

Abstract
Water permeation and electrostatic interactions between water and channel are investigated in the Escherichia coli glycerol uptake facilitator GlpF, a member of the aquaporin water channel family, by molecular dynamics simulations. A tetrameric model of the channel embedded in a 16:0/18:1c9-palmitoyloleylphosphatidylethanolamine membrane was used for the simulations. During the simulations, water molecules pass through the channel in single file. The movement of the single file water molecules through the channel is concerted, and we show that it can be described by a continuous-time random-walk model. The integrity of the single file remains intact during the permeation, indicating that a disrupted water chain is unlikely to be the mechanism of proton exclusion in aquaporins. Specific hydrogen bonds between permeating water and protein at the channel center (at two conserved Asp-Pro-Ala “NPA” motifs), together with the protein electrostatic fields enforce a bipolar water configuration inside the channel with dipole inversion at the NPA motifs. At the NPA motifs water-protein electrostatic interactions facilitate this inversion. Furthermore, water-water electrostatic interactions are in all regions inside the channel stronger than water-protein interactions, except near a conserved, positively charged Arg residue. We find that variations of the protein electrostatic field through the channel, owing to preserved structural features, completely explain the bipolar orientation of water. This orientation persists despite water translocation in single file and blocks proton transport. Furthermore, we find that for permeation of a cation, ion-protein electrostatic interactions are more unfavorable at the conserved NPA motifs than at the conserved Arg, suggesting that the major barrier against proton transport in aquaporins is faced at the NPA motifs.
INTRODUCTION
All life-forms need to continuously and precisely regulate water content of their cells and organs through exchange of water with their surroundings. Although all cell membranes are to some extent water-permeable, water transport through lipid bilayers is slow and inadequate for exchange of large volumes of water, e.g., as occurring in kidneys, or for a rapid response of cells to osmotic stress, as in red blood cells. The high water permeability of certain cell membranes was explained by the discovery of aquaporins (AQPs), integral membrane proteins that are specialized for selective transport of water (Preston et al., 1992; Agre et al., 1998; Borgnia et al., 1999; Yasui et al., 1999).
AQPs are present in all life-forms, and more than 100 have been characterized (Borgnia et al., 1999). Eleven human AQPs have been identified, and their impaired function is implicated in pathological situations, such as nephrogenic diabetes insipidus and congenital cataract (Borgnia et al., 1999; Kozono et al., 2002). All members of the AQP family provide a passive means of water transport. A subfamily of AQPs, aquaglyceroporins, possesses the extended capability of stereo-selectively conducting small, linear carbohydrates in addition to water (Heller et al., 1980; Fu et al., 2000; Borgnia et al., 1999; Jensen et al., 2001, 2002). The most extensively studied aquaglyceroporin is the Escherichia coli glycerol uptake facilitator GlpF, which is used by the organism to accelerate absorption of glycerol from the environment, especially at low glycerol concentrations (Richey and Lin, 1972).
AQPs form tetramers in the membrane; the biological significance of oligomerization was not clear until recent reports implicated that the central pore allows ion transport (Yool and Weinstein, 2002; Hazama et al., 2002). Atomic resolution models of human aquaporin-1 (AQP1) based on electron microscopy of two-dimensional crystals provided the first insight into the tetrameric architecture and folding of AQPs (Murata et al., 2000; Ren et al., 2001). The first molecular dynamics (MD) simulations performed on these models, however, indicated that the models needed further improvement (Zhu et al., 2001; de Groot et al., 2001). GlpF was the first AQP whose high-resolution structure (2.2 Å) was determined by x-ray crystallography (Fu et al., 2000). The first GlpF structure included three glycerol molecules, a natural substrate for GlpF, in each monomer. MD simulations of this glycerol-saturated GlpF (GlpF+G) resulted in a detailed description of protein-substrate interactions and conduction pathway, which for the first time explained why two half-membrane spanning (re-entrant) loops with unusual secondary structures are conserved in the architecture of AQPs (Jensen et al., 2001), as sketched for a single monomer in Fig. 1. In each monomer, the transmembrane segment of the channel is formed by six helices and the re-entrant loops that meet each other at the center of the channel. Conservation of the re-entrant loops, which are related by quasi-twofold symmetry, is of structural and functional importance (Murata et al., 2000; Ren et al., 2001; Fu et al., 2000; Zhu et al., 2001; Jensen et al., 2001; Sui et al., 2001). Approximately one-half of each loop is nonhelical and defines a curve-linear conduction pathway, constituted by backbone carbonyl groups that are exposed toward the channel interior forming here hydrogen bonds with the permeant substrate (Jensen et al., 2001). The otherwise energetically unfavorable secondary structure of the nonhelical parts is stabilized through multiple hydrogen bonds of backbone N-H groups to two highly conserved glutamate residues (Fu et al., 2000; Jensen et al., 2001; Nollert et al., 2001).
FIGURE 1.

Schematic representation of a single GlpF monomer. The six membrane-spanning helices are drawn as green cylinders. The two re-entrant, half-membrane spanning repeats meeting in the center of the channel at the position of the NPA motifs are drawn in red with the M3 and M7 α-helices represented as cylinders and the conduction pathway represented as red threads.
The other half of each loop, known as M3 and M7 in GlpF, and HB and HE in AQP1 (Murata et al., 2000; Ren et al., 2001; Sui et al., 2001), is α-helical. These helices terminate at the channel center, where two conserved (Asn-Pro-Ala) NPA motifs (Fu et al., 2000; Murata et al., 2000; Ren et al., 2001; Sui et al., 2001) meet each other. Multiple hydrogen bonds between the NPA motifs (Zhu et al., 2001; Jensen et al., 2001) result in a configuration in which one of the amide hydrogen atoms of each asparagine points toward the pore. Together they provide two very close hydrogen-bonding sites for the permeating substrate at the center of the channel.
These structural details of GlpF (Fu et al., 2000) were used to refine AQP1 models (de Groot et al., 2001; Zhu et al., 2001), before a 2.7 Å resolution structure of AQP1 was determined by x-ray crystallography (Sui et al., 2001). In addition to a better description of the channel interior, the high-resolution structure of AQP1 revealed the position of four water molecules inside the channel. Three-dimensional structures of GlpF have also been solved under low glycerol concentration conditions, resulting in water-filled, glycerol-free GlpF (GlpF-G) and revealing the presence of a single file of water molecules inside the channel (Tajkhorshid et al., 2002). Comparison of the GlpF+G and GlpF-G structures shows a remarkable similarity between the conformations of side chains in the two channels, indicating that the channel can accommodate glycerol with only minor conformational changes occurring mainly at its narrowest part, denoted as the selectivity filter (SF) (Fu et al., 2000; Jensen et al., 2001; Tajkhorshid et al., 2002; Grayson et al., 2003).
Substrate selectivity is crucial in the function of AQPs; these proteins should provide a facilitatory mechanism for transmembrane water flow without affecting the electro-chemical properties of the membrane. Water pores in AQPs are impermeable to charged species. The narrow size of the channel combined with its hydrophobic interior seems to be an effective means of preventing ions from entering the channel; hydrated ions are too large to enter the pore, and, besides an unfavorable increase in entropy, the solvation energy provided by the channel interior is too weak to dehydrate ions. Selectivity against protons, essential in conservation of the transmembrane proton gradient and in the bioenergetics of the cell, may, however, not be explained as easily, since protons can, be transported very fast through a chain of water molecules according to the Grotthuss mechanism (de Grotthuss, 1806; Schulten and Schulten, 1986; Agmon, 1995; Pomès and Roux, 1996, 2002; Brewer et al., 1999; Smondyrev and Voth, 2002).
Proton exclusion is indeed effectively accomplished by AQPs through tuning of the orientation of water molecules inside the channel (Tajkhorshid et al., 2002) that prevents them from adopting a uniform arrangement required for a proton wire, e.g., as observed in gramicidin A (Pomès and Roux, 1996, 2002). This mechanism of proton exclusion has been investigated most recently for water conducting nanotubes (Zhu and Schulten, 2003). An alternative mechanism of proton exclusion, based on the disruption of the single file water in the SF region, i.e., the narrowest part of the channel, or in the NPA region was suggested by others (de Groot et al., 2001; Ren et al., 2001).
Since substrates of AQPs, e.g., water and linear sugar molecules, are neutral, electrostatic forces might not seem to be critical for the function of the channel. It turns out, however, that such forces, which have proven crucial in the function of ion channels, e.g., the KcsA potassium channel (Roux and MacKinnon, 1999; Bernèche and Roux, 2001), also play important roles in the selectivity mechanisms employed by AQPs (Jensen et al., 2001; Tajkhorshid et al., 2002; Grayson et al., 2003). We report here a detailed analysis of water dynamics and electrostatic interaction of permeating water molecules with the GlpF channel using trajectories from multinanosecond MD simulations. MD has been applied to study water and glycerol permeation in AQPs (Zhu et al., 2001, 2002; Jensen et al., 2001, 2002; de Groot et al., 2001; Tajkhorshid et al., 2002), as well as water transport in nanotubes (Hummer et al., 2001; Zhu and Schulten, 2003). To investigate in further detail how water conduction and proton exclusion are established by conserved features of the protein architecture of AQPs, we analyze here the dynamics of water inside the channel, and investigate and decompose the electrostatic forces acting on water in various regions of the channel during its permeation.
METHODS
In this section we describe the modeling of tetrameric GlpF in a hydrated lipid membrane, the details of simulation protocols, and the procedures applied for trajectory analysis.
Modeling
The crystal structure of a GlpF monomer in its glycerol-bound form (GlpF+G) (Fu et al., 2000) was obtained from the Research Collaboratory for Structural Bioinformatics Data Bank (PDB) (Bernstein et al., 1977), PDB entry code 1FX8 (Fu et al., 2000). Missing atoms of Arg257 and hydrogen atoms were added using Insight II (Insight, 1998) and XPLOR (Brünger, 1992), respectively. The tetrameric structure of GlpF was generated with VMD (Humphrey et al., 1996) using the coordinate transformation matrices provided in the PDB file.
