

Abstract
Molecular simulations of hydrated dipalmitoylphosphatidylcholine lipid bilayers have been performed for temperatures in the range of 250–450 K. The area per headgroup increases with temperature from 58 to 77 Å2. Other properties such as hydration number, alkyl tail order parameter, diffusion coefficients, and radial distribution functions exhibit a clear dependence on temperature. Simulations of bilayers have also been performed in the presence of two disaccharides, namely trehalose and sucrose, at concentrations of up to 18 wt % (lipid-free basis). The simulated area per headgroup of the bilayer is not affected by the presence of the disaccharides, suggesting that the overall structure of the bilayer remains undisturbed. The results of simulations reveal that the interaction of disaccharide molecules with the bilayer occurs at the surface of the bilayer, and it is governed by the formation of multiple hydrogen bonds to specific groups of the lipid. Disaccharide molecules are observed to adopt specific conformations to fit onto the surface topology of the bilayer, often interacting with up to three different lipids simultaneously. At high disaccharide concentrations, the results of simulations indicate that disaccharides can serve as an effective replacement for water under anhydrous conditions, which helps explain their effectiveness as lyophilization agents for liposomes and cells.
INTRODUCTION
It has been established experimentally that disaccharides have a stabilizing effect on biological membranes (Crowe et al., 1987, 1988, 2001). Trehalose, a disaccharide of glucose, and sucrose (which consists of a fructose and a glucose ring connected by a glycosidic bond) can be viewed as naturally occurring stabilizing agents. Trehalose is found in animals capable of enduring cold temperatures, whereas sucrose is found in plants (Crowe et al., 1988). Numerous experimental studies have assessed their efficacy as protective additives for freezing (as cryoprotectants) and freezedrying (as lyoprotectants) (Crowe et al., 2001). Studies on liposome suspensions, in particular, have shown that addition of disaccharides prevents leakage and fusion during drying and freezedrying (Madden et al., 1985; Womersley et al., 1986). The stabilizing effect of trehalose has led to a number of recent applications in the food, biomedical, pharmaceutical, and cosmetics industries (Matlouthi, 1999; O'Brien Nabors, 2001).
The precise mechanism by which disaccharides act to preserve biological systems during freezing and drying is not well understood. It is currently believed that effective cryoprotectants for proteins or enzymes in solution are excluded from the immediate vicinity of these biological molecules. This argument has also been extended to membranes (Crowe et al., 2001). This mode of action should be contrasted with that proposed to explain the effectiveness of disaccharides for lyophilization of liposomes and cellular organisms; for such systems it has been argued that, in the absence of water, disaccharides lower the melting temperature of bilayer membranes, thereby preventing leakage during freezing, drying, and rehydration (Crowe et al., 2001).
Cell membranes consist largely of phospholipid molecules. A number of additional inclusions (e.g., proteins) complicates considerably their theoretical and experimental characterization. Pure dipalmitoylphosphatidylcholine (DPPC) bilayers provide a simple but useful model for understanding the interaction of cellular membranes with extracellular media. In fact, in the general area of cryopreservation and lyophilization, considerable insights have been obtained from experimental and theoretical studies of phospholipid monolayers and bilayers.
The mechanistic models proposed to explain cryopreservation and lyophilization have been largely phenomenological. The purpose of this work is to use molecular simulations to provide an in-depth analysis of the atomic-level interactions that arise between disaccharides and a model lipid bilayer membrane in slightly anhydrous environments.
A number of molecular dynamics simulations have been performed to examine self-assembled structures of lipid molecules in water. For recent reviews see work by Tieleman et al. (1997), Tobias et al. (1997), and Bandyopadhyay et al. (1998). Early pioneering work on these systems focused on the structure of water near a monolayer (Alper et al., 1993), and on the structure of the gel state of DPPC (Egberts et al., 1994); it was found that the dipoles in the headgroups were inclined at an angle to the surface (Egberts et al., 1994). Feller et al. (1995) determined the dependence of the surface area of the bilayer on the surface tension and identified important differences between bilayer and monolayer; they showed that monolayers are only of limited use as models for bilayers. More recently, the focus of a number of studies has shifted toward other phospholipid bilayers. For a number of these, researchers have been able to establish good agreement with selected experimental results. For liquid crystalline (Shinoda et al., 1997) and gel-phase DMPC (Tu et al., 1996), for example, direct comparisons of simulations to experiment (x-ray and nuclear magnetic resonance, i.e., NMR) show good agreement. The gel study showed that the two tail chains of a phospholipid are not necessarily equivalent, and that the phosphate group is typically well hydrated; water, however, is not able to penetrate the aliphatic region (Tu et al., 1996). Studies on DPhPC (Husslein et al., 1998), DOPC (Mashl et al., 2001), and SDPC (Saiz and Klein, 2002b) elucidated the structure of bilayers of these different lipids. Studies with cholesterol showed that it increases the order in DPPC (Smondryev and Berkowitz, 2000) and DMPC (Rog and Pasenkiewicz-Gierula, 2001) bilayers. Molecular dynamics has been the standard tool for investigations of bilayers. In a few cases, long simulation runs (on the order of tens of nanoseconds) have permitted calculation of diffusion coefficients (Essmann and Berkowitz, 1999; Moore et al., 2001).
In contrast to research on pure bilayers, computational studies of saccharide-lipid interactions have been limited to energy minimizations in the absence of water (Gaber et al., 1986; Rudolph et al., 1990); these calculations suggested that the bilayer area per headgroup increases in the presence of trehalose (Gaber et al., 1986), and that the interaction energy of the saccharide-lipid complex becomes less stable in the order trehalose < glucose < sucrose (Rudolph et al., 1990). Despite the frequent use of saccharides in cryopreservation and lyophilization of biological systems, full-blown simulations of their interaction with lipid bilayers in either the dry or hydrated state have not appeared in the literature. This work presents results from simulations of pure DPPC bilayers and aqueous disaccharide DPPC bilayers in the liquid-crystalline state over a wide range of temperatures.
SIMULATION METHODS AND DETAILS
Molecular dynamics simulations were performed on systems containing a total of 128 DPPC molecules arranged in a bilayer structure (64 molecules on each side) in the presence of water. Simulations were also performed with low-to-intermediate concentrations of two disaccharides, namely sucrose and trehalose. Fig. 1 shows a snapshot of the simulation box and Fig. 2 provides an illustration of the molecules of interest. The initial conformation of a pure lipid bilayer was provided by the Tieleman group (Tieleman, 2002). To erase any memory of the initial configuration, the structure was initially cycled between 300 and 600 K. The simulation temperature ranged from 250 to 450 K. The initial configuration of the lipid bilayer with disaccharides was obtained by random insertion of disaccharide molecules into the aqueous region, and subsequent removal of overlapping water molecules; additional water molecules were then added or removed to achieve a target composition.
FIGURE 1.

Representative configuration of simulated lipid bilayer/water system.
FIGURE 2.

