

Abstract
Overexpression of HER2, a receptor-like tyrosine kinase and signaling partner for the epidermal growth factor receptor (EGFR), has been implicated in numerous experimental and clinical studies as promoting the progression of many types of cancer. One avenue by which HER2 overexpression may dysregulate EGFR-mediated cell responses, such as proliferation and migration, downstream of EGF family ligand binding, is by its modulation on EGFR endocytic trafficking dynamics. EGFR signaling is regulated by downregulation and compartmental relocalization arising from endocytic internalization and endosomal sorting to degradation versus recycling fates. HER2 overexpression influences both of these processes. At the endosomal sorting stage, increased HER2 levels elicit enhanced EGFR recycling outcomes, but the mechanism by which this transpires is poorly understood. Here, we determine whether alternative mechanisms for HER2-mediated enhancement of EGFR recycling can be distinguished by comparison of corresponding mathematical models to experimental literature data. Indeed, we find that the experimental data are clearly most consistent with a mechanism in which HER2 directly competes with EGFR for a stoichiometrically-limited quantity of endosomal retention components (ERCs), thereby reducing degradation of ERC-coupled EGFR. Model predictions based on this mechanism exhibited qualitative trends highly similar to data on the fraction of EGF/EGFR complexes sorted to recycling fate as a function of the amount of internalized EGF/EGFR complexes. In contrast, model predictions for alternative mechanisms—blocking of EGFR/ERC coupling, or altering EGF/EGFR dissociation—were inconsistent with the qualitative trends of the experimental data.
INTRODUCTION
Elevated expression of the epidermal growth factor receptor (EGFR) and/or human epidermal growth factor receptor 2 (HER2) has been implicated in the development of cancer by contributing to aberrant cell behavior including increased motility, increased sensitivity to mitogenic stimuli, anchorage independence, and cell transformation (Brandt et al., 1999; Chazin et al., 1992; DiFiore et al., 1987a,b; Ignatoski et al., 1999; Spencer et al., 2000). The quantity and intracellular localization of these receptors is able to influence cell behavior by dictating both the strength and quality of signals generated. Thus, understanding the regulatory mechanisms involved in controlling the number of EGFR and/or HER2 is of prime importance in dissecting how elevated receptor expression is able to alter cell signaling that manifests itself in tumorigenesis.
After EGF binding, the EGFR family of receptor protein tyrosine kinases (EGFR, HER2, HER3, and HER4) interact and transphosphorylate to form a wide array of homo- and heterodimers, each with distinct signaling abilities (Alroy and Yarden, 1997; Muthuswamy et al., 1999; Olayioye et al., 1998; Yarden and Sliwkowski, 2001). Activated receptors recruit cascades of intracellular signaling molecules, including members of the Ras/MAPK and PLC-γ pathways that control a diverse range of cell responses. The signals that are recruited depend heavily on receptor location. For example, PLC-γ, calpain, and Grb2, are primarily activated or recruited by surface EGFR, while Eps8 is associated with only intracellular receptors and the Ras pathway may be activated by both surface and intracellular EGFR (Burke et al., 2001; Glading et al., 2001; Haugh et al., 1999a,b).
Signaling through EGFR is negatively regulated via intracellular trafficking (Sorkin and Waters, 1993; Wiley and Burke, 2001), a process that attenuates growth factor signaling via both the short- and long-term downregulation of receptor number. Overexpression or mutation of the EGFR has been shown to impair downregulation, as a consequence of altered trafficking, indicating the importance of proper trafficking for the normal regulation of cell growth (Vieira et al., 1996; Wells et al., 1990).
EGF binding initiates the rapid internalization of EGF-EGFR complexes via clathrin-coated-pit endocytosis to early endosomal compartments. This process can be saturated in cases where surface complex number exceeds the capacity of the adaptor proteins involved in receptor-mediated endocytosis (Lund et al., 1990; Wiley, 1988). Dimerization with other EGFR family members is also thought to slow this process as HER2, HER3, and HER4 all exhibit some degree of endocytic impairment (Baulida et al., 1996; Hendriks et al., 2003b; Sorkin et al., 1993; Wang et al., 1999).
After internalization, occupied EGFR have been shown to be selectively retained within the endosome, whereas empty receptors follow the default recycling pathway back to the surface (French et al., 1994; Herbst et al., 1994). The endosomal retention of occupied receptors has been demonstrated to be both specific and saturable, requiring cytoplasmic sequences for efficient retention and lysosomal targeting, but independent of intrinsic tyrosine kinase ability (French et al., 1994; Herbst et al., 1994; Opresko et al., 1995; Wiley et al., 1991). The selective retention of occupied EGFR is mediated through via a di-leucine motif in the juxtamembrane region (Kil and Carlin, 2000; Kil et al., 1999). Two regions of the EGFR, residues 1022–1063 and, to a lesser extent, 1063–1123, are believed to contribute in targeting receptors to the degradation pathway (Kornilova et al., 1996). Additionally, EGFR residues 943–957 are known to interact with SNX1, a putative endosomal sorting protein believed to be involved in targeting EGFR to degradative fates (Kurten et al., 1996; Zhong et al., 2002). EGFR deactivation and degradation also vary with the sorting behavior of different ligands (EGF vs. TGF) (French et al., 1995).
Ligand stimulus also results in EGFR-mediated phosphorylation of c-Cbl, a protein involved in the ubiquitization and degradation of EGFR. Overexpression of c-Cbl enhances ubiquitination and degradation of EGFR, whereas oncogenic viral Cbl interferes with the sorting function of c-Cbl, directing EGFR to recycling fates (Levkowitz et al., 1999, 1998). Interestingly, c-Cbl does not interact with other EGFR family members, including HER2 (Levkowitz et al., 1996).
HER2 is an almost ubiquitously expressed EGFR family member that does not bind any ligands and therefore must rely on dimerization with another EGFR family member for complete activation (Graus-Porta et al., 1997; Hynes and Stern, 1994; Karunagaran et al., 1996; Worthylake and Wiley, 1997). Overexpression of HER2 has been demonstrated to inhibit downregulation of the EGFR and of itself, as well as increase the recycling rate of EGF (Hendriks et al., 2003a; Worthylake et al., 1999). HER2 expression has been shown to shunt ligand-activated receptors to recycling fates suggesting that receptor heterodimer species may have a superior signaling potency as a consequence of their intracellular routing (Lenferink et al., 1998; Waterman et al., 1998). Receptor heterodimerization has been shown to affect the dissociation rate of EGF or heregulin and may also do so inside of endosomal compartments (Karunagaran et al., 1996; Lenferink et al., 1998; Lewis et al., 1996; Wada et al., 1990). Although it is apparent that HER2 expression influences the endosomal sorting of EGF and EGFR, the dominant mechanism(s) by which it occurs remain unclear.
Theoretical models of endosomal sorting have examined the biophysical requirements for molecular transport out of a central endosomal vesicle into recycling tubules and spawned the development of a mechanistic model (French and Lauffenburger, 1996; Lauffenburger and Linderman, 1993; Linderman and Lauffenburger, 1986). In this model, the endosomal sorting of the EGFR and its ligands were mathematically modeled using a compartmental analysis incorporating endosomal retention components (ERCs) and a representation of endosomal architecture. By detailing the mechanistic and biophysical basis for endosomal sorting, one unified model is able to account for a wide range of experimentally observed sorting results. However, this model does not account for the effects of HER2 on EGF sorting.
The goal of this work is to build upon the French ERC model of sorting to understand how different EGFR and HER2 interactions could contribute to qualitative trends in experimental sorting curves. Specifically, we seek to discriminate between three different mechanisms by which HER2 may disrupt the sorting process through the comparison of experimental and modeling outcomes. Our results suggest that HER2 is able to alter EGF sorting primarily through a competitive mechanism wherein it competes for a limited number of ERCs, rather than by blocking EGF-EGFR interaction with ERCs or by altering the affinity of EGF.