A 16:0/18:1c9-palmitoyloleylphosphatidylethanolamine (POPE) lipid bilayer was used to mimic the E. coli membrane. Initially 480 POPE lipids, all in cis-conformation were placed on a regular hexagonal lattice with a lattice vector of 8.1 Å. The membrane plane was in the xy plane, and the membrane normal was along the z-direction. The GlpF tetramer including channel-bound water and glycerol molecules resolved crystallographically was inserted into the membrane using the location of tyrosine residues and the proposed mid-membrane residues (Thr18, Gly49, Asn68, Pro69, Ala70, Gly96, Thr156, Gly184, and Gly243) (Fu et al., 2000) to adjust the normal position of the membrane relative to the protein. An optimal position, where equal distances from the lipid headgroups to the above residues resulted, was obtained by separately adjusting the two monolayers relative to the protein. As a result the hydrophobic surface of the protein was embedded by the hydrophobic core of the membrane, with a vertical phosphorus-phosphorus separation between the two monolayers initially measuring 39.6 Å. Upon visual inspection in VMD (Humphrey et al., 1996), 83 lipids overlapping with the protein were removed.
The program SOLVATE (Grubmüller, 1996) was used to add a 20 Å ellipsoidal solvation shell around the membrane and the solvent-exposed parts of the protein. From this system a rectangular box, based on the positions of the nitrogen atoms of the ethanolamine headgroups of the lipids, was cut out. In the resulting box, all water molecules located inside the hydrophobic core of the membrane were discarded. The normal dimension of the rectangular system ensured ∼10 Å hydration of each POPE monolayer. Four water molecules were replaced by chloride ions, to neutralize the net charge of the system.
For the study of water conduction, we modified the GlpF+G/POPE/water system described above by replacing three glycerol molecules (per monomer), as present in GlpF+G (1FX8) (Fu et al., 2000), by water. Water insertion using DOWSER (Hermans et al., 1998) placed in the constriction region of each monomer eight water molecules in a single file in close accord with the later reported crystal structures of GlpF-G (PDB entry codes 1LDI and 1LDA) (Tajkhorshid et al., 2002) and with the resulting equilibrium distribution of water inside the channel as obtained from MD simulations (Tajkhorshid et al., 2002).
The resulting water-saturated tetramer (GlpF-G) was substituted into a hydrated POPE membrane, pre-equilibrated for 100 ps with fixed protein at constant temperature and pressure (310 K, 1 atm) (Jensen et al., 2001). The total GlpF-G/POPE/water system counted 106,245 atoms including the GlpF tetramer (15,356 atoms), 317 POPE lipid molecules (39,625 atoms), 17,068 water molecules (51,204 atoms), and four glycerol molecules (56 atoms; harmonic constraints with a force constant of 5 kcal/mol/Å2 were applied to all glycerol carbon atoms to ensure that glycerol molecules remained in the bulk water region) and four chloride ions in the bulk region, with initial dimensions of 122.0 × 112.7 × 77.0 Å3. The corresponding dimensions of the pure GlpF tetramer are 73.4 × 73.4 × 57.0 Å3. The system was subsequently minimized and subjected to MD for another 100 ps with fixed protein. The protein was then released, the full system was minimized, and the simulation was conducted for 5 ns in the NPT ensemble with the first nanosecond considered equilibration, and the last 4 ns used for analysis.
The program NAMD (Kalé et al., 1999) and the CHARMM27 parameter set (Schlenkrich et al., 1996; MacKerell Jr. et al., 1998; Feller and MacKerell, 2000) were used for all simulations. Full periodic boundary conditions were imposed. The particle mesh Ewald method (Darden et al., 1993) was used for computation of electrostatic forces without truncation. Langevin dynamics was used to maintain a constant pressure of 1 atm and a constant temperature of 310 K (Feller et al., 1995). The applied temperature represents the natural environment of E. coli, and is above the gel-liquid crystalline phase transition temperature of POPE. A W48F/F200T double mutant of GlpF with increased water permeability (Tajkhorshid et al., 2002) was prepared from the GlpF-G/POPE system (equilibrated for 1 ns) by mutating the appropriate residues. The mutant was simulated for 5 ns in the NPT ensemble using the same protocol as for the wild-type (WT), while again considering the first 1-ns equilibration (Tajkhorshid et al., 2002; the mutant structure is accessible with PDB entry code 1LDF).
To investigate the electrostatic influence of the protein on the structure and dynamics of water, in addition to the native WT system above denoted (I), we carried out simulations on four charge-modified systems denoted (II)–(V): in (II) the charges of the NH2 groups of Asn68 and Asn203 of the NPA motifs were turned off; in (III) charge contributions from the backbone atoms of the M3 (residues 70–79) and M7 (residues 205–217) helices were turned off; (II) and (III) were combined in (IV); and in (V) a “hydrophobic” channel was created by combining (IV) with turning off the charges of the carbonyl groups of residues 64–67 and 196–201, which constitute the conduction pathway (Jensen et al., 2001). Due to the (charge) group concept of the CHARMM27 parameter set (Schlenkrich et al., 1996; MacKerell Jr. et al., 1998; Feller and MacKerell, 2000), we also set the charges of the Gly209 side chain and of Cδ and Hδ of Pro210 to zero, thereby avoiding introduction of net charges. In systems (III)–(V), all atoms of the M3 and M7 helices with charges turned off were fixed, as were the carbonyl groups in (V). The initial configuration used in systems (II)–(V) was the one of system (I) already equilibrated for 1 ns. We simulated systems (II)–(V) for 0.5 ns under the same conditions as used for simulating system (I).
Analysis
All trajectories were analyzed with the molecular graphics and analysis program VMD (Humphrey et al., 1996). Numeric values presented for a protein monomer, or atom subsets thereof, as emerging from the analysis procedures described below, are all averaged over the four monomers of the tetramer. Furthermore, various properties are calculated for water molecules occupying different regions of the channel. In assigning the calculated values to different regions of the channel, the position of the oxygen atom of water is used. When a pair of water molecules is considered, e.g., in calculation of pair correlations, the midpoint of the two oxygen atoms is adopted.
Water permeation
Water diffusion inside the channel occurs in single file and is highly correlated. Such a concerted translocation of water in single file, as also encountered in carbon nanotubes (Hummer et al., 2001; Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003), can be described by a continuous-time random-walk model (Berezhkovskii and Hummer, 2002). In that model, the hopping rate k is the frequency of water file translocation by a distance equal to that of the average water-water spacing 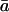 .
. 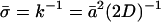 is the mean hopping time, i.e., the average time between successive single file translocations (Berezhkovskii and Hummer, 2002). From 2Dt ≈ 〈Δz2(t)〉 (for t → ∞) we obtain the one-dimensional diffusion coefficient D with 〈Δz2(t)〉 being the center of mass mean square displacement along the channel axis z:
is the mean hopping time, i.e., the average time between successive single file translocations (Berezhkovskii and Hummer, 2002). From 2Dt ≈ 〈Δz2(t)〉 (for t → ∞) we obtain the one-dimensional diffusion coefficient D with 〈Δz2(t)〉 being the center of mass mean square displacement along the channel axis z:
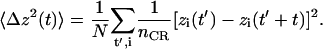 |
(1) |
N is here the number of gliding time origins and nCR is the number of water molecules within the constriction region (CR ≡ −6 Å < z < 14 Å) between t′ and t′ + t. Any water molecule diffusing outside the constriction region within a given time window starting at t′ is discarded in the remaining part of the calculation within that time window. With  being the average number of single-file water molecules in the constriction region we obtain the bi-directional conduction rate k±, i.e., the number of permeating water molecules per channel per time, as
being the average number of single-file water molecules in the constriction region we obtain the bi-directional conduction rate k±, i.e., the number of permeating water molecules per channel per time, as 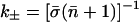 (Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003; Zhu et al., 2003). We note in passing that the uni-directional conduction rate equals k±/2).
(Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003; Zhu et al., 2003). We note in passing that the uni-directional conduction rate equals k±/2).
Water-water correlation
To quantify the correlated motion of water within the constriction region of the channel, we, as others (de Groot et al., 2001) define, for each position z along the channel axis, a nearest-neighbor cross correlation coefficient c(z), as an average over all pairs of adjacent water molecules, i and j visiting z (meaning that the midpoint of the two correlated oxygen atoms is z):
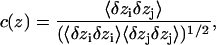 |
(2) |
where
 |
 |
and
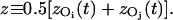 |
Single file disruption
For each position z along the channel axis, a single file disruption ratio, dr(z), based on the distance between any two adjacent water molecules i and j visiting z, was calculated as
 |
(3) |
Again,  and NDWP (z) and NWP (z) are the total number of disrupted water pairs and the total number of water pairs, respectively, counted at position z within the constriction region. If the distance
and NDWP (z) and NWP (z) are the total number of disrupted water pairs and the total number of water pairs, respectively, counted at position z within the constriction region. If the distance 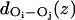 between the oxygen atoms of the i,jth nearest-neighbor water pair exceeded 3.75 Å, the single file was considered disrupted at z, i.e., at the midpoint of that water pair.
between the oxygen atoms of the i,jth nearest-neighbor water pair exceeded 3.75 Å, the single file was considered disrupted at z, i.e., at the midpoint of that water pair.
Water orientation
The equilibrium water orientation inside the channel was characterized by the Legendre polynomials: P1(z) = 〈cos(θ)z〉 and P2(z) = 0.5 〈3cos2(θ)z − 1〉. θ is the angle between the membrane normal, directed along the z-axis (Fig. 2), and the dipole moment of water. z is the position of the oxygen atom of water along the channel axis. The orientation of water was further analyzed by calculating the probability distribution p[cos(θ),z] yielding the likelihood for observing a given water orientation cos(θ) at position z along the channel axis.
FIGURE 2.

The simulated system. Direction of periplasm is upwards and cytoplasm is downwards. GlpF monomers are rendered in cartoon representation in different colors. Aliphatic tails of lipid molecules are rendered in dark yellow with some lipid molecules omitted for clarity; headgroups are shown in sphere representation. Water molecules are shown in red on the two sides of the membrane. The single file of water molecules in one monomer is shown as vdW spheres in red and white.
Electrostatic interaction energy
The electrostatic interaction energy U(z) between a water molecule W located at position z along the channel axis and the environment (E; the membrane, other water molecules, the protein, or subsets of the latter) was evaluated as a time-averaged sum of the nontruncated Coulomb interaction between the three atoms of the water molecule and all atomic charges of the environment E in question:
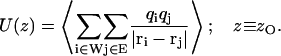 |
(4) |
Averaging involved 400 configurations separated by 10 ps and collected over the last 4 ns of the simulation of system (I), i.e., WT GlpF. zO is the z-coordinate of the oxygen of the water molecule. The channel axis was divided into slabs of ∼0.8 Å width.