Schematic of phospholipid and disaccharides used in study. The alkyl chains of DPPC are modeled with a united-atom force field. For the disaccharides, all atoms are explicitly described; for clarity, some of the hydrogens are not shown.
The force field employed in our simulations was assembled from various sources: the GROMOS force field was used (van Gunsteren et al., 1996) for the headgroups of DPPC, and for the aliphatic tails the NERD force field (Nath et al., 1998, 2001; Nath and de Pablo, 2000) was employed (note, however, that the force constants corresponding to the bond potential of the NERD force-field were strengthened by two orders of magnitude to be consistent with the GROMOS parameters). The OPLS force field (Damm et al., 1997) was used for the sugars, and the SPC/E model (Berendsen et al., 1987) was adopted for water.
The compositions of the systems considered in this work are given in Table 1. At each temperature and composition, the system was allowed to equilibrate for at least 1 nanosecond; equilibrium properties were accumulated over simulations of at least 10 nanoseconds. A time-step of 2 femtoseconds was used for all simulations with a leap-frog integration algorithm (Allen and Tildesley, 1987). Nonbonded interactions (Lennard-Jones and Coulombic) were cut off beyond 9 Å. A reaction-field correction was used to account for long-range electrostatic interactions, with a dielectric constant (ɛRF) equal to 80 (Allen and Tildesley, 1987). Comparisons between different long-range correction techniques (large cutoff, reaction-field correction, Ewald summation, switched-potentials) commonly used in simulations of biological systems (Faraldo-Gómez et al., 2002; Nina and Simonson, 2002; Norberg and Nilsson, 2000; Tobias, 2001) suggest that the results of reaction-field and Ewald summation simulations of lipid bilayers yield equivalent results. In the interest of computational efficiency, we have chosen to use the reaction-field correction. For completeness, we have also performed our own set of calculations comparing the reaction-field correction to smooth particle-mesh Ewald (Essman et al., 1995) for several bilayer systems, and we do not find significant differences between them in nanosecond-long simulations. The temperature and pressure of the simulation box were kept constant using the weak coupling technique (Berendsen et al., 1984), with correlation times τT = 0.2 ps and τp = 2.0 ps for temperature and pressure, respectively. For constant-pressure simulations, the three Cartesian directions were independently coupled to an ambient pressure of p = 101.3 kPa with a compressibility κ = 1.12 × 10−6 kPa–1, thereby allowing the area of the bilayer and the distance between the interfaces to fluctuate independently.
TABLE 1.
Composition of simulated lipid bilayer systems; for all cases, 128 DPPC molecules are used
| Water | Disaccharide | Composition* | Temperature (K) |
|---|---|---|---|
| 3655 | — | — | 250,325,350,400,450 |
| 4335 | 8 | 3.40 wt % / 0.18 mol % | 350,400 |
| 2322 | 27 | 18.1 wt % / 1.15 mol % | 350,400 |
Lipid-free basis.
RESULTS AND DISCUSSION
Pure DPPC bilayers
As mentioned earlier, several simulations of the DPPC/water system have appeared recently in the literature (Essmann and Berkowitz, 1999; Pastor et al., 2002; Saiz and Klein, 2002a; Scott, 2002; Shinoda et al., 1997; Tieleman et al., 1997; Tu et al., 1998, 1996). However, to the best of our knowledge, the influence of temperature on bilayer structure has not been examined before. A first goal of this work was to fill this gap by considering the temperature range between 250 and 450 K.
Fig. 3 A shows the density profile for the pure DPPC bilayer at several temperatures. The density profiles have been shifted for clarity (except that at T = 350 K). Three distinct domains can be identified in those figures. The flat region between ∼|z| = 20 Å and |z| = 30 Å represents the aqueous phase, undisturbed from its bulk value. The adjacent area (on both sides), where the density is clearly higher than that of bulk water, corresponds to the interface region, containing lipid headgroups and water. The region where the local density drops well below the water density corresponds to the inner part of the bilayer, where the alkyl chains of the phospholipid reside. The middle of the bilayer, where the ends of the tails of both lipid layers meet, is the plane of lowest density. If the local density is resolved according to different components, water and lipid, we estimate the width of the interface (between water and the aliphatic chains—see Fig. 3 B) as the distance over which the water density rises from 10 to 90% of the bulk value. The width of this interface becomes slightly broader with decreasing temperature. This distance drops from 15.2 to 9.8 Å between 250 and 450 K. Moreover, the bilayer itself becomes thicker with decreasing temperature. This result appears counterintuitive, as we have a negative heat expansion coefficient; however, it can be explained by the fact that the bilayer structure changes with temperature. The thickness of the bilayer (measured between the maximum density peaks of the density profile) drops from 34.3 to 30.0 Å between 250 and 450 K. This decrease in thickness is attributed to the stiffness and conformation of the lipid alkyl tails. Fig. 4 shows the probability distribution of the torsion angles for the lipid alkyl tails (180° correspond to the trans conformation). As the temperature is increased, the alkyl tails are able to curl and deviate from the predominant straight orientation, thereby reducing the spacing between the bilayer interfaces. At the lowest temperature of 250 K, a double-peak structure appears in the density profile at the interface, which suggests an incipient structural change in the bilayer.
FIGURE 3.


(A) Density profile of fully hydrated DPPC bilayer; (B) Density profile resolved by molecular type at 350 K (numbers in parentheses indicate order of magnification of profile).
FIGURE 4.

Probability distribution of torsion angle for lipid alkyl tails. An angle of 180° corresponds to the trans conformation. Lines correspond to the probability distribution at 250 K (solid), 350 K (dashed), and 450 K (dot-dashed).
An important, experimentally accessible observable is the average area per headgroup; it is defined by the x- and y-dimensions of the simulation cell over the number of phospholipids per layer, i.e., 64 in our case. Fig. 5 summarizes the results of our simulations as well as simulation values (Tieleman and Berendsen, 1996) and experimental data (Nagle, 1993; Nagle et al., 1996) from the literature. Although the scatter in the data is significant, our results appear to be consistent with experimental and other simulations using various force fields.
FIGURE 5.

