | 160 | −9 | 6 | −6 | ||
| AcA | 14,592 (685) | – | −10 | 194 | −5 | 18 | |||
| A0A | 14,592 (685) | – | 3 | 31 | 5 | ||||
| A0B | 14,592 (685) | – | 5 | 65 | |||||
| cChlA | 14,652 (682.5) | – | 3 | ||||||
| cChlB | 14,652 (682.5) | – | |||||||
For six RC Chls and two connecting Chls, there are shown site energies taken to fit theoretical data to experimentally observed positions of the ΔA bands (see Fig. 7).
FIGURE 6.

Arrangement and interaction energies between six ETC Chls, two connecting Chls (cChlA and cChlB), and two Chl dimers, A38-A39 and B37-B38, interacting with the two connecting Chls. Full set of interaction energies between the eight central Chls, are presented in Table 1. Interaction energies between any of the six ETC Chls and remaining antenna Chls do not exceed 19 cm−1. Interaction energies between the connecting Chls/the two dimers, and the remaining antenna Chls do not exceed 16 cm−1 except for the Chl A39 that interact with Chl A01 more strongly (−24 cm−1; this interaction is shown as a dashed line).
FIGURE 7.

Positions of experimentally measured and theoretically calculated, based on PSI structure, negative transient absorption bands after excitation at 700 nm. In the case of calculations for the set of eight Chls (WT PSI, HL(A730)/HL(B714) mutant PSI, A0 mutants PSI, and WT PSI with oxidized primary donor), adjustments of site energies of all eight Chls were performed to fit the experimentally measured bands (see Table 1). In the case of calculations for the set of 12 Chls, site energies of all Chls are the same (683 nm). See text for details.
TABLE 2.
The energy levels, oscillator strengths, and distributions of eight exciton states calculated for the system of six RC Chls and two connecting Chls in WT PSI
|
Di,j
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Exciton level [cm−1]([nm]) | Oscillator strength [a.u.] | PA | PB | AcB | AcA | A0A | A0B | cChlA | cChlB |
| 14,099 (709) | 1.96 | 0.484 | 0.483 | 0.014 | 0.013 | 0.003 | 0.003 | 0.000 | 0.000 |
| 14,387 (695) | 1.09 | 0.025 | 0.003 | 0.037 | 0.436 | 0.032 | 0.454 | 0.000 | 0.013 |
| 14,429 (693) | 1.02 | 0.002 | 0.028 | 0.445 | 0.028 | 0.445 | 0.039 | 0.006 | 0.005 |
| 14,631 (683) | 0.59 | 0.006 | 0.001 | 0.000 | 0.105 | 0.002 | 0.033 | 0.054 | 0.798 |
| 14,647 (683) | 1.25 | 0.003 | 0.004 | 0.053 | 0.011 | 0.018 | 0.001 | 0.869 | 0.041 |
| 14,751 (678) | 0.83 | 0.012 | 0.028 | 0.393 | 0.000 | 0.491 | 0.003 | 0.070 | 0.003 |
| 14,802 (676) | 0.84 | 0.023 | 0.010 | 0.008 | 0.353 | 0.004 | 0.462 | 0.000 | 0.140 |
| 14,999 (667) | 0.42 | 0.445 | 0.444 | 0.050 | 0.054 | 0.002 | 0.006 | 0.000 | 0.000 |
The strongest argument for including the connecting Chls into the exciton calculations comes from experiments with the HL(A730)/HL(B714) mutant. Replacing the histidine ligands to the connecting Chls with leucines is expected to, at least, modify the position and/or orientation of these Chls and, consequently, change their interaction energies with their neighbors. Thus, the effect of this mutation may be an “elimination” of the connecting Chls from the excitonically coupled system. This approach was applied to the calculation of exciton levels in this mutant by adjusting the interaction energies between the connecting Chls and A0 Chls (and all remaining ETC Chls) to zero. Because the connecting Chls do not interact very strongly with A0 even in WT PSI, excluding them from the excitonically coupled system in calculations does not significantly affect the remaining bands. Thus, the theoretically calculated band positions (Fig. 7, Table 3) agree with those measured in the experiment (Fig. 1 B).
TABLE 3.
