

Abstract
Photosystem II catalyzes photosynthetic water oxidation in plants, green algae, and cyanobacteria. The manganese-containing active site cycles through a series of five oxidation states, Sn, where n refers to the number of oxidizing equivalents stored. In this report, reaction-induced Fourier transform infrared and electron paramagnetic resonance spectra of the S1-to-S2 transition are presented. These data suggest that changes in carboxylate ligation to manganese, changes in secondary structure, and/or changes in polarity occur during dark adaptation in the S1 state. These spontaneous structural changes are attributed to a S1′ intermediate, at the same oxidation level as S1, in the process of photosynthetic water oxidation.
INTRODUCTION
Photosystem II (PSII) catalyzes the oxidation of water and the reduction of plastoquinone. Oxygen is produced as a product of the water oxidation reaction. This process requires a tetra-nuclear Mn cluster at the oxygen-evolving catalytic site (OEC). Upon photoexcitation, the primary chlorophyll donor, P680, donates an electron to QA, which is a plastoquinone-9 molecule that acts as a single electron acceptor. QA− in turn reduces a second plastoquinone-9 molecule, QB, which functions as a two-electron and two-proton acceptor. P680+ oxidizes a redox-active tyrosine, Z, which then abstracts an electron from the Mn cluster (Britt, 1996). Early studies established that four photo-oxidation reactions are required for oxygen production and postulated that the OEC cycles through at least five different intermediate states, S0–4 (Joliot and Kok, 1975). These photoconverted states correspond to different oxidation states of the catalytic site. Oxygen is produced from the S4 state, which is unstable and resets to the S0 state. The S0 state slowly converts to the S1 state in the dark through electron transfer to a second tyrosyl radical, tyrosyl D•. S1 is thus the dark-stable state of the OEC. Recently, x-ray-diffraction-derived structures of cyanobacterial PSII have been presented (Kamiya and Shen, 2003; Zouni et al., 2001). However, the resolution of the structural models is not yet adequate to define the position of amino-acid side chains (Kamiya and Shen, 2003; Zouni et al., 2001).
Proposals for the structure of the Mn cluster have been suggested, based on magnetic resonance studies of PSII (see Peloquin et al., 2000, and references therein). The cluster is known to consist of four manganese atoms, which are linked by oxobridges. Coordination is provided by amino-acid residues in PSII subunits and by substrate water (reviewed in Barry et al., 1994; Jansson and Maeenpaeae, 1997; Vermaas, 1998). At least one histidine coordinates manganese (Tang et al., 1994). Other ligands are probably provided by carboxylate side chains of aspartate and glutamate residues (for examples, see Boerner et al., 1992; Pujols-Ayala and Barry, 2002; and references therein).
Many different mechanisms have been proposed to explain the chemistry of photosynthetic oxygen evolution (for examples, see Haumann and Junge, 1999; Hoganson and Babcock, 1997; Messinger, 2000; Pecoraro et al., 1998; Vrettos et al., 2001). Spectroscopic techniques, such as magnetic resonance, x-ray absorption/fluorescence, and optical spectroscopies, have been applied to understanding the OEC, but have not yet differentiated among the proposed mechanisms (for review see Britt, 1996). Many of these spectroscopic studies have focused on the S1-to-S2 transition. Fourier transform infrared spectroscopy (FT-IR) is particularly well suited for studies of water oxidation mechanism, because it is sensitive to alterations in coordination and protonation of the Mn cluster (for representative examples, see Hillier and Babcock, 2001; Hutchison et al., 1999; Kimura and Ono, 2001; Noguchi and Sugiura, 2001; Zhang et al., 1998; and references therein).
Proposed mechanisms and previous spectroscopic studies leave open the possibility of other intermediates in photosynthetic oxygen evolution. Previous studies have suggested a rearrangement of the Mn cluster during dark adaptation of PSII to give a resting state (Beck et al., 1985; Koulougliotis et al., 1992). Later studies showed that several, distinct electron paramagnetic resonance (EPR) signals originate from the S1 state (Campbell et al., 1998; Dexheimer and Klein, 1992, Nugent et al., 2002). Two distinct EPR signals are also observed from the S2 state (reviewed in Miller and Brudvig, 1991). In addition, two distinct FT-IR spectra have been assigned to the S1-to-S2 transition (Noguchi et al., 1995; Steenhuis and Barry, 1997). Taken together, these data could be consistent with additional intermediates between the S1 and S2 states. In this report, we have used reaction-induced FT-IR spectroscopy to probe the identity of these intermediate states. We find evidence for the existence of more than one S1 dark state. Our data suggest that multiple S1 states interconvert in the dark, possibly through changes in carboxylate ligation to Mn.
MATERIALS AND METHODS
PSII was isolated from market spinach (Berthold et al., 1981), and the preparations employed had an average oxygen evolution rate of 1000 μmol O2/mg chl-hr, when assayed as previously described (Barry, 1995). In some experiments, the OEC was inactivated with alkaline Tris to give manganese-depleted PSII (Anderson et al., 2000).
In Figs. 1 and 2, the PSII samples, 3–4 mg chl/ml, were mixed with the exogenous electron acceptors, 0.12 mM potassium ferricyanide and 0.02 mM recrystallized 2,6-dichloro-p-benzoquinone (DCBQ), and were pelleted at 20,000 rpm for 5 min. In Fig. 4, the samples, 3–4 mg chl/ml, were mixed with 0.12 mM potassium ferricyanide, 0.02 mM recrystallized 2,6-DCBQ, and 0.1 mM 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU), and were pelleted at 20,000 rpm for 5 min. The pellet was then placed on a CaF2 window, concentrated by evaporation with a stream of nitrogen, and sealed with a second CaF2 window. A water-tight seal was achieved with a thin bead of vacuum grease, which did not contact the sample. All samples were in a sucrose buffer, containing 0.4 M sucrose, 50 mM 2-(n-morpholino)ethanesulfonic acid-NaOH, pH 6.0, and 15 mM NaCl.
FIGURE 1.