Area per headgroup for a DPPC bilayer. A linear dependence is found over the temperature range studied. Solid circles are results from this work; unfilled squares are experimental data (Nagle, 1993; Nagle et al., 1996); and unfilled triangles are previous simulation results (Tieleman and Berendsen, 1996).
The lipid tail order parameter, SCD (Tieleman et al., 1997), provides a measure of the alignment of the phospholipid tails in the bilayer. It is given by
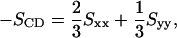 |
(1) |
 |
(2) |
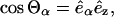 |
(3) |
where 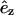 is a unit vector in the laboratory z-direction and
is a unit vector in the laboratory z-direction and 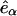 is a unit vector in the local coordinate system of the tails, defined in Fig. 6 A. This quantity is accessible by NMR measurements and provides another means for quantitative comparisons between experiments and simulations. Fig. 6 B shows SCD as a function of the position of the carbon vector along the lipid tail (carbon number one is the carbonyl carbon closest to the headgroup). At 325 K and above, the order parameter profiles are qualitatively similar, displaced to lower absolute values with increasing temperature. Our results are in qualitative agreement with NMR measurements (Tu et al., 1996) and earlier simulation studies at 350 K (Tieleman et al., 1997). The order parameter for the system at 250 K is clearly different from that obtained at higher temperatures. This behavior provides additional evidence supporting a possible structural transition between 250 and 325 K. The observed experimental temperature for a phase transition from a liquid to a gel phase is 315 K (Gennis, 1989).
is a unit vector in the local coordinate system of the tails, defined in Fig. 6 A. This quantity is accessible by NMR measurements and provides another means for quantitative comparisons between experiments and simulations. Fig. 6 B shows SCD as a function of the position of the carbon vector along the lipid tail (carbon number one is the carbonyl carbon closest to the headgroup). At 325 K and above, the order parameter profiles are qualitatively similar, displaced to lower absolute values with increasing temperature. Our results are in qualitative agreement with NMR measurements (Tu et al., 1996) and earlier simulation studies at 350 K (Tieleman et al., 1997). The order parameter for the system at 250 K is clearly different from that obtained at higher temperatures. This behavior provides additional evidence supporting a possible structural transition between 250 and 325 K. The observed experimental temperature for a phase transition from a liquid to a gel phase is 315 K (Gennis, 1989).
FIGURE 6.


(A) Description of the local coordinate system for calculation of order parameter (each sphere represents a carbon atom from the alkyl chain). (B) Order parameter SCD for phospholipid tails: lines are results from this work, unfilled circles are experimental NMR results (Tu et al., 1996), and unfilled squares are previous simulation results (Tieleman et al., 1997) at 325 K.
Figs. 7 and 8 show several relevant radial distribution functions (RDF). The two graphs in Fig. 7 depict the radial distribution function between the nitrogen and phosphorus atoms in the headgroups. A nearest-neighbor peak is clearly visible at ∼7 and 6 Å for the nitrogen and phosphorus groups, respectively. In the phosphorus case, the second nearest-neighbor is also strongly pronounced, at ∼8.5 Å. The clear second peak conforms better to a square packing than a hexagonal packing. In the planar square packing case, the distance to the second neighbor is 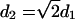 , where d1 = 6 Å and the distance to the second neighbor is d2 = 8.4 Å. In the hexagonal case, the distance to the second neighbor is d2 = 2 × sin(60°) × d1 = 10.4 Å. For phosphorus, even a third peak at d3 ≈ 11.5 Å is apparent. For both square and hexagonal packing, d3 = 2d1, which is consistent with our results. We therefore conclude that DPPC exhibits a slightly distorted square packing.
, where d1 = 6 Å and the distance to the second neighbor is d2 = 8.4 Å. In the hexagonal case, the distance to the second neighbor is d2 = 2 × sin(60°) × d1 = 10.4 Å. For phosphorus, even a third peak at d3 ≈ 11.5 Å is apparent. For both square and hexagonal packing, d3 = 2d1, which is consistent with our results. We therefore conclude that DPPC exhibits a slightly distorted square packing.
FIGURE 7.

(a) Nitrogen-nitrogen and (b) phosphorus-phosphorus radial distribution functions for the DPPC/water system. Different lines correspond to different temperatures: 250 K (solid), 325 K (short-dash), 350 K (long-dash), 400 K (dot-dashed), and 450 K (dot-dash-dash).
FIGURE 8.

Structure of the binding of water molecules to the lipid headgroups. The radial distribution functions correspond to water interacting with (A) lipid nitrogen, (B) lipid phosphorus, and (C) lipid carbonyl oxygen. The lines correspond to different temperatures: 250 K (solid), 325 K (short-dash), 350 K (long-dash), 400 K (dot-dashed), and 450 K (dot-dash-dash).
The fact that the phosphorus RDF exhibits more structure than the nitrogen RDF can be explained by the higher mobility of the choline group, which is able to penetrate the aqueous region more readily than the phosphate group. If we compare the solvation of the choline (nitrogen) and phosphate (phosphorus) groups, we recognize that the phosphorus is more closely approached by water molecules and its hydration is pronounced (see Fig. 8 and Table 2). The choline group extends deeper into the aqueous region and the water around it is loosely bound (the nitrogen atom is shielded by three neighboring methyl groups). The phosphate group is exposed to fewer water molecules but it exhibits specific interactions with them. The well-defined structure of the phosphorus-water RDFs stems from the fact that water molecules form hydrogen-bonded hydration shells around the phosphate. The first solvation shell contains ∼4 water molecules (see Table 2), all of them hydrogen-bonded to the four oxygen atoms of the phosphate group. Note that a hydrogen bond is defined in this work using the criteria suggested by Brady and Schmidt (1993) (the oxygen-oxygen distance should be <3.5 Å and the O–HċċċO angle >120°).
TABLE 2.
Average number of water molecules in first solvation shell for choline (N) and average hydration number for phosphate (P), and carbonyl oxygens (Oc) in the lipid headgroup in the DPPC/water system
| T (K) | Choline | Phosphate | Carbonyl oxygen* |
|---|---|---|---|
| 250 | 24.19 | 4.89 | 1.64 / 0.95 |
| 325 | 19.95 | 4.29 | 1.44 / 1.01 |
| 350 | 19.38 | 4.12 | 1.42 / 1.02 |
| 400 | 17.94 | 4.00 | 1.38 / 0.98 |
| 450 | 16.98 | 3.92 | 1.35 / 0.94 |
Value for carbonyl oxygen in each chain.
From the RDFs in Fig. 8, we also see that water molecules are able to penetrate the bilayer as far as the carbonyl groups of DPPC. The RDFs and hydration numbers of the carbonyl oxygen show that the number of water molecules is slightly larger for one of the two carbonyl oxygens of DPPC (the position of the carbonyl oxygen is not symmetric—see Fig. 2). As seen in Table 2, on average the hydration of the carbonyl oxygens is around unity.
The main influence of temperature on the structure is a quantitative decrease of the peak heights and the hydration number. The only salient feature is the different peak shape in the N–N RDF and relative peak heights of the P–P RDF at the lowest temperature (250 K). Here the shape of the N–N peak is well defined and the solvation of the choline groups is substantially increased (see Table 2). In addition, the first and second peaks of the P–P RDF have similar height, reversing the trend observed at higher temperatures. These features are again suggestive of an incipient phase transition at temperatures somewhere below 325 K.
Lipid bilayers and disaccharides
Both trehalose and sucrose are considered in this work; the concentrations are listed in Table 1. The simulated areas per headgroup for the bilayers with disaccharide are given in Table 3. For the concentrations and conditions considered in this work, the presence of the disaccharides does not alter the area per headgroup. Experiments have only been performed on monolayers (Crowe et al., 1984; Lambruschini et al., 2000) and, in contrast to our findings, these report an increase in the area per headgroup upon addition of trehalose. Bilayers and monolayers are known to behave differently but, unfortunately, experimental data for disaccharide bilayer systems are not available.
TABLE 3.
Lipid area per headgroup for bilayer system with disaccharides; units are Å2
| System | T = 350 K | T = 400 K |
|---|---|---|
| no sugar | 66.8 ± 0.5 | 72.2 ± 0.9 |
| 8 trehalose | 66.6 ± 0.6 | 72.1 ± 0.6 |
| 27 trehalose | 66.6 ± 1.0 | 72.3 ± 0.6 |
| 8 sucrose | 67.3 ± 0.8 | 73.1 ± 0.6 |
| 27 sucrose | 66.8 ± 0.9 | 72.5 ± 0.8 |
The presence of trehalose in the system has no effect on the spacing between the lipid headgroups. Our simulations indicate that trehalose has the flexibility to expand and contract its glucose rings and adjust to the proper dimensions between the lipid headgroups. Fig. 9 shows the trajectory of two trehalose molecules as a function of their distance to the interface. Also shown in the graph are the distances between the various oxygen atoms belonging to opposite rings of trehalose. We see that as the trehalose molecule approaches the interface, the distance between the trehalose O6 oxygen doubles in distance, from ∼5 to 10 Å, indicating that the conformation of the trehalose molecule changes from its state in the bulk aqueous region. Trehalose molecules in the aqueous region are well solvated by water (see Fig. 9); the population of conformations comprises both contracted and extended states (Conrad and de Pablo, 1999; Ekdawi-Sever et al., 2001). As they approach the lipid headgroups, trehalose molecules adjust their conformation without disrupting the arrangement of the lipids. Fig. 10, a and b, illustrates the change in conformation of a trehalose molecule from a distance near the interface to the state when it is “bonded” to the bilayer surface.
FIGURE 9.