The energy levels, oscillator strengths, and distributions of eight exciton states calculated for the system of six RC Chls and two connecting Chls in the HL(A730)/HL(B714) mutant PSI
|
Di,j
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Exciton level [cm−1] ([nm]) | Oscillator strength [a.u.] | PA | PB | AcB | AcA | A0A | A0B | cChlA | cChlB |
| 14,099 (709) | 1.96 | 0.484 | 0.483 | 0.014 | 0.013 | 0.003 | 0.003 | 0 | 0 |
| 14,390 (695) | 1.17 | 0.025 | 0.003 | 0.051 | 0.445 | 0.045 | 0.431 | 0 | 0 |
| 14,432 (693) | 1.30 | 0.003 | 0.028 | 0.441 | 0.046 | 0.432 | 0.051 | 0 | 0 |
| 14,652 (682.5) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| 14,652 (682.5) | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| 14,743 (678) | 0.43 | 0.017 | 0.033 | 0.428 | 0.007 | 0.503 | 0.011 | 0 | 0 |
| 14,778 (677) | 0.72 | 0.026 | 0.009 | 0.016 | 0.436 | 0.014 | 0.499 | 0 | 0 |
| 14,999 (667) | 0.41 | 0.445 | 0.441 | 0.050 | 0.053 | 0.002 | 0.005 | 0 | 0 |
In WT PSI, the ΔA bands peaking at ∼675 and ∼695 nm are, according to our model, associated mainly with the Ac-A0 dimers in branches A and B (see Fig. 7 and distribution of exciton states in Table 2). However, the 695-nm band observed when exciting at 700 nm is contributed largely by a species that is independent from the excitonically coupled system, as concluded from the fact that excitation at 705 or 710 nm results in a significant decrease in the amplitude of this band (Fig. 4). In fact, this independent origin of the majority of the 695-nm band allows treating it as a good reference for comparing the amplitudes of the remaining bands in WT and A0 mutants (assuming that the amplitude of the 695-nm band is not influenced by the mutations). In the case of all three A0 mutants, after excitation at 700 nm, the amplitudes of the 675- and 683-nm bands are about two times lower relative to the 695-nm bands and compared to the WT PSI (Figs. 1 C and 3 B). Independently, this drop in the amplitudes of these bands is visualized in Fig. 1 F, where subpicosecond DAS amplitudes (at 675 and 683 nm) are again about two times smaller, relative to the ND component and when compared to the WT PSI (Fig. 1 D). The decrease in amplitude of the 675- and 683-nm bands is interpreted in terms of a disturbance in the normal excitonic interactions in branch B in the A0 mutants. In particular, interactions between A0 and its closest neighbors, the accessory Chl and connecting Chl, that are responsible in our model for 675- and 683-nm bands, are expected to be affected by the mutations. As proposed for the HL(A730)/HL(B714) mutant, a change in the position/orientation of the A0 Chl in branch B reduces its interaction energies with neighbors and thus the components of branch B are no longer excitonically coupled. In consequence, the observed ΔA signals at 675 and 683 nm are proposed to come from branch A only, and this explains the approximate twofold drop of the amplitudes. This interpretation is illustrated in Fig. 7. In the case of the A0 mutants, the exciton calculations were performed assuming that the interaction energy between A0 in the B branch and all other Chls is zero (Table 4). As seen from Fig. 7, this does not influence significantly the positions of the bands resulting from excitonic interactions in branch A in line with experimental observations.
TABLE 4.
The energy levels, oscillator strengths, and distributions Di,j of eight exciton states calculated for the system of six RC Chls and two connecting Chls in the A0 mutants PSI
|
Di,j
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Exciton level [cm−1] ([nm]) | Oscillator strength [a.u.] | PA | PB | AcB | AcA | A0A | A0B | cChlA | cChlB |
| 14,100 (709) | 1.84 | 0.491 | 0.484 | 0.014 | 0.009 | 0.003 | 0 | 0.000 | 0.000 |
| 14,426 (693) | 1.15 | 0.000 | 0.029 | 0.482 | 0.005 | 0.478 | 0 | 0.005 | 0.001 |
| 14,576 (686) | 0.94 | 0.041 | 0.007 | 0.001 | 0.886 | 0.003 | 0 | 0.007 | 0.054 |
| 14,592 (685) | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| 14,646 (683) | 0.64 | 0.003 | 0.004 | 0.050 | 0.013 | 0.019 | 0 | 0.885 | 0.028 |
| 14,657 (682) | 1.24 | 0.001 | 0.000 | 0.004 | 0.046 | 0.000 | 0 | 0.032 | 0.917 |
| 14,751 (678) | 0.84 | 0.009 | 0.025 | 0.399 | 0.002 | 0.495 | 0 | 0.070 | 0.000 |
| 14,997 (667) | 0.35 | 0.456 | 0.452 | 0.051 | 0.039 | 0.003 | 0 | 0.000 | 0.000 |
Interactions of the primary donor
Fig. 5 shows that the chemical oxidation of the primary donor to P+ does not result in any significant alterations in the transient ΔA spectrum compared to the experiment under neutral conditions. Oxidation of P is expected to strongly decrease the interaction energy between the two Chls forming the primary pair (Beddard, 1998) and, in consequence, to change the excitonic coupling in the ETC Chls system. This effect should be observable in the experiment if P has contributed to the ΔA spectrum, despite the fact that even under neutral conditions, at low temperatures, ∼50% of the RCs are expected to be in the P+ state (Schlodder et al., 1998; Gibasiewicz et al., 2002). Thus, the similarity between the initial ΔA spectra under neutral and oxidizing conditions may indicate that the primary donor is only weakly coupled to the rest of the ETC Chls and is not significantly represented in the transient spectra after excitation at ≥700 nm. Alternatively, as we have no direct way to monitor the redox state of P, the observed structured ΔA signal may come mainly from closed RC (P+) even under “neutral” conditions. This possibility, however, seems to be less likely in view of the earlier observations that even at low temperatures, P remains in the neutral state in ∼50% of the RCs due to fast charge recombination between primary donor and acceptors (≪1 ms; Schlodder et al., 1998; Gibasiewicz et al., 2002). An additional argument against this possibility is that at RT, when all RCs remains open, a similar, although spectrally less well-resolved transient ΔA signal, was observed (Gibasiewicz et al., 2001).