Electric field
The electric field E(z) generated at position z along the channel axis by the partial charges of the environment E was computed using, as trial field points, the position of the oxygen atoms of water molecules inside the channel:
 |
(5) |
where rO and ri are the position of the oxygen atom of the water and the position of the partial charge from the environment, respectively. Averaging and binning of E(z) were performed as in the calculation of U(z) (Eq. 4).
To examine the electrostatic interaction between the environment (or parts thereof) and a trial point dipole moment μ of magnitude 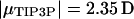 (Jorgensen et al., 1983), representing a water molecule inside the channel, normal (Uz(z)) and in-plane (Uxy(z)) interaction energies were calculated as
(Jorgensen et al., 1983), representing a water molecule inside the channel, normal (Uz(z)) and in-plane (Uxy(z)) interaction energies were calculated as
 |
(6) |
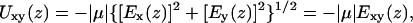 |
(7) |
where Ex(z), Ey(z), and Ez(z) are the x, y, and z components of the electric field E(z) as obtained from Eq. 5, respectively, and Exy(z)=|Exy(z)| is the in-plane field amplitude. Binning and averaging were again performed as in Eqs. 4 and 5, and the positions of the oxygen atoms of water molecules were used for positioning the trial point dipole along the channel axis z. In calculating Uz and Uxy, the dipole moment of water μ is assumed to be aligned favorably with Ez and Exy, respectively.
RESULTS AND DISCUSSION
In this section, we first briefly describe the conformational fluctuation of the channel during the simulations. Then we discuss water permeation, water translocation mechanism, and water equilibrium configurational behavior. A detailed analysis of the electric field generated by the channel and water-protein electrostatic interaction, which constitutes the most important basis of selectivity in AQPs, is finally provided along with a suggestion of barriers raised against ion conduction through the water pore.
Conformational fluctuations of the channel
Fig. 3 a presents the radius of the water pore along with a snapshot (Fig. 3 b) from the simulation of WT GlpF-G. The latter, drawn at a scale matching the pore radius profile, illustrates the structural details of the channel interior, including Asn68 and Asn203 of the conserved NPA motifs, Arg206 at the SF, and backbone carbonyl groups of the re-entrant loops, which participate in substrate conduction.
FIGURE 3.

(a) Channel radius calculated along the channel axis by the program HOLE (Smart et al., 1996) for crystal structures of GlpF+G (PDB code 1FX8) and GlpF-G (PDB code 1LDI) and for the simulated WT GlpF-G. For the simulated channel, the fluctuations (standard deviations) are shown as dashed lines. In all structures the narrowest part of the channel is located at the SF region (−5 Å < z < −2 Å). Removal of glycerol from the channel results in a narrowing of the SF and, in the case of the simulated system, a narrowing of the region near the NPA motifs. (b) Snapshot from the MD simulation of WT GlpF-G drawn at a scale matching the pore-radius profile in a. The bipolar orientation of the hydrogen bonded water file precluding proton conduction in GlpF is discernible in b, i.e., opposite orientation of water molecules in the two half channels is observed. This pattern propagates from the central water molecule hydrogen bonded to Asn68/Asn203 at the two NPA motifs (residues 68–70/201–203). These motifs are shown in green with the side chains of Asn68/Asn203 displayed in ball and stick representation. Recognizable are also hydrogen bonds between the water molecules and between water molecules and the carbonyl groups in the nonhelical parts of the two re-entrant loops, which are shown in gray. The exposed carbonyl oxygen atoms of residues 65–66 and 198–200 are displayed as red spheres. The carbonyl groups of Ala201 and His66 are those closest to the NPA motifs. The carbonyl groups of Met202 and Leu67, which participate in hydrogen bonding to the Hδs of Asn68 and Asn203, are not shown. The α-helical parts of the loops are shown in blue. Residues Trp48, Phe200, and Arg206, constituting the SF, appear in light blue; for clarity, nonpolar hydrogen atoms in these residues are omitted. The polarity of the electrostatic dipoles generated by the M3/M7 half-helices is indicated by + (blue) and − (red). Water molecules in the constriction region of the channel are marked by red stars.
Comparison of MD simulations of GlpF+G (Jensen et al., 2001; Tajkhorshid et al., 2002) with WT GlpF-G (Tajkhorshid et al., 2002, and present study) indicates that removal of glycerol from the channel results in a slight narrowing of the pore at the SF region. Calculating the radius of the channel in different regions by the program HOLE (Smart et al., 1996) indicates that the narrowest region of the channel in WT GlpF-G is 10–15% smaller than that in GlpF+G and is shifted ∼2.0 Å along the channel axis toward the channel center (see Fig. 3 a), a finding confirmed when comparing the crystal structure of GlpF+G (PDB-code 1FX8) (Fu et al., 2000) with those of WT GlpF-G (PDB-codes 1LDI and 1LDA) (Tajkhorshid et al., 2002). After removing glycerol, the narrowest part of the channel is still located in the SF region, and the length of the constriction region (20 Å) where water molecules move concertedly, is preserved. Although the crystal structures of GlpF+G and WT GlpF-G are very similar in the remainder of the channel, we notice in simulated WT GlpF-G, i.e. after removal of glycerol another narrowing of the channel right above the NPA motifs ( Å).
Å).
The RMSD values between the crystal structure of GlpF+G (PDB-code 1FX8) and those of WT GlpF-G are 0.26 Å (1FX8 vs. 1LDI) and 0.51 Å (1FX8 vs. 1LDA) (Tajkhorshid et al., 2002). The RMSD between the backbone atoms of the simulated WT GlpF-G and the crystal structure converges in each of the four monomers to 0.9–1.5 Å.
Water translocation
In all WT simulations, i.e., systems (I)–(V), and in simulations of the double mutant system, water molecules in the constriction region of the channel moved due to thermal fluctuations. Water molecules form a single file inside the channel and translocate concertedly. This is discernible from Fig. 3 b where a representative snapshot illustrates the highly constrained water configuration in WT GlpF-G. Trajectories of all water molecules visiting the constriction region of each of the four monomers during the 5 ns of the simulation of WT GlpF-G are presented in Fig. 4. Although several features of water motion in the channel are analyzed and quantified in this article, Fig. 4 is the most transparent way of presenting water dynamics in AQPs. The movements of neighboring water molecules in the single file along the channel axis are highly correlated. Water permeation through the channel, therefore, can be viewed as the translocation of the whole single file through the channel, which occurs in hops (shifts) of ∼2–3 Å (Fig. 4). The movement of the single file involves simultaneous exchange of protein-water hydrogen bonds, which is a consequence of a narrow pore and has also been observed during glycerol-water co-translocation in GlpF+G (Jensen et al., 2001). Defects (water-water interchanges or crossings) in the single file occasionally occur, whereas no persistent disruption of the single file in any part of the channel is evident from the trajectories (Fig. 4).
FIGURE 4.

Water translocation in the constriction region of WT GlpF-G during the 5-ns time course of the simulation. The direction of cytoplasm and periplasm is along increasing and decreasing z, respectively. A running average of 50 ps is given to improve clarity. Individual water molecules are assigned different colors each time they enter or re-enter the channel.
No preferential direction of water movement inside the channel was observed, i.e., water moved in either direction and no net transmembrane flux occurred, although local density fluctuations together with thermal fluctuations caused 18 water molecules to completely permeate the channel during the 4 ns of analysis. Considering that AQPs are passive water channels, the absence of net diffusion is expected in equilibrium MD simulations with constant isotropic pressure and periodic boundary conditions. For a study of pressure-induced water transport in AQPs, which readily leads to a net water flow across the membrane allowing for appropriate comparison with water flux induced by osmotic or hydrostatic pressure differences in experiments, we refer the reader to the work of Zhu and co-workers (Zhu et al., 2002, 2003; Zhu and Schulten, 2003).
Water-water interchanges, i.e., single file defects, observed in the simulation of WT GlpF are quantified in Table 1. Although water dynamics inside the channel is largely coupled to the motion of groups lining the pore, the radius of the pore seems to be the main structural determinant of such interchanges. Interchanges in the SF (z ≅ −2 Å), i.e., the narrowest region of the channel, are essentially rare. Not surprisingly, interchanges are more likely to occur in wider and more fluctuating regions of the channel ( Å; compare to Fig. 3). Thus one expects that in AQP1, which has a narrower pore (Sui et al., 2001), interchanges would be fewer than in GlpF. The large standard deviation found at
Å; compare to Fig. 3). Thus one expects that in AQP1, which has a narrower pore (Sui et al., 2001), interchanges would be fewer than in GlpF. The large standard deviation found at 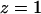 Å is due to the fact that monomers 1 and 4 exhibit a large number of interchanges in this region (184 and 281, respectively), whereas monomers 2 and 3 do not (46 and 35, respectively).
Å is due to the fact that monomers 1 and 4 exhibit a large number of interchanges in this region (184 and 281, respectively), whereas monomers 2 and 3 do not (46 and 35, respectively).
TABLE 1.
Water-water interchanges per nanosecond Xww along the channel axis observed over the last 4 ns of the simulation of WT GlpF
| Periplasmic half
|
Cytoplasmic half
|
||||
|---|---|---|---|---|---|
| z(Å) | Xww | σ(Xww) | z(Å) | Xww | σ(Xww) |
| −4 | 9.1 | 8.3 | 6 | 27.1 | 12.1 |
| −3 | 1.7 | 1.2 | 7 | 35.4 | 16.2 |
| −2 | 0.4 | 0.3 | 8 | 73.7 | 22.5 |
| −1 | 2.4 | 1.5 | 9 | 36.8 | 12.7 |
| 0 | 48.6 | 36.6 | 10 | 49.0 | 12.0 |
| 1 | 136.3 | 101.8 | 11 | 73.1 | 19.1 |
| 2 | 63.8 | 36.4 | |||
| 3 | 16.8 | 8.9 | |||
| 4 | 21.4 | 13.9 | |||
| 5 | 24.6 | 14.5 | |||
Interchanges were assigned to position z along the channel axis (compare to Fig. 3), taken as the position of the O atom of one of the interchanging water molecules using a bin-width resolution of 1 Å. Standard deviations σ are obtained as fluctuations from the total average calculated over the four monomers.