Trajectory for two trehalose molecules in the DPPC/water/trehalose system. The zero position corresponds to the lipid bilayer center, and the average position of the phosphorus atoms in one layer is given for reference. The lower lines correspond to the distance between oxygens in opposite rings of trehalose: O2–O′2 (solid), O3–O′3 (dashed), O4–O′4 (dotted), and O6–O′6 (bold solid). The arrows indicate the time for the configurations of the trehalose molecule shown in Fig. 10.
FIGURE 10.

Snapshot of conformation of trehalose molecule as it approaches the lipid headgroups. The molecule shown in this figure corresponds to the trajectory in Fig. 9 A. Conformation A, at 1070-ps, is as shown in Fig. 9 A; conformation B occurred 360-ps later. Hydrogens on the carbon atoms are not shown for clarity.
Within the timescale of our simulations, trehalose molecules are able to approach the interface and eventually leave it; lipid molecules, on the other hand, are constrained to the bilayer region. Our simulations indicate that trehalose molecules move freely in the bulk aqueous region. When they approach the interface, depending on how they bind to the lipid groups, their interaction can be short-lived (∼0.5 ns in Fig. 9 A) or persistent, lasting over several nanoseconds (Fig. 9 B).
Our results show that disaccharide molecules do not penetrate the bilayer to any extent (see Fig. 11), even at temperatures as high as 450 K. Note, however, that our simulations were carried out over ∼10 ns, and it is conceivable that longer simulations could yield a penetration of disaccharide molecules into the bilayer. The spacing between the lipid interfaces remains unaltered; the interaction of disaccharide molecules and lipids only occurs at the surface of the interface. At infinite dilution, a single disaccharide molecule remains in the middle of the aqueous region (results not shown). At a concentration of 3.4 wt %, disaccharide molecules exhibit a preference for the lipid headgroups, near the interface. This can be inferred from the higher disaccharide density or peaks observed at ∼|z| = 10 Å in Fig. 11. Both trehalose and sucrose interact with the bilayer in a similar manner (see Fig. 11). At this concentration, the highest trehalose density occurs 5.2 Å from the plane of highest density for phosphorus. For sucrose the highest concentration occurs 6.0 Å away from the phosphorus peak.
FIGURE 11.




Density profile of the lipid/water/disaccharide ternary system resolved according to molecular types. (A) 8 trehalose, (B) 27 trehalose, (C) 8 sucrose, and (D) 27 sucrose. All profiles shown are at 400 K. The density profiles have been shifted so that the origin corresponds to the middle of the aqueous phase.
At higher concentrations (∼18 wt %), the behavior of sucrose is slightly different from that of trehalose (Fig. 11, B and D). The sucrose density is highest in the middle of the aqueous region; in contrast, trehalose exhibits a more uniform distribution throughout the aqueous region. Note that for higher concentrations, we opted to study a system with less water than at lower concentrations. This simply reflects our interest in understanding how disaccharides interact with cell membranes in low moisture environments.
Several radial distribution functions can be examined to gain some insights into the structure of the resulting lipid-disaccharide “complexes.” Analyses of the N–N RDF for the choline group and the P–P RDF for the phosphate group confirm that the arrangement of the lipid headgroups is unaltered by the presence of trehalose or sucrose (the RDFs are not shown because their appearance is similar to those shown in Fig. 7 for the pure bilayer system).
Table 4 shows the hydration number for the choline, phosphate, and carbonyl groups of the lipid headgroup, calculated using the geometric criteria described earlier. At low disaccharide concentrations, the hydration number is unchanged from that observed in pure bilayers. At higher concentrations (∼18 wt %), however, we see a decrease in the hydration of the lipid headgroups, suggesting that disaccharides replace some of the water around the lipid headgroups, particularly the water hydrogen-bonded to the phosphate group.
TABLE 4.
Average number of water molecules in first solvation shell for choline (N) and average hydration number for phosphate (P), and carbonyl oxygens (Oc) in the lipid headgroup in the ternary DPPC/water/disaccharide system
| System | T (K) | Choline | Phosphate | Carbonyl oxygen* |
|---|---|---|---|---|
| 8 trehalose | 350 | 18.66 | 4.09 | 1.41 / 0.98 |
| 400 | 17.41 | 3.95 | 1.38 / 0.98 | |
| 27 trehalose | 350 | 18.19 | 3.87 | 1.39 / 0.94 |
| 400 | 17.42 | 3.84 | 1.33 / 0.90 | |
| 8 sucrose | 350 | 19.33 | 4.22 | 1.44 / 0.98 |
| 400 | 17.71 | 4.03 | 1.39 / 0.98 | |
| 27 sucrose | 350 | 17.88 | 3.83 | 1.38 / 0.92 |
| 400 | 16.73 | 3.71 | 1.26 / 0.88 |
Value for carbonyl oxygen in each chain.
As mentioned above, trehalose molecules change conformation as they approach the bilayer interface; the data shown in Figs. 9 and 10, however, only provide limited insights on how these molecules interact with the lipid headgroups. Additional information can be gained by studying the various RDFs between the lipid headgroups and sites in trehalose. Each glucose ring in trehalose has four hydroxyl groups, each being a hydrogen-bond donor and acceptor. The RDFs shown in Figs. 12 and 13 indicate that the phosphate group is the principal interaction site between the lipid and trehalose; the O3 and O4 sites of trehalose approach the phosphate, whereas the highly mobile O6 site is mostly inactive. This last site may be efficiently hydrated by water, and therefore exhibits a lesser propensity to bind to the layer. The O2 site also exhibits a reasonably strong interaction with the phosphate group, even though it is relatively close to the glycosidic oxygen. The RDFs between the nitrogen in the choline group and trehalose do not show a pronounced structure, largely because the nitrogen atom is shielded by three methyl groups and there are no favorable interaction sites to bind with trehalose. It is also interesting to note that trehalose interacts with the ester groups of each lipid chain. The RDFs in Figs. 12 and 13 show some interaction between the carbonyl oxygen and mainly the O3 and O4 sites of trehalose. The findings above suggest that multiple interaction sites of each trehalose ring bind simultaneously to the lipid. The phosphate group has two exposed oxygens that are ∼2.6 Å apart, a distance that is well within the range in which the O2 and O3 or O3 and O4 sites in trehalose could align and hydrogen bond to the oxygens in the phosphate group. This may also explain why the O6 site of trehalose does not interact with the lipid headgroup; if it were to hydrogen-bond to the bilayer, the trehalose molecule would not be in a favorable position to align the other hydroxyl groups and bind to the lipid. Fig. 14 provides an illustration of how a trehalose molecule can actually bind to two phospholipids. In this particular configuration, the O2 and O3 sites of trehalose are bound to the phosphate and carbonyl groups of the lipids.
FIGURE 12.