In our original model, the two bands peaking at 667 and 709 nm are associated almost exclusively with the primary donor (see Table 2). The oscillator strength of the 709-nm band is predicted to be the largest and, in particular, ∼5 times bigger than that of the band at 667 nm. However, as seen from analysis of the exciton states distribution (Table 2; Fig. 8 (see below)), the primary donor, despite having significant interaction energies with the accessory Chls, is only weakly coupled to the rest of the ETC Chls. This may be the reason why in the experiment, at least after 700-nm excitation, there is no photobleaching at 709 nm. This relative isolation of the primary donor from the rest of ETC Chls is also in line with the experimental observation of the insensitivity of the initial ΔA spectrum to the redox state of the primary donor.
FIGURE 8.

Distributions of 12 exciton states over 12 Chls presented in Fig. 6 (except for A01). Each panel presents distribution of one particular exciton state over 12 Chls. In a corner of each panel there is information on which Chl monomer or dimer the particular exciton state is mainly localized. The letters H and L refer to higher and lower excitonic states associated with each Chl dimer, if treating the system as a set of five dimers plus two monomers.
The assignment of the hypothetical 709-nm ΔA band to the primary donor is based on the assumption that the experimentally observed 667-nm band is due to the upper exciton state of the primary donor and on the calculated amount of energy splitting between the upper and lower exciton states. Actually, the 709-nm band is quite unusually red shifted. The (P+–P) difference spectrum for PSI from C. reinhardtii was measured at RT to peak at 696 nm on the millisecond timescale and at 691–693 nm on the picosecond timescale (Melkozernov et al., 1997; Gibasiewicz et al., 2001). So at 20 K, one could expect to observe photobleaching from excited P at a wavelength significantly shorter than 709 nm. One possible way to solve this problem in a model calculation is to down shift the site energies of the P components by, for e.g., from 16 nm to ∼672 nm. As the primary donor is not strongly coupled to the rest of the ETC Chls, this would not significantly change the predictions about the positions of the other ΔA negative bands associated with the other ETC Chls and connecting Chls. If this is correct, the two bands associated with P would peak at 651 and 693 nm, and the experimentally observed band at 667 nm would not be associated with the primary donor. This possibility is supported by the observation that this small band is not sensitive to the oxidation state of P (Fig. 5). Alternatively, the energy splitting for P calculated here to be 900 cm−1 (Table 2) may be overestimated. Actually, any small variation in the position of the primary pair Chls would introduce significant changes in the interaction energy between these two Chls and, consequently, in the energy splitting. It is likely that the RC structure details are a little bit but sufficiently different in PSI from S. elongatus and C. reinhardtii to account for such changes. In fact, a simple example that this may be the case is the different absorption spectrum of (P+–P) in cyanobacteria and Chlamydomonas. Similarly, calculations based on extended dipole approximation and other models instead of point dipole approximation result in a significantly reduced value of the interaction between the primary pair components (Byrdin et al., 2002; Damjanović et al., 2002). Therefore, it is still possible that the 667-nm band together with another band at, for e.g., ∼693 nm, belong to the primary donor. This explanation, however, does not explain the insensitivity of the 667-nm band to the different redox states of P. It should be noticed that the independent species excited at 700 nm and peaking at 695 nm is most probably not associated with P, as it is clearly not sensitive to the oxidation state of the primary donor (Fig. 5) and that most of its oscillator strength comes from independently excited red-shifted antenna Chls (see below).
