

Abstract
The equilibrium unfolding of dimeric yeast glutathione reductase (GR) by guanidine hydrochloride (GdnHCl) was investigated. Unfolding was monitored by a variety of techniques, including intrinsic fluorescence emission, anisotropy and iodide quenching measurements, far-ultraviolet circular dichroism and thiol reactivity measurements. At 1 M GdnHCl, one thiol group of GR became accessible to modification with 5,5′-dithiobis-(2-nitrobenzoic) acid (DTNB), whereas no changes could be detected in the spectroscopic properties (fluorescence, circular dichroism) of the protein. Between 2 and 3 M GdnHCl, two partially folded intermediate states possessing flexible tertiary structures (revealed by fluorescence data) but compact secondary structures (as indicated by circular dichroism measurements) were identified. The quaternary structure of GR in the presence of GdnHCl was also investigated by size-exclusion liquid chromatography. These results indicated the presence of an expanded predissociated dimer at 2.5 M GdnHCl and partially folded monomers at 3 M GdnHCl. Taken together, these results suggest the existence of two molten-globule-like intermediate species (one dimeric and one monomeric) in the unfolding of GR. The results are discussed in terms of the mechanism of GR folding and dimerization.
INTRODUCTION
Understanding the thermodynamic and structural basis of protein folding is one of the key issues in current research in biochemistry. Thermodynamic data on the conformational stability of proteins can be obtained using several chemical or physical perturbants, including chaotropic agents (guanidine hydrochloride, urea), hydrostatic pressure, and temperature. Folding/unfolding can be followed by measurements of biological activity and/or by a variety of spectroscopic methods. Using these procedures, the energetics and pathways of folding, as well as the existence of transient intermediate states between unfolded and fully folded polypeptides, have been characterized for a number of proteins (for a collection of reviews, see Creighton, 1992; Pain, 1994). The discovery and characterization of partially folded states known as molten globules (MG) were important to our understanding of protein folding mechanisms (Dolgikh et al., 1981, 1984, 1985; Kim and Baldwin, 1990). The MG concept was introduced to describe the properties of partially folded intermediate states sharing similar structural characteristics (i.e., nativelike secondary structure and fluctuating tertiary structure) in different proteins.
In the present study, we have investigated the equilibrium unfolding of yeast glutathione reductase (GR) by guanidine hydrochloride (GdnHCl). GR is a 108-kDa, homodimeric flavoenzyme that catalyzes the β-nicotinamide-adenine dinucleotide phosphate (NADPH)-dependent reduction of oxidized glutathione (GSSG) to its reduced form (GSH), thus participating in cellular defense mechanisms against oxidative stress. By using a combination of fluorescence and circular dichroism spectroscopies, thiol group titration, and size-exclusion chromatography we have detected the existence of partially folded intermediate states in the unfolding of GR. Two intermediate states were populated at 2–3 M GdnHCl, as indicated by a combination of spectroscopic and chromatographic methods. These folding intermediates exhibited flexible tertiary structures and nativelike secondary structures. In addition, size-exclusion chromatography analysis indicated that the intermediates populated at 2.5 and 3 M GdnHCl corresponded to expanded predissociated dimers and molten globule monomers, respectively. Fully unfolded monomers were obtained at higher concentrations of GdnHCl. These results indicate a multistate folding pathway for GR and suggest a critical role of molten globule monomers in the correct dimerization of GR.
MATERIALS AND METHODS
Materials
GR (type IV, from Baker's yeast), GSSG, NADPH, DTNB, GdnHCl, and HEPES were from Sigma Chemical (St. Louis, MO). All other reagents were of the highest analytical grade available. The purity of stock GR preparations was routinely verified by SDS-PAGE, followed by Coomassie-Blue staining. This analysis showed a single protein band in the GR preparations used, indicating that the protein was essentially pure. Protein concentration was determined by the Lowry method (Lowry et al., 1951).
Fluorescence measurements
Fluorescence measurements were carried out on an ISS PC1 (ISS Inc., Champaign, IL) photon-counting spectrofluorometer. Samples were excited at 270 nm (16-nm bandpasses for excitation and emission). Yeast GR presents a quite blue-shifted fluorescence emission spectrum (maximal emission wavelength at 317 nm for the native protein; see Fig. 1 A). Thus, to record adequate emission spectra for GR, we routinely started acquisition of the spectra at 295 nm (as shown in Fig. 1 A). This led us to shift the excitation to a lower wavelength (270 nm) to minimize the contribution from Rayleigh scattering in the emission spectra. Experiments were carried out at room temperature in 30 mM HEPES, at pH 7.6, using 0.54 μM GR. All spectra were corrected for background contribution by subtracting appropriate blanks containing only buffer and GdnHCl.