Structure of the binding of trehalose molecules to the lipid headgroups. System with 27 trehalose molecules at 400 K. The radial distribution functions correspond to (A) lipid nitrogen, (B) lipid phosphorus, and (C) lipid carbonyl oxygen with the following hydroxyl oxygen in trehalose (see Fig. 2): O2 (solid); O3 (dotted); O4 (dashed); and O6 (dot-dashed).
FIGURE 13.

Structure of the binding of sucrose molecules to the lipid headgroups. System with 27 sucrose molecules at 400 K. The radial distribution functions correspond to (A) lipid nitrogen, (B) lipid phosphorus, and (C) lipid carbonyl oxygen with the following hydroxyl oxygen in sucrose (see Fig. 2): O2 (solid); O3 (dotted); O4 (dashed); and O6 (dot-dashed). For the carbonyl oxygen radial distribution functions, the bottom set corresponds to the oxygens in the fructose ring, and the displaced set to the glucose ring.
FIGURE 14.

Snapshot of a configuration showing a trehalose molecule binding to two lipids. The dashed lines represent the hydrogen bonds between trehalose and the lipid headgroups. Water molecules and other lipids are not shown for clarity.
In an attempt to quantify our dynamical observations of the system, we present in Fig. 15 the time evolution of individual sites of two trehalose molecules with respect to their hydrogen-bonding to the lipids. That figure was generated by analyzing the structure of the system at regular intervals (every 10 ps); for a trehalose molecule in each one of these snapshots, we determined whether any one of the hydroxyl groups was hydrogen-bonded (based on the distance and angle) to either the phosphate or ester groups of the lipids (both shown because of the asymmetry of chains) and, if so, an entry was assigned for that interaction (shown as circles in Fig. 15). We next tried to determine whether this same trehalose molecule was bound to another lipid; if this was true, another entry was assigned for that interaction (shown as squares in Fig. 15). This process was continued until all lipids directly interacting with this one trehalose molecule were identified (third-lipid interactions are shown as diamonds in the Fig. 15). A number of interesting features and conclusions can be drawn from the graphs. First, we notice that trehalose is not always hydrogen-bonded to the lipid surface; it can approach and leave the surface in the scale of nanoseconds. Second, there is a pronounced tendency for both glucose rings of the trehalose molecule to bind to the bilayer (when this is not the case, the nonbonded ring dangles in solution). Most of the time, trehalose simultaneously hydrogen-bonds to more than one lipid. The most prevalent trehalose oxygens hydrogen-bonded to the lipid headgroup are the O2, O3, and O4 and, as discussed earlier, they usually hydrogen-bond in pairs, that is, O2 and O3 or O3 and O4 hydrogen-bond to the oxygens in the phosphate group. As mentioned above, the O6 in trehalose is not a predominant interaction site with the lipids; this is confirmed in Fig. 15 by the relatively few times this interaction shows up. When a trehalose molecule interacts with two lipids we see that, generally, each glucose ring interacts with a different lipid. These data provide support to our hypothesis that trehalose molecules do not alter the lipid structure, but are able to stabilize the structure by serving as bridges between adjacent lipids, thereby preventing them from aggregating or collapsing in low moisture environments. We also see from the figure that, occasionally, the same glucose ring interacts with two different lipids. In such configurations, the glucose ring fills the void space between adjacent headgroups.
FIGURE 15.


Time evolution of binding between trehalose and lipids. Each graph represents the path of two different trehalose molecules. The lower part of the graph shows the binding of the different hydroxyl oxygens (for each ring) in trehalose to the lipid. Each entry in the graph indicates that the site is hydrogen-bonded to the lipid at that instant in time. The circles represent the binding of the trehalose to one lipid, squares to a second lipid, and diamonds to a third lipid. The upper part of the graph shows which lipid headgroup the trehalose molecule interacts with; Phos is the phosphate group and Est is each of the ester groups.
In the process of determining the binding sites of trehalose, we also collected information about which lipid headgroups are directly involved. As before, we see that trehalose and phosphate interact, and that there is also a significant interaction with the ester groups. A careful analysis of the upper portion of the graphs shows that there is a greater tendency for trehalose to bind to the ester groups of the second or third lipid, and this may be because these groups become more accessible or exposed once the trehalose initially binds to another, adjacent lipid. The interaction of a single trehalose molecule with three lipids was also observed; interactions with a fourth lipid were rarely noticed.
Most of the results and discussion presented so far pertain to systems with trehalose. The results of similar analyses with sucrose do not reveal any major difference between these two disaccharides. The structural differences between these two molecules are not significant enough to discern a structural stabilization effectiveness of one molecule over the other.
Fig. 16 shows the lipid tail order parameter in the presence of disaccharides; as we can see, there are no major changes compared to the pure bilayer systems. Results for the systems with eight disaccharides are similar to those shown in the Fig. 16. These additional observations are consistent with the proposition that, since the disaccharides do not penetrate into the bilayer, the structure and properties of the aliphatic chains remain intact.
FIGURE 16.