Light-minus-dark difference FT-IR spectra, associated with S2QB−-minus-S1QB at 4°C. Difference spectra were constructed from data acquired immediately before the actinic flash and data acquired 0–15 s (A); 15–30 s (B); 30–45 (C); and 45–60 s (D) after the actinic flash. Samples were preflashed 2 h before the actinic flash to preset PSII centers in the S1 state. E shows a control spectrum constructed with 5 s of data obtained before the actinic flash. F shows a control spectrum constructed with 15 s of data obtained before the actinic flash. To correct for the difference in data acquisition time relative to the light-minus-dark spectra, the control in E was scaled by a factor of 1.7. On the y-axis, the tick marks represent 2 × 10−4 absorbance units.
FIGURE 2.

Light-minus-dark difference FT-IR spectra, associated with S2QB−-minus-S1QB at 4°C. Difference spectra were constructed from data acquired immediately (0–30 s) after the actinic flash and data acquired immediately before the flash. In A, a preflash was given, followed by a 2-h dark adaptation before the actinic flash. In B, a preflash was given, followed by a 1-h dark adaptation before the actinic flash. In C, a preflash was given, followed by a 20-min dark adaptation before the actinic flash. D, solid line, shows a control spectrum constructed with data acquired before each actinic flash. D, dotted line, shows a control light-minus-dark spectrum acquired from manganese-depleted PSII. On the y-axis, the tick marks represent 2 × 10−4 absorbance units.
FIGURE 4.

Light-minus-dark difference FT-IR spectra, associated and S2QA−-minus-S1QA at 4°C. Difference spectra were constructed from data acquired immediately (0–30 s) after the actinic flash and data acquired immediately before the flash. In A, a preflash was given, followed by a 2-h dark adaptation before the actinic flash. In B, a preflash was given, followed by a 1-h dark adaptation before the actinic flash. In C, a preflash was given, followed by a 20-min dark adaptation before the actinic flash. D shows a control spectrum constructed with data acquired before each actinic flash. On the y-axis, the tick marks represent 2 × 10−4 absorbance units.
The low concentrations of exogenous acceptors, 0.12 mM potassium ferricyanide and 0.02 mM recrystallized 2,6-DCBQ, were chosen on the basis of several criteria. First, samples showed rapid and reproducible oxidation of QA−, as determined from fluorescence experiments (data not shown). Second, samples showed high retention of oxygen evolution activity after the measurement (70 ± 12%). Third, the DCBQ and ferricyanide concentrations were low enough that no significant spectral contribution was observed from acceptor-based redox reactions. Higher concentrations of potassium ferricyanide (20 mM) gave a faster QA− oxidation reaction, by a factor of 7, but a much lower retention of oxygen evolution activity (38 ± 18%) after the measurement.
Actinic and preflashes were provided by a frequency-doubled, 532-nm output from a Surelight I or III Nd:YAG laser (Continuum, Santa Clara, CA). The pulse width was ∼7 ns, and the pulse energy was 20–30 mJ cm−2. FT-IR data were collected on a Bruker (Billerica, MA) IFS-66v/S spectrometer, equipped with a MCT detector and a Harrick (Ossining, NY) temperature controller. The temperature was 4°C, and the spectral resolution was 8 cm−1. A Happ-Genzel apodization function and four levels of zero filling were employed. A germanium filter blocked illumination of the sample by the internal HeNe laser of the FT-IR spectrometer. FT-IR data acquisition began 40 ms after the actinic flash, and the data were collected in 5-s data sets (34 mirror scans) for a minimum of 60 s. These data sets were ratioed to data recorded before the laser flash to give individual difference FT-IR spectra. Data collected either over 15 s (Fig. 1) or over 30 s (Figs. 2, 4, and 5) were then averaged. Dark-minus-dark controls were constructed from 5-s or 15-s data sets recorded before the actinic flash. All spectra were normalized to an amide II amplitude of 0.5 absorbance units. In Fig. 1 and Fig. 2, the spectra are an average of 25–27 difference spectra acquired from 9–16 samples. In Fig. 4, spectra are an average of 7–8 difference spectra acquired on 3–7 different samples.
FIGURE 5.

Double difference spectra, reflecting the spectral changes induced by an increase in dark adaptation time. In A, the spectrum was generated by a one-to-one subtraction of Fig. 4 C (20-min dark adaptation) from Fig. 4 A (2-h dark adaptation). In B, the spectrum was generated by a one-to-one subtraction of Fig. 2 C (20-min dark adaptation) from Fig. 2 A (2-h dark adaptation). C shows a control double difference spectrum constructed by subtracting one-half of the 2-h data set from the other half of the data set and dividing by a factor of 1.4. D shows a control spectrum constructed with data acquired before each actinic flash. On the y-axis, the tick marks are 1 × 10−4 absorbance units.
EPR samples were prepared as the FT-IR samples, except that the EPR samples were concentrated by evaporation on Mylar strips, instead of CaF2 windows. Actinic and preflashes were provided as described above. Data was collected at 10 K on a Bruker EMX spectrometer, equipped with an Oxford cryostat. Parameters were: 32 G modulation amplitude; 10 mW, microwave power; 82 ms, time constant; 168 s, scan time; and eight total scans.
RESULTS
In Fig. 1, reaction-induced FT-IR spectra, associated with S2QB− -minus-S1QB, are presented. PSII samples were given a preflash, dark adapted for 2 h, and then given an actinic flash to photo-oxidize the S1 state. Although there may be heterogeneity in QB occupancy (see de Wijn and van Gorkom, 2001, and references therein), we assume that QB is the terminal endogenous electron acceptor in the majority of PSII centers. QB− will then be oxidized by the exogeneous acceptors added to the sample. However, the concentration of these acceptors is low, and no spectral contribution from the electron acceptors is evident in these data. For example, the CN vibrational band associated with ferricyanide reduction is not observed (Kim and Barry, 1998).