FIGURE 1.

Changes in intrinsic fluorescence emission of GR induced by GdnHCl. (A) Emission spectra of GR (0.54 μM) in the absence (continuous line) or in the presence (dashed line) of 3 M GdnHCl. Spectra are normalized to the maximal fluorescence emission intensities. (B) Spectral centers of mass (calculated as described in Materials and Methods) of the fluorescence emission of GR as a function of GdnHCl concentration. Samples were excited at 270 nm and emission was measured between 295 and 420 nm. Measurements were carried out after incubation for 90 min in the presence of the indicated GdnHCl concentration.
Spectral centers of mass (λav, average emission wavelength) were calculated with software provided by ISS Inc., as
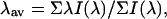 |
(1) |
where I(λ) is the fluorescence intensity at wavelength λ.
Fluorescence anisotropy measurements were performed in the same instrument with polarizers in the excitation and emission ports. The excitation wavelength was 285 nm and emission was measured at 315 nm.
For fluorescence quenching measurements, KI was added to the cuvette from a freshly prepared 3 M stock solution. Data were analyzed according to the Stern-Volmer equation,
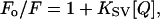 |
(2) |
where KSV is the Stern-Volmer quenching constant, Q is the molar concentration of quencher, and F and Fo are the fluorescence intensities in the presence and in the absence of quencher, respectively.
Circular dichroism
Far-UV CD spectra of GR were measured on a JASCO (Tokyo, Japan) J-715 spectropolarimeter (kindly made available by Dr. J. L. Silva) using 0.1-cm-pathlength quartz cells. Spectra were averaged over three scans for each sample containing 3 μM GR in 30 mM HEPES, at pH 7.6, in the absence or in the presence of GdnHCl, as indicated below in Results.
Titration of reactive thiol groups
This was performed by following the increase in 2-nitro-5-thiobenzoic acid (TNB) absorbance at 412 nm, according to Collier (1973). GR (5.4 μM) was dissolved in 0.1 M Tris-Cl, at pH 8.0, containing 1 mM EDTA and the indicated concentrations of GdnHCl. Reaction was started by addition of 1.4 mM DTNB.
HPLC analysis
GR samples (5.4 μM) were incubated for 90 min in the absence or in the presence of different GdnHCl concentrations and applied onto a Superdex 200 HR column (Pharmacia, Uppsala, Sweden), previously equilibrated with 20 mM Tris-HCl, at pH 7.6, containing 150 mM NaCl, at 23°C. Runs were carried out at 23°C on a Shimadzu (Kyoto, Japan) LC-10AS HPLC at a flow rate of 0.9 ml/min−1, with intrinsic fluorescence detection at 315 nm (excitation at 280 nm). Column calibration was done with a set of eight proteins of known molecular weights (thyroglobulin, 669 kDa; ferritin; 440 kDa; alcohol dehydrogenase, 150 kDa; bovine serum albumin, 66 kDa; ovalbumin, 45 kDa; carbonic anhydrase, 29 kDa; trypsinogen, 24 kDa; and cytochrome C, 12 kDa).
RESULTS
Fig. 1 A shows fluorescence emission spectra of GR in the absence or in the presence of GdnHCl. The fluorescence emission of native GR is quite blue-shifted (λmax at 317 nm), indicating that, on average, the four tryptophan residues in each GR subunit are well-protected from exposure to the aqueous medium in the folded protein. After equilibration for 90 min in the presence of 3 M GdnHCl, major changes in fluorescence emission were observed. A distinct shoulder (λmax ∼ 305 nm) corresponding to tyrosine emission (18 Tyr residues in each GR subunit) appeared in the spectrum. In addition, the peak corresponding to tryptophan emission displayed a significant red-shift to λmax ∼ 334–339 nm. In native proteins containing both tyrosine and tryptophan residues, tyrosine emission is not commonly observed due to efficient energy transfer from tyrosine to neighboring tryptophan residues (Lakowicz, 1983). However, protein unfolding may cause an increase in the average distance between tyrosine and tryptophan residues, thus minimizing energy transfer and allowing detection of tyrosine emission. On the other hand, tryptophan emission is quite sensitive to the polarity of the surrounding environment, and fluorescence red-shifts are indicative of increased exposure of tryptophan residues to the aqueous medium (Lakowicz, 1983). Thus, both the appearance of tyrosine emission and the red-shift of tryptophan fluorescence indicated extensive unfolding of GR at 3 M GdnHCl.