Alkane chain order parameter as a function of disaccharide concentration: (A) trehalose and (B) sucrose. Solid lines are results at 350 K and dashed lines at 400 K.
Dynamic properties
Fig. 17 A shows the mean-squared displacement of the phosphorus atom in the lipid headgroup. Phosphorus is the heaviest atom in the headgroup and approximates its center of mass. A diffusion coefficient D can be calculated from
 |
(4) |
where r(t) is the position vector at time t, and the brackets denote an ensemble average. The factor d is equal to the number of dimensions considered (d = 1 for linear, d = 2 for lateral, and d = 3 for bulk diffusion). Table 5 shows the long-time diffusion coefficient for the phosphorus atom in the lipid headgroup. Only the results for those temperatures at which diffusion was attained are shown. Several studies have examined the dynamics of lipids in bilayer structures. Marrink et al. (1993) calculated short-time diffusion coefficients (Dlipid = 10 ± 1 × 10−6 cm2/s) from short DPPC trajectories at 350 K. Lindahl and Edholm (2001) performed a 100-ns simulation of DPPC at 323 K and calculated a long-time diffusion coefficient for lateral motion of the lipid (Dlipid = 0.12 × 10−6 cm2/s). Essmann and Berkowitz (1999) inferred a lateral diffusion coefficient (Dlipid = 0.3 ± 0.06 × 10−6 cm2/s) from a 10-ns simulation of DPPC at 333 K. All the reported values calculated from simulations are in good agreement with experimental measurements for DPPC at 323 K: Dlipid = 0.095 × 10−6 cm2/s from pulsed NMR (Kuo and Wade, 1979), and Dlipid = 0.125 × 10−6 cm2/s from fluorescence recovery after photobleaching (Vaz et al., 1985). The long-time diffusion coefficient for the lipids from our simulations at 350 K is Dlipid = 0.33 ± 0.03 × 10−6 cm2/s, which is consistent with previously reported values from simulations and experiments. Table 5 lists the long-time diffusion for lipid and water at other temperatures. It should be noted that long simulation runs (at least 10 ns) are necessary to attain a diffusive regime in these systems.
FIGURE 17.