MODEL DEVELOPMENT
Our model of endosomal sorting is an extension of the mechanistic sorting model based on ERC sorting model originally proposed by French and Lauffenburger (1996). We add HER2 to the model and consider three distinct mechanisms by which HER2 interaction may augment EGF sorting.
The framework of the ERC sorting model is briefly presented here; its development, assumptions, and validation are explained in detail elsewhere (French and Lauffenburger, 1996, 1997). This model (illustrated schematically in Fig. 1) simulates the quasi-steady-state sorting of EGFRs (R1) and ligands (L) as they pass through the endosomal pathway. The cell interior is separated into four compartments: an endosomal vesicular compartment (v), an endosomal tubular compartment (t), a postsorting recycling compartment (r), and a postsorting degradation compartment (d). Ligand may bind to receptors to form complexes and subsequently dissociate at rates kon and koff, respectively. Internalized ligand-receptor complexes (R1L) enter the vesicular compartment of the endosome. Within the vesicular compartment, complexes and unoccupied receptors may diffuse into the tubular compartment with transport rate γ. Complexes may interact with ERCs (E), at rate kc,R1E (or at rate kc,R1E,het for the case of heterodimers), in the vesicular compartment to form ternary complexes (R1LE) which have a negligible rate of transport into the tubular compartment. ERCs only bind occupied receptors; their total quantity is assumed to be at steady state, and all ERC-containing species are restricted to the vesicular compartment of the endosome. Ligands may dissociate from ternary complexes at rate koff leaving binary complexes (R1E) that either rebind ligand at rate kon or uncouple at rate ku,R1E to form free receptors and ERCs. Free ligand may bind unoccupied receptors at rate kon and is assumed to be in equilibrium between the tubular and vesicular lumen, related by partition coefficient, κ, accounting for excluded volume due to ligand size. Vesicular receptor and ligand species are targeted for the postsorting degradation compartment at rate ksv, whereas tubular receptor and ligand species are targeted for the postsorting recycling compartment at rate kst. The input into the model is the flux of ligand-receptor complexes (IR1L).
FIGURE 1.




(A) Endosomal sorting model, proposed by French and Lauffenburger. The cell is divided into four compartments: endosomal vesicle, endosomal tubule, recycling, and degradation compartments. Internalized ligand-receptor complexes enter vesicular compartment where they diffuse into the tubular compartment or are selectively retained in the central vesicle by ERCs. The vesicular compartment of the endosome targets species for degradation while the tubular compartment of the endosome targets species for recycling. (B) Within endosome, occupied EGFR (Y-shaped species) are selectively retained in vesicular portion by interaction with ERCs (solid pentagons). Species not bound by ERCs are free to diffuse into tubular compartments for recycling. (C) Additional receptor interactions are added as a result of HER2 (T-shaped species) presence. HER2 may dimerize with occupied EGFRs. EGF may dissociate from receptor heterodimers yielding unoccupied heterodimers which instantaneously break apart into unoccupied EGFR and free HER2. (D) The addition of HER2 to the model drives the formation of heterodimers and shifts the model input from 100% EGF-EGFR complexes to a combination of complexes and heterodimers to 100% heterodimers.
The general changes brought about by HER2 presence are presented here and the details unique to each model follow below (Fig. 1 c). The addition of HER2 to the sorting model adds a few additional species. Free HER2 (R2) is permitted to heterodimerize with occupied EGFR at rate kc to form occupied heterodimers (R1LR2) and uncouple at rate ku,R1LR2. Free HER2 and occupied heterodimers move from the vesicular compartment to the tubular compartment at transport rate γ. Ligand may dissociate from heterodimers at rate koff,het. Unoccupied heterodimers are assumed to be sufficiently unstable that they instantaneously break apart to yield free EGFR and free HER2. The input to the model consists of the flux of ligand-receptor complexes (IR1L), as well as the flux of occupied heterodimers (IR1LR2), as cartooned in Fig. 1 d. Elevated HER2 expression drives the formation of heterodimers and shifts the model input from 100% EGF-EGFR complexes to a combination of complexes and heterodimers to 100% heterodimers. The interactions between free HER2 and heterodimers with ERCs are unique to each model, and presented below.
We propose three distinct, although not mutually exclusive, mechanisms by which HER2 may disrupt the normal endosomal sorting of EGF and the EGFR. Each mechanism is considered separately for clarity and ease of interpretation. The complete set of equations encompassing all models is listed in the Appendix.
Blockade model
In this model, we propose that EGF-EGFR complex heterodimerization with HER2 may impair EGFR interaction with ERCs, cartooned in Fig. 2 a. When in the heterodimerized state, complexes are no longer able to bind ERCs (kc,R1E,het is set to zero). As such, HER2 is able to block the selective endosomal retention of EGF-EGFR complexes.
FIGURE 2.

(A) Blockade model. Heterodimerization with HER2 prevents EGFR-ERC and (EGF-EGFR)-ERC interaction. (B) Competition model. HER2 competes with EGFR for ERC interaction. Both free and heterodimerized HER2 species may interact with ERCs. (C) Affinity model. EGF-EGFR heterodimerization with HER2 alters the dissociation rate of EGF.
Competition model
Here, in addition to occupied EGFR, both free HER2 and HER2 that is heterodimerized with EGFRs can bind ERCs. All HER2-containing species bind ERCs at rate kc,R2E or kc,R2E,het to form species R2E or R1LR2E or ER1LR2E (depending on whether HER2 has heterodimerized and whether the EGF-EGFR complex has an ERC bound or not), and uncouple at rate ku,R2E. The model is cartooned in Fig. 2 b.
Affinity model
In this model, EGF-EGFR complex heterodimerization with HER2 alters the endosomal affinity of EGF for its receptor by altering its rate of dissociation from heterodimers (koff,het is different from koff). The presence of HER2 and heterodimerization does not affect the ability of occupied EGFR to bind ERCs and HER2 does not bind ERCs itself. This model is cartooned in Fig. 2 c.
Model inputs
The input to the model is a specified flux of ligand-receptor complexes (IR1L) and the flux of ligand-bound heterodimers (IR1LR2) (see Fig. 1 d). These parameters represent the rates of complex and heterodimer internalization, respectively. Although the internalization rate and the number of surface receptors has experimentally been shown to vary with ligand concentration and time, internalization fluxes are held constant for simplicity so that the effects on endosomal sorting may be isolated from effects due to differences in internalization (Wiley et al., 1991).
An increase in HER2 expression level should result in a higher degree of EGF-EGFR/HER2 heterodimerization by simple mass action kinetics. When no HER2 is present, the model input is only IR1L (with IR1LR2 set to 0). In the other extreme, when HER2 is in great excess, the model input is only IR1LR2 (with IR1L set to 0). The cases in between, where neither ke,R1L nor ke,R1LR2 are 0, reflect modest degrees of heterodimerization and directly reflect the receptor expression levels and their affinities for homo- versus heterodimerization.
Parameter determination
While there exists a great deal of cellular data and rate constants in the literature, it is scattered across many cell types. Because of this we have chosen to adopt the physiological reasonable parameters values, based on the ranges used in the original ERC sorting model, shown in Tables 1 and 2. The uncoupling rate of occupied heterodimers (ku,R1LR2) is estimated based on previous work and is set to 0.1 min−1 (Hendriks et al., 2003b). Parameters with no estimate or those believed to be of particular importance in determining the system output (ku,R1LR2, ku,R2E, and koff,het) are varied over wide ranges as shown in Results. A complete list of parameter values are shown in Tables 1 and 2.
TABLE 1.