The equilibrium unfolding of GR by GdnHCl was monitored by measuring fluorescence emission spectra at various denaturant concentrations. The fluorescence spectral centers of mass showed a marked (∼13–14 nm) red-shift between 1 M and 3 M GdnHCl (Fig. 1 B). Spectral changes were found to be reversible upon removal of the denaturant by dialysis (data not shown).
To characterize in more detail the structural changes induced by GdnHCl, we have investigated the accessibility of thiol groups of GR (which contains six Cys residues per subunit) to modification with DTNB (Fig. 2). Massey and Williams (1965) found 3.5 sulfhydryl (SH) groups/monomer in urea-denatured yeast GR. Under reducing conditions, the number of SH groups in urea-denatured yeast GR increases to six SH groups/monomer (Massey and Williams, 1965), indicating full exposure of all the Cys residues in the protein. Here, we detected a maximum of ∼0.3 mol of reactive SH group per mol of monomer under native conditions, indicating that one of the thiol groups of GR is partially accessible to DTNB in the folded protein. Incubation with 1 M GdnHCl, a concentration that caused no changes in the fluorescence emission of GR (Fig. 1 B), resulted in the full exposure of one SH group per subunit. Control experiments revealed a similar increase in thiol reactivity in the presence of 1 M KCl (data not shown). This suggests that the exposure of one SH group in the presence of 1 M GdnHCl is due to an ionic strength effect.
FIGURE 2.

Titration of thiol groups of GR by DTNB. The figure shows reactions carried out in the absence (•) or in the presence of 1 M GdnHCl (▪), as described in Materials and Methods.
Unfolding of GR by GdnHCl was further characterized by intrinsic fluorescence anisotropy measurements (Fig. 3). No changes in anisotropy were observed up to 2 M GdnHCl. Between 2 and 5 M GdnHCl, a significant drop in anisotropy was observed, indicating faster rotational mobility of tryptophan residues upon unfolding of GR. Since spectral shift measurements showed a sharp transition between 1 and 3 M GdnHCl (Fig. 1 B), and most of the changes in anisotropy only took place between 2 and 5 M GdnHCl (Fig. 3), we used fluorescence quenching to further investigate the solvent accessibility of tryptophan residues upon unfolding. Fig. 4 shows quenching of GR fluorescence by iodide. Iodide is a negatively charged quencher expected to quench the fluorescence emission of solvent-exposed tryptophan residues (Lakowicz, 1983). Compared to native GR (circles), addition of 3 M GdnHCl (triangles) led to a significant increase in quenching efficiency, as indicated by an increase in KSV from 12.5 to 20 M−1, respectively. A further increase in quenching efficiency was observed at 7 M GdnHCl (squares, KSV = 40 M−1), indicating that even at 3 M GdnHCl the tryptophan residues of GR were still significantly protected from iodide quenching relative to the fully unfolded protein. It is interesting to compare the efficiencies obtained for iodide quenching of the fluorescence of GR and of free Trp in aqueous solution. The values of KSV for Trp residues in proteins and for free Trp in aqueous solution are not directly comparable. Since KSV equals the product of the bimolecular quenching rate constant (kq) and the excited-state lifetime of the fluorophore (τ), the value of KSV obtained from steady-state fluorescence measurements is dependent on the average lifetime. For indole in aqueous solution, kq for iodide quenching is 6.4 × 109 M−1 s−1 (Eftink and Ghiron, 1981). Assuming an average lifetime of 3 ns for Trp in aqueous solution (Ferreira, 1989), the resulting value of KSV for iodide quenching of Trp is 19.2 M−1. For Trp residues in proteins, it is not uncommon to find average lifetimes that are significantly longer than for free Trp. Thus, the value for KSV will depend on the particular average intrinsic fluorescence lifetime of the protein, and may be higher than the value for free Trp. Therefore, the KSV of 40 M−1 obtained for iodide quenching of GR in the presence of 7 M GdnHCl suggests that the Trp residues of the protein are significantly, if not completely, exposed to the solvent.