(A) Mean-squared displacement of phosphorus atom in a pure bilayer and in the presence of disaccharides. (B) Directionally resolved mean-squared displacement for the phosphorus atom in a pure bilayer at 450 K.
TABLE 5.
Simulated long-time two-dimensional diffusion coefficient of the phosphorus atom in the lipid headgroup
| T (K) | Dlipid | Dwater |
|---|---|---|
| 350 | 0.33 | 45.5 |
| 400 | 0.88 | 86.7 |
| 450 | 1.9 | 134.3 |
All values reported as D × 106 cm2/s.
Our simulations with disaccharides were run over 10 ns, but this was insufficient to reach the diffusive regime for the lipid. Nevertheless, it can be seen in Fig. 17 A that, in the presence of disaccharides, the motion of the phosphorus atom in the lipid headgroups is reduced significantly with respect to that observed in the absence of disaccharides.
An analysis of the individual components of the mean-squared displacement for the phosphorus atom in terms of the distinct spatial directions (the bilayer spreads in the xy-plane—see Fig. 1) reveals that, at short times (t < 50 ps), the displacement along the z-direction is the largest, meaning that the bilayer headgroups move more rapidly in the direction normal to the layer interface. However, as the layer structure cannot be disrupted, the normal displacement attains an upper limit (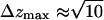 Å), and the lateral diffusion along the layer plane takes over at longer timescales. The motion is never truly three-dimensional; it is two-dimensional at intermediate and long timescales.
Å), and the lateral diffusion along the layer plane takes over at longer timescales. The motion is never truly three-dimensional; it is two-dimensional at intermediate and long timescales.
Also shown in Tables 5 and 6 are the self-diffusion coefficients of the water molecules. Experimental measurements of the water self-diffusion coefficient in the bulk at 318 K give Dwater = 35.8 × 10−6 cm2/s (Mills, 1973), and recent measurements at 358 K give a value of Dwater = 64.6 × 10−6 cm2/s (Ekdawi-Sever et al., 2003). Our calculated value at 350 K is in good agreement with the measured value at 358 K, suggesting that water molecules in the aqueous phase behave as bulk water, essentially undisturbed by the bilayer. The self-diffusion coefficients of water in the presence of the disaccharides are considerably lower than in the pure systems.
TABLE 6.
Simulated long-time two-dimensional diffusion coefficients of disaccharides in lipid bilayer at 350 and 400 K
| System | T (K) | Dsugar | Dwater |
|---|---|---|---|
| 8 trehalose | 350 | 3.5 | 30.1 |
| 400 | 9.7 | 51.0 | |
| 358 | 15.6 | 59.9 | |
| 27 trehalose | 350 | 0.8 | 25.0 |
| 400 | 1.7 | 34.5 | |
| 358 | 10.1 | 42.9 | |
| 8 sucrose | 350 | 4.1 | 39.6 |
| 400 | 6.3 | 64.3 | |
| 358 | 21.2 | 59.0 | |
| 27 sucrose | 350 | 0.7 | 16.1 |
| 400 | 2.7 | 34.5 | |
| 358 | 10.5 | 41.7 |
For comparison, diffusion coefficients at 358 K were extrapolated from experimental measurements (Ekdawi-Sever et al., 2003). All values reported as D × 106 cm2/s.
The diffusion coefficients of the disaccharide molecules in the aqueous phase are given in Table 6. Also shown in the table are experimental values of recent NMR data for aqueous disaccharide solutions (Ekdawi-Sever et al., 2003). The diffusion coefficients of the disaccharides in the bilayer systems are significantly lower than those measured in bulk aqueous solutions; this is indicative of the fact that the molecules are more constrained, largely through the binding to the surface of the bilayer. As shown and discussed in Fig. 9, the hydrogen-bonding of the disaccharides to the bilayer can be long-lived, thereby restricting their motion. At the high disaccharide concentrations, the diffusion of the molecules is substantially lower than that observed in free solution.
CONCLUSIONS
Simulations of pure, hydrated DPPC bilayers indicate that the area per headgroup increases linearly with temperature. This area increase is accompanied by a decrease of the lipid tail order parameter.
The addition of disaccharides (trehalose or sucrose) to these systems does not alter the bilayer structure; the interactions between disaccharides and the bilayer occur along the surface of the model membrane, and disaccharide molecules do not penetrate the aliphatic region to any measurable extent. Close inspection of the trajectory of individual disaccharide molecules reveals that they hydrogen-bond to the phosphate and ester groups of the lipids. Up to three different lipids are often observed to interact simultaneously with a single trehalose molecule. The conformations adopted by trehalose molecules on the surface of the bilayer can conform to the topology of the nearest lipids, thereby acting as a bridge unit between adjacent lipids.
Disaccharides prevent lipid bilayers from collapsing and fusing during freezedrying of liposomes and cells. It has been argued that a disaccharide matrix provides a scaffold for the bilayer, but the nature of the anchoring between that scaffold and a membrane's phospholipids has not been determined. The simulations presented in this work have revealed many of the details of that anchoring. Furthermore, it has been shown that, even in relatively dilute solutions (e.g., 3.4 wt %), the concentration of trehalose or sucrose molecules in the near vicinity of the bilayer is higher than in the bulk aqueous phase. This observation should be contrasted with the “exclusion principle” proposed to explain the action of osmo- or cryoprotectants for solutions of biological molecules, which postulates that effective stabilizing agents are excluded from the near vicinity of a biological macromolecule (Timasheff, 1998).
Experimental data for DPPC monolayers with and without trehalose indicate that the area per headgroup increases considerably upon addition of the disaccharide. The results of simulations indicate that for DPPC bilayers the area per headgroup remains unaltered. At this point we cannot determine whether this discrepancy is due to a shortcoming of our model or force field, or to a fundamental difference between the behavior of monolayers and bilayers. We hope that the results presented in this work will stimulate additional experiments on disaccharide-bilayer systems.
The simulations presented here have also demonstrated that the dynamics of disaccharide-bilayer systems are slowed down considerably by the presence of trehalose and sucrose. On the timescales of the simulations presented in this work (∼10 ns), the binding of the disaccharides to the lipid headgroups arrests the motion of the lipids significantly. Moreover, due to the strong interaction of the disaccharides with the lipid interface, a substantial decrease in the diffusivity of the disaccharides and the water is observed compared to that encountered in bulk solutions.
Acknowledgments
We are very grateful to John Crowe and Fern Tablin for sharing their valuable experimental insight with us.
We acknowledge financial support by the United States National Science Foundation (CTS-0218357) and Defense Advanced Research Projects Agency (DARPA).
References
- Allen, M. P., and D. J. Tildesley. 1987. Computer Simulation of Liquids. Clarendon Press, Oxford.
- Alper, H. E., D. Bassolino, and T. R. Stouch. 1993. Computer simulation of a phospholipid monolayer-water system: the influence of long-range forces on water structure and dynamics. J. Chem. Phys. 98:9798–9807. [Google Scholar]
- Bandyopadhyay, S., M. Tarek, and M. L. Klein. 1998. Computer simulation studies of amphiphilic interfaces. Curr. Opin. Coll. Interf. Sci. 3:242–246. [Google Scholar]
- Berendsen, H. J. C., J. R. Grigera, and T. P. Straatsma. 1987. The missing term in effective pair potentials. J. Phys. Chem. 91:6269–6271. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external heat bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Brady, J. W., and R. K. Schmidt. 1993. The role of hydrogen bonding in carbohydrates: molecular dynamics simulations of maltose in aqueous solution. J. Phys. Chem. 97:958–966. [Google Scholar]
- Conrad, P., and J. J. de Pablo. 1999. Computer simulation of the cryoprotectant α,α-trehalose in aqueous solution. J. Phys. Chem. A. 103:4049–4055. [Google Scholar]
- Crowe, J. H., L. M. Crowe, J. F. Carpenter, and C. Aurell Wistrom. 1987. Stabilization of dry phospholipid bilayers and proteins by sugar. Biochem. J. 242:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe, J. H., L. M. Crowe, J. F. Carpenter, A. S. Rudolph, C. Aurell Wistrom, B. J. Spargo, and T. J. Anchordoguy. 1988. Interactions of sugars with membranes. Biochim. Biophys. Acta. 947:367–384. [DOI] [PubMed] [Google Scholar]
- Crowe, J. H., L. M. Crowe, A. E. Oliver, N. Tsvetkova, W. Wolkers, and F. Tablin. 2001. The trehalose myth revisited: introduction to a symposium on stabilization of cells in the dry state. Cryobiology. 43:89–105. [DOI] [PubMed] [Google Scholar]