Interchanges of water molecules inside the channel are associated with a slight steric repulsion between the interchanging water molecules (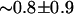 kcal/mol) and between the water molecules and their surroundings (
kcal/mol) and between the water molecules and their surroundings ( kcal/mol). It has to be mentioned, however, that interchanging water molecules have attractive electrostatic interactions with each other (∼−3.9 ± 3.7 kcal/mol) and with their surroundings (∼−22.8 ± 6.3 kcal/mol), which can easily overcome steric barriers introduced by the repulsive van der Waals (vdW) interactions. Despite occasions of water interchange, single file arrangement of water is favored by the channel, and in most parts of the simulation time, water molecules avoid crossing each other inside the channel of GlpF.
kcal/mol). It has to be mentioned, however, that interchanging water molecules have attractive electrostatic interactions with each other (∼−3.9 ± 3.7 kcal/mol) and with their surroundings (∼−22.8 ± 6.3 kcal/mol), which can easily overcome steric barriers introduced by the repulsive van der Waals (vdW) interactions. Despite occasions of water interchange, single file arrangement of water is favored by the channel, and in most parts of the simulation time, water molecules avoid crossing each other inside the channel of GlpF.
Channel occupancy
The water occupancy in WT GlpF-G over the 5-ns period of the simulation is shown for each monomer in Fig. 5. The average occupancy n in each monomer is fluctuating closely around the average in monomers 1 and 2, whereas monomers 3 and 4 display more dramatic deviations from the average at certain time instances. However, in all four monomers the average occupancy number n is the same as the overall average, 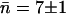 water molecules.
water molecules.
FIGURE 5.

Water occupancy within the constriction region of each of the four monomers (M1–M4) in WT GlpF-G during the 5-ns time course of the simulation. For each monomer, the average occupancy n given in each panel leads to the overall average occupancy of 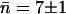 water molecules.
water molecules.
In the simulation of system (V), in which the channel interior is almost completely hydrophobic, at times the channel is almost instantaneously emptied (but later refilled). This observation illustrates the profound role of carbonyl groups in continuously maintaining the water file inside a narrow channel with an otherwise hydrophobic interior. For a larger pore radius, even a purely hydrophobic channel exhibits rapid water conduction, as demonstrated by simulations of water conduction through carbon nanotubes (Hummer et al., 2001; Zhu and Schulten, 2003). The sensitivity of a channel's water occupancy to vdW interaction between water and the channel in nanotubes (Hummer et al., 2001; Beckstein et al., 2001) and the channel emptying observed in system (V) of the present study suggest that a balance between channel radius and hydrophobicity is required for accommodation of water (Beckstein and Sansom, 2003).
A largely hydrophobic channel interior in AQPs keeps direct hydrogen bonding with the permeant substrate at a minimum, and ensures rapid water diffusion through the channel, as in hydrophobic carbon nanotubes (Hummer et al., 2001; Zhu and Schulten, 2003). A slightly more polar interior is found in AQP1 (Sui et al., 2001) than in GlpF (Fu et al., 2000; Tajkhorshid et al., 2002; de Groot et al., 2003). Presumably, the more polar interior in AQP1 is necessary since the narrower channel in AQP1 relative to that of GlpF has to be compensated by more polar residues to attract water into the channel. An amphipathic channel lining in GlpF, on the other hand, is a prerequisite for efficient conduction of amphipathic substrates like glycerol (Fu et al., 2000; Jensen et al., 2001, 2002; Grayson et al., 2003).
Conduction rates
For correlated water transport in single file, the single parameter determining the bi-directional conduction rate, k±, is the hopping rate k or, equivalently, the mean hopping time  describing, respectively, frequency and average time of single file translocations by an amount equal to that of the average water-water spacing (Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003). From knowledge of the diffusion coefficient D and of the average water-water separation
describing, respectively, frequency and average time of single file translocations by an amount equal to that of the average water-water spacing (Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003). From knowledge of the diffusion coefficient D and of the average water-water separation 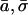 can be calculated (Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003; see also Methods). The average diffusion coefficient of water for WT GlpF-G, as calculated from linear regression of a plot of the mean square displacement vs. time for water molecules within the constriction region of the channel (compare to Eq. 1), is 0.42 × 10−5 cm2 s−1, with lower and upper bounds of 0.23 × 10−5 cm2 s−1 and 0.55 × 10−5 cm2 s−1, respectively. These values for the diffusion constants and an average water spacing
can be calculated (Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003; see also Methods). The average diffusion coefficient of water for WT GlpF-G, as calculated from linear regression of a plot of the mean square displacement vs. time for water molecules within the constriction region of the channel (compare to Eq. 1), is 0.42 × 10−5 cm2 s−1, with lower and upper bounds of 0.23 × 10−5 cm2 s−1 and 0.55 × 10−5 cm2 s−1, respectively. These values for the diffusion constants and an average water spacing 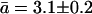 Å lead to an average hopping time
Å lead to an average hopping time  with lower and upper bounds of
with lower and upper bounds of  and
and 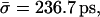 respectively. Using the average channel occupancy reported in the previous section, i.e.,
respectively. Using the average channel occupancy reported in the previous section, i.e., 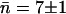 , the corresponding bidirectional conduction rate (per monomer) is k± = 1.1 ns−1, with lower and upper bounds of k± = 0.5 ns−1 and k± = 1.6 ns−1, respectively.
, the corresponding bidirectional conduction rate (per monomer) is k± = 1.1 ns−1, with lower and upper bounds of k± = 0.5 ns−1 and k± = 1.6 ns−1, respectively.
For WT GlpF-G, the 18 complete permeation events we observed in 4 ns correspond to k± = 1.1 ns−1 which is identical to the average value for k± derived from the continuous-time random-walk model over the same time interval. This demonstrates the validity of applying the latter model, which was developed for analysis of water permeation through carbon nanotubes (Berezhkovskii and Hummer, 2002; Zhu and Schulten, 2003), to single file transport through AQPs despite defects (water-water interchanges) that arise occasionally in the single file arrangement (compare to Fig. 4 and Table 1). Our average value for the bi-directional conduction rate and its lower and upper bounds agree well with the experimentally deduced values (Borgnia and Agre, 2001; Haymann and Engel, 1999; Borgnia et al., 1999). However, the average hopping time 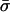 in WT GlpF-G is 9× longer than
in WT GlpF-G is 9× longer than  (Berezhkovskii and Hummer, 2002) and 3× longer than
(Berezhkovskii and Hummer, 2002) and 3× longer than 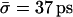 (Zhu and Schulten, 2003) as found from similar analysis of single file water conduction through carbon nanotubes (Hummer et al., 2001; Zhu and Schulten, 2003; both nanotube simulations were at 300 K, but used AMBER and CHARMM force fields, respectively). The slower conduction in WT GlpF-G is well-explained by the fact that the channel lining of this protein is one-half hydrophilic and one-half hydrophobic, whereas purely hydrophobic carbon nanotubes are less strongly interacting with water.
(Zhu and Schulten, 2003) as found from similar analysis of single file water conduction through carbon nanotubes (Hummer et al., 2001; Zhu and Schulten, 2003; both nanotube simulations were at 300 K, but used AMBER and CHARMM force fields, respectively). The slower conduction in WT GlpF-G is well-explained by the fact that the channel lining of this protein is one-half hydrophilic and one-half hydrophobic, whereas purely hydrophobic carbon nanotubes are less strongly interacting with water.
Water distribution
The water file inside the channel extends along the conduction pathway, which is formed by the exposed backbone carbonyl groups of re-entrant loops (residues 64–67 and 198–202) and the side chains of Arg206, Asn68, and Asn203 (Jensen et al., 2001; Tajkhorshid et al., 2002), as illustrated by the snapshot in Fig. 3 b. Only a few polar hydrogen atoms are involved in stabilizing water during permeation; besides Asn68:Hδ and Asn203:Hδ of the NPA motifs, Arg206:Hη and Arg206:Hɛ of the SF contribute. Arg206:Hη marks the beginning of the one-dimensional constriction region of the channel at 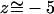 Å (approaching here from the periplasmic side; see Fig. 3).
Å (approaching here from the periplasmic side; see Fig. 3).
The equilibrium distribution of water inside the channel is essentially of overlapping Gaussians-type and correlates closely with the position of exposed carbonyl groups and hydrogen donor groups lining the channel interior (Tajkhorshid et al., 2002). A very similar arrangement of water molecules inside the channel was reported in the crystal structure of WT GlpF-G (Tajkhorshid et al., 2002).
The average protein-water hydrogen bonding interactions inside the channel and the water-water hydrogen bonds together defining the integrity of the single file are evident from the atomic radial distribution functions between the channel and water, r(GlpF:O–H2O:H) and r(GlpF:O–H2O:O), and between water, r(H2O:O–H2O:H) and r(H2O:O–H2O:O), respectively (Fig. 6). For systems (I)–(IV), the peaks of r(GlpF:O–H2O:H) and r(H2O:O–H2O:H) are consistently located at 1.9 Å (data for systems (II)–(IV) are very similar to system (I) and are not shown). This demonstrates nearly ideal hydrogen bond distances between the channel lining carbonyls and water and between the nearest-neighbor water molecules. The average hydrogen bonding characteristics are not particularly sensitive to the electrostatic modifications invoked in systems (II)–(IV) and r(GlpF:O–H2O:H) and r(GlpF:O–H2O:O) obtained for these systems are virtually identical to those of system (I). However, after turning off the charges of the channel lining carbonyls in system (V), no structure is featured in r(GlpF:O–H2O:H) and the peak in r (GlpF:O–H2O:O) shifts to a larger value (Fig. 6, bottom panels). This demonstrates that the water-channel interactions define the conduction pathway in GlpF and, at the same time, confirms that the electrostatic interactions between water and the channel lining carbonyls help structuring water inside the channel.
FIGURE 6.

Atom-atom radial distribution functions between the carbonyl oxygen atoms of the channel and water (channel-water) and between nearest-neighboring water molecules (water-water) located within the constriction region (compare to Fig. 3). The distribution functions are shown for WT GlpF-G (I) in the top panels, and for system (V) in the bottom panels. Because the spatial arrangement of the single file is linear, no radial normalization was invoked. The distribution function of systems (II)–(IV) are very similar to those of system (I), and are not shown.