FIGURE 3.

Intrinsic fluorescence anisotropy measurements of GR. Samples (0.54 μM GR) were incubated for 90 min at room temperature with the indicated concentrations of GdnHCl and fluorescence anisotropy was measured as described in Materials and Methods. Excitation was at 285 nm and emission was measured at 315 nm. Symbols correspond to means ± standard deviations of at least six independent measurements at each GdnHCl concentration.
FIGURE 4.

Iodide quenching of GR fluorescence. Quenching was measured in the absence (•) or in the presence of 3 M (▴) or 7 M (▪) GdnHCl. Samples (0.54 μM GR) were incubated for 90 min at room temperature with the indicated concentrations of GdnHCl before measurements.
Changes in secondary structure of GR induced by GdnHCl were examined by CD measurements (Fig. 5). Addition of up to 2.5 M GdnHCl caused only a minor decrease in ellipticity at 222 nm (Fig. 5, inset). At 3 M GdnHCl, however, a significant drop in ellipticity was observed. Addition of 7 M GdnHCl completely abolished the CD signal at 222 nm, indicating extensive or complete unfolding of GR.
FIGURE 5.

Circular dichroism spectra of GR. Samples (3 μM GR) were incubated for 90 min in the absence or in the presence of the indicated GdnHCl concentrations. Spectra are labeled according to the molar concentrations of GdnHCl used. (Inset) Plot of the ellipticity at 222 nm as a function of GdnHCl concentration.
Size exclusion chromatography was used to investigate the quaternary structure of GR (Fig. 6). Native dimeric GR eluted at a volume corresponding to an apparent molecular weight of ∼85 kDa in the calibrated column, indicating a fairly compact dimer structure. No changes in the elution volume of GR were observed when the sample contained 1 M GdnHCl. However, when the concentration of GdnHCl in the sample was raised to 2.5 M, the GR dimer eluted at a volume corresponding to ∼120 kDa. Interestingly, when GR was incubated in the presence of 3 M GdnHCl the area of the 120 kDa peak decreased markedly, and a second peak corresponding to a molecular weight of ∼60 kDa appeared. This indicates that dissociation of the GR dimer takes place in a narrow range of GdnHCl concentrations between 2.5 and 3 M, and that a predissociated state with an expanded hydrodynamic radius compared to the native dimer is stabilized at 2.5 M GdnHCl. An additional experiment in which the incubation time of the protein in the presence of 3 M GdnHCl was increased from 90 min to 24 h showed that the small dimer fraction observed in Fig. 6 (lower chromatogram) persists even after overnight incubation (data not shown), indicating that a small proportion of dimers is still in equilibrium with monomers at this concentration of denaturant.
FIGURE 6.

Size-exclusion HPLC analysis of GR. From top to bottom, GR samples were incubated for 90 min in the absence or in the presence of 1, 2.5, and 3 M GdnHCl before injection into the column. The HPLC elution buffer did not contain GdnHCl (see Materials and Methods). Apparent molecular weights of each peak obtained from the calibrated column are indicated.
DISCUSSION
Molten globule (MG) folding intermediates have been characterized for a number of proteins (for reviews, see Kuwajima, 1989; Ptitsyn, 1992, 1995). Classical properties of the MG state include increased molecular volume caused by an increase in internal water content (Kuwajima, 1989), preservation of nativelike secondary structure (Nozaka et al., 1978; Bolotina, 1987) and fluctuating tertiary structure. Several functions for MG intermediates have been proposed, including binding to chaperones for correct folding (Bochkareva et al., 1988; Rothman, 1989; Rawat and Rao, 1998; Trevino et al., 1999), easier translocation of the molten globule through membranes compared to fully folded globular proteins (Bychkova et al., 1988) and facilitation of protein lysosomal degradation (Ptitsyn, 1992).
In this study, we characterize in detail some of the physicochemical properties of partially denatured states of yeast glutathione reductase. The data support the presence of at least two intermediate states in the equilibrium unfolding of GR by GdnHCl. According to the definitions mentioned above, such folding intermediates of GR can possibly be described as MG states.