Parameter values used in endosomal sorting model that are common to all models
| Parameter | Description | Base value |
|---|---|---|
| Nav | Avogadro's number | 6.023 × 1023 #/mol |
| Vtotal | Total endosomal volume | 3 × 10−14 L |
| η | Ratio of volume in tubular compartments to vesicular compartments | 0.67 |
| kst | Tubular sorting rate constant | 0.53 min−1 |
| ksv | Vesicular rate constant | 0.06 min−1 |
| κ | Partition coefficient accounting for excluded volume in tubules due to ligand size | 0.81 |
| ξ | Fraction of internalized ligand nonspecifically endocytosed | 0 |
| γ | Transport rate constant of receptors out of vesicular compartment into tubular compartment | 1 min−1 |
| kh | Degradation rate constant | 0.09 min−1 |
| kx | Recycling rate constant | 0.15 min−1 |
| kon | EGF binding rate constant | 5 × 107 M−1 min−1* |
| koff | EGF dissociation rate constant from EGFR | 0.5 min−1* |
| kc | EGF-EGFR/HER2 dimerization rate constant | 1 × 10−3 (#/cell)−1 min−1 |
| ku,R1LR2 | EGF-EGFR/HER2 uncoupling rate constant | 0.1 min−1† |
| kc,R1E | EGF-EGFR/ERC coupling rate constant | 1 × 10−3 (#/cell)−1 min−1 |
| ku,R1E | EGFR-ERC uncoupling rate constant | 0.1 min−1 |
| ERCtotal | Total number of ERCs | 10,000 #/cell |
| IR1L | Input flux of EGF-EGFR complexes | Varied |
| IR1LR2 | Input flux of EGF-EGFR/HER2 | Varied |
All values come from the original ERC sorting model (French and Lauffenburger, 1996), except as noted.
Experimentally measured (data not shown).
Parameter value from Hendriks et al. (2003a).
TABLE 2.
Parameter values that are specific to individual models
| Value | ||||
|---|---|---|---|---|
| Parameter | Description | Blockade model | Competition model | Affinity model |
| koff_het | EGF dissociation rate constant from heterodimers | 0.5 min−1 | 0.5 min−1 | 0.1–2.5 min−1 |
| kc,R1E,het | EGF-EGFR/ERC coupling rate constant when in a heterodimer | 0 | 1 × 10−3 (#/cell)−1 min−1 | 1 × 10−3 (#/cell)−1 min−1 |
| kc,R2E | HER2/ERC coupling rate constant | 0 | 1 × 10−3 (#/cell)−1 min−1 | 0 |
| kc,R2E,het | HER2/ERC coupling rate constant when in a heterodimer | 0 | 1 × 10−3 (#/cell)−1 min−1 | 1 × 10−3 (#/cell)−1 min−1 |
| ku,R2E | HER2/ERC uncoupling rate constant | 0 | 0.1 min−1 | 0 |
Computations
All model equations are simultaneously coded into Matlab, ver. 6.5 (Mathworks, Natick, MA) and solved at steady state. Individual models are examined by setting appropriate parameters to zero and/or varying parameters of interest before evaluation. Each simulation is run with a specified flux of ligand-receptor complexes and occupied heterodimers. After 120 min of simulation time, when steady state has been reached, sorting fractions and intracellular ligand concentrations were determined as described in Results. By varying the magnitude of the input fluxes of ligand-receptor complexes and occupied heterodimers, holding their ratio constant, sorting curves relating sorting fraction to intracellular ligand concentration were generated.
RESULTS
Experimental sorting outcomes
The motivation for this work comes from the experimental observations of the endosomal sorting of EGF as a function of HER2 expression level originally published in Hendriks et al. (2003a) (reprinted in Fig. 3 a). These results describe steady-state sorting outcomes for 184A1 human mammary epithelial cells for varying EGF concentrations as a function of HER2 expression level. Each cell clone shown has a comparable level of EGFR expression (∼2 × 105) and HER2 expression levels of 3 × 104 and 6 × 105 for the parental line, and HER2 clone 24H, respectively (Hendriks et al., 2003a). In the parental cell line, increasing intracellular EGF resulted in a downward slope in the fraction of EGF recycled. This is consistent with other work demonstrating the selective retention of EGF-EGFR complexes within the endosome (French et al., 1994). Elevated HER2 expression, as seen in clone 24H, demonstrated an increase in the fraction of EGF recycled relative to the parental cell line. The shallow positive relationship between intracellular EGF and the fraction of EGF recycled for clone 24H suggests that the endosomal cargo is starting to exceed the capacity of the sorting apparatus.
FIGURE 3.

Experimental EGF sorting data. (A) Fraction of EGF recycled as a function of intracellular EGF concentration for cell lines expressing increasing levels of HER2 (reprinted from Hendriks et al., 2003a). Circles and squares represent the parental cell line and HER2 clone 24H, expressing roughly 3 × 104 and 6 × 105 HER2 per cell, respectively. (B) Addition of heterodimerization-blocking antibodies (2C4) abrogates HER2 effect on EGF recycling. Steady-state sorting assays were conducted with (open symbols), or without (solid symbols), overnight pretreatment of saturating amounts of 2C4 antibody on the parental cell line (circles), and HER2 clone 24H (squares).
In addition, the role of heterodimerization was examined in steady-state sorting experiments after overnight pretreatment with saturating amounts (10 μg/ml) of monoclonal antibody 2C4. 2C4 binds to an extracellular epitope on HER2 and has been shown to block both its transactivation and heterodimerization with the EGFR (Agus et al., 2002; Baselga, 2002; Fendly et al., 1990; Lewis et al., 1996). As shown in Fig. 3 b (reprinted from Hendriks et al., 2003a), blocking heterodimerization was sufficient to reverse effect of elevated HER2 expression on sorting fraction. The sorting curve for HER2 clone 24H after 2C4 treatment closely resembled that of the parental cell line. As expected, the addition of 2C4 had no effect on sorting for the parental cell line (Hendriks et al., 2003a).
Sorting fractions
The degree to which internalized ligands are recycled toward the surface versus targeted for endosomal degradation can be described by a sorting fraction. This fraction represents the ratio of ligand molecules that leave the endosomal tubules and enter the recycling compartment to the total amount of ligand molecules that leave the endosomes through either the tubular or vesicular compartment and enter the recycling or degradative compartments, respectively. Ligand molecules may transit through the system either as unbound ligand that is free in the endosomal lumen or as bound ligand that is complexed with EGFR in the form of receptor-ligand complexes or as part of a bound EGF-EGFR/HER2 heterodimer.
When the sorting process is at steady state, the sorting fractions can be defined as follows:
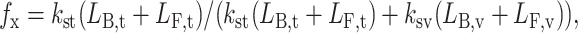 |
with
 |
where kst is the tubular sorting rate constant; ksv is the vesicular sorting rate constant; LF,i and LB,i are the free and bound ligand concentrations in compartment i, respectively; where compartment i is either the tubular (t) or vesicular (v) compartment of the endosome; Vv is the vesicular volume; NA is Avogadro's number; R1Li is the concentration of ligand-receptor complexes (#/cell) in compartment i; R1LR2i is the concentration of ligand bound heterodimers in compartment i; R1LEv is the concentration of ternary ligand-receptor-endosomal retention component complexes; and ER1LR2v and R1LR2Ev are bound heterodimers with an ERC bound to the EGFR or HER2, respectively.
The differences in the definitions of the sorting fraction for each model directly reflect each model's construction. Since the differences in each model lie only in the receptor interactions allowed, the expression for free ligand concentration is identical in each case. For each model the bound ligand in the tubular compartment consists of all ligand bound species allowed (R1Lt and R1LR2t). The bound ligand in the vesicular compartment consists of all ligand-bound species allowed by the model including those that contain ERCs.
For comparison with experimental results, sorting fractions are plotted as a function of total intracellular ligand concentration at steady state. Examination of steady state sorting helps to decouple recycling from the effects of internalization. Total intracellular ligand concentration (Ci) is given by
 |
where the intracellular ligand concentration is the sum of the free (F) and bound (B) ligands in each compartment, and the tubular (t), vesicular (v), recycling (r), and degradation (d) compartments.