Finally, the less favorable case of the oxidized primary donor even under “neutral” conditions, should shortly be considered. The simplified exciton calculations for that case were done assuming a strong decrease in the interaction energy between the primary donor components (from 426 cm−1 for P to 12 cm−1 for P+ as proposed by Beddard, 1998) and keeping the other interactions unchanged. This results solely in elimination of the P energy levels splitting whereas the other exciton levels remain unchanged (Fig. 7). The exact position of the exciton levels associated mainly with P+ depends on site energies of the primary donor's components.
Interactions of the connecting chlorophylls
A difficult point in our structure-based model is that the coupling between the connecting Chls and their neighbors (A0) are the weakest among the ETC Chls (31 cm−1 and 65 cm−1; Fig. 6), yet the band ascribed to the exciton state at 683 nm (localized mainly on the connecting Chls) is the largest transient ΔA signal (Fig. 1 A; Table 2). At this time, we do not have a satisfactory explanation for this, although again interaction energies between A0 and the connecting Chls are calculated based on the available structure and may be underestimated. Also, the oscillator strength distributions, both in the WT and mutant PSI, do not fit the amplitudes of the particular bands measured experimentally (Tables 2–4; Fig. 1, A–C).
Excitation wavelength (in)dependence
It is not clear why excitation at 700, 705, and 710 nm can result in reproducible photobleaching at 675, 683, and 695 nm. This is similar to the observation made with PSI from Synechocystis sp. PCC 6803, where excitation at 700, 710, and 720 nm also gave resonant photobleaching at 683 nm and a partly resolved band at 675 (678) nm (Melkozernov et al., 2000b, 2001). There, excitation at 700, 710, and 720 nm was additionally followed by photobleaching at 700 nm (ascribed to the primary donor), 708 nm (ascribed to one of the red Chl pool), and at 714 nm (ascribed to another red Chl pool), respectively. In consequence, the species being photobleached at 683 nm (and 675 nm) was proposed to be coupled both to the primary donor and to the long wavelength forms of Chls, likely two dimers located close to the RC. Based on our study, we propose that the long-wavelength bands (peaking at >690 nm) of both early ΔA (Δt = 0.32 ps) and ND spectra (Δt = 2 ps) (Fig. 4) come from Chls not coupled to the ETC Chls. The profiles of these long-wavelength bands are so much different with 700-, 705-, and 710-nm excitation (also with 700-, 710-, and 720-nm excitation in Melkozernov et al. (2000b, 2001)) that it is hard to believe that they are all so strongly coupled to the 675/683-nm bands and not at all to each other. We suggest that the similar bands at 675, 683 (and 695) nm at different excitation wavelengths are due to excitation of a very wide and low amplitude absorption band that cannot be resolved in our studies (due to noise) and that is strongly coupled to the 675/683/695-nm bands. Actually, there are many strongly coupled dimers in PSI far away from the RC that may be responsible for the photobleaching at >700 nm shown in Fig. 4 (Byrdin et al., 2002).
Chlorophyll dimers neighboring the ETC chlorophylls
For the sake of completeness and to further test the hypothesis that the Chl dimers that are close neighbors of the RC may contribute to the observed ΔA spectra, we extended our exciton calculation to include the two Chl dimers, A38-A39 and B37-B38 (Fig. 6). Because these dimers are relatively weakly coupled to the ETC Chls via the connecting Chls (−39 cm−1, −41 cm−1) the new system is essentially composed of five dimers plus two connecting Chls. It turns out that the interaction energies and, consequently, the energy splitting in these additional dimers is comparable to those in the Ac-A0 pairs. Thus, it is possible that the additional dimers contribute to the 675- and 695-nm bands observed in the experiment (Fig. 7) but there is no particular reason to claim that just these dimers are red Chls absorbing at 703, 708, or 714 nm. Actually, the coupling between the primary pair components significantly exceeds coupling between components of the four remaining dimers (Fig. 6, Table 1) and still its absorption band probably does not peak above 700 nm.
In Fig. 8, the distribution of all 12 exciton states localized over the 12 Chls is presented (compare to Fig. 6). Most of the observations from this figure are the same as in the simpler case of the set of eight Chls (see Table 2). The interesting and unexpected new feature is that some exciton states are quite evenly distributed between the two antenna dimers and the neighboring Ac-A0 pairs (via connecting Chls), especially in the B branch (Fig. 8, B, C, J, and K). This means, that these two antenna dimers may indeed contribute to the observed initial ΔA spectra.