The fluorescence emission of native GR is quite blue shifted, suggesting a compact native structure (Fig. 1), and undergoes a progressive red-shift with increasing concentrations of GdnHCl. Interestingly, no significant spectral changes were detected up to 1 M GdnHCl, whereas an increase in reactivity of one thiol group of GR to DTNB was observed at this concentration of GdnHCl (Fig. 2). This indicates that subtle conformational changes are induced by low concentrations of GdnHCl (≤1M), resulting in the full exposure of a previously buried thiol group to the aqueous medium. This effect is likely due to the ionic strength of GdnHCl solutions, as addition of 1 M KCl also caused the exposure of 1 SH group of GR (data not shown). A previous investigation of the unfolding of Spirulina maxima glutathione reductase by GdnHCl reported similar thiol reactivity (Rendon et al., 1995). At 1 M GdnHCl, a partially non-accessible free sulfhydryl group of S. maxima GR became fully exposed. Early studies showed that the reactivity of sulfhydryl groups of yeast GR was significantly higher (∼3.5 SH reactive groups per subunit) in concentrated urea solutions (Massey and Williams Jr., 1965). This indicates that the conformation of GR obtained at 1 M GdnHCl retained most of the overall folded structure of the protein, a conclusion that is supported by the lack of changes in fluorescence and CD at this concentration of GdnHCl.
The existence of partially folded intermediate states in the unfolding of GR is indicated by a comparison of fluorescence spectral shift, anisotropy, iodide quenching, and CD data as a function of GdnHCl concentration. The fluorescence emission of GR undergoes a marked red-shift between 1 and 3 M GdnHCl (with a transition midpoint at ∼2.5 M GdnHCl), reflecting exposure of tryptophan residues to the aqueous medium (Fig. 1 B). On the other hand, fluorescence anisotropy and circular dichroism are practically unaffected up to 2–2.5 M GdnHCl (Figs. 3 and 5). Furthermore, fluorescence spectral shift data suggested that the unfolding of GR was complete at 3 M GdnHCl (Fig. 1 B), whereas iodide quenching and CD measurements revealed significant preservation of GR structure at 3 M GdnHCl. CD measurements suggested that unfolding of GR was complete at 7 M GdnHCl, a result that was consistent with the iodide quenching data. Moreover, the existence of MG folded states in the unfolding of GR was indicated by HPLC studies (Fig. 6), which revealed the presence of dimers and monomers with expanded hydrodynamic radii at intermediate concentrations of GdnHCl. Investigation of the properties of these partially folded states using probes such as ANS or bis-ANS (which bind to exposed hydrophobic domains in proteins) was not possible due to the significant overlap between the emission spectra of these probes when bound to proteins and the excitation spectrum of the prosthetic group, FAD (data not shown). In line with these results, the presence of MG intermediate states in the unfolding of S. maxima GR by GdnHCl has been proposed (Rendon et al., 1995). Molten globule monomers have been proposed as intermediates in the dissociation of some dimeric proteins, including human superoxide dismutase (Silva et al., 1993) and rabbit muscle creatine kinase (Couthon et al., 1995). In addition, the existence of a partially denatured predissociated state has been proposed in the unfolding of the dimeric DNA-binding protein Arc repressor (Peng et al., 1994). Taken together, these results suggest that at least two intermediate states can be identified in the equilibrium unfolding of GR, namely a partially unfolded predissociated state and a molten globule monomer. Both of these states are characterized by expanded hydrodynamic radii, loosening of tertiary structure, and preservation of secondary structure. It is worth noting that a multi-event equilibrium unfolding/dissociation of human GR by GdnHCl has been previously reported (Nordhoff et al., 1997), suggesting a high degree of intersubunit cooperativity in the reactivation of the enzyme.