Difference spectra were constructed from data collected 0–15 s (Fig. 1 A), 15–30 s (Fig. 1 B), 30–45 s (Fig. 1 C), or 45–60 s (Fig. 1 D) after an actinic flash and from data collected immediately before the actinic flash. Difference spectra in Fig. 1 exhibit significant signals, compared to control spectra, constructed from data acquired before each actinic flash (Fig. 1, E and F). Fig. 1 A resembles a previously reported S2QB− -minus-S1QB spectrum (Zhang et al., 1998). EPR controls (data not shown) show there is a significant tyrosyl D• contribution to this spectrum. The assignments of spectral features have been discussed (Kim and Barry, 1998; Kim et al., 2000; Noguchi et al., 1995; Zhang et al., 1998).
Fig. 2 compares S2QB− -minus-S1QB data acquired in the first 30 s after the actinic flash. The spectra were obtained with different dark adaptation times between a preflash and an actinic flash. In Fig. 2 A, PSII was dark-adapted for 2 h after the preflash, and in Fig. 2 B, PSII was dark-adapted for 1 h after the preflash. In these samples, ∼100% of the reaction centers will be preset in the S1 state (Roelofs et al., 1996). In Fig. 2 C, PSII was dark-adapted for 20 min after the preflash. In this sample, our EPR control experiments will show that ∼100% of the centers are also preset in the S1 state (see Fig. 3 below).
FIGURE 3.
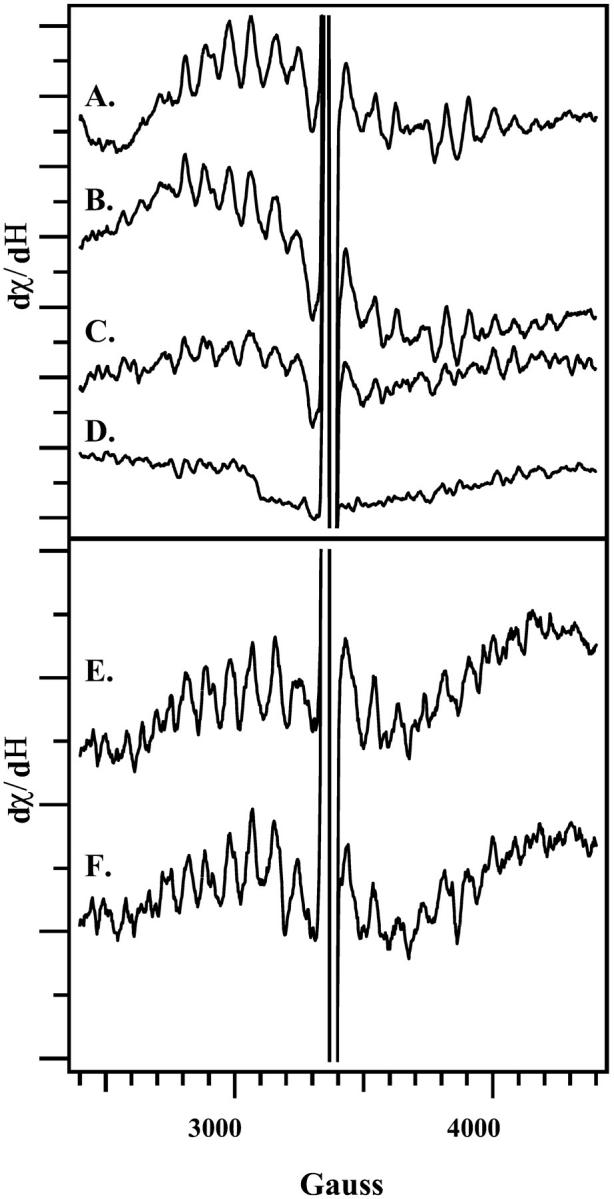
Light-minus-dark EPR spectra, reflecting the decay of the S2 multiline signal after an actinic flash. Spectra reflect the amount of the S2 signal remaining immediately (A); 30 s (B); 5 min (C); or 20 min (D) after an actinic flash. In E and F, PSII was dark-adapted for 1 h and then preflashed at 4°C. The sample was then incubated at 4°C in the dark either for 1 h (E) or 20 min (F), flashed, and then immediately frozen. EPR samples were concentrated on Mylar and were prepared in the same manner as the FT-IR samples employed in Figs. 1 and 2.
When the spectra in Fig. 2, A–C, are compared, alterations are observed in the 1430–1360 cm−1 region as a function of dark adaptation time (Fig. 2, A–C). For example, spectral shifts are observed between 1405 and 1344 cm−1 that cause changes in the intensity and width of vibrational bands. This 1430–1360 cm−1 region may contain contributions from the symmetric stretching vibrations of aspartate and glutamate residues in proteins (see Bellamy, 1980, and references therein). Accompanying spectral contributions are also observed in the 1690–1630 cm−1 region (Fig. 2, A–C). These spectral changes are significant, compared to control data obtained before the actinic flash (Fig. 2 D, solid line) or to data obtained on manganese-depleted PSII (Fig. 2 D, dotted line). Other spectral changes also occur and will be discussed in more detail below.
To investigate the origin of these dark adaptation-induced spectral changes, control experiments were performed. Changes in water content have the potential to alter the difference FT-IR spectra, associated with S-state advancement (Noguchi and Sugiura, 2002). Therefore, we assessed the water content of our samples by calculating the ratio of the absorbance at 3290 cm−1 (water plus protein contributions) to the absorbance at 1545 cm−1 (protein contributions). The ratio was derived from each FT-IR absorption spectrum (Patzlaff et al., 2000). The average ratios for the 2-h, 1-h, and 20-min data sets were indistinguishable and were 2.15 + 0.35, 2.22 + 0.40, and 2.13 + 0.39, respectively. These values are in a hydration range in which no inhibitory effects on PSII activity are expected (Noguchi and Sugiura, 2002). There was also little change in water content in a single sample over the course of data acquisition. For example, in a representative 6-h experiment, the initial 3290/1545 cm−1 ratio was 2.34, and the final 3290/1545 cm−1 ratio was 2.26. Therefore, we conclude that alterations in PSII hydration state do not cause the spectral changes observed here.