Melkozernov et al. (2000b) initially put forward the hypothesis that the 683-nm (and 675-nm) ΔA band may be the upper excitonic state of one of the dimers located close to the RC. In view of our observation that the 683-nm band disappears in the HL(A730)/HL(B714) connecting Chls mutant, this interpretation seems to be not correct. Later, Melkozernov et al. (2001) proposed that the 678- (675) and 683-nm bands originate from borrowing of the oscillator strength from the excited state absorption of a Chl dimer to a nearby Chl monomer (Valkunas et al., 1999; van Amerongen et al., 2000). Our studies on the WT and the HL(A730)/HL(B714) mutant do not exclude this interpretation. Moreover, if it is correct, they indicate that the “monomeric” Chl is a connecting Chl and the Chl dimer that interacts with the monomer is either an Ac-A0 pair or the A38-A39 (B37-B38) dimer in one or both branches.
The origin of the subpicosecond dynamics
The fast decay of both positive and negative bands of the ΔA spectrum (Fig. 2) could be interpreted in three ways. First, the fast decay could be due to the charge separation. Kumazaki et al. (2001), who studied PSI particles with 12–13 Chls per primary donor, observed a subpicosecond ΔA decay component that they ascribed to immediate charge separation. However, observation of similar dynamics of the spectrum under oxidizing conditions, when charge separation from the primary donor cannot occur, seems to exclude this possibility, unless charge separation occurs from the accessory Chl forming the state P+Ac+A0−. Interestingly, charge separation between the accessory Chl and the primary acceptor has been observed in PSII-type RC (Diner et al., 2001; van Brederode et al., 1999). This hypothesis could further be elucidated by studying PSI with mutations affecting the accessory Chls. Alternatively, the subpicosecond component could be due to exciton localization on a smaller subset (or one) of the initially coupled Chls, likely on the primary donor, or be due to the escape of excitation to the antenna. Both these interpretations encounter difficulties, as one could expect that initial excitation of the same group of Chls should result in exciton localization on the same pool of Chls, and the actual 2-ps spectra are clearly excitation wavelength dependent (Fig. 4). Finally, the fast dynamics may be due to exciton quenching by the oxidized primary donor in this fraction of RCs that remain closed.
The model of excitonically coupled Chls that we propose in this paper deals only with the negative part of the ΔA spectrum. However, the positive bands have the same kinetics (Fig. 2), and are interpreted as previously reported (Gibasiewicz et al., 2002) as excited-state absorption from one-exciton states to two-exciton states. The quality of the experimental data in mutants or at excitation at >700 nm does not allow for a detailed analysis of this part of the ΔA spectrum. The observation that the red edge of the positive part of the ΔA spectrum shifts to the red together with the excitation wavelength, may be explained by the contribution of additional excited-state absorption from different Chls that are excited at 700, 705, and 710 nm (see above).
CONCLUSIONS
Excitation of the red edge of the QY absorption band of Chls in PSI causes transient ΔA spectra in a wide blue-shifted spectral range. Photobleaching at 675 and 683 nm is a universal feature of PSI observed both in eukaryotic C. reinhardtii and prokaryotic Synechocystis, although differences in (P+–P) ΔA spectra, (A0−–A0) ΔA spectra, and the amount of red Chls in these two organisms have been reported (Hastings et al., 1995; Gibasiewicz et al., 2001, 2002). The 683-nm ΔA band was ascribed to the connecting Chls, and the wide transient ΔA spectrum was associated with an excitonically coupled group of chlorophylls: the ETC Chls, connecting Chls and possibly two nearby antenna Chl dimers. The primary donor does not significantly contribute to the observed ΔA spectra, likely due to a relatively weak interaction with the rest of the ETC Chls compared to the interaction between the two chlorophylls constituting P. The wide, multiband structure of the ΔA spectra associated with the ETC Chls and their closest antenna neighbors reveals wide steady-state absorption spectrum of this excitonically coupled system. This wide spectrum may be of biological significance as it is expected to improve spectral overlap between RC Chls and different spectral forms of antenna Chls and thus increase efficiency of energy trapping in photosystem 1, as previously suggested (Gibasiewicz et al., 2002).
Acknowledgments
We thank Professor A. Freiberg for a valuable discussion.
This work was supported by the U. S. Department of Energy (grant DE-FG03-99ER20349 to A.W. and N.W.).