ERC sorting model fundamentals
From a foundation of previous modeling efforts, we have a useful basis for understanding of how different experimental outcomes reflect various molecular-level interactions in the sorting process (French and Lauffenburger 1996, 1997). The typical sorting curve can be broken into three regimes (see French and Lauffenburger, 1996), as shown in Fig. 4. In regime I, at low intracellular EGF, sorting outcomes are the result of fluid phase sorting. The majority of ligands dissociate from their receptors and the recycling fraction reflects the fluid phase partitioning of ligands between the endosomal lumen and recycling tubules. In regime II, at intermediate EGF concentration, occupied receptors are selectively retained by ERCs and targeted for degradation; hence, the downward slope of the sorting curve. Here, endosomal ligand concentration is high enough to force receptor occupancy, but low enough so that the ERCs are not saturated. In regime III, at high intracellular EGF, the ERCs become saturated and there is a sharp increase in fraction recycled, reflecting the fact that recycling is the default pathway for the EGFR (French et al., 1994). It should be noted that experimental results usually do not contain all three regimes due to limitations in 125I-EGF detection and/or limitations at the level of internalization, including limited internalization capacity and/or EGFR number. Based on the experimental data, it is apparent that the parental cell line falls entirely within regime II, whereas clone 24H displays the onset of saturation, as seen in the beginning of regime III. The area of interest for our experimental data is outlined in Fig. 4. All further model results will focus within this region.
FIGURE 4.

Typical sorting curve from the original ERC sorting model. The curve can be broken into three regimes: I, fluid-phase sorting; II, EGFR complex interaction with ERCs, decreasing the fraction of EGF recycled; and III, saturation of a limited quantity of ERCs. Experimental data shown in Fig. 3 fall primarily within regime II as indicated by the rectangle.
Blockade model
Some recent experimental work has indicated potential differential signaling abilities of heterodimers versus homodimers. Controlled homo- and heterodimerization of EGFR and HER2 has shown that heterodimerization with HER2 impedes the ability of the EGFR to recruit c-Cbl, possibly by failing to phosphorylate a key tyrosine residue on the cytoplasmic domain of the EGFR (Muthuswamy et al., 1999). In this model, we propose that EGF-EGFR complex heterodimerization with HER2 may impair EGFR interaction with ERCs (see Fig. 2 a). Fig. 5 a illustrates how the sorting fraction of EGF is predicted to change for the blockade model as the input ratio is varied from 100% EGF-EGFR complexes to 100% heterodimers.
FIGURE 5.

Simulated sorting curves for the blockade model, in which heterodimerization blocks EGFR/ERC interaction. (A) Model input is varied from 100% EGF-EGFR complexes/0% heterodimers (solid line), to 50% EGF-EGFR complexes/50% heterodimers (dot-dashed line), to 0% EGF-EGFR complexes/100% heterodimers (dotted line). (B) Bound heterodimer uncoupling rate constant (ku,R1LR2) is varied from 0.1 to 100× the base value (0.1 min−1), for a model input of 0% EGF-EGFR complexes/100% heterodimers.
As the ratio of heterodimers to complexes increases, there is an immediate effect on the sorting fraction, particularly at low intracellular ligand concentrations. An input of 100% heterodimers elicits an increase in sorting fraction of up to 0.4 when compared to an input of 100% complexes, at an intracellular ligand concentration of only 104 #/cell. In the regime before ERC saturation, the entire curve is shifted upward so that the effect of adding HER2 to the system is immediate and is readily observed. In this model, a single molecule of HER2 is able to elicit a direct difference in sorting fraction, particularly at low intracellular EGF concentrations.
Effects of the basic parameters of the original endosomal sorting model have already been explored in detail (French and Lauffenburger, 1996, 1997). As such, we constrain ourselves to examination of the parameters whose response is affected as a consequence of HER2 expression. In particular, the effect of HER2 expression on endosomal sorting can be modulated by changes in the affinity for heterodimerization. As many of the membrane-level receptor interactions are likely to be diffusion-limited, we choose to examine changes in heterodimerization affinity by altering the heterodimer uncoupling rate (ku,R1LR2) (Shea et al., 1997). As shown in Fig. 5 b, using a model input of 100% heterodimers, a decrease in the heterodimer uncoupling rate increases the efficiency with which a given HER2 expression level is able to enhance EGF sorting toward recycling.
Competition model
Based on the high degree sequence similarity of the cytoplasmic domains of the EGFR and HER2 it is possible that the presence of HER2 may compete with the EGFR for interaction with ERCs (Earp et al., 1995; Schechter et al., 1985; Ullrich et al., 1984). In this model's construction, both free HER2 and HER2 that is heterodimerized with EGFRs can bind ERCs. Consequently, HER2 competes with the EGFR for a limited quantity of available ERCs (see Fig. 2 b), accelerating the onset of saturation of endosomal sorting.
Fig. 6 a shows the predicted effect of increasing HER2 expression level on the endosomal sorting of EGF-EGFR complexes when HER2 competes for interaction with ERCs. The model input is varied from 100% EGF-EGFR complexes to 50% EGF-EGFR complexes, 50% heterodimers to 100% heterodimers. At very low intracellular EGF concentrations (<10<103 #/cell) the three curves merge and are indistinguishable (not shown on graph). As intracellular EGF increases, the curves diverge and ultimately converge again (this portion is not shown on the graph) as the sorting machinery becomes saturated. HER2 expression has its greatest impact just before ERC saturation at intermediate intracellular EGF concentrations (∼104 #/cell). At this EGF concentration, an input of 100% heterodimers elicits an increase in sorting fraction of ∼0.1 over that of an input of 100% complexes. The addition of HER2 accelerates the onset of saturation of the sorting machinery by effectively titrating out the number of ERCs. Consequently, at low EGF concentrations, where the number of ERCs greatly exceeds the number of internal receptors, HER2 is unable to induce any effect on EGF sorting.
FIGURE 6.

Simulated sorting curves for the competition model, in which both free and heterodimerized HER2 competes with EGFR for interaction with ERCs. (A) Model input is varied from 100% EGF-EGFR complexes/0% heterodimers (solid line), to 50% EGF-EGFR complexes/50% heterodimers (dot-dashed line), to 0% EGF-EGFR complexes/100% heterodimers (dotted line). (B) HER2/ERC uncoupling rate constant (ku,R2E) is varied from 0.1 to 100× the base value (0.1 min−1), for a model input of 0% EGF-EGFR complexes/100% heterodimers.
In this model, the system interaction is governed by the affinities of HER2 and EGF-EGFR complexes for ERCs. For a model input of 100% heterodimers, decreasing the HER2/ERC uncoupling rate resulted in an increase in sorting fraction (Fig. 6 b). Thus, increasing the expression of HER2 has the same effect as decreasing the HER2/ERC uncoupling rate.
Affinity model
The endosomal sorting of ligands is strongly controlled by their binding properties at endosomal pH. There are many reports of HER2 increasing EGFR affinity for EGF by as much as sixfold (Karunagaran et al., 1996; Lenferink et al., 1998; Lewis et al., 1996; Wada et al., 1990; Worthylake et al., 1999). Under certain conditions, heightened EGF affinity within the endosome has been shown to enhance ligand recycling (French and Lauffenburger 1996).
Conversely, Lenferink and co-workers demonstrated an increase in EGF dissociation from heterodimers at endosomal pH (Lenferink et al., 1998). The increased dissociation of TGFα at pH 6.0, relative to EGF, results in an increase in recycling (French et al., 1995). Thus, it is conceivable that EGF-EGFR heterodimerization with HER2 may increase EGF recycling by promoting the dissociation of EGF from EGFR.