Possible functions of nativelike states of proteins have been proposed, but not clearly demonstrated. For example, Rawat and Rao (1998) showed that α-crystallin, a chaperone, interacts with the MG state of dimeric xylose reductase from Neurospora crassa, suggesting a possible role of molten globules in molecular recognition in the process of protein folding. For both Escherichia coli and human GR, it has been demonstrated that structural elements from both subunits in the dimer are important for catalysis, leading to the conclusion that the monomers are inactive (Schulz et al., 1978; Arscott et al., 1989). The stability of GR dimers is also largely generated by the interface domain, since double or even single mutations in this region are sufficient to lead to the assembly of less stable dimers in the E. coli protein (Scrutton et al., 1992; Bashir et al., 1995), and a single mutation is sufficient to prevent dimerization of human GR (Nordhoff et al, 1993). There is considerable sequence homology among yeast, human, and E. coli GR: comparison of human and yeast GR yields ∼68% of similarity and ∼52% of sequence identity between the two proteins. Essentially the same levels of sequence similarity and identity are obtained in the comparison between E. coli GR and yeast GR. Thus, it seems reasonable to expect that the monomers of yeast GR may also be catalytically inactive and that dimerization may be critical for enzyme activity. On the basis of our results, we propose that assembly of the native GR dimer proceeds via initial formation of MG monomers that dimerize to form a partially folded, expanded dimer. Structural rearrangements then take place at the dimer interface (and presumably also in other domains of each subunit), leading to a final, compact dimer structure. In the final structure, the interface domain is hidden in a hydrophobic, packed core surrounded by the other domains, as demonstrated by crystallographic studies of human GR (Schulz et al., 1978; Karplus and Schulz, 1987). According to this view, the tight and specific association of subunits in the native GR dimer may be facilitated by contacts between MG monomers. Since the MG conformation is more flexible than a fully folded polypeptide, this could lead to better packing at the dimer interface, leading to more stable dimers. Indeed, the significant stability of GR dimers is illustrated by the fact that no changes in fluorescence emission of this protein take place even in the presence of 2 M urea and at a hydrostatic pressure of 3.5 kbar (data not shown), a pressure at which a large number of protein dimers and higher oligomers have been found to undergo subunit dissociation (Gross and Jaenicke, 1994; Heremans and Smeller, 1998).
It is interesting to consider what the possible impact of FAD and NADPH would be on the folding of GR. Nordhoff et al. (1997) have shown that the rate of reactivation of GR that had been previously unfolded by GdnHCl is not altered by addition of FAD. This is similar to previous observations with another FAD-dependent enzyme, pyruvate oxidase (Risse et al., 1992). These observations suggest that FAD binding is unlikely to play a critical role in the process of GR folding/assembly. The inactivation of E. coli GR by NADPH has been previously described (Arscott et al., 1989), and the enzyme concentration-dependence observed in the process suggests that the nucleotide affects the dimer (active form)/monomer (inactive form) equilibrium of GR. However, those authors propose that this phenomenon is unlikely to have physiological relevance, since the inactivation of GR is very slow at cellular NADPH concentrations. Besides, complex formation between GR and NADPH was only detected in the dimeric form of the enzyme, which would argue that NADPH does not participate directly in the process of GR folding/assembly.
Finally, given the role of GR in the maintenance of cellular redox state, preservation of a stable enzymatic capacity is likely to be very important for cell survival (under stress situations, in particular). As previously pointed out (Rietveld and Ferreira, 1996; 1998; Ferreira and De Felice, 2001), dimerization may lead to a significant increase in stability against chemical modification of GR, including thiol oxidation, de-amidation, or proteolytic digestion.
Acknowledgments
This work was supported by grants from the Howard Hughes Medical Institute, The John Simon Guggenheim Memorial Foundation, Conselho Nacional de Desenvolvimento Científico e Tecnológico, and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro.
Abbreviations used: CD, circular dichroism; DTNB, 5,5′-dithiobis-(2-nitrobenzoic) acid; EDTA, ethylenediaminetetracetic acid; GdnHCl, guanidine hydrochloride; GR, yeast glutathione reductase; GSH, reduced glutathione; GSSG, oxidized glutathione; HEPES, n-(2-hydroxyethyl) piperazine-n′-(2-ethanesulfonic acid); HPLC, high performance liquid chromatography; MG, molten globule.