The spectral differences, observed when Fig. 2, A–C, are compared, could arise from incomplete decay of the S2 state with a 20-min dark adaptation. If the S2 state has not completely decayed in the 20 min after the preflash, then the actinic flash will produce a mixture of S2 and S3 states, which can alter the FT-IR spectrum (Hillier and Babcock, 2001; Noguchi and Sugiura, 2001). To measure the lifetime of the S2 state in these samples, EPR spectroscopy was employed. The S2 state gives rise to a multiline signal centered at g = 2.0, which can be used to monitor S2 state decay under the conditions employed for FT-IR spectroscopy. For EPR experiments, PSII samples were dark-adapted for 1 h and given an actinic flash at 4°C. Acceptor concentrations were the same as the concentrations employed for the FT-IR experiments, and EPR samples did not contain DCMU. Fig. 3 A demonstrates that the S2 multiline signal is observed when samples are frozen immediately after the flash. Comparison of Fig. 3, B–D, with Fig. 3 A, indicates that the S2 state has decayed by ∼50% after 5 min and has completely decayed after 20 min. This control experiment suggests that variation in S-state composition does not underlie the observed variation in the FT-IR spectra (Fig. 2), because the S2 state decays completely to S1 in the shortest dark adaptation time employed. Note that the reaction-induced FT-IR spectrum, assigned here and previously (Zhang et al., 1998) to S2QB− -minus-S1QB, decays more rapidly (Fig. 1) than the EPR signal from the S2 state (Fig. 3). This discrepancy seems to indicate that protein-based conformational changes at the Mn cluster have a shorter lifetime than the lifetime of the paramagnetic, EPR-detected state.
In another EPR control experiment, the effect of dark adaptation on the EPR properties of the S2 state was probed. As shown in Fig. 3, varying the time between the preflash and the actinic flash caused no significant alteration in the S2 EPR multiline signal, given the signal-to-noise ratio (Fig. 3, E and F). No other EPR signal was detected from the S2 state (data not shown). This similarity suggests the FT-IR spectral differences arise from different forms of the S1 state and not from alterations in the S2 state.
EPR controls (data not shown) also show there is a significant (∼30%) change in tyrosyl D• content, when the 20-min and 1-h protocols are compared. These data suggest that vibrational modes of D• may also contribute to observed spectral alterations in Fig. 2. The vibrational spectrum of D• has been reported and assigned by isotopic labeling (see Kim and Barry, 1998; Pujols-Ayala et al., 2003).
Difference FT-IR spectra were also obtained in the presence of DCMU, an inhibitor that prevents reduction of QB (Velthuys and Amesz, 1974). This inhibitor limits PSII to QA as the terminal endogenous electron acceptor. Structural changes arising from dark adaptation in the S1 state should be in common when S2QB− -minus-S1QB and S2QA− -minus-S1QA spectra are compared. Accordingly, S2QA− -minus-S1QA difference spectra were constructed from data recorded immediately after the actinic flash or immediately before the actinic flash (Fig. 4). In Fig. 4, A–C, samples were preflashed and dark-adapted for 2 h, 1 h, and 20 min, respectively. In these samples, ∼100% of the reaction centers will be preset in the S1 state (Roelofs et al., 1996). The spectra acquired with DCMU resemble spectra previously assigned to S2QA− -minus-S1QA (Noguchi et al., 1995). The decay of QA− is expected to be a factor of ∼10–20 faster than the decay of QB (Rutherford and Inoue, 1984). This is consistent with our data and assignments, because in the presence of DCMU, we observed faster decay of the FT-IR signals (data not shown).
Fig. 4, A–C, provide evidence that difference FT-IR spectra, acquired in the presence of DCMU, also show a spectral dependence on dark adaptation time. These changes are larger than the deviations observed in a control spectrum, constructed from data acquired before the actinic flash (Fig. 4 D). With 20 min of dark adaptation (Fig. 4 C), bands at (−) 1403/1388, (+) 1372, (−) 1345, and (+) 1192 cm−1 are observed. With 1 or 2 h of dark adaptation (Fig. 4, A and B), these bands are observed to broaden and to shift in frequency. These changes are accompanied by a change in the intensity of bands between 1679 and 1630 cm−1. Other spectral changes are also observed and will be discussed below.
The presence or absence of DCMU is observed to have an overall effect on the spectrum (compare Figs. 2 and 4, which are slightly different). This may be due to differential QA−/QA and QB−/QB contributions to the spectra. QA−/QA contributions to the difference FT-IR spectrum have been identified by isotopic labeling of plastoquinone (Razeghifard et al., 1999) and time-resolved spectroscopy (Zhang et al. 1998). Another possible source of this spectral difference is that recombination of S2 with QB− is much slower than recombination with QA− (see discussion above). Therefore, our 30-s data acquisition may average over different kinetic events in the two cases. In addition, differential contributions of tyrosine D• may also occur under the two conditions, and binding of DCMU may have an effect on PSII structure, which is detectable on the donor side.
Despite these considerations, bands with similar frequencies (within 6 cm−1) are observed, when DCMU-containing and DCMU-lacking samples are compared. This is evident in Fig. 5, which presents double difference spectra constructed from the S2QA−-minus-S1QA (Fig. 5 A) and S2QB−-minus-S1QB (Fig. 5 B) data sets. These double difference spectra show the effect of increasing dark adaptation on the vibrational spectra and were constructed by subtraction of a 20-min from a 2-h data set. Acceptor side spectral contributions should cancel out in these double difference spectra, which differ only in the length of dark adaptation after a preflash. Not all spectral features are in common when Fig. 5 A is compared to Fig. 5 B, perhaps due to a DCMU-induced structural change in PSII or to a difference in tyrosine D• contribution to the two data sets.