The interaction between the channel lining and water molecules perturbs to some extent the hydrogen bonding between neighboring water molecules (Tajkhorshid et al., 2002; de Groot and Grubmüller, 2001). To examine this effect, we calculated for each position along the channel axis a disruption ratio 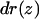 and the corresponding average distance
and the corresponding average distance  between the oxygen atoms of the neighboring water molecules for systems (I)–(V) and for the W48F/F200T double mutant (see Methods section). The results for WT GlpF-G, system (I), and for the double mutant are shown in Fig. 7. To also provide a dynamic description of the relation of the neighboring water molecules inside the channel, the correlation of their motion
between the oxygen atoms of the neighboring water molecules for systems (I)–(V) and for the W48F/F200T double mutant (see Methods section). The results for WT GlpF-G, system (I), and for the double mutant are shown in Fig. 7. To also provide a dynamic description of the relation of the neighboring water molecules inside the channel, the correlation of their motion 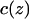 was calculated and is also shown in Fig. 7. The results for systems (II)–(V) do not differ significantly from the results obtained for system (I), and are consequently not shown.
was calculated and is also shown in Fig. 7. The results for systems (II)–(V) do not differ significantly from the results obtained for system (I), and are consequently not shown.
FIGURE 7.

Average configuration of the single file through the channel for WT GlpF-G (I) (top panels) and for the W48F/F200T double mutant (Tajkhorshid et al., 2002) (bottom panels): (left) distance between oxygen atoms of nearest-neighbor water molecules dO–O(z), and disruption ratio dr(z) (dashed lines) resolved along the channel axis; (right) correlation coefficient c(z) between nearest-neighbor water molecules.
In general, disruption is very rare, indicating that water molecules predominantly remain joined in single file. In system (I) the disruption reaches a minimum in the SF region coinciding with c(z) reaching a maximum in this region (Fig. 7). In the SF, the side chain of the conserved Arg206 interacts strongly with water molecules. However, this has a minimal effect on the interaction of the water with neighboring water molecules. Other side chains in this region, Phe200 and Trp48, are nonpolar and cannot interact with water strongly. Therefore, neighboring water molecules will not be constrained by hydrogen bonds to multiple sites that could keep them apart from each other. At the exit of the SF toward the center of the channel, however, water can form hydrogen bonds with Arg206:Hɛ (at z = −2.5 ± 0.5 Å) and with Phe200:O (at z = −2.5 ± 0.3 Å), where the first peak in dr(z) and dO–O(z) is observed for WT GlpF-G. The next major disruption peak in system (I) is observed at the NPA region where water molecules hydrogen-bond through their O atom, rather than their H atoms, to the polar side chains of Asn68/Asn203. In the W48F/F200T double mutant both the size and the polarity of the SF are increased. Together, these effects result in the presence of multiple water molecules in the SF region of this mutant, which, in turn, changes dr(z) and c(z) (Fig. 7).
The average nearest-neighbor cross-correlation coefficient c(z) along the channel axis (Fig. 7) quantifies the concertedness of water translocation shown in Fig. 4. The correlation curves in Fig. 7 show that, with no exception, water translocation inside the channel is correlated and that the correlation is highest in the periplasmic half of the channel. Interestingly, for WT GlpF-G, the maximum correlation is exhibited in the SF region. In contrast, for the W48F/F200T double mutant the correlation in this region is reduced due to stronger hydrogen bonding interactions owing to the presence of multiple water molecules at the entrance of the channel, and, possibly, due to the higher polarity of residues introduced by the mutations.
Water orientation
The equilibrium water orientation inside the channel of WT GlpF-G (I), characterized by the order parameters P1(z) = 〈cos(θ)z〉 and P2(z) = 0.5〈3cos2(θ)z −1〉, is presented in Fig. 8, b and a, respectively. In Fig. 8 b, P1(z) is furthermore shown for systems (II)–(IV). The probability p[cos(θ),z] of a given water orientation cos(θ) as a function of position z inside the channel of WT GlpF-G is shown in Fig. 8 c. Fig. 8 a illustrates that a high degree of water ordering exists inside the channel of WT GlpF-G, whereas disorder of water is approached at the channel outlets as revealed by P2(z). Inside the channel, P2(z) reaches a minimum in the channel center, i.e., at the NPA region around which P2(z) is fairly symmetric. Together, this is indicating a similar degree of ordering in both the periplasmic and the cytoplasmic regions of the channel. Within the channel, the order parameter profile P1(z) in Fig. 8 b is roughly sigmoidal and fairly similar in systems (I)–(III). P1(z) exhibits an inflection point at 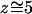 Å, i.e., at the midpoint between the two Hδ atoms of Asn68 and Asn203 [z(Asn68:Hδ) = 6.1 Å ± 0.3 Å; z(Asn203:Hδ) = 4.7 Å ± 0.4 Å]. The shape of P1(z) reflects the well-known (Tajkhorshid et al. 2002) equilibrium, bipolar water orientation in the two halves of the channel with a hydrogen donor/hydrogen acceptor pattern and, hence, the water dipole inversion around the NPA motifs (compare to Fig. 3 b).
Å, i.e., at the midpoint between the two Hδ atoms of Asn68 and Asn203 [z(Asn68:Hδ) = 6.1 Å ± 0.3 Å; z(Asn203:Hδ) = 4.7 Å ± 0.4 Å]. The shape of P1(z) reflects the well-known (Tajkhorshid et al. 2002) equilibrium, bipolar water orientation in the two halves of the channel with a hydrogen donor/hydrogen acceptor pattern and, hence, the water dipole inversion around the NPA motifs (compare to Fig. 3 b).
FIGURE 8.

Water orientation in the channel. (a) Order parameter P2(z) = 0.5 〈3 cos2(θ) − 1〉 for WT GlpF-G (I). (b) Order parameter P1(z) = 〈cos(θ)〉 for systems (I)–(V). θ is the angle between the unit vector approximately aligned along the channel axis/membrane normal,  and the water dipole vector. Fluctuations in P2(z) and P1(z) are shown in a and b for WT GlpF-G (I). (c) Histogram p[cos(θ),z] displaying the probability p for observing a given water orientation cos(θ) at position z along the channel axis of WT GlpF-G (I).
and the water dipole vector. Fluctuations in P2(z) and P1(z) are shown in a and b for WT GlpF-G (I). (c) Histogram p[cos(θ),z] displaying the probability p for observing a given water orientation cos(θ) at position z along the channel axis of WT GlpF-G (I).
A polarized water configuration, in which the dipole moment of all water molecules in the channel are aligned along the channel axis in the same direction, allows proton conduction through the water wire by the Grotthuss mechanism (Agmon, 1995; Tajkhorshid et al., 2002; Zhu and Schulten, 2003). As shown in Fig. 8 b(IV), to induce such a configuration of water, one would need to simultaneously eliminate both the electric fields generated by helices M3 and M7 and the hydrogen bonding of the conserved Asn68/Asn203 side chains (Tajkhorshid et al., 2002). This illustrates a profound role of these conserved residues and of the architecture of the protein in the selectivity of the channel against protons. Additional reduction of the electric influence of the channel on permeating water, realized by neutralizing the channel lining carbonyl groups, results in a faster formation of the polarized water chain, which in the case of present simulations shows an opposite polarity; see Fig. 8 b(V). This finely tuned water orientation therefore effectively blocks proton transport in AQPs (Tajkhorshid et al., 2002; Zhu and Schulten, 2003), preserving the electrochemical gradient across the cell membrane while allowing rapid water permeation through the pore.
The configuration of water is illustrated in more detail by the probability distribution 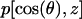 in Fig. 8 c, indicating that there is essentially vanishing probability for locating the proton conducting orientation in either half of the channel, and only the proton blocking orientation is allowed. The observed bipolar orientation of water inside the channel might be used by a hydroxyl ion defect to propagate to the center of the channel. This defect, however, cannot reach the other side of the channel either, and is destined to return and exit the channel from the same side.
in Fig. 8 c, indicating that there is essentially vanishing probability for locating the proton conducting orientation in either half of the channel, and only the proton blocking orientation is allowed. The observed bipolar orientation of water inside the channel might be used by a hydroxyl ion defect to propagate to the center of the channel. This defect, however, cannot reach the other side of the channel either, and is destined to return and exit the channel from the same side.
The polar hydrogen atoms in the SF, Arg206:Hη and Arg206:Hɛ, interact strongly with the permeating water molecules, thereby increasing P1(z) at −7.5 Å < z < −2.5 Å (Fig. 8 b), and in Fig. 8 c shifting the center of p[cos(θ), z ≅ −5 Å] toward cos(θ) = 0. Hence Arg206 makes P1(z) deviate from the order parameter profile that would result if the water molecules formed hydrogen bonds only with the channel lining carbonyl groups. The exact control of the water orientation is a result of precisely optimized electrostatic forces between water and the channel interior. For instance, when eliminating the electrostatic dipoles of the M3/M7 α-helices (compare to Fig. 3 b) in systems (III)–(V), the deviation of P1(z) at z ≅ 5 Å becomes even more pronounced, since, Arg206:Hη and Arg206:Hɛ, in a similar way to the Hδs of Asn68/Asn203, tend to orient water molecules with their dipole moment perpendicular to the channel axis, whereas the M7 dipole tends to align the dipole moment along z. However, the net electrostatics of the Arg206-M3/M7 competition implies that only in the channel center can water be oriented perpendicularly to the channel axis. We demonstrate in the following sections why this is the case.
Electrostatic interaction energies
Protein-water interactions are critical in controlling the dynamics and configurational behavior of water in AQPs (Tajkhorshid et al., 2002; de Groot et al., 2001). In this and the following two sections, protein electric field effects are analyzed in terms of 1), electrostatic stabilization energy resulting from interaction between the protein and the permeating water; 2), orientational constraints imposed onto the water file due to the vectorial nature of the protein electric field; and 3), Coulomb blockade of ion permeation through the water pore.
The electrostatic interaction energy U(z) between water molecules moving along the channel axis and the environment, decomposed into contributions from the protein tetramer, the protein monomer, bulk water within a 20 Å distance of the protein (exclusive of other water molecules in the single file), and the lipid bilayer, are shown in Fig. 9. The dominant part of the interaction energy calculated in Fig. 9 a is naturally due to direct hydrogen bonds between water molecules and the nearby groups, such as the channel lining carbonyl groups, as we discuss further below.
FIGURE 9.

Electrostatic interaction energy between a water molecule in the constriction region and its surroundings. (a) Interaction with protein tetramer and embedding monomer. (b) Interaction with bulk water and membrane. (c) Interaction with embedding monomer (same as in a), with other water molecules of the water file in the same monomer, and their sum.