In this model, EGFR/HER2 heterodimerization acts to alter the dissociation rate of EGF from the EGFR (see Fig. 2 c). Two cases are considered. In the first case we consider the possibility that heterodimerization increases the dissociation rate of EGF, modeled by an increased dissociation rate from 0.5 to 2.5 min−1. Secondly, we consider the case where heterodimerization enhances EGFR affinity for EGF, modeled by a dissociation rate decreased from 0.5 to 0.1 min−1. HER2 does not interact with ERCs, and heterodimerization does not affect the ability of the EGFR to bind ERCs whereas EGF is still bound. Similar to the blockade model, altering EGF dissociation results in an immediate effect on EGF sorting such that the entire sorting curve (in the regime before saturation) is shifted up in the case where the dissociation rate is increased (Fig. 7 a) and is shifted down in the case where the dissociation rate is decreased (Fig. 7 b). Given the relatively high affinity of human EGF at endosomal pH, we are unable to reproduce the phenomena in which increasing affinity increases recycling (results not shown).
FIGURE 7.

Simulated sorting curves for the affinity model, in which heterodimerization alters the dissociation rate of EGF. (A) The dissociation rate of EGF from heterodimers (koff,het) is increased to 2.5 min−1 and the model input is varied from 100% EGF-EGFR complexes/0% heterodimers (solid line), to 50% EGF-EGFR complexes/50% heterodimers (dot-dashed line), to 0% EGF-EGFR complexes/100% heterodimers (dotted line). (B) koff,het is decreased to 0.1 min−1, and model input is varied from 100% EGF-EGFR complexes/0% heterodimers (solid line), to 50% EGF-EGFR complexes/50% heterodimers (dot-dashed line), to 0% EGF-EGFR complexes/100% heterodimers (dotted line).
DISCUSSION
Elevated HER2 expression and its interactions with EGFR family members have been demonstrated to be of great importance in tumor progression (Hynes and Stern, 1994). HER2 amplifies the magnitude of EGFR signaling through the recruitment of additional signaling molecules and also increases the duration of EGFR signaling via the disruption of the normal trafficking and downregulation of the EGFR (Karunagaran et al., 1996; Worthylake et al., 1999). Impaired EGFR trafficking has been linked to tumor formation in mice and as such, we have chosen to concern ourselves with processes involved in receptor downregulation, specifically, endosomal sorting (Wells et al., 1990). Recent work has quantitatively demonstrated the importance of endosomal sorting in determining the distribution and downregulation of EGFR (Hendriks et al., 2003a). Endosomal sorting represents a critical regulatory point in EGFR trafficking by controlling the fraction of receptors and ligands that are targeted for degradation. In this study, we have utilized computational modeling techniques to gain insight into the receptor interactions that govern the qualitative aspects of the observed endosomal sorting outcomes.
From the experimental data, the parental cell line is clearly operating in the regime where EGFR complex interaction with ERCs dominates and mediates EGF degradation. Secondly, over the experimentally accessible range of intracellular EGF concentration we do not see the onset of ERC saturation, as the slope of the sorting curve remains negative. For HER2 clone 24H, however, the shallow positive slope suggests the onset of ERC saturation. Thus, elevated HER2 expression appears to accelerate the onset of endosomal sorting saturation.
To account for this result, we have expanded the ERC sorting model to include HER2 and investigated how different HER2 interactions affect the sorting process. Qualitatively, our three models give us one of two possible results, with the differences manifesting themselves at low intracellular EGF concentrations. In the blockade and affinity model (in which heterodimerization decreases affinity), recycling is increased at the low intracellular EGF concentrations, where the leftmost portion of the sorting curve is shifted upwards. The increase in recycling with HER2 expression simply reflects the fraction of EGF-EGFR complexes that are in heterodimers and each individual heterodimer directly affects the fraction of EGF recycled. The competition model, by contrast, is a titration effect, where increased presence of HER2 inside the sorting endosome is unable to alter EGF recycling until its quantity is on the same order of magnitude as the number of ERCs. Consequently, we observe a result where there is no difference at low intracellular EGF concentration for the different levels of HER2 expression. As intracellular EGF increases the curves begin to diverge and elevated HER2 expression expedites the point at which endosomal saturation begins.
While the three models proposed are not mutually exclusive, comparison with experimental results suggests that the competitive mechanism is dominant. Experimental sorting outcomes for varying HER2 expression levels (see in Fig. 3 a) converge at low intracellular EGF, qualitatively similar to the competition model results (see Fig. 6 a). However, it is possible that different mechanisms may dominate in different cell types. Data from Worthylake and Wiley show that increased HER2 expression shifted the entire sorting curve upward (Worthylake et al., 1999). This may be indicative of the blockade or affinity models being dominant, or it is possible that the sorting curves may still converge if experiments were carried out at sufficiently low EGF concentrations.
We remind the reader that the purpose of this work is not to quantitatively fit the data, but rather to understand how various molecular-level interactions are translated into qualitative trends in the experimental data. A number of the model parameters are based upon estimates from other cell types and may not necessarily be optimal choices to reflect our experimental setup. Nonetheless, the qualitative nature of the model results is quite robust, and is insensitive to reasonable parameter variations. Further, the model assumes a constant input flux of receptors and ligands for ease of interpretation. Experimentally, the internalization flux may not be constant since internalization rates have been shown to be a function of the number of surface complexes and vary with the surface expression levels of HER2 as well (Hendriks et al., 2003a,b; Wiley et al., 1991). For these reasons, a direct, quantitative comparison of the experimental and modeling results is not appropriate.
Our models contain several simplifications including the fact that EGF-EGFR complex homodimerization is not explicitly included. In the original ERC model the EGF-EGFR complex is the functional unit in terms of interaction within the endosome. Conceptually, this unit could be thought of either as a single EGF-EGFR complex or an EGF-EGFR homodimer with no effect on the sorting results. When HER2 is added to the model it is best to conceptualize the EGF-EGFR representation as a homodimer and the process of heterodimerization with HER2 simply reflects trading an EGF-EGFR complex for a HER2 within the dimer. If one explicitly includes all possible EGFR-HER2 interactions the results are indistinguishable from those presented here (results not shown). These simplifications serve to simplify the computations and do not affect the characteristic qualities of each model—the blockade and affinity models still show immediate influence from HER2 expression, while the competition model requires sufficient HER2 present before any effect is apparent.
If HER2 affects EGF recycling through a competitive mechanism then, at first glance, one would expect heterodimerization to have no effect on the sorting process. As such, the addition of monoclonal antibody 2C4 would be predicted to have no effect on EGF sorting. However, Fig. 3 b shows this is not the case. If one considers the trafficking process as a whole, we find that this is still consistent with HER2 acting via a competitive mechanism. As a side effect from blocking heterodimerization, one would predict that 2C4 prevents the EGF-induced internalization of HER2 (Hendriks et al., 2003a,b). Thus, addition of 2C4 prevents the internalization of HER2 into the endosomal sorting compartment so that it is unable to compete with EGFR for ERC interaction. Heterodimerization itself is not predicted to have any effect on the sorting process; however, effects at the level of internalization affect the sorting process by dictating the receptor composition within endosomal compartments.
The hypothesis that HER2 is able to alter EGFR sorting through a competitive mechanism suggests that there must be some sequence similarity between the two receptors in the carboxy-terminal domain regions interacting with the sorting apparatus. The precise identity of the endosomal retention component remains unknown at present, but evidence suggests that SNX1 may play such a role (Kurten et al., 1996; Zhong et al., 2002). SNX1 was identified via its interaction with EGFR residues 943–957, and has been shown to localize to endosomal compartments. Its inhibition decreases the rate of ligand-induced EGFR degradation, consistent with the behavior of ERCs in the ERC sorting model (French and Lauffenburger, 1996; Kurten et al., 1996; Zhong et al., 2002). EGFR residues 943–957 are known to interact with SNX1 and share 80% identity with HER2 residues 951–965, suggesting that HER2 may also be able to interact with SNX1.