- Crowe, J. H., M. A. Whittam, D. Chapman, and L. M. Crowe. 1984. Interactions of phospholipid monolayers with carbohydrates. Biochim. Biophys. Acta. 769:151–159. [DOI] [PubMed] [Google Scholar]
- Damm, W., A. Frontera, J. Tirado-Rives, and W. L. Jorgensen. 1997. OplS all-atom force field for carbohydrates. J. Comput. Chem. 18:1955–1970. [Google Scholar]
- Egberts, E., S.-J. Marrink, and H. J. C. Berendsen. 1994. Molecular dynamics simulation of a phospholipid membrane. Eur. Biophys. J. 22:423–436. [DOI] [PubMed] [Google Scholar]
- Ekdawi-Sever, N. C., P. B. Conrad, and J. J. de Pablo. 2001. Molecular simulation of sucrose solutions near the glass transition temperature. J. Phys. Chem. A. 105:734–742. [Google Scholar]
- Ekdawi-Sever, N. C., J. J. de Pablo, E. Feick, and E. von Meerwall. 2003. Diffusion of sucrose and α,α-trehalose in aqueous solutions. J. Phys. Chem. A. 107:936–943. [Google Scholar]
- Essman, U., L. Perera, M. L. Berkwitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle-mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Essmann, U., and M. L. Berkowitz. 1999. Dynamical properties of phospholipid bilayers from computer simulation. Biophys. J. 76:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraldo-Gómez, J. D., G. R. Smith, and M. S. P. Sansom. 2002. Setting up and optimization of membrane protein simulations. Eur. Biophys. J. 31:217–227. [DOI] [PubMed] [Google Scholar]
- Feller, S. E., Y. H. Zhang, and R. W. Pastor. 1995. Computer-simulation of liquid/liquid interfaces. II. Surface-tension area dependence of a bilayer and a monolayer. J. Chem. Phys. 103:10267–10276. [Google Scholar]
- Gaber, B. P., I. Chandrasekhar, and N. Pattabiraman. 1986. The interaction of trehalose with the phospholipid bilayer: a molecular study. In Membranes, Macromolecules and Stability in the Dry State. C. Leopold, editor. Cornell University Press, Ithaca, NY. 231–241.
- Gennis, R. B. 1989. Biomembranes: Molecular Structure and Function. Springer-Verlag, New York.
- Husslein, T., D. M. Newns, P. C. Pattnaik, Q. Zhong, P. B. Moore, and M. L. Klein. 1998. Constant pressure and temperature molecular-dynamics simulation of the hydrated diphytanolphosphatidylcholine lipid bilayer. J. Chem. Phys. 109:2826–2832. [Google Scholar]
- Kuo, A.-L., and C. Wade. 1979. Lipid lateral diffusion by pulsed nuclear magnetic resonance. Biochemistry. 18:2300–2308. [DOI] [PubMed] [Google Scholar]
- Lambruschini, C., A. Relini, A. Ridi, L. Cordone, and A. Gliozzi. 2000. Trehalose interacts with phospholipid polar heads in Langmuir monolayers. Langmuir. 16:5467–5470. [Google Scholar]
- Lindahl, E., and O. Edholm. 2001. Molecular dynamics simulation of NMR relation rates and slow dynamics in lipid bilayers. J. Chem. Phys. 115:4938–4950. [Google Scholar]
- Madden, T. D., M. B. Bally, M. J. Hope, P. R. Curtis, H. P. Schieren, and A. S. Janoff. 1985. Protection of large unilamellar vesicles by trehalose during dehydration—retention of vesicle contents. Biochim. Biophys. Acta. 817:67–74. [DOI] [PubMed] [Google Scholar]
- Marrink, S.-J., M. Berkowitz, and H. J. C. Berendsen. 1993. Molecular dynamics simulation of a membrane/water interface: the ordering of water and its relation to the hydration force. Langmuir. 9:3122–3131. [Google Scholar]
- Mashl, R. J., H. L. Scott, S. Subramaniam, and E. Jakobsson. 2001. Molecular simulation of dioleoylphosphatidylcholine lipid bilayers at differing levels of hydration. Biophys. J. 81:3005–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlouthi, M. (editor). 1994. Food Packaging and Preservation. Aspen Publishers, New York, NY.
- Mills, R. 1973. Self-diffusion in normal and heavy water in the range 1–45°. J. Phys. Chem. 77:685–688. [Google Scholar]
- Moore, P. B., C. F. Lopez, and M. L. Klein. 2001. Dynamical properties of a hydrated lipid bilayer from a multinanosecond molecular dynamics simulation. Biophys. J. 81:2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F. 1993. Area/lipid of bilayers from NMR. Biophys. J. 64:1476–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F., R. T. Zhang, S. Tristram-Nagle, W. J. Sun, H. I. Petrache, and R. M. Suter. 1996. X-ray structure determination of fully hydrated l(α) phase dipalmitoylphosphatidylcholine bilayers. Biophys. J. 70:1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath, S. K., B. J. Banaszak, and J. J. de Pablo. 2001. A new united atom force field for α-olefins. J. Chem. Phys. 114:3612–3616. [Google Scholar]
- Nath, S. K., and J. J. de Pablo. 2000. Simulation of vapour-liquid equilibria for branched alkanes. Mol. Phys. 98:231–238. [Google Scholar]
- Nath, S. K., F. A. Escobedo, and J. J. de Pablo. 1998. On the simulation of vapor-liquid equilibria for alkanes. J. Chem. Phys. 108:9905–9911. [Google Scholar]
- Nina, M., and T. Simonson. 2002. Molecular dynamics of the tRNAala acceptor stem: comparison between continuum reaction field and particle-mesh Ewald electrostatic treatments. J. Phys. Chem. B. 106:3696–3705. [Google Scholar]
- Norberg, J., and L. Nilsson. 2000. On the truncation of long-range electrostatic interactions in DNA. Biophys. J. 79:1537–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien Nabors, L. (editor). 2001. Alternative Sweeteners, Vol. 112, Food Science and Technology Series. Marcel Dekker, New York, NY.
- Pastor, R. W., R. M. Venable, and S. E. Feller. 2002. Lipid bilayers, NMR relaxation, and computer simulations. Acc. Chem. Res. 35:438–446. [DOI] [PubMed] [Google Scholar]
- Rog, T., and M. Pasenkiewicz-Gierula. 2001. Cholesterol effects on the phosphatidylcholine bilayer nonpolar region: a molecular dynamics study. Biophys. J. 81:2190–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph, B. R., I. Chandrasekhar, B. P. Gaber, and M. Nagumo. 1990. Molecular modelling of saccharide–lipid interactions. Chem. Phys. Lipids. 53:243–261. [Google Scholar]
- Saiz, L., and M. L. Klein. 2002a. Computer simulation studies of model biological membranes. Acc. Chem. Res. 35:482–489. [DOI] [PubMed] [Google Scholar]
- Saiz, L., and M. L. Klein. 2002b. Electrostatic interactions in a neutral model phospholipid bilayer by molecular dynamics. J. Chem. Phys. 116:3052–3057. [Google Scholar]
- Scott, H. L. 2002. Modeling the lipid component of membranes. Curr. Opin. Struct. Biol. 12:495–502. [DOI] [PubMed] [Google Scholar]
- Shinoda, W., N. Namiki, and S. Okazaki. 1997. Molecular dynamics study of a lipid bilayer: convergence, structure, and long-time dynamics. J. Chem. Phys. 106:5731–5743. [Google Scholar]
- Smondryev, A. M., and M. L. Berkowitz. 2000. Molecular dynamics simulation of dipalmitoylphosphatidylcholine membrane with cholesterol sulfate. Biophys. J. 78:1672–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieleman, D. P., and H. J. C. Berendsen. 1996. Molecular dynamics simulations of fully hydrated DPPC with different macroscopic boundary conditions and parameters. J. Chem. Phys. 105:4871–4880. [Google Scholar]
- Tieleman, D. P., S. J. Marrink, and H. J. C. Berendsen. 1997. A computer perspective of membranes: molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta-Rev. Biomem. 1331:235–270. [DOI] [PubMed] [Google Scholar]
- Tieleman, P. 2002. University of Calgary, Department of Biological Sciences. http://moose.bio.ucalgary.ca/download.html.
- Timasheff, S. N. 1998. Control of protein stability and reactions by weakly interacting cosolvents: the simplicity of the complicated. In Advances in Protein Chemistry, Vol. 51. Academic Press, New York, NY. 356–428. [DOI] [PubMed]
- Tobias, D. J. 2001. Electrostatics calculations: recent methodological advances and applications to membranes. Curr. Opin. Struct. Biol. 11:253–261. [DOI] [PubMed] [Google Scholar]
- Tobias, D. J., K. Tu, and M. L. Klein. 1997. Atomic-scale molecular dynamics simulations of lipid membranes. Curr. Opin. Coll. Interf. Sci. 2:15–27. [Google Scholar]
- Tu, K., M. L. Klein, and D. J. Tobias. 1998. Constant-pressure molecular dynamics investigation of cholesterol effects in a dipalmitoylphosphatidylcholine bilayer. Biophys. J. 75:2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, K., D. J. Tobias, J. K. Blasie, and M. L. Klein. 1996. Molecular dynamics investigation of the structure of a fully hydrated gel-phase dipalmitoylphosphatidylcholine bilayer. Biophys. J. 70:596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gunsteren, W. F., S. R. Billeter, A. A. Eising, P. H. Hünenberger, P. Krüger, A. E. Mark, W. R. P. Scott, and I. G. Tironi. 1996. Biomolecular Simulation: The GROMOS Manual and User Guide. Vdf, Zürich, Switzerland.
- Vaz, W. L. C., R. M. Clegg, and D. Hallman. 1985. Translational diffusion of lipids in liquid-crystalline phase phosphatidylcholine multibilayers—a comparison of experiment with theory. Biochemistry. 24:781–786. [DOI] [PubMed] [Google Scholar]
- Womersley, C., P. S. Uster, A. S. Rudolph, and J. H. Crowe. 1986. Inhibition of dehydration-induced fusion between liposomal membranes by carbohydrates as measured by fluorescence energy-transfer. Cryobiology. 23:245–254. [DOI] [PubMed] [Google Scholar]