The electrostatic interaction of permeating water and the full protein tetramer is essentially identical to that between water and the embedding monomer only (Fig. 9 a). Therefore, the tetrameric architecture of AQPs does not reinforce electrostatic stabilization of the water file. Since other monomers do not influence the permeating water, the interaction with the embedding monomer suffices for our further discussion. A maximal (20 kcal/mol) stabilization of water is encountered at the SF in the periplasmic half of the channel (Fig. 9 a). The location of the energy minimum (z ≅ −3 Å) corresponds to the position of the charged guanidinium group of the conserved Arg206 (z(Arg206:Hɛ) = −2.5 ± 0.4 Å).
Despite the polarity of lipid headgroups and the interfacial water layers, bulk water and the lipid bilayer hardly interact with the water file and, consequently, do not contribute to its stabilization. For instance, the stabilizing effect of the membrane amounts to only 1 kcal/mol (Fig. 9 b). Cross-interactions between water files in different monomers are also negligible, and amount to <0.1 kcal/mol (not shown), which is somewhat surprising since water dipoles in different monomers are favorably aligned relative to each other. In contrast, self-stabilizing electrostatic interactions between the water molecules within the same single file are large, −5 to −17 kcal/mol and, therefore, very important (Fig. 9 c). Interestingly, Fig. 9 c reveals that the interaction between a single water molecule and other water molecules of the same single file is the weakest at the SF region. This observation can be explained by the fact that water interacts most strongly with the protein at this region; the positively charged side chain of the conserved Arg206 has a very strong interaction with a water molecule located in this region, as indicated by the location of the maximal protein stabilization effect in the same region (Fig. 9 c). Apart from the SF region, water-water interaction is stronger than water-protein interaction in all parts of the channel, as clearly demonstrated in Fig. 9 c, and accordingly permits fast water permeation in single file.
The observed strong interaction of water with the protein and its weaker interaction with neighboring water molecules at the SF region are somewhat in line with the proposed disruption of the water-hydrogen-bonded network at this position (de Groot et al., 2001). Monitoring the intermolecular distance of adjacent water molecules, however, we find very little disruption at this position (compare to Fig. 7). The observed differences might in part be related to the different force fields applied, particularly with respect to the water models used (the GROMACS force field and the SPC water model were used by the authors in de Groot et al., 2001; here we use the CHARMM27 parameter set with the TIP3 water model). However, it should be noted that even transient (ps) association of two fragments of a disrupted water file would permit significant proton conduction, since charge translocation occurs on a subpicosecond timescale (Schulten and Schulten, 1986; Pomès and Roux, 1996; Roux, 2002; Zhu and Schulten, 2003). In contrast, propagation of an orientational defect in the proton conduction cycle is orders-of-magnitudes slower (Schulten and Schulten, 1986; Pomès and Roux, 1996; Roux, 2002; Zhu and Schulten, 2003), and clearly not possible with the bipolar water configuration found in AQPs.
To examine the importance of the protein architecture and the role of conserved residues in the electrostatic tuning of AQPs, we studied the electrostatic interaction of permeating water with various subsets of the protein, namely the side chains of Arg206, Asn68/Asn203, the channel lining carbonyl groups of the re-entrant loops, and the half-helices M3 and M7 (Fig. 10).
FIGURE 10.

(a) Electrostatic stabilization energy of the water molecules along the channel due to the side chains of Asn68 and Asn203, the channel lining carbonyl groups of residues 64–67 and 196–202, and the Arg206 side chain. (b) Backbone contribution of the M3/M7 helices and full residue contribution of these helices, exclusive of the contribution of the side chain of Arg206. (c) Channel with the key groups displayed in colors matching the results for the electrostatic interaction energy in a and b. Hydrogen atoms are omitted for clarity.
Direct hydrogen bonds to the carbonyl groups of the re-entrant loops, which constitute the conduction pathway (Jensen et al., 2001; Tajkhorshid et al., 2002; compare to Figs. 3 b and 6), stabilize individual permeating water molecules by 4–7 kcal/mol in the two halves of the channel (Fig. 10 a). Near the NPA motifs at the center of the channel, the contribution of these carbonyl groups becomes minimal, since no direct hydrogen bond between them and water can arise in this region. Instead of interacting with the substrate, carbonyl groups of residues in this region are engaged in important hydrogen bonds that stabilize the structure of the channel interior (Zhu et al., 2001; Jensen et al., 2001). At the NPA motifs, water gains a smaller, yet significant, stabilization (2 kcal/mol) through hydrogen bonds with the side chains of Asn68/Asn203 (Fig. 10 a). The most pronounced effect is, however, due to the charged side chain of Arg206 reaching 15 kcal/mol in the periplasmic half of the channel (Fig. 9 c). Near the channel center, Arg206 causes a slight destabilizing effect, which is compensated by the side chains of Asn68/Asn203 stabilizing water at this location (Fig. 10 a).
The subtle balance of electrostatic contributions inside the channel ensures a stable bipolar water configuration in the pore. Furthermore, a minimal interaction between the centrally located water molecule and all of the protein but the Asn68/Asn203 side chains in the channel center ensures unhindered and fast dipole inversion of water at this position. Consequently, simultaneous proton blockage and fast water permeation, not limited by the crucial dipole inversion, is efficiently accomplished. Along these lines, as shown in Fig. 10 b, the resultant electric field due to the backbone atoms (Cα–Hα, C=O, and N–H) of the M3/M7 helices also stabilize the centrally (NPA) located water molecule only marginally, namely by <1 kcal/mol.
The electrostatic interaction energy between water and the helices (mainly M7) in the periplasmic region is positive, yet small. This is due to the unfavorable interaction of water dipoles, which are fixed by strong hydrogen bonding to Arg206, and the field generated by backbone atoms of M7 (compare to Fig. 8, b and c). After inclusion of side chains, except that of Arg206 whose very strong interaction can mask the effect of the others, the interaction of water with M3/M7 in this region becomes attractive (Fig. 10 b). Still though, it is <1 kcal/mol per water molecule since no direct hydrogen bonding interactions are present.
Electrostatic field components
To further analyze the electrostatic tuning of permeating water molecules by the channel, normal (Ez) and in-plane (Exy) components of the electric field E of the protein along the channel axis were calculated. The results are summarized in Fig. 11, a and b. Similar to electrostatic interaction energies (Fig. 10), the electric field was further decomposed in terms of group contributions from the protein. We note in passing that due to the fourfold symmetry of the channel, averaging of the in-plane (Exy) component of the electric field over four monomers cannot be done, and, therefore, we report in Fig. 11 b only the amplitude average of the in-plane field component, Exy=|Exy| (see also Eqs. 5–7).
FIGURE 11.

Electric field components calculated along the channel axis. (a) Group contributions to the normal component of the field (Ez). (b) Group contributions to the average amplitude of the in-plane component of the field (|Exy|; see text for details). (c) Group contributions to the electrostatic energy along the channel axis between Ez and a dipole of magnitude 2.35 D aligned perfectly along ±z. A relative free energy profile for water, G(z) = −lnPw(z), expressed in kcal/mol, with Pw(z) being the equilibrium distribution of the oxygen atoms of water obtained from the simulations (dotted line) is included in c for comparison of energy (barrier) magnitudes. The zero-point of G(z) is arbitrarily chosen at the cytoplasmic outlet, i.e., at z ≅ −10 Å. (Inset) One monomer with the appropriate groups of GlpF color-coded to match the results for the electric field and the electrostatic interaction energy in a–c. Hydrogen atoms are omitted for clarity.
As shown in Fig. 11 a, the normal component (Ez) of the total electric field varies in close accord with the order parameter profile presented in Fig. 8 b; a sign change at the center of the channel, where the NPA motifs are located, indicates that Ez favors opposite orientations of a dipole, such as water, in the two halves of the channel. In the periplasmic half, Arg206 is a major contributor to Ez (Fig. 11 a). The sign change of 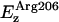 at
at  Å corresponds to the observed perturbation of the water orientation in the SF region (Fig. 8, b and c). As shown in Fig. 11 a, the contribution of other groups to Ez are significantly smaller; however, they all consistently feature a sign change at the NPA region and, thus, collectively enhance the dipole inversion of water at this position.
Å corresponds to the observed perturbation of the water orientation in the SF region (Fig. 8, b and c). As shown in Fig. 11 a, the contribution of other groups to Ez are significantly smaller; however, they all consistently feature a sign change at the NPA region and, thus, collectively enhance the dipole inversion of water at this position.
Regarding the in-plane component of the electric field Exy (Fig. 11 b), the most pronounced component is 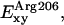 which exceeds in magnitude the normal contribution of this residue
which exceeds in magnitude the normal contribution of this residue 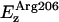 (Fig. 11 a). This strong field rationalizes the almost perpendicular orientation of water to the channel axis at the SF. Exy contributions from channel lining dipolar groups (Cα–Hα, C=O, and N–H) are essentially symmetric around the NPA region and all feature two maxima, one in each half of the channel. At the NPA region, these contributions reach minimal values, whereas the contributions of Asn68/Asn203 and M3/M7 to Exy feature peak values in this region. The overall in-plane field
(Fig. 11 a). This strong field rationalizes the almost perpendicular orientation of water to the channel axis at the SF. Exy contributions from channel lining dipolar groups (Cα–Hα, C=O, and N–H) are essentially symmetric around the NPA region and all feature two maxima, one in each half of the channel. At the NPA region, these contributions reach minimal values, whereas the contributions of Asn68/Asn203 and M3/M7 to Exy feature peak values in this region. The overall in-plane field 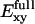 in the NPA region features a local maximum, which can stabilize an orientation of water dipoles perpendicular to the channel axis. Together with the sign change of Ez and hydrogen bonds of water to Asn68 and Asn203, the strong Exy at the NPA region ensures the inversion of water dipoles at the center of the channel.
in the NPA region features a local maximum, which can stabilize an orientation of water dipoles perpendicular to the channel axis. Together with the sign change of Ez and hydrogen bonds of water to Asn68 and Asn203, the strong Exy at the NPA region ensures the inversion of water dipoles at the center of the channel.