Another candidate for relevant involvement in the sorting process is c-Cbl. Overexpression of c-Cbl stimulates ligand-induced EGFR degradation (Levkowitz et al., 1999, 1998). Further, c-Cbl associates only with EGF-EGFR homodimers, but not with EGF-EGFR/HER2 heterodimers, HER2, HER3, or HER4 (Levkowitz et al., 1996; Muthuswamy et al., 1999). A current model has c-Cbl transiently associating with kinase-active EGFR to mediate ubiquitination, with ubiquitin-tagged EGFR then exhibiting increased affinity for the sorting apparatus resulting in enhanced degradation (Wiley and Burke, 2001). The fact that heterodimerization impedes this process is suggestive of a blockade-type mechanism; however, based on our results its role in generating the experimentally-observed sorting outcomes is not obvious.
Our integrative systems approach toward EGFR trafficking has gained us interesting insight into the trafficking process as a whole, especially in the context of a hierarchy of receptor trafficking models (Hendriks et al., 2003a,b). The sorting process is tightly regulated and its output (sorting fraction) is heavily dependent on the composition of its input (complexes vs. heterodimers). In the case of the EGFR-HER2 system it appears that the highest level of control is exerted at the surface since this determines the input into the sorting compartment. It is at the surface where the distribution of complexes and heterodimers is determined. Increased formation of heterodimers results in a reduced rate of EGF internalization in addition to an increase in the fraction recycled toward the surface (Hendriks et al., 2003a,b). These two processes work in concert to maintain EGFR expression on the surface and presumably maintain signaling through surface-activated signaling pathways, such PLC-γ and calpain, both involved in cell migration. Whether or not the distribution of dimer species reshuffles once inside of internal compartments due to a different receptor composition is unclear. The degree to which these internal species participate in signaling once internalized is also in need of further investigation. Because trafficking is an iterative process, however, we would still expect sorting to play an important role in the dictating long-term behavior after successive rounds of internalization and recycling.
Acknowledgments
The authors are grateful to A. Wells, W. H. Deen, and K. D. Wittrup for helpful insights.
This work was funded in part by a National Cancer Institute grant to D.A.L., a National Institute of Child Health and Human Development grant to D.A.L. (G. Kruger, Principal Investigator), and a graduate Whitaker fellowship to B.S.H.
APPENDIX
Endosomal sorting model equations
The following equations are used to simulate the various possible effects of HER2 on the steady state endosomal sorting of EGF. The model is solved at steady state for varying inputs of complexes (IR1L) and heterodimers (IR1LR2). Each model (blockade, competition, or affinity) is independently simulated by changing the appropriate parameters as described in Model Development.
Central vesicle
 |
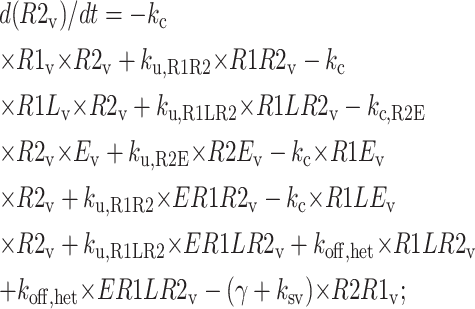 |
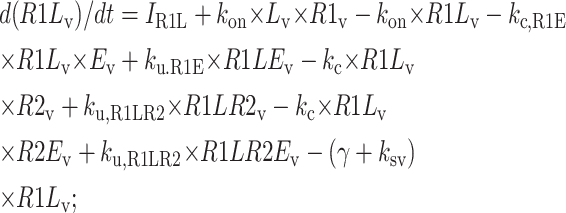 |
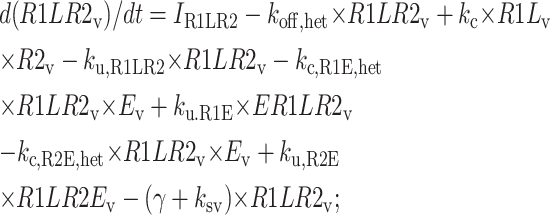 |
 |
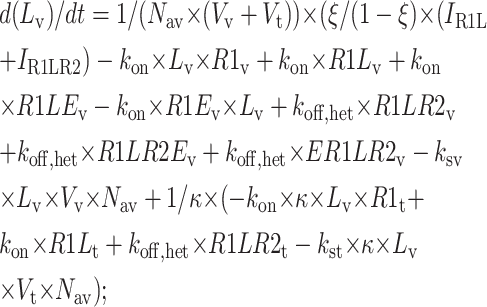 |
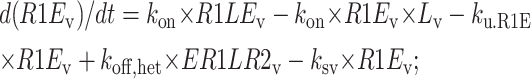 |
 |
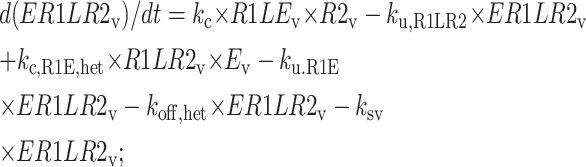 |
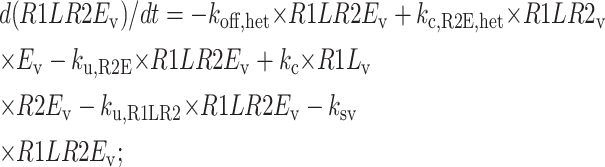 |
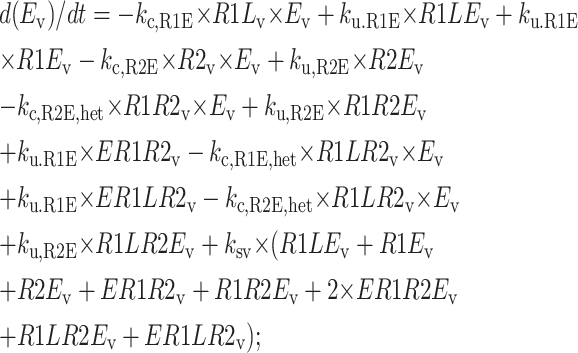 |
Vesicle tubule compartment
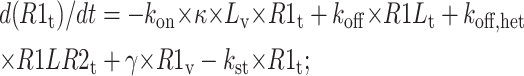 |
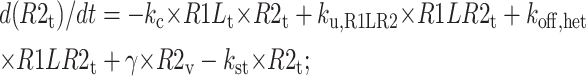 |
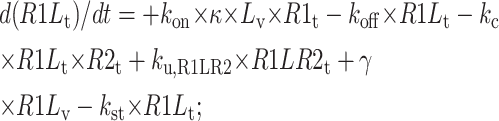 |
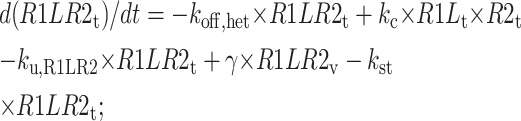 |
Degradation compartment
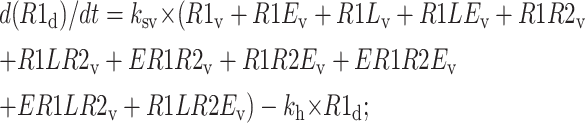 |
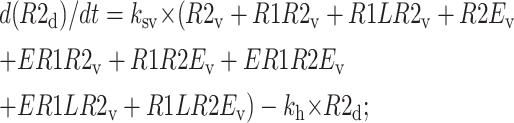 |
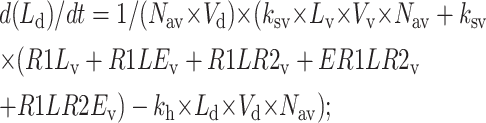 |
Recycling compartment
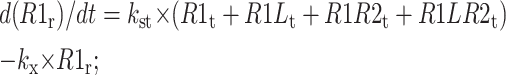 |
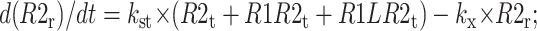 |
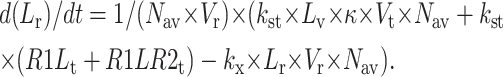 |
References
- Agus, D. B., R. W. Akita, W. D. Fox, G. D. Lewis, B. Higgins, P. I. Pisacane, J. A. Lofgren, C. Tindell, D. P. Evans, K. Maiese, H. I. Scher, and M. X. Sliwkowski. 2002. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2:127–137. [DOI] [PubMed] [Google Scholar]
- Alroy, I., and Y. Yarden. 1997. The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand-receptor interactions. Fed. Eur. Biol. Sci. Lett. 410:83–86. [DOI] [PubMed] [Google Scholar]
- Baselga, J. 2002. A new anti-ErbB2 strategy in the treatment of cancer: prevention of ligand-dependent ErbB2 receptor heterodimerization. Cancer Cell. 2:93–94. [DOI] [PubMed] [Google Scholar]
- Baulida, J., M. H. Kraus, M. Alimandi, P. P. DiFiore, and G. Carpenter. 1996. All ErbB receptors other than the epidermal growth factor receptor are endocytosis impaired. J. Biol. Chem. 271:5251–5257. [DOI] [PubMed] [Google Scholar]
- Brandt, B. H., A. Roetger, T. Dittmar, G. Nikolai, M. Seeling, A. Merschjann, J.-R. Nofer, G. Dehmer-Moller, R. Junker, G. Assmann, and K. S. Zaenker. 1999. c-ErbB-2/EGFR as dominant heterodimerization partners determine a motogenic phenotype in human breast cancer cells. FASEB J. 13:1939–1950. [DOI] [PubMed] [Google Scholar]
- Burke, P. M., K. Schooler, and H. S. Wiley. 2001. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol. Biol. Cell. 12:1897–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazin, V. R., M. Kaleko, A. D. Miller, and D. J. Slamon. 1992. Transformation mediated by the human HER-2 gene independent of the epidermal growth factor receptor. Oncogene. 7:1859–1866. [PubMed] [Google Scholar]