References
- Beddard, G. S. 1998. Exciton coupling in the photosystem I reaction center. J. Phys. Chem. B. 102:10966–10973. [Google Scholar]
- Boudreaux, B., F. MacMillan, C. Teutloff, R. Agalarov, F. Gu, S. Grimaldi, R. Bittl, K. Brettel, and K. Redding. 2001. Mutations in both sides of the photosystem I reaction center identify the phylloquinone observed by electron paramagnetic resonance spectroscopy. J. Biol. Chem. 276:37299–37306. [DOI] [PubMed] [Google Scholar]
- Byrdin, M., P. Jordan, N. Krauss, P. Fromme, D. Stehlik, and E. Schlodder. 2002. Light harvesting in photosystem I: modeling based on 2.5-Å structure of photosystem I from Synechococcus elongatus. Biophys. J. 83:433–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanović, A., H. M. Vaswani, P. Fromme, and G. R. Fleming. 2002. Chlorophyll excitations in photosystem I of Synechococcus elongatus. J. Phys. Chem. B. 106:10251–10262. [Google Scholar]
- Diner, B. A., E. Schlodder, P. J. Nixon, W. J. Coleman, F. Rappaport, J. Lavergne, W. F. J. Vermaas, and D. A. Chisholm. 2001. Site-directed mutations of D1–198 and D2–197 of photosystem II in Synechocystis PCC 6803: sites of primary charge separation and cation and triplet stabilization. Biochemistry. 40:9265–9281. [DOI] [PubMed] [Google Scholar]
- Durrant, J. R., D. R. Klug, S. L. S. Kwa, R. van Grondelle, G. Porter, and J. P. Dekker. 1995. A multimer model for P680, the primary electron donor of photosystem II. Proc. Natl. Acad. Sci. USA. 92:4798–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiberg, A., S. Lin, K. Timpmann, and R. E. Blankenship. 1997. Exciton dynamics in FMO bacteriochlorophyll protein at low temperatures. J. Phys. Chem. 101:7211–7220. [DOI] [PubMed] [Google Scholar]
- Freiberg, A., K. Timpmann, S. Lin, and N. W. Woodbury. 1998. Exciton relaxation and transfer in the LH2 antenna network of photosynthetic bacteria. J. Phys. Chem. 102:10974–10982. [Google Scholar]
- Freiberg, A., K. Timpmann, R. Ruus, and N. W. Woodbury. 1999. Disordered exciton analysis of linear and nonlinear absorption spectra of antenna bacteriochlorophyll aggregates: LH2–only mutant chromatophores of Rhodobacter sphaeroides at 8 K under spectrally selective excitation. J. Phys. Chem. 103:10032–10041. [Google Scholar]
- Frese, R. N., M. A. Palacios, A. Azzizi, I. H. M. van Stokkum, J. Kruip, M. Rögner, N. V. Karapetyan, E. Schlodder, R. van Grondelle, and J. P. Dekker. 2002. Electric field effects on red chlorophylls, β-carotenes and P700 in cyanobacterial photosystem I complexes. Biochim. Biophys. Acta. 1554:180–191. [DOI] [PubMed] [Google Scholar]
- Fromme, P., H. T. Witt, W. D. Schubert, O. Klukas, W. Saenger, and N. Krauss. 1996. Structure of photosystem I at 4.5 Å resolution: a short review including evolutionary aspects. Biochim. Biophys. Acta. 1275:76–83. [Google Scholar]
- Fromme, P., Jordan, P., Krauβ, N. 2001. Structure of photosystem I. Biochim. Biophys. Acta. 1507:5–31. [DOI] [PubMed] [Google Scholar]
- Gibasiewicz, K., V. M. Ramesh, A. N. Melkozernov, S. Lin, N. W. Woodbury, R. E. Blankenship, and A. N. Webber. 2001. Excitation dynamics in the core antenna of PS I from Chlamydomonas reinhardtii CC2696 at room temperature. J. Phys. Chem. B. 105:11498–11506. [Google Scholar]
- Gibasiewicz, K., V. M. Ramesh, S. Lin, N. W. Woodbury, and A. N. Webber. 2002. Excitation dynamics in eukaryotic PS I from Chlamydomonas reinhardtii CC269 at 10 K. Direct detection of the reaction center exciton states. J. Phys. Chem. B. 106:6322–6330. [Google Scholar]
- Gobets, B., H. van Amerongen, R. Monshouwer, J. Kruip, M. Rögner, R. van Grondelle, and J. P. Dekker. 1994. Polarized site-selected fluorescence spectroscopy of isolated photosystem I particles. Biochim. Biophys. Acta. 1188:75–85. [Google Scholar]
- Gobets, B., and R. van Grondelle. 2001. Energy transfer and trapping in photosystem I. Biochim. Biophys. Acta. 1507:80–99. [DOI] [PubMed] [Google Scholar]
- Golbeck, J. H. 1994. In Molecular Biology of Cyanobacteria. D. A. Bryant, editor. Kluwer Academic Publishers, Dordrecht, The Netherlands. 179–220.