To further quantify the preference for a bipolar water configuration in GlpF, we have also calculated the interaction energy between the normal field Ez and a test point dipole representing water (μ) at different positions along the channel axis. Assuming that the dipole is aligned along z, i.e., with opposite directions in the two halves of the channel, the interaction energy can be calculated as 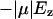 (Fig. 11 c). One can recognize that the interaction energy is maximal in the SF region due to the positively charged Arg206. For comparison, the free energy profile, G(z) = −kBTln[Pw(z)], derived from the water distribution Pw(z) in the simulations, is also presented in Fig. 11 c. G(z) shows a wide barrier at the SF but the barrier heights are relatively similar along the entire channel and roughly one order-of-magnitude smaller than the electrostatic interaction energies.
(Fig. 11 c). One can recognize that the interaction energy is maximal in the SF region due to the positively charged Arg206. For comparison, the free energy profile, G(z) = −kBTln[Pw(z)], derived from the water distribution Pw(z) in the simulations, is also presented in Fig. 11 c. G(z) shows a wide barrier at the SF but the barrier heights are relatively similar along the entire channel and roughly one order-of-magnitude smaller than the electrostatic interaction energies.
The interaction energy of water dipole and Ez along the channel axis (Fig. 11 c) corresponds well to the electrostatic interaction energies of water and the channel calculated at geometries taken from MD simulations (Fig. 9), both demonstrating that a bipolar water configuration is favorable. Deviations are due to the fact that in the simulation water molecules interact with the channel, and their dipole moments deviate from a perfect alignment along z, which is assumed in the calculation of 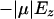 .
.
At the NPA region the interaction of water dipoles with Ez is almost zero since Ez vanishes here (Fig. 11 a). However, the interaction energy between the point dipole and the in-plane field strongly stabilizes, by ∼8 kcal/mol, the perpendicular (to the channel axis) orientation of the water dipole, also in accordance with the observed orientation of water at this position (Figs. 3 b and 8). On each side of the NPA motifs, the channel lining dipoles, the carbonyl groups in particular, yield high interaction energies between the dipole and Ez, whereas contributions from the M3/M7 helices and from the side chains of Asn68 and Asn203 are small.
From Ez and 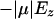 in Fig. 11, a and c, respectively, we can also estimate the energy cost of inverting the orientation of half of the water molecules, which would result in a polarized chain of water inside the channel, i.e., a proton wire composed of water molecules uniformly oriented through the channel. Knowing that the average occupancy is
in Fig. 11, a and c, respectively, we can also estimate the energy cost of inverting the orientation of half of the water molecules, which would result in a polarized chain of water inside the channel, i.e., a proton wire composed of water molecules uniformly oriented through the channel. Knowing that the average occupancy is 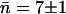 water molecules (Fig. 5), we realize that formation of a proton wire requires the inversion of at least three water molecules in either half-channel. The total energy cost of such an inversion would be ∼3 × 10 kcal/mol, indicating a low probability for formation of a proton wire in GlpF and for propagating an orientational defect as required in the proton conduction cycle (Schulten and Schulten, 1986; Pomès and Roux, 1996; Brewer et al., 1999; Pomès and Roux, 2002; Zhu and Schulten, 2003).
water molecules (Fig. 5), we realize that formation of a proton wire requires the inversion of at least three water molecules in either half-channel. The total energy cost of such an inversion would be ∼3 × 10 kcal/mol, indicating a low probability for formation of a proton wire in GlpF and for propagating an orientational defect as required in the proton conduction cycle (Schulten and Schulten, 1986; Pomès and Roux, 1996; Brewer et al., 1999; Pomès and Roux, 2002; Zhu and Schulten, 2003).
Ion permeation
Water pores in AQPs are impermeable to ions (Borgnia et al., 1999). Clearly, the energy cost of partial dehydration of an ion when transferred from aqueous solution into a narrow channel cannot be compensated by interaction of the ion with the relatively hydrophobic interior of AQPs. However, insight into the origin of a Coulomb blockade of ion permeation in AQP water pores can be gained by examining electrostatic barriers raised against fictitious permeation of a monovalent positive/negative point charge (ion) through the water pore. To determine these energy barriers, we associate at each time t the positions of the oxygen atoms of water molecules inside the channel with a monovalent ion of charge −1e, and calculate the resulting electrostatic interaction energy between the ion and the protein. We realize that the position of the oxygen atom may not precisely reflect the true position of an anion inside the channel if it was to enter at all; however, the position of the negatively charged O atoms of water is a reasonable approximation for the hypothetical position of an anion for our estimation. The electrostatic interaction energies for a negative ion are shown in Fig. 12 a with the corresponding residue contributions in color. The ion positions are illustrated in Fig. 12 b. The negative of the curves shown in Fig. 12 a can be viewed as the interaction of the channel with a cation, except that the positions chosen for the cation in the channel may not be optimal. It should be kept in mind that for both anions and cations, partial dehydration would introduce high barriers at channel entrances, which should also block the entrance of the ion to the channel from either side.
FIGURE 12.

Electrostatic interaction energies for a virtual monovalent anion. (a) Energies along the channel axis at the positions of the water oxygen atoms. The total interaction energy between the ion and the protein (monomer) is shown (solid line). Contributions of key residues (Arg206, Lys211, Asn68/Asn203, Glu43, Asp207, and Glu152) and groups (C=O, NH/Cα-Hα, and M3/M7, with Arg206 excluded) to the energy are shown as colored lines; the contribution from the remaining part of the protein (denoted other) is shown as a dashed line. (b) Channel with key groups displayed and color-coded to match the results for the electrostatic interaction energy in a. Hydrogen atoms are omitted for clarity. The fictitious ion is schematically represented at the locations sampled, through gray vdW spheres.
Despite our crude approximations a few interesting points can be made. The largest negative interaction energy resulting from permeation of an anion is located at the center of the channel (Fig. 12 a), i.e., at the NPA motifs. In other words, in addition to dehydration penalty at the entrance of the channel, the main obstacles against passage of a cation, protons included, should be expected at the NPA region. It is surprising that the key obstacle does not arise at the SF where the positively charged Arg206 is located.
We note that the strong interaction of the ion with the protein at the NPA region is due to the conserved residues Asn68/Asn203. In the periplasmic half of the channel, the large negative interaction energy between the anion and Arg206 is essentially canceled by several repulsive interactions between the ion and other groups of the protein, including the conserved, negatively charged residues Glu43 and Glu152, and the structurally preserved M3/M7 helices. The two other charged residues in this region, namely Asp207 and Lys211, which are both located in the M7 helix, form a salt bridge, and therefore do not raise a barrier against ion permeation, as is evident from their equal, but opposite contributions to the interaction energies (Fig. 12). The channel lining dipoles, i.e., carbonyl groups and N–H dipoles, could stabilize positive ions, as to be expected upon comparison with, e.g., the selectivity filter in the KcsA potassium channel, which is lined with carbonyl groups that coordinate the dehydrated K+ ion (Roux and MacKinnon, 1999). A detailed study of the overall ion conduction process in AQPs is presently conducted by the authors.
CONCLUSION
Water translocation in aquaporins occurs via a concerted displacement of a single file of water, containing on average seven molecules. The structure of the single file is ensured by water-water hydrogen bonds and water-channel hydrogen bonds, the latter predominantly via water interactions with exposed carbonyl oxygen atoms of the nonhelical parts of two re-entrant, half-membrane spanning loops that constitute the conduction pathway.
Proton exclusion is ensured by a bipolar water arrangement in the two halves of the channel, which is induced by electrostatic interaction of water with the channel. At the inversion point of this arrangement, the oxygen atom of a centrally located water molecule is saturated by two hydrogen bonds to side chains of Asn68 and Asn203 of the conserved NPA motifs, and, therefore, cannot act as a proton acceptor (Tajkhorshid et al., 2002). The electric field of the protein, mainly from helices M3 and M7, stabilizes the bipolar configuration that propagates from this central water molecule along the single file. Simultaneously turning off the charges of the Asn68/Asn203 side chains and the charges of the M3/M7 backbone atoms induces the water molecules to orient uniformly and form a proton wire, i.e., to conduct protons.
In the bipolar configuration, water molecules are electrostatically stabilized through specific interactions with the protein as well as with neighboring water molecules, by 15–30 kcal/mol per water molecule. An absolutely conserved Arg residue located in the selectivity filter, i.e., at the beginning of the constriction region, and carbonyl groups of the two inverted helices lining the channel are major contributors to this stabilization, which allows water to enter a narrow, fairly hydrophobic channel. The M3/M7 helices and the NPA motifs contribute only ∼1 kcal/mol and 2 kcal/mol, respectively, to the electrostatic stabilization, mainly to stabilize the centrally located water molecule at the position between the NPA motifs where the dipole moment of the bipolar configuration inverts. The self-stabilizing effect due to water-water hydrogen bonds in the single file is larger than the electrostatic energy gained by water-protein interactions in all parts of the channel, except at the selectivity filter where water interacts very strongly with the positively charged side chain of Arg206. The latter interaction, however, is not strong enough to disrupt the water file as previously proposed (de Groot et al., 2001). The detailed electrostatic balance between water interactions with the channel lining residues and the interactions between the water molecules themselves ensures that the water file is stable inside the protein in a proton filtering configuration, while at the same time being held together in a highly correlated network. This balance permits rapid water diffusion across biological membranes.
Being able to accurately model water permeation, water structure and water dynamics, and to tie simulations to experimental observations, is a critical step toward a thorough understanding of different aquaporins presently investigated, e.g., human aquaporins AQP0, AQP1, … , AQP10. Insight gained may prove valuable in tracking origins of clinical disorders associated with dysfunctional aquaporins but also in resolving ion-conducting and gating mechanisms as well as nucleotide and pH regulation of these channels, issues recently debated (Yool and Weinstein, 2002; Hazama et al., 2002; Pohl et al., 2001; Fotiadis et al., 2002). Aquaporin channels also serve to improve our understanding of membrane channels in general.
Acknowledgments
All molecular images have been made with VMD (Humphrey et al., 1996).
The work is supported by the National Institutes of Health (PHS 5 P41RR05969 and R01 GM067887), and computer time provided by the National Resource Allocations Committee (MCA93S028). M.Ø.J. acknowledges postdoctoral support from the Danish National Research Foundation through a grant to the Quantum Protein Centre.
M. Ø. Jensen and E. Tajkhorshid participated equally in this study.
Morten Ø. Jensen's permanent address is Quantum Protein Centre, Department of Physics, Technical University of Denmark, DK-2800 Lyngby, Denmark.