References
- Arscott, L. D., D. M. Drake, and C. H. Williams, Jr. 1989. Inactivation-reactivation of two-electron reduced Escherichia coli glutathione reductase involving a dimer-monomer equilibrium. Biochemistry. 28:3591–3598. [DOI] [PubMed] [Google Scholar]
- Bashir, A., R. N. Perham, N. S. Scrutton, and A. Berry. 1995. Altering kinetic mechanism and enzyme stability by mutagenesis of the dimer interface of glutathione reductase. Biochem. J. 312:527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochkareva, E. S., N. M. Lissin, and A. S. Girsovich. 1988. Transient association of newly synthesized unfolded proteins with the heat-shock GroEL protein. Nature. 336:254–257. [DOI] [PubMed] [Google Scholar]
- Bolotina, I. A. 1987. Secondary structure of proteins from circular dichroism spectra. V. Secondary structure of proteins in a “molten globule” state. Mol. Biol. (Mosk). 21:1625–1635. [PubMed] [Google Scholar]
- Bychkova, V. E., R. H. Pain, and O. B. Ptitsyn. 1988. The “molten globule” state is involved in the translocation of proteins across membranes? FEBS Lett. 238:231–234. [DOI] [PubMed] [Google Scholar]
- Collier, H. B. 1973. A note on the molar absorptivity of reduced Ellman's reagent, 3-carboxylato-4-nitrothiophenolate. Anal. Biochem. 56:310–311. [DOI] [PubMed] [Google Scholar]
- Couthon, F., E. Clottes, C. Ebel, and C. Vial. 1995. Reversible dissociation and unfolding of dimeric creatine kinase isoenzyme MM in guanidine hydrochloride and urea. Eur. J. Biochem. 234:160–170. [DOI] [PubMed] [Google Scholar]
- Creighton, T. E. 1992. Protein Folding. W. H. Freeman and Co., New York.
- Dolgikh, D. A., A. P. Kolomiets, I. A. Bolotina, and O. B. Ptitsyn. 1984. “Molten-globule” state accumulates in carbonic anhydrase folding. FEBS Lett. 165:88–92. [DOI] [PubMed] [Google Scholar]
- Dolgikh, D. A., L. V. Abaturov, I. A. Bolotina, E. V. Brazhnikov, V. E. Bychkova, V. N. Bushuev, R. I. Gilmanshin, Y. O. Lebedev, G. V. Semisotnov, E. I. Tiktopulo, and O. B. Ptitsyn. 1985. Compact state of a protein molecule with pronounced small-scale mobility: bovine alpha-lactalbumin. Eur. Biophys. J. 13:109–121. [DOI] [PubMed] [Google Scholar]
- Dolgikh, D. A., R. I. Gilmanshin, E. V. Brazhnikov, V. E. Bychkova, G. V. Semisotnov, S. Y. Venyaminov, and O. B. Ptitsyn. 1981. Alpha-lactalbumin: compact state with fluctuating tertiary structure? FEBS Lett. 136:311–315. [DOI] [PubMed] [Google Scholar]
- Eftink, M. R., and C. A. Ghiron. 1981. Fluorescence quenching studies with proteins. Anal. Biochem. 114:199–227. [DOI] [PubMed] [Google Scholar]
- Ferreira, S. T. 1989. Fluorescence studies of the conformational dynamics of parvalbumin in solution: lifetime and rotational motions of the single tryptophan residue. Biochemistry. 28:10066–10072. [DOI] [PubMed] [Google Scholar]
- Ferreira, S. T., and F. G. De Felice. 2001. Protein dynamics, folding and misfolding: from basic physical chemistry to human conformational diseases. FEBS Lett. 498:129–134. [DOI] [PubMed] [Google Scholar]
- Gross, M., and R. Jaenicke. 1994. Proteins under pressure. The influence of high hydrostatic pressure on structure, function and assembly of proteins and protein complexes. Eur. J. Biochem. 221:617–630. [DOI] [PubMed] [Google Scholar]
- Heremans, K., and L. Smeller. 1998. Protein structure and dynamics at high pressure. Biochim. Biophys. Acta. 1386:353–370. [DOI] [PubMed] [Google Scholar]
- Karplus, P. A., and G. E. Schulz. 1987. Refined structure of glutathione reductase at 1.54 Å resolution. J. Mol. Biol. 195:701–729. [DOI] [PubMed] [Google Scholar]
- Kim, P. S., and R. L. Baldwin. 1990. Intermediates in the folding reactions of small proteins. Annu. Rev. Biochem. 59:631–660. [DOI] [PubMed] [Google Scholar]
- Kuwajima, K. 1989. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins. 6:87–103. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1983. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- Lowry, D. H., N. J. Rosebrough, A. L. Farr, and R. J. Randall. 1951. Protein measurement with the folin phenol reagent. J. Biol. Chem. 193:265–275. [PubMed] [Google Scholar]
- Massey, V., and C. H. Williams, Jr. 1965. On the reaction mechanism of yeast glutathione reductase. J. Biol. Chem. 240:4470–4480. [PubMed] [Google Scholar]
- Nordhoff, A., C. Tziatzios, J. A. Van der Broeck, M. K. Schott, H. Kalbitzer, K. Becker, D. Schubert, and R. H. Schirmer. 1997. Denaturation and reactivation of dimeric human glutathione reductase. Eur. J. Biochem. 245:273–282. [DOI] [PubMed] [Google Scholar]
- Nordhoff, A., U. S. Bücheler, D. Werner, and R. H. Schirmer. 1993. Folding of the four domains and dimerization are impaired by the Gly446-Glu exchange in human glutathione reductase. Implications for the design of antiparasitic drugs. Biochemistry. 32:4060–4066. [DOI] [PubMed] [Google Scholar]
- Nozaka, M., K. Kuwajima, K. Nitta, and S. Sugai. 1978. Detection and characterization of the intermediate on the folding pathway of human alpha-lactalbumin. Biochemistry. 17:3753–3758. [DOI] [PubMed] [Google Scholar]
- Pain, R. 1994. Mechanisms of Protein Folding. IRL Press, Oxford, England.