- Hastings, G., S. Hoshina, A. N. Webber, and R. E. Blankenship. 1995. Universality of energy and electron transfer processes in photosystem I. Biochemistry. 34:15512–15522. [DOI] [PubMed] [Google Scholar]
- Holzwarth, A. R., G. Schatz, H. Brock, and E. Bittersman. 1993. Energy transfer and charge separation kinetics in photosystem I. Part 1: picosecond transient absorption and fluorescence study of cyanobacterial photosystem I particles. Biophys. J. 64:1813–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson, S. 1994. The light-harvesting chlorophyll a/b-binding proteins. Biochim. Biophys. Acta. 1184:1–19. [DOI] [PubMed] [Google Scholar]
- Jordan, P., P. Fromme, H. T. Witt, O. Klukas, W. Saenger, and N. Krauβ. 2001. Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature. 411:909–917. [DOI] [PubMed] [Google Scholar]
- Karapetyan, N. V., A. R. Holzwarth, and M. Rögner. 1999. The photosystem I trimer of cyanobacteria: molecular organization, excitation dynamics and physiological significance. FEBS Lett. 460:395–400. [DOI] [PubMed] [Google Scholar]
- Krabben, L., E. Schlodder, R. Jordan, D. Carbonera, G. Giacometti, H. Lee, A. N. Webber, and W. Lubitz. 2000. Influence of the axial ligands on the spectral properties of P700 of photosystem I: a study of site-directed mutants. Biochemistry. 39:13012–13025. [DOI] [PubMed] [Google Scholar]
- Kumazaki, S., I. Ikegami, H. Furusawa, S. Yasuda, and K. Yoshihara. 2001. Observation of the excited state of the primary electron donor chlorophyll (P700) and the ultrafast charge separation in the spinach Photosystem I reaction center. J. Phys. Chem. B. 105:1093–1999. [Google Scholar]
- Mathis, P., I. Ikegami, and P. Sétif. 1988. Nanosecond flash studies of the absorption spectrum of the photosystem I primary acceptor A0. Photosynth. Res. 16:203–210. [DOI] [PubMed] [Google Scholar]
- McDermott, G., S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell, and N. W. Isaacs. 1995. Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature. 374:517–521. [Google Scholar]
- Melkozernov, A. N., H. Su, S. Lin, S. E. Bingham, A. N. Webber, and R. E. Blankenship. 1997. Specific mutation near the primary donor in photosystem I from Chlamydomonas reinhardtii alters the trapping time and spectroscopic properties of P700. Biochemistry. 36:2898–2907. [DOI] [PubMed] [Google Scholar]
- Melkozernov, A. N., J. M. Olson, Y. F. Li, J. P. Allen, and R. E. Blankenship. 1998a. Orientation and excitonic interactions of the Fenna–Matthews–Olson bacteriochlorophyll a protein in membranes of the green sulfur bacterium Chlorobium tepidum. Photosynth. Res. 56:315–328. [Google Scholar]
- Melkozernov, A. N., V. H. R. Schmid, G. W. Schmidt, and R. E. Blankenship. 1998b. Energy redistribution in heterodimeric light-harvesting complex LHCI-730 of Photosystem I. J. Phys. Chem B. 102:8183–8189. [Google Scholar]
- Melkozernov, A. N., S. Lin, V. H. R. Schmid, H. Paulsen, G. W. Schmidt, and R. E. Blankenship. 2000a. Ultrafast excitation dynamics of low energy pigments in reconstituted peripheral light-harvesting complexes of photosystem I. FEBS Lett. 471:89–92. [DOI] [PubMed] [Google Scholar]
- Melkozernov, A. N., S. Lin, and R. E. Blankenship. 2000b. Femtosecond transient spectroscopy and excitonic interactions in Photosystem I. J. Phys. Chem. B. 104:1651–1656. [DOI] [PubMed] [Google Scholar]
- Melkozernov, A. N., S. Lin, and R. E. Blankenship. 2000c. Excitation dynamics and heterogeneity of energy equilibration in the core antenna of photosystem I from cyanobacterium Synechocystis sp. PCC 6803. Biochemistry. 39:1489–1498. [DOI] [PubMed] [Google Scholar]
- Melkozernov, A. N., S. Lin, R. E. Blankenship, and L. Valkunas. 2001. Spectral inhomogeneity of photosystem I and its influence on excitation and trapping in the cyanobacterium Synechocystis sp. PCC6803 at 77 K. Biophys. J. 81:1144–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechushtai, R., A. Eden, Y. Cohen, and J. Klein. 1996. In Oxygenic Photosynthesis: The Light Reactions. D. R. Ort, C. F. Yocum, editors. Kluwer Academic Publishers, Dordrecht. 289–311.