References
- Agmon, N. 1995. The Grotthuss mechanism. Chem. Phys. Lett. 224:456–462. [Google Scholar]
- Agre, P., M. Bonhivers, and M. J. Borgnia. 1998. The aquaporins, blueprints for cellular plumbing systems. J. Biol. Chem. 273:14659–14662. [DOI] [PubMed] [Google Scholar]
- Beckstein, O., P. C. Biggin, and M. S. P. Sansom. 2001. A hydrophobic gating mechanism for nanopores. J. Phys. Chem. B. 105:12902–12905. [Google Scholar]
- Beckstein, O., and M. S. P. Sansom. 2003. Liquid-vapor oscillations of water in hydrophobic nanopores. Proc. Natl. Acad. Sci. USA. 100:7063–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezhkovskii, A., and G. Hummer. 2002. Single-file transport of water molecules through a carbon nanotube. Phys. Rev. Lett. 89:064503. [DOI] [PubMed] [Google Scholar]
- Bernèche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- Bernstein, F. C., T. F. Koetzle, G. J. Williams, E. F. Meyer, M. D. Brice, J. R. Rogers, O. Kennard, T. Shimanouchi, and M. Tasumi. 1977. The Protein Data Bank: a computer-based archival file for macromolecular structures. J. Mol. Biol. 112:535–542. [DOI] [PubMed] [Google Scholar]
- Borgnia, M., S. Nielsen, A. Engel, and P. Agre. 1999. Cellular and molecular biology of the aquaporin water channels. Annu. Rev. Biochem. 68:425–458. [DOI] [PubMed] [Google Scholar]
- Borgnia, M. J., and P. Agre. 2001. Reconstitution and functional comparison of purified GlpF and AqpZ, the glycerol and water channels from Escherichia coli. Proc. Natl. Acad. Sci. USA. 98:2888–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer, M. L., U. W. Schmitt, and G. A. Voth. 1999. The formation and dynamics of proton wires in channel environments. Biophys. J. 80:1691–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A. T. 1992. X-PLOR, Version 3.1: A System for X-Ray Crystallography and NMR. The Howard Hughes Medical Institute and Department of Molecular Biophysics and Biochemistry, Yale University Press.
- Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald. An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- de Groot, B. L., A. Engel, and H. Grubmüller. 2001. A refined structure of human aquaporin-1. FEBS Lett. 504:206–211. [DOI] [PubMed] [Google Scholar]
- de Groot, B. L., A. Engel, and H. Grubmüller. 2003. The structure of the Aquaporin-1 water channel: a comparison between cryo-electron microscopy and x-ray crystallography. J. Mol. Biol. 325:485–493. [DOI] [PubMed] [Google Scholar]
- de Groot, B. L., and H. Grubmüller. 2001. Water permeation through biological membranes: mechanism and dynamics of Aquaporin-1 and GlpF. Science. 294:2353–2357. [DOI] [PubMed] [Google Scholar]
- de Grotthuss, C. J. T. 1806. Sur la décomposition de l'eau et des corps qu'elle tient en dissolution à l'aide de l'électricité galvanique. Ann. Chim. LVIII:54–74. [Google Scholar]
- Feller, S. E., and A. MacKerell. 2000. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B. 104:7510–7515. [Google Scholar]
- Feller, S. E., Y. H. Zhang, R. W. Pastor, and B. R. Brooks. 1995. Constant pressure molecular dynamics simulation—the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- Fotiadis, D., K. Suda, P. Tittmann, P. Jenö, A. Phillipsen, D. J. Müller, H. Gross, and A. Engel. 2002. Identification and structure of a putative Ca2+ binding domain at the C-terminus of AQP1. J. Mol. Biol. 318:1381–1394. [DOI] [PubMed] [Google Scholar]
- Fu, D., A. Libson, L. J. W. Miercke, C. Weitzman, P. Nollert, J. Krucinski, and R. M. Stroud. 2000. Structure of a glycerol conducting channel and the basis for its selectivity. Science. 290:481–486. [DOI] [PubMed] [Google Scholar]
- Grayson, P., E. Tajkhorshid, and K. Schulten. 2003. Mechanisms of selectivity in channels and enzymes studied with interactive molecular dynamics. Biophys. J. 85:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmüller, H. 1996. SOLVATE 1.0. http://www.mpibpc.gwdg.de/abteilungen/071/solvate/docu.html.
- Haymann, J. B., and A. Engel. 1999. Aquaporins: phylogeny, structure and physiology of water channels. News Physiol. Sci. 14:187–194. [DOI] [PubMed] [Google Scholar]
- Hazama, A., D. Kozono, W. B. Guggino, P. Agre, and M. Yasui. 2002. Ion permeation of AQP6 water channel protein-single-channel recording after Hg2+ activation. J. Biol. Chem. 277:29224–29230. [DOI] [PubMed] [Google Scholar]
- Heller, K. B., E. C. Lin, and T. H. Wilson. 1980. Substrate specificity and transport properties of the glycerol facilitator of Escherichia coli. J. Bacteriol. 144:274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans, J., X. Xia, and D. Cavanaugh. 1998. DOWSER. Department of Biochemistry and Biophysics School of Medicine University of North Carolina at Chapel Hill, NC. http://femto.med.unc.edu/DOWSER/.
- Hummer, G., J. C. Rasaiah, and J. P. Noworyta. 2001. Water conduction through the hydrophobic channel of a carbon nanotube. Nature. 414:188–190. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Insight. 1998. INSIGHT II, Version 98.0—Molecular Modeling System. Molecular Simulations Inc., San Diego, CA.
- Jensen, M. Ø., S. Park, E. Tajkhorshid, and K. Schulten. 2002. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc. Natl. Acad. Sci. USA. 99:6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, M. Ø., E. Tajkhorshid, and K. Schulten. 2001. The mechanism of glycerol conduction in aquaglyceroporins. Structure. 9:1083–1093. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kalé, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comp. Phys. 151:283–312. [Google Scholar]
- Kozono, D., M. Yasui, L. S. King, and P. Agre. 2002. Aquaporin water channels: atomic structure and molecular dynamics meet clinical medicine. J. Clin. Invest. 109:1395–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-hydrogen empirical potential for molecular modeling and dynamics studies of proteins using the CHARMM22 force field. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Murata, K., K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J. B. Heymann, A. Engel, and Y. Fujiyoshi. 2000. Structural determinants of water permeation through aquaporin-1. Nature. 407:599–605. [DOI] [PubMed] [Google Scholar]
- Nollert, P., W. E. C. Harries, D. Fu, L. J. W. Miercke, and R. M. Stroud. 2001. Atomic structure of a glycerol channel and implications for substrate permeation in aqua(glycero)porins. FEBS Lett. 504:112–117. [DOI] [PubMed] [Google Scholar]
- Pohl, P., S. M. Saparov, M. J. Borgnia, and P. Agre. 2001. Highly selective water channel activity measured by voltage clamp: analysis of planar lipid bilayers reconstituted with purified AqpZ. Proc. Natl. Acad. Sci. USA. 98:9624–9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomès, R., and B. Roux. 1996. Structure and dynamics of a proton wire: A theoretical study of H+ translocation along the single-file water chain in the gramicidin A channel. Biophys. J. 71:19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomès, R., and B. Roux. 2002. Molecular mechanism of H+ conduction in the single-file water chain of the gramicidin channel. Biophys. J. 82:2304–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston, G. M., P. Piazza-Carroll, W. B. Guggino, and P. Agre. 1992. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 water channel. Science. 256:385–387. [DOI] [PubMed] [Google Scholar]
- Ren, G., V. S. Reddy, A. Cheng, P. Melnyk, and A. K. Mitra. 2001. Visualization of a water-selective pore by electron crystallography in vitreous ice. Proc. Natl. Acad. Sci. USA. 98:1398–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richey, D. P., and E. C. C. Lin. 1972. Importance of facilitated diffusion for effective utilization of glycerol by Escherichia coli. J. Bacteriol. 112:784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, B. 2002. Computational studies of the gramicidin channel. Acc. Chem. Res. 35:366–375. [DOI] [PubMed] [Google Scholar]
- Roux, B., and R. MacKinnon. 1999. The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science. 285:100–102. [DOI] [PubMed] [Google Scholar]
- Schlenkrich, M., J. Brickmann, A. D. MacKerell, Jr., and M. Karplus. 1996. Empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz, and B. Roux, editors. Birkhauser, Boston, MA. 31–81.
- Schulten, Z., and K. Schulten. 1986. Proton conduction through proteins: an overview of theoretical principles and applications. Meth. Enzymol. 127:419–438. [DOI] [PubMed] [Google Scholar]
- Smart, O. S., J. G. Neduvelil, X. Wang, B. A. Wallace, and M. S. P. Sansom. 1996. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 14:354–360. [DOI] [PubMed] [Google Scholar]
- Smondyrev, A. M., and G. A. Voth. 2002. Molecular dynamics simulation of proton transport through the influenza-A virus M2 channel. Biophys. J. 1987–1996. [DOI] [PMC free article] [PubMed]
- Sui, H., B.-G. Han, J. K. Lee, P. Walian, and B. K. Jap. 2001. Structural basis of water-specific transport through the AQP1 water channel. Nature. 414:872–878. [DOI] [PubMed] [Google Scholar]
- Tajkhorshid, E., P. Nollert, M. Ø. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud, and K. Schulten. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 296:525–530. [DOI] [PubMed] [Google Scholar]
- Yasui, M., T. H. Kwon, M. A. Knepper, S. Nielsen, and P. Agre. 1999. Aquaporin-6: an intracellular vesicle water channel protein in renal epithelia. Proc. Natl. Acad. Sci. USA. 96:5808–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yool, A. J., and A. M. Weinstein. 2002. New roles for old holes: ion channel function in aquaporin-1. News Physiol. Sci. 17:68–72. [DOI] [PubMed] [Google Scholar]
- Zhu, F., and K. Schulten. 2003. Water and proton conduction through carbon nanotubes as models for biological channels. Biophys. J. 85:236–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, F., E. Tajkhorshid, and K. Schulten. 2001. Molecular dynamics study of aquaporin-1 water channel in a lipid bilayer. FEBS Lett. 504:212–218. [DOI] [PubMed] [Google Scholar]
- Zhu, F., E. Tajkhorshid, and K. Schulten. 2002. Pressure-induced water transport in membrane channels studied by molecular dynamics. Biophys. J. 83:154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, F., E. Tajkhorshid, and K. Schulten. 2003. Theory and simulation of water permeation in aquaporin-1. Biophys. J. In press. [DOI] [PMC free article] [PubMed]