- Peng, X., J. Jonas, and J. L. Silva. 1994. High-pressure NMR study of the dissociation of Arc repressor. Biochemistry. 33:8323–8329. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O. B. 1992. The molten globule state. In Protein Folding. T. E. Creighton, editor. W. H. Freeman and Co., New York.
- Ptitsyn, O. B. 1995. How the molten globule became. Trends Biochem. Sci. 20:376–379. [DOI] [PubMed] [Google Scholar]
- Rawat, U., and M. Rao. 1998. Interactions of chaperone alpha-crystallin with the molten globule state of xylose reductase. Implications for reconstitution of the active enzyme. J. Biol. Chem. 273:9415–9423. [DOI] [PubMed] [Google Scholar]
- Rendon, J. L., J. P. Pardo, G. Mendoza-Hernandez, A. Rojo-Dominguez, and A. Hernandez-Arana. 1995. Denaturing behavior of glutathione reductase from cyanobacterium Spirulina maxima in guanidine hydrochloride. Arch. Biochim. Biophys. 318:264–270. [DOI] [PubMed] [Google Scholar]
- Rietveld, A. W. M., and S. T. Ferreira. 1996. Deterministic pressure dissociation and unfolding of triose phosphate isomerase: persistent heterogeneity of a protein dimer. Biochemistry. 35:7743–7751. [DOI] [PubMed] [Google Scholar]
- Rietveld, A. W. M., and S. T. Ferreira. 1998. Kinetics and energetics of subunit dissociation/unfolding of TIM: the importance of oligomerization for conformational persistence and chemical stability of proteins. Biochemistry. 37:933–937. [DOI] [PubMed] [Google Scholar]
- Risse, B., G. Stempfer, R. Rudolph, H. Möllering, and R. Jaenicke. 1992. Stability and reconstitution of pyruvate oxidase from Lactobacillus plantarum: dissection of the stabilizing effects of coenzyme binding and subunit interaction. Protein Sci. 1:1699–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman, J. E. 1989. Polypeptide chain binding proteins: catalysts of protein folding and related processes in cells. Cell. 59:591–601. [DOI] [PubMed] [Google Scholar]
- Schulz, G. E., R. H. Schirmer, W. Sachsenheimer, and E. F. Pai. 1978. The structure of the flavoenzyme glutathione reductase. Nature. 273:120–124. [DOI] [PubMed] [Google Scholar]
- Scrutton, N. S., M. P. Deonarain, A. Berry, and R. N. Perham. 1992. Cooperativity induced by a single mutation at the subunit interface of a dimeric enzyme: glutathione reductase. Science. 258:1140–1143. [DOI] [PubMed] [Google Scholar]
- Silva, N., Jr., E. Gratton, G. Mei, N. Rosato, R. Rusch, and A. Finazzi-Agro. 1993. Molten globule monomers in human superoxide dismutase. Biophys. Chem. 48:171–182. [DOI] [PubMed] [Google Scholar]
- Trevino, R. J., F. Gliubich, R. Berni, M. Cianci, J. M. Chirgwin, G. Zanotti, and P. M. Horowitz. 1999. NH2-terminal sequence truncation decreases the stability of bovine rhodanese, minimally perturbs its crystal structure, and enhances interaction with GroEL under native conditions. J. Biol. Chem. 274:13938–13947. [DOI] [PubMed] [Google Scholar]