

Abstract
The kinetics of formation and transformation of oxygen complexes of two heme-thiolate proteins (the F393H mutant of cytochrome P450 BM3 and the oxygenase domain of endothelial nitric oxide synthase, eNOS) were studied under high pressure. For BM3, oxygen-binding characteristics (rate and activation volume) matched those measured for CO-binding. In contrast, pressure revealed a different CO- and oxygen-binding mechanism for eNOS, suggesting that it is hazardous to take CO-binding as a model for oxygen-binding. With eNOS, a ferric NO complex is formed as an intermediate in the second reaction cycle. Here we report the pressure stability of this compound. Furthermore, in the presence of 4-amino-tetrahydrobiopterin (ABH4), an analog to the natural second electron donor tetrahydrobiopterin (BH4), biphasic pressure profiles of the oxygen-binding rates were observed, both in the first and the second reaction cycles, indicative of the formation of an additional reaction intermediate. This was confirmed by experiments where ABH4 was replaced by ABH2, a cofactor which cannot deliver an electron. Altogether, high pressure appears to be a useful tool to characterize elementary steps in the reaction cycle of heme-thiolate proteins.
INTRODUCTION
Cytochromes P450 and nitric oxide synthases are among the most intensively studied of all enzymes. The many hundred known forms of cytochrome P450 are notably involved in detoxification of xenobiotics and steroid biosynthesis, but also in adverse reactions such as activation of procarcinogens (Lewis, 2001; Lange, 2003). The three forms of nitric oxide synthase (neuronal, endothelial, and inducible) participate in diverse physiological processes, such as neuromodulation, vasodilatation, and immune response (Marletta et al., 1998; Pfeiffer et al., 1999; Stuehr, 1999). Depending on the circumstances, their action can be beneficial for the organism or lead to severe pathologies (Grisham et al., 1999; Davis et al., 2001). Of course, any attempt to modulate the activity of these enzymes requires an understanding of the catalytic steps at the molecular level. In both enzymes catalysis takes place at a thiolate-ligated heme. This structural property is reflected by a similar reaction mechanism with oxygen, in which a ferrous oxygen complex must be “activated” by the input of a second electron, leading finally to the incorporation of one oxygen atom into an organic substrate and to the formation of a water molecule. Several intermediate states, including higher valence iron oxygen complexes, are hypothesized (Ortiz de Montellano, 1995).
The interaction with oxygen is a key feature of the reaction mechanism of these enzymes. Oxygen-binding has been studied for a number of cytochrome P450 forms and nitric oxide synthase, both spectroscopically (at subzero temperatures), and by stopped-flow. However, little is known about the mechanism of this reaction in terms of elementary steps. For a better understanding of the steps involved, we undertook their kinetic analysis under high pressure. The effect of pressure is of interest since it can be used as a complementary thermodynamic parameter to temperature: it is a relatively mild structural perturbant specifically affecting electrostatic and hydrophobic interactions (Mozhaev et al., 1996). These perturbation properties have been used with success to explore structural and dynamic properties of the heme pocket (Bancel et al., 2002). Furthermore, the high pressure stopped-flow method (Balny et al., 1984) allowed us to relate kinetic aspects of hemoproteins to the nature of their proximal axial ligands (Lange et al., 1994). In this study we compare the pressure dependence of oxygen- and CO-binding rates for heme-thiolate enzymes. From the pressure dependence of the rate constants the activation volume, ΔV‡, is then determined. The latter parameter informs us about structural features of the transition state of the reaction, where its sign and absolute value reflect type and extent of protein conformational changes and protein hydration changes within an elementary reaction step.
The high pressure approach is applied here to the study of the F393H mutant of cytochrome P450 BM3 (BM3) and the oxygenase domain of endothelial nitric oxidase (eNOS). These flavocytochrome enzymes operate similar electron transport chains, with electrons delivered from NADPH by a diflavin reductase (a P450 reductase-like FAD- and FMN-containing enzyme) fused to the heme-containing oxygenase domain. The strong reactivity of wild-type P450 BM3 with oxygen means that the complex is transient, collapsing rapidly to generate ferric heme and superoxide. However, the F393H mutant has heme thermodynamic features altered from the wild-type; specifically, a more positive heme iron reduction potential and a stabilized ferrous-oxy form (Ost et al., 2001). The properties of the F393H mutant enable the kinetics of the binding reaction between P450 and oxygen to be analyzed and compared with the reaction with carbon monoxide (which forms a stable ferrous complex with both wild-type and F393H BM3).
The oxygenase domain of endothelial NOS is the part of nitric oxide synthase that resembles most closely a cytochrome P450. Our previous studies on the eNOS oxygenase domain have shown it to be active (in the formation of product). For an input of electrons, it does not specifically require the presence of the reductase domain. The kinetics of the formation of the NOS oxycomplex have been reported by Stuehr's group (Abu-Soud et al., 1997, 2000; Boggs et al., 2000). Our previous spectroscopic work at subzero temperatures has shown the possibility of stabilizing the oxygen complex of these enzymes. One important difference between P450 and NOS is that NOS requires the presence of tetrahydrobiopterin (BH4) as cofactor. We have recently shown that BH4 is a redox partner of NOS in the first and the second reaction cycles (Bec et al., 1998; Gorren et al., 2000). An analogous compound, 4-amino-tetrahydrobiopterin (ABH4), is an inhibitor of the NOS reaction. However, as we observed recently, its presence significantly affects the absorbance spectrum of NOS oxygen intermediate complexes (Gorren et al., 2000). In our present experiments, we have compared the effects of BH4, ABH4, and ABH2 (the oxidized form of ABH4) on the reaction of eNOS with oxygen. The high pressure stopped-flow kinetic approach is shown to help in the mechanistic understanding of these physiologically important reactions.
MATERIALS AND METHODS
Enzymes
The F393H heme domain of P450 BM3 was prepared from TG1 transformants of the mutant clone, as described previously (Ost et al., 2001). Escherichia coli cell growth and the extraction and purification of protein were largely as described in previous publications (Ost et al., 2001; Noble et al., 1999). Transformant cells (typically 5 liters of cell culture) were induced (with 1 mM IPTG) at an OD600 = 1, and growth was continued for 6–12 h before harvesting cells. Cells were disrupted by two passes through a French press (950 psi) followed by sonication (10 × 15 s bursts on a Sonopuls sonicator (Bandelin, Berlin, Germany) set at 90% power, with cooling on ice between bursts). Enzyme was purified using ion exchange chromatography on DEAE-Sephacel and hydroxyapatite, as described previously (Noble et al., 1999). Phenylmethylsulfonyl fluoride (1 mM) was added to all buffers to minimize proteolysis. Enzyme was concentrated to ∼1 mM by ultrafiltration and dialyzed into 50 mM Tris·HCl at pH 7.0, containing 50% (v/v) glycerol before storage at −80°C. Enzyme was used within 1 month of purification. Enzyme concentration and integrity was determined by preparation of the ferrous-CO complex, as described previously. Complete conversion to the P450 form (with absorption maximum at 445 nm for the F393H mutant) was observed, with negligible absorption in the region of 420 nm, indicating homogeneous native enzyme. The heme domain of flavocytochrome P450 BM3 was predominantly monomeric in solution.
The eNOS oxygenase domain was cloned and purified from E. coli cells as described previously (Gorren et al., 2000). Its concentration was determined from the absorption coefficient of the ferrous-CO complex according to Sono et al. (1995).
Reagents
BH4 and NOHLA were from Alexis Biochemicals (Lausen, Switzerland); 4-aminotetrahydrobiopterin [