- DiFiore, P. P., J. H. Pierce, T. P. Fleming, R. Hazan, A. Aullrich, C. R. King, J. Schlessinger, and S. A. Aaronson. 1987a. Overexpression of the human EGF receptor confers an EGF-dependent transformed phenotype to NIH 3T3 cells. Cell. 51:1063–1070. [DOI] [PubMed] [Google Scholar]
- DiFiore, P. P., J. H. Pierce, M. H. Kraus, O. Segatto, C. R. King, and S. A. Aaronson. 1987b. ErbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. Science. 237:178–182. [DOI] [PubMed] [Google Scholar]
- Earp, H. S., T. L. Dawson, X. Li, and H. Yu. 1995. Heterodimerization and function interaction between EGF receptor family members: a new signaling paradigm with implications for breast cancer research. Breast Cancer Res. Treat. 35:115–132. [DOI] [PubMed] [Google Scholar]
- Fendly, B. M., M. Winget, R. M. Hudziak, M. T. Lipari, M. A. Napier, and A. Ullrich. 1990. Characterization of murine monoclonal antibodies reactive to either the human epidermal growth factor receptor or HER2/neu gene product. Cancer Res. 50:1550–1558. [PubMed] [Google Scholar]
- French, A. R., and D. A. Lauffenburger. 1996. Intracellular receptor/ligand sorting based on endosomal retention components. Biotechnol. Bioeng. 51:281–297. [DOI] [PubMed] [Google Scholar]
- French, A. R., and D. A. Lauffenburger. 1997. Controlling receptor/ligand trafficking: effects of cellular and molecular properties on endosomal sorting. Ann. Biomed. Eng. 25:690–707. [DOI] [PubMed] [Google Scholar]
- French, A. R., G. P. Sudlow, H. S. Wiley, and D. A. Lauffenburger. 1994. Postendocytic trafficking of epidermal growth factor-receptor complexes is mediated through saturable and specific endosomal interactions. J. Biol. Chem. 269:15749–15755. [PubMed] [Google Scholar]
- French, A. R., D. K. Tadaki, S. K. Niyogi, and D. A. Lauffenburger. 1995. Intracellular trafficking of epidermal growth factor family ligands is directly influenced by the pH sensitivity of the receptor/ligand interaction. J. Biol. Chem. 270:4334–4340. [DOI] [PubMed] [Google Scholar]
- Glading, A., F. Uberall, S. M. Keyse, D. A. Lauffenburger, and A. Wells. 2001. Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J. Biol. Chem. 276:23341–23348. [DOI] [PubMed] [Google Scholar]
- Graus-Porta, D., R. R. Beerli, J. M. Daly, and N. E. Hynes. 1997. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. Eur. Mol. Biol. Org. J. 16:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugh, J. M., A. C. Huang, H. S. Wiley, A. Wells, and D. A. Lauffenburger. 1999a. Internalized epidermal growth factor receptors participate in the activation of p21ras in fibroblasts. J. Biol. Chem. 274: 34350–34360. [DOI] [PubMed] [Google Scholar]
- Haugh, J. M., K. Schooler, A. Wells, H. S. Wiley, and D. A. Lauffenburger. 1999b. Effect of epidermal growth factor receptor internalization on regulation of the phospholipase C-γ1 signaling pathway. J. Biol. Chem. 274:8958–8965. [DOI] [PubMed] [Google Scholar]
- Hendriks, B. S., L. K. Opresko, H. S. Wiley, and D. A. Lauffenburger. 2003a. Co-regulation of EGFR/HER2 levels and locations: quantitative analysis of HER2 overexpression effects. Cancer Res. 63:1130–1137. [PubMed] [Google Scholar]
- Hendriks, B. S., L. K. Opresko, H. S. Wiley, and D. A. Lauffenburger. 2003b. Quantitative analysis of HER2-mediated effects on HER2 and EGFR endocytosis: distribution of homo- and hetero-dimers depends on relative HER2 levels. J. Biol. Chem. 278:23343–23351. [DOI] [PubMed] [Google Scholar]
- Herbst, J. J., L. K. Opresko, B. J. Walsh, D. A. Lauffenburger, and H. S. Wiley. 1994. Regulation of postendocytic trafficking of the epidermal growth factor receptor through endosomal retention. J. Biol. Chem. 269:12865–12873. [PubMed] [Google Scholar]
- Hynes, N. E., and D. F. Stern. 1994. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim. Biophys. Acta. 1198:165–184. [DOI] [PubMed] [Google Scholar]
- Ignatoski, K. M. W., A. J. Lapointe, E. H. Radany, and S. P. Ethier. 1999. ErbB-2 overexpression in human mammary epithelial cells confers growth factor independence. Endocrinology. 140:3615–3622. [DOI] [PubMed] [Google Scholar]
- Karunagaran, D., E. Tzahar, R. R. Beerli, X. Chen, D. Graus-Porta, B. J. Ratzkin, R. Seger, N. E. Hynes, and Y. Yarden. 1996. ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. Eur. Mol. Biol. Org. ET J. 15:254–264. [PMC free article] [PubMed] [Google Scholar]
- Kil, S. J., and C. Carlin. 2000. EGF receptor residues Leu679, Leu680 mediate selective sorting of ligand-receptor complexes in early endosomal compartments. J. Cell. Physiol. 185:47–60. [DOI] [PubMed] [Google Scholar]
- Kil, S. J., M. Hobert, and C. Carlin. 1999. A leucine-based determinant in the epidermal growth factor receptor juxtamembrane domain is required for the efficient transport of ligand-receptor complexes to lysosomes. J. Biol. Chem. 274:3141–3150. [DOI] [PubMed] [Google Scholar]
- Kornilova, E. S., T. Sorkina, L. Beguinot, and A. Sorkin. 1996. Lysosomal targeting of epidermal growth factor receptors via a kinase-dependent pathway is mediated by the receptor carboxyl-terminal residues 1022–1123. J. Biol. Chem. 271:30340–30346. [DOI] [PubMed] [Google Scholar]
- Kurten, R. C., D. L. Cadena, and G. N. Gill. 1996. Enhanced degradation of EGF receptors by a sorting nexin, SNX1. Science. 272:1008–1010. [DOI] [PubMed] [Google Scholar]
- Lauffenburger, D. A., and J. J. Linderman. 1993. Receptors. Oxford University Press, New York.