- Palacios, M. A., F. L. de Weerd, J. A. Ihalainen, R. van Grondelle, and H. van Amerongen. 2002. Superradiance and exciton (de)localization in light-harvesting complex II from green plants? J. Phys. Chem B. 106:5782–5787. [Google Scholar]
- Pålsson, L.-O., J. P. Dekker, E. Schlodder, R. Monshouwer, and R. van Grondelle. 1996. Polarized site-selective fluorescence spectroscopy of the long-wavelength emitting chlorophylls in isolated photosystem I particles of Synechococcus elongates. Photosynth. Res. 48:239–246. [DOI] [PubMed] [Google Scholar]
- Redding, K., F. MacMillan, W. Leibl, K. Brettel, K. Hanley, A. Rutherford, J. Breton, and J. D. Rochaix. 1998. A systematic survey of conserved histidines in the core subunits of Photosystem I by site-directed mutagenesis reveals the likely axial ligands of P700. EMBO J. 17:50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlodder, E., K. Falkenberg, M. Gergeleit, and K. Brettel. 1998. Temperature dependence of forward and reverse electron transfer from A1−, the reduced secondary electron acceptor in photosystem I. Biochemistry. 37:9466–9476. [DOI] [PubMed] [Google Scholar]
- Schubert, W.-D., O. Klukas, N. Krauβ, W. Saenger, P. Fromme, and H. T. Witt. 1997. Photosystem I of Synechococcus elongatus at 4 Å resolution: comprehensive structure analysis. J. Mol. Biol. 272:741–769. [DOI] [PubMed] [Google Scholar]
- Shipman, L. L. 1977. Oscillator and dipole strength for chlorophyll and related molecules. Photochem. Photobiol. 26:287–292. [Google Scholar]
- Stewart, D. H., A. Cua, D. F. Bocian, and G. W. Brudvig. 1999. Selective Raman scattering from the core chlorophylls in photosystem I. J. Phys. Chem B. 103:3758–3764. [Google Scholar]
- Sundström, V., T. Pullerits, and R. van Grondelle. 1999. Photosynthetic light-harvesting: reconciling dynamics and structure of purple bacterial LH2 reveals function of photosynthetic unit. J. Phys. Chem B. 103:2327–2346. [Google Scholar]
- Trissl, H.-W. 1993. Long wavelength absorbing antenna pigments and heterogeneous absorption bands concentrate excitons and increase absorption cross section. Photosynth. Res. 35:247–263. [DOI] [PubMed] [Google Scholar]
- Valkunas, L., V. Cervinskas, G. Trinkunas, M. G. Muller, and A. R. Holzwarth. 1999. Effects of excited state mixing on transient absorption spectra in dimmers: application to photosynthetic light-harvesting complex II. J. Chem. Phys. 111:3121–3132. [Google Scholar]
- Van Amerongen, H., L. Valkunas, and R. van Grondelle. 2000. Photosynthetic Excitons. World Scientific, Singapore-New Jersey-London-Hong Kong.
- Van Brederode, M. E., F. van Mourik, I. H. M. van Stokkum, M. R. Jones, and R. van Grondelle. 1999. Multiple pathways for ultrafast transduction of light energy in the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 96:2054–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Grondelle, R., J. P. Dekker, T. Gillbro, and V. Sundström. 1994. Energy transfer and trapping in photosynthesis. Biochim. Biophys. Acta. 1187:1–65. [Google Scholar]
- Webber, A. N., and S. E. Bingham. 1998. In The Molecular Biology of Chloroplasts and Mitochondria in Chlamydomonas. J.-D. Rochaix, M. Goldschmidt-Clermont, S. Merchant, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 323–348.
- Wendling, M., M. A. Przyjalgowski, D. Gullen, S. I. E. Vulto, T. J. Aartsma, R. van Grondelle, and H. van Amerongen. 2002. The quantitative relationship between structure and polarized spectroscopy in the FMO complex of Prosthecochloris aestuarii: refining experiments and simulations. Photosynth. Res. 71:99–123. [DOI] [PubMed] [Google Scholar]
- White, N. T. H., G. S. Beddard, J. R. G. Thorne, T. M. Feehan, T. E. Keyes, and P. Heathcote. 1996. Primary charge separation and energy transfer in the photosystem I reaction center of higher plants. J. Phys. Chem. 100:12086–12099. [Google Scholar]