Spectral features in common, when Fig. 5 A is compared to Fig. 5 B, are likely to be caused by a dark-adaptation-induced structural change on the PSII donor side. Features with similar frequencies include bands at (−) 1661/(+) 1653; (−) 1554/(+) 1540/(−) 1531/(+) 1517; (−)1454; and (−)1425/(+) 1407 cm−1 (Fig. 5 B, gray fill). A spectral feature at ∼1478 cm−1 in both data sets may arise from a small tyrosine D• contribution (Kim and Barry, 1998). Derivative-shaped features in these double difference spectra can be caused by frequency shifts of vibrational bands during dark adaptation. The observed spectral features in Fig. 5, A and B, are significant, as compared either to a dark control (Fig. 5 D) or to a control double difference spectrum, constructed by subtracting one-half of the 2-h data set from the other half (Fig. 5 C). In addition, a statistical treatment of the data, using 95% confidence intervals, confirms that the assigned spectral changes are significant relative to Fig. 5, C and D. The amplitudes of spectral features in the double difference spectrum were compared to the amplitude of the (−) 1401/ (+) 1365 cm−1 band in Fig. 2 A. This comparison suggests that ∼40–100% of the centers, giving rise to the S2QB−-minus-S1QB difference spectrum, are involved in a conformational rearrangement in the dark.
DISCUSSION
In this report, we present evidence that the length of dark adaptation in the S1 state alters the difference FT-IR spectra, assigned either to S2QB−-minus-S1QB or to S2QA−-minus-S1QA. In our experiments, the length of dark adaptation varied from 20 min to 2 h. Control experiments showed that variation in S2/S3 composition or in hydration state did not underlie the observed spectral variation. Also, the length of dark adaptation did not alter the EPR properties of the S2 state. Therefore, we hypothesize that these spectral differences are due to spontaneous structural changes, which occur in the dark in the S1 state.
In these data sets, alterations are evident in the 1430 cm−1 spectral region in which symmetric stretching vibrations of carboxylates may contribute. To contribute to the spectrum, aspartate or glutamate residues may be perturbed when Mn is photo-oxidized. Therefore, the perturbed aspartate and glutamate residues that contribute to the spectrum may be manganese ligands. Unidentate, bridging, and chelating ligands all contribute symmetric CO stretching frequencies in the 1450–1300 cm−1 region (Nakamoto, 1986; Smith et al., 1997). The exact frequencies depend on the oxidation state and nuclearity of the Mn cluster.
Alternatively, aspartate and glutamate residues may protonate, when Mn is photo-oxidized, and thus contribute to the difference spectrum (Bellamy, 1980; Hutchison et al., 1999). The dark adaptation-induced spectral alterations do not seem consistent with protonation of glutamate and aspartate side chains alone. Model studies show that carboxylate protonation should lead to two negative bands at ∼1570 and ∼1400 cm−1 and two positive bands at ∼1720 and 1220 cm−1 (Bellamy, 1980; Hutchison et al., 1999). The negative bands are the asymmetric and symmetric stretching vibration of the anion, and the positive bands are the C=O and C-O stretching vibration of the carboxylic acid. Whereas Fig. 5 A shows bands in the 1720 and 1220 cm−1 regions, these lines are not in common between Fig. 5, A and B. Therefore, we do not attribute the dark adaptation-induced structural alteration to a protonation change. A final possibility is that photo-oxidation of Mn induces changes in polarity, and these changes in polarity perturb carboxylate and carboxylic acid vibrational energies (see Bellamy, 1980; Steenhuis and Barry, 1997; and references therein). Such a spectral contribution from free carboxylates cannot be ruled out.
Bridging/chelating ligands to higher valence Mn, which are relevant for the S1-to-S2 transition, have stretching vibrations in the 1600–1540 and the 1430–1360 cm−1 regions (Smith et al., 1997). Mn photo-oxidation is expected to shift the frequencies of all carboxylate ligands (Nakamoto, 1986; Smith et al., 1997). Therefore, some of the changes in the 1550–1515 and 1420–1370 cm−1 regions, which are in common to our S2QA−-minus-S1QA and S2QB−-minus-S1QB spectra, may be assignable to structural changes involving ligands to Mn. A structural change involving unidentate ligands (Nakamoto, 1986; Smith et al., 1997) is also possible. Because these changes occur in the dark S1 state, the structural alterations must correspond to a spontaneous reaction. This change in carboxylate ligation may serve to stabilize the Mn cluster in the dark or under low light intensities.
Supporting assignment to carboxylate ligands, site-directed mutagenesis has suggested that aspartate and glutamate residues ligate manganese (Boerner et al., 1992; Chu et al., 1994; Vermaas et al., 1990). Also, previous work has assigned bands in the 1450–1300 cm−1 region of the S2QA−-minus-S1QA FT-IR spectrum to ligating carboxylates (Noguchi et al., 1995; Steenhuis and Barry, 1997). In addition, previous FT-IR studies have identified carboxylate shifts in the DE190-D1 mutant, which is active, but impaired, in water oxidation (Steenhuis et al., 1999).
Other interpretations of the spectra are also possible. Bands between 1680 and 1620 cm−1 may be assigned to amide I contributions from the peptide bond. This interpretation also predicts peptide bond contributions in the amide II (1550 cm−1) region (Krimm and Bandekar, 1986). Bands in common between the two double difference spectra in the ∼1440 cm−1 region may also be associated with in-plane NH bending vibrations of the peptide bond (Krimm and Bandekar, 1986). Bands in the 1650 and 1550 cm−1 region of the S2QA− -minus-S1QA spectrum have been assigned to amide I and II bands previously (see Noguchi and Sugiura, 2001, and references therein). Because the carboxyl terminus of the D1 polypeptide is believed to be a ligand to Mn (Kamiya and Shen, 2003; Nixon et al., 1992), changes in Mn coordination might be expected to give amide I and II contributions in the FT-IR difference spectrum.
Our data suggest that the scheme for photosynthetic water oxidation be modified to include an equilibration between states at the same oxidation level, S′1 and S1. These states differ in manganese coordination, in polarity, and/or in protein structure surrounding the Mn cluster. The dark interconversion of two different S1 states may help to explain the attribution of two different FT-IR spectra to the S2QA− -minus-S1QA transition (Noguchi et al., 1995; Steenhuis and Barry, 1997; Zhang et al., 1998). Some of the bands, with amplitudes increased by dark adaptation, have frequencies in common with bands in the 200 K S2QA−-minus-S1QA spectrum (Steenhuis and Barry, 1997). For example, bands at 1265, 1300, and 1400 cm−1 in Fig. 5 A may correspond to 200 K bands previously reported at 1267, 1307, and 1390 cm−1 (Steenhuis and Barry, 1997). These published 200 K studies were conducted with long dark adaptation times, which may account for the corresponding bands.