- Lenferink, A. E. G., R. Pinkas-Kramarski, M. L. M. van der Poll, M. J. H. von Vugt, L. N. Klapper, E. Tzahar, H. Waterman, M. Sela, E. J. J. von Zoelen, and Y. Yarden. 1998. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. Eur. Mol. Biol. Org. J. 17:3385–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkowitz, G., L. N. Klapper, E. Tzahar, A. Freywald, M. Sela, and Y. Yarden. 1996. Coupling of the c-Cbl protooncogene product to ErbB-1/EGF-receptor but not to other ErbB proteins. Oncogene. 12:1117–1125. [PubMed] [Google Scholar]
- Levkowitz, G., H. Waterman, S. A. Ettenberg, M. Katz, A. Y. Tsygankov, I. Alroy, S. Lavi, K. Iwai, Y. Reiss, A. Ciechanover, S. Lipkowitz, and Y. Yarden. 1999. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell. 4:1029–1040. [DOI] [PubMed] [Google Scholar]
- Levkowitz, G., H. Waterman, E. Zamir, Z. Kam, S. Oved, W. Y. Langdon, L. Beguinot, B. Geiger, and Y. Yarden. 1998. c-Cbl/sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev. 12:3663–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, G. D., J. A. Lofgren, A. E. McMurtrey, A. Nuijens, B. M. Fendly, B. D. Bauer, and M. X. Sliwkowski. 1996. Growth regulation of human breast and ovarian tumor cells by heregulin: evidence of the requirement of ErbB2 as a critical component in mediating heregulin responsiveness. Cancer Res. 56:1457–1465. [PubMed] [Google Scholar]
- Linderman, J. J., and D. A. Lauffenburger. 1986. Analysis of intracellular sorting: calculation of mean surface and bulk diffusion times within a sphere. Biophys. J. 50:295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund, K. A., L. K. Opresko, C. Starbuck, B. J. Walsh, and H. S. Wiley. 1990. Quantitative analysis of the endocytic system involved in hormone-induced receptor internalization. J. Biol. Chem. 265:15713–15723. [PubMed] [Google Scholar]
- Muthuswamy, S., M. Gilman, and J. S. Brugge. 1999. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol. Cell. Biol. 19:6845–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olayioye, M. A., D. Graus-Porta, R. R. Beerli, J. Rohrer, B. Gay, and N. E. Hynes. 1998. ErbB-1 and ErbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol. Cell. Biol. 18:5042–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opresko, L., C. Chang, B. Will, P. M. Burke, G. N. Gill, and H. S. Wiley. 1995. Endocytosis and lysosomal targeting of epidermal growth factor receptors are mediated by distinct sequences independent of tyrosine kinase domain. J. Biol. Chem. 270:4325–4333. [DOI] [PubMed] [Google Scholar]
- Schechter, A. L., M.-C. Hung, L. Vaidyanathan, R. A. Weinberg, T. L. Yang-Feng, U. Francke, A. Ullrich, and L. Coussens. 1985. The neu gene: an ErbB-homologous gene distinct from and unlinked to the gene encoding the EGF receptor. Science. 229:976–978. [DOI] [PubMed] [Google Scholar]
- Shea, L. D., G. M. Omann, and J. J. Linderman. 1997. Calculation of diffusion-limited kinetics for the reactions in collision coupling and receptor cross-linking. Biophys. J. 73:2949–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkin, A., P. P. DiFiore, and G. Carpenter. 1993. The carboxyl terminus of epidermal growth factor receptor/ErbB-2 chimerae is internalization impaired. Oncogene. 8:3021–3028. [PubMed] [Google Scholar]
- Sorkin, A., and C. M. Waters. 1993. Endocytosis of growth factor receptors. Bioessays. 15:375–382. [DOI] [PubMed] [Google Scholar]
- Spencer, K. S. R., D. Graus-Porta, J. Leng, N. Hynes, and R. L. Klemke. 2000. ErbB2 is necessary for induction of carcinoma cell invasion by ErbB family receptor tyrosine kinases. J. Cell Biol. 148:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich, A., L. Coussens, J. S. Hayflick, T. J. Dul, A. Gray, A. W. Tam, J. Lee, Y. Yarden, T. A. Libermann, J. Schlessinger, J. Downward, J. Bye, N. Whittle, M. Waterfield, and P. Seeburg. 1984. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carncinoma cells. Nature. 309:418–425. [DOI] [PubMed] [Google Scholar]
- Vieira, A. V., C. Lamaze, and S. L. Schmid. 1996. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 274:2086–2089. [DOI] [PubMed] [Google Scholar]
- Wada, T., X. Qian, and M. I. Greene. 1990. Intermolecular association of the p185neu protein and EGF receptor modulates EGF receptor function. Cell. 61:1339–1347. [DOI] [PubMed] [Google Scholar]
- Wang, Z., L. Zhang, T. K. Yeung, and X. Chen. 1999. Endocytosis deficiency of epidermal growth factor (EGF) receptor-ErbB2 heterodimers in response to EGF stimulation. Mol. Biol. Cell. 10:1621–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman, H., I. Sabanai, B. Geiger, and Y. Yarden. 1998. Alternative intracellular routing of ErbB receptors may determine signaling potency. J. Biol. Chem. 273:13819–13827. [DOI] [PubMed] [Google Scholar]
- Wells, A., J. B. Welsh, C. S. Lazar, H. S. Wiley, G. N. Gill, and M. G. Rosenfeld. 1990. Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science. 247:962–964. [DOI] [PubMed] [Google Scholar]
- Wiley, H. S. 1988. Anomalous binding of epidermal growth factor to A431 cells is due to the effect of high receptor densities and a saturable endocytic system. J. Cell Biol. 107:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, H. S., and P. M. Burke. 2001. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2:12–18. [DOI] [PubMed] [Google Scholar]
- Wiley, H. S., J. J. Herbst, B. J. Walsh, D. A. Lauffenburger, M. G. Rosenfeld, and G. N. Gill. 1991. The role of tyrosine kinase activity in endocytosis, compartmentation, and down-regulation of the epidermal growth factor receptor. J. Biol. Chem. 266:11083–11094. [PubMed] [Google Scholar]
- Worthylake, R., L. K. Opresko, and H. S. Wiley. 1999. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J. Biol. Chem. 274:8865–8874. [DOI] [PubMed] [Google Scholar]
- Worthylake, R., and H. S. Wiley. 1997. Structural aspects of the epidermal growth factor receptor required for transmodulation of ErbB-2/neu. J. Biol. Chem. 272:8594–8601. [DOI] [PubMed] [Google Scholar]
- Yarden, Y., and M. X. Sliwkowski. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2:127–137. [DOI] [PubMed] [Google Scholar]
- Zhong, Q., C. S. Lazar, H. Tronchere, T. Sato, T. Meerloo, M. Yeo, Z. Songyang, S. D. Emr, and G. N. Gill. 2002. Endosomal localization and function of sorting nexin 1. Proc. Natl. Acad. Sci. USA. 99:6767–6772. [DOI] [PMC free article] [PubMed] [Google Scholar]