However, note that the spectra acquired at 4°C (this work) and 200 K (Steenhuis and Barry, 1997) are not identical. One difference between the 4°C and 200 K spectra is that the 200 K data exhibit fewer, but broader spectral features in the 1400–1300 cm−1 region. This spectral width was attributed to homogeneous broadening caused by movement of carboxylate groups under illumination (Steenhuis and Barry, 1997). Heterogeneous broadening, due to different positions of carboxylate groups in different PSII reaction centers, was also presented as a possible explanation (Steenhuis and Barry, 1997). Taken together, that previously published work and the results presented here suggest photo-oxidation-induced dynamics in the rearrangement of carboxylate ligands to manganese. The increase in spectral complexity with long dark adaptations, reported here, may be due to heterogeneity in the position of one or more mobile carboxylate ligand(s).
Alternatively, the conditions employed for the 200 K experiment, in which high glycerol concentrations were used to produce a glass, may also contribute to spectral alterations. Glycerol/sucrose effects on the S2QB−-minus-S1Qb FT-IR spectrum have been reported (Halverson and Barry, 2003). Finally, at 200 K, the water oxidizing complex is known to be partially inhibited (Styring and Rutherford, 1988), perhaps by a difference in donor-side protonation state (Sachs, Halverson, and Barry, unpublished results). Therefore, the S2 state produced at 200 K may differ from the S2 state produced at higher temperatures, where all S-state transitions can occur. Spectral differences between the 200 K and 277 K data are likely to involve a contribution from all these factors.
Previous work has defined active and resting states of PSII by variation in dark adaptation time (Beck et al., 1985; Koulougliotis et al., 1992). In some of those experiments (Beck et al., 1985), changes in the EPR properties of the S2 state were observed. Those observations are distinct from the results presented here, in which we find that the S2 EPR amplitude and lineshape are unaffected by the length of dark adaptation. This discrepancy is probably caused by differences in experimental conditions. Despite these experimental differences, this work supports the overall conclusions of Beck et al. (1985) and Koulougliotis et al. (1992), in which a spontaneous conformational change at the Mn cluster was postulated to occur during dark adaptation.
Redox-linked changes in protein structure are known to occur in the di-iron centers of methane mono-oxygenase and ribonucleotide reductase (Nordlund and Eklund, 1993; Rosenweig et al., 1995). These binuclear enzymes catalyze different reactions, but the active sites are similar and have flexible carboxylate ligation. This flexibility has been postulated to be important for function, in that the binuclear site can have bidentate carboxylate ligation, when saturation of the first coordination sphere is optimal, or unidentate carboxylate ligation, when additional coordination sites are required for catalysis. Experimental and theoretical investigations have demonstrated that a carboxylate shift is a facile process both thermodynamically and kinetically (see Torrent et al., 2002, and references therein). Our results may suggest a relationship between the di-iron-containing enzymes and PSII, because we have presented evidence that PSII may contain a flexible Mn cluster. We propose that these structural changes define a new S1 intermediate, S′1, in the process of photosynthetic water oxidation. The S′1 state may stabilize the Mn cluster during long dark adaptations and may be an accessible, intermediate state under low light intensities.
Acknowledgments
The authors thank Prof. Roseann Sachs, and Drs. Colette Sacksteder and Idelisa Pujols-Ayala, for helpful comments on the manuscript.
This work was supported by National Science Foundation grant MCB 0134968 to B.A.B.
Kelly M. Halverson's present address is Bacteriology Division, USAM-RIID, 1425 Porter St., Ft. Detrick, MD 21702.
Bridgette A. Barry's present address is School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332.
References
- Anderson, L. B., A. J. A. Ouellette, and B. A. Barry. 2000. Probing the structure of photosystem II with amines and phenylhydrazine. J. Biol. Chem. 275:4920–4927. [DOI] [PubMed] [Google Scholar]
- Barry, B. A. 1995. Tyrosyl radicals in photosystem II. Methods Enzymol. 258:303–319. [DOI] [PubMed] [Google Scholar]
- Barry, B. A., R. J. Boerner, and J. C. de Paula. 1994. The use of cyanobacteria in the study of the structure and function of photosystem II. In The Molecular Biology of the Cyanobacteria. Vol. 1. D. Bryant, editor. Kluwer Academic Publishers, Dordrecht, The Netherlands. pp.215–257.
- Beck, W. F., J. C. de Paula, and G. W. Brudvig. 1985. Active and resting states of the O2-evolving complex of photosystem II. Biochemistry. 24:3035–3043. [DOI] [PubMed] [Google Scholar]
- Bellamy, L. J. 1980. The Infrared Spectra of Complex Molecules. Chapman and Hall, London, UK.
- Berthold, D. A., G. T. Babcock, and C. F. Yocum. 1981. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 134:231–234. [Google Scholar]
- Boerner, R. J., A. P. Nguyen, B. A. Barry, and R. J. Debus. 1992. Evidence from directed mutagenesis that aspartate 170 of the D1 polypeptide influences the assembly and/or stability of the manganese cluster in the photosynthetic water-splitting complex. Biochemistry. 31:6660–6672. [DOI] [PubMed] [Google Scholar]
- Britt, R. D. 1996. Oxygen evolution. In Oxygenic Photosynthesis: The Light Reactions. Vol. 4. D. R. Ort, and C. F. Yocum, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. pp.137–164.
- Campbell, K. A., J. M. Peloquin, D. P. Pham, R. J. Debus, and R. D. Britt. 1998. Parallel polarization EPR detection of an S1-state “multiline” EPR signal in photosystem II particles from Synechocystis sp. PCC 6803. J. Am. Chem. Soc. 120:447–448. [Google Scholar]
- Chu, H. A., A. P. Nguyen, and R. J. Debus. 1994. Site-directed photosystem II mutants with perturbed oxygen-evolving properties. 1. Instability or inefficient assembly of the manganese cluster in vivo. Biochemistry. 33:6137–6149. [DOI] [PubMed] [Google Scholar]
- de Wijn, R., and H. J. van Gorkom. 2001. Kinetics of electron transfer from QA to QB in Photosystem II. Biochemistry. 40:11912–11922. [DOI] [PubMed] [Google Scholar]
- Dexheimer, S. L., and M. P. Klein. 1992. Detection of a paramagnetic intermediate in the S1 state of the photosynthetic oxygen-evolving complex. J. Am. Chem. Soc. 114:2821–2826. [Google Scholar]
- Halverson, K. M., and B. A. Barry. 2003. Sucrose and glycerol effects on photosystem II. Biophys. J. 85:99059–99067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haumann, M., and W. Junge. 1999. Photosynthetic water oxidation: a simplex-scheme of its partial reactions. Biochim. Biophys. Acta. 1411:86–91. [DOI] [PubMed] [Google Scholar]
- Hillier, W., and G. T. Babcock. 2001. S-state dependent Fourier transform infrared difference spectra for the photosystem II oxygen evolving complex. Biochemistry. 40:1503–1509. [DOI] [PubMed] [Google Scholar]
- Hoganson, C. W., and G. T. Babcock. 1997. A metalloradical mechanism for the generation of oxygen from water in photosynthesis. Science. 277:1953–1956. [DOI] [PubMed] [Google Scholar]
- Hutchison, R. S., J. J. Steenhuis, C. F. Yocum, M. R. Razeghifard, and B. A. Barry. 1999. Deprotonation of the 33-kDa, extrinsic, manganese-stabilizing subunit accompanies photo-oxidation of manganese in photosystem II. J. Biol. Chem. 274:31987–31995. [DOI] [PubMed] [Google Scholar]
- Jansson, C., and P. Maeenpaeae. 1997. III. Mutation: Site-directed mutagenesis for structure-function analyses of the photosystem II reaction center protein D1. Prog. Botany. 58:252–267. [Google Scholar]
- Joliot, P., and B. Kok. 1975. Oxygen evolution in photosynthesis. In Bioenergetics of Photosynthesis. Govindjee, editor. Academic Press, New York. pp.388–412.
- Kamiya, N., and J.-R. Shen. 2003. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7 Å resolution. Proc. Natl. Acad. Sci. USA. 100:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S., and B. A. Barry. 1998. The vibrational spectrum associated with the reduction of tyrosyl radical, D•: a comparative biochemical and kinetic study. Biochemistry. 37:13882–13892. [DOI] [PubMed] [Google Scholar]
- Kim, S., J. Patzlaff, T. Krick, I. Ayala, R. K. Sachs, and B. A. Barry. 2000. Isotope-based discrimination between the infrared modes of plastosemiquinone anion radicals and neutral tyrosyl radicals in photosystem II. J. Phys. Chem. B. 104:9720–9727. [Google Scholar]
- Kimura, Y., and T. A. Ono. 2001. Chelator-induced disappearance of carboxylate stretching vibrational modes in Sa/S1 FTIR spectrum in oxygen-evolving complex of photosystem II. Biochemistry. 40:14061–14068. [DOI] [PubMed] [Google Scholar]
- Koulougliotis, D., D. J. Hirsh, and G. W. Brudvig. 1992. The oxygen-evolving center of photosystem II is diamagnetic in the S1 resting state. J. Am. Chem. Soc. 114:8322–8323. [Google Scholar]
- Krimm, S., and J. Bandekar. 1986. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. In Advances in Protein Chemistry. Vol. 38. C. B. Anfinsen, J. T. Edsall, and F. M. Richards, editors. Academic Press, New York. pp.181–364. [DOI] [PubMed]
- Messinger, J. 2000. Towards understanding the chemistry of photosynthetic oxygen evolution: dynamic structural changes, redox states and substrate water binding of the Mn cluster in photosystem II. Biochim. Biophys. Acta. 1459:481–488. [DOI] [PubMed] [Google Scholar]
- Miller, A.-F., and G. W. Brudvig. 1991. A guide to electron paramagnetic resonance spectroscopy of photosystem II membranes. Biochim. Biophys. Acta. 1056:1–18. [DOI] [PubMed] [Google Scholar]
- Nakamoto, K. 1986. Infrared and Raman spectra of inorganic and coordination compounds. John Wiley & Sons, New York.
- Nixon, P. J., J. T. Trost, and B. A. Diner. 1992. Role of the carboxy terminus of polypeptide D1 in the assembly of a functional water-oxidizing manganese cluster in photosystem II of the cyanobacterium Synechocystis sp. PCC 6803: assembly requires a free carboxyl group at C-terminal position 344. Biochemistry. 31:10859–10871. [DOI] [PubMed] [Google Scholar]
- Noguchi, T., T. A. Ono, and Y. Inoue. 1995. A carboxylate ligand interacting with water in the oxygen-evolving center of photosystem II as revealed by Fourier transform infrared spectroscopy. Biochim. Biophys. Acta. 1232:59–66. [Google Scholar]
- Noguchi, T., and M. Sugiura. 2001. Flash-induced Fourier transform infrared detection of the structural changes during the S-state cycle of the oxygen-evolving complex in photosystem II. Biochemistry. 40:1497–1502. [DOI] [PubMed] [Google Scholar]
- Noguchi, T., and M. Sugiura. 2002. Flash-induced FTIR difference spectra of the water oxidizing complex in moderately hydrated photosystem II core films: effect of hydration extent on S-state transitions. Biochemistry. 41:2322–2330. [DOI] [PubMed] [Google Scholar]
- Nordlund, P., and H. Eklund. 1993. Structure and function of the Escherichia coli ribonuclease reductase protein R2. J. Mol. Biol. 232:123–164. [DOI] [PubMed] [Google Scholar]
- Nugent, J. H. A., I. P. Muhiuddin, and M. C. W. Evans. 2002. Electron transfer from the water oxidizing complex at cryogenic temperatures: the S1 to S2 step. Biochemistry. 41:4117–4126. [DOI] [PubMed] [Google Scholar]
- Patzlaff, J. S., R. J. Brooker, and B. A. Barry. 2000. A reaction-induced Fourier transform-infrared spectroscopic study of the lactose permease: a transmembrane potential perturbs carboxylic acid residues. J. Biol. Chem. 275:28695–28700. [DOI] [PubMed] [Google Scholar]
- Pecoraro, V. L., M. J. Baldwin, M. T. Caudle, W. Y. Hsieh, and N. A. Law. 1998. A proposal for water oxidation in photosystem II. Pure Appl. Chem. 70:925–929. [Google Scholar]
- Peloquin, J. M., K. A. Campbell, D. W. Randall, M. A. Evanchik, V. L. Pecoraro, W. H. Armstrong, and R. D. Britt. 2000. Mn-55 ENDOR of the S2-state multiline EPR signal of photosystem II: implications on the structure of the tetranuclear Mn cluster. J. Am. Chem. Soc. 122:10926–10942. [Google Scholar]
- Pujols-Ayala, I., and B. A. Barry. 2002. His 190–D1 and Glu 189–D1 provide structural stabilization in photosystem II. Biochemistry. 41:11456–11465. [DOI] [PubMed] [Google Scholar]
- Pujols-Ayala, I., C. A. Sacksteder, and B. A. Barry. 2003. Redox-active tyrosine residues: role for the peptide bond in electron transfer. J. Am. Chem. Soc. 125:7536–7538. [DOI] [PubMed] [Google Scholar]
- Razeghifard, M. R., S. Kim, J. S. Patzlaff, R. S. Hutchison, T. Krick, I. Ayala, J. J. Steenhuis, S. E. Boesch, R. A. Wheeler, and B. A. Barry. 1999. The in vivo, in vitro, and calculated vibrational spectra of plastoquinone and the plastosemiquinone anion radical. J. Phys. Chem. B. 103:9790–9800. [Google Scholar]
- Roelofs, T. A., W. Liang, M. L. Latimer, R. M. Cinco, A. Rompel, J. C. Andrews, K. Sauer, V. K. Yachandra, and M. P. Klein. 1996. Oxidation states of the manganese cluster during the flash-induced S-state cycle of the photosynthetic oxygen-evolving complex. Proc. Natl. Acad. Sci. USA. 93:3335–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig, A. C., P. Nordlund, P. M. Takahara, C. A. Frederick, and S. J. Lippard. Geometry of the soluble methane monooxygenase catalytic di-iron center in two oxidation states. 1995. Chem. Biol. 2:409–418. [PubMed] [Google Scholar]
- Rutherford, A. W., and Y. Inoue. 1984. Oscillation of delayed luminescence from PSII: recombination of S2QB− and S3QB−. FEBS Lett. 165:163–170. [Google Scholar]
- Smith, J. C., E. Gonzalez-Vergara, and J. B. Vincent. 1997. Detection of structural changes upon oxidation in multinuclear Mn-oxo-carboxylate assemblies by Fourier transform infrared spectroscopy: relationship to photosystem II. Inorg. Chim. Acta. 255:99–103. [Google Scholar]
- Steenhuis, J. J., and B. A. Barry. 1997. The protein and ligand environment of the S2 state in photosynthetic oxygen evolution: a difference FT-IR study. J. Phys. Chem. 101:6652–6660. [Google Scholar]
- Steenhuis, J. J., R. S. Hutchison, and B. A. Barry. 1999. Alterations in carboxylate ligation at the active site of photosystem II. J. Biol. Chem. 274:14609–14616. [DOI] [PubMed] [Google Scholar]
- Styring, S., and A. W. Rutherford. 1988. Deactivation kinetics and temperature dependence of the S-state transitions in the oxygen-evolving system of photosystem II measured by EPR spectroscopy. Biochim. Biophys. Acta. 933:378–387. [Google Scholar]
- Tang, X.-S., B. A. Diner, B. S. Larsen, M. L. Gilchrist, G. A. Lorigan, and R. D. Britt. 1994. Identification of histidine at the catalytic site of the photosynthetic oxygen evolving complex. Proc. Natl. Acad. Sci. USA. 91:704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent, M., D. G. Musaev, H. Basch, and K. Morokuma. 2002. Computational studies of reaction mechanisms of methane mono-oxygenase and ribonucleotide reductase. J. Comput. Chem. 23:59–76. [DOI] [PubMed] [Google Scholar]
- Velthuys, B. R., and J. Amesz. 1974. Charge accumulation at the reducing side of system 2 of photosynthesis. Biochim. Biophys. Acta. 333:85–94. [DOI] [PubMed] [Google Scholar]
- Vermaas, W., J. Charite, and G. Shen. 1990. Glu-69 of the D2 protein in photosystem II is a potential ligand to Mn involved in photosynthetic oxygen evolution. Biochemistry. 29:5325–5332. [DOI] [PubMed] [Google Scholar]
- Vermaas, W. F. J. 1998. Gene modifications and mutation mapping to study the function of photosystem II. Methods Enzymol. 297:293–310. [DOI] [PubMed] [Google Scholar]
- Vrettos, J. S., J. Limburg, and G. W. Brudvig. 2001. Mechanism of photosynthetic water oxidation: combining biophysical studies of photosystem II with inorganic model chemistry. Biochim. Biophys. Acta. 1503:229–245. [DOI] [PubMed] [Google Scholar]
- Zhang, H., G. Fischer, and T. Wydrzynski. 1998. Room-temperature vibrational difference spectrum for S2QB−/S1QB of photosystem II determined by time-resolved Fourier transform infrared spectroscopy. Biochemistry. 37:5511–5517. [DOI] [PubMed] [Google Scholar]
- Zouni, A., H. T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger, and P. Orth. 2001. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Ångstrom resolution. Nature. 409:739–743. [DOI] [PubMed] [Google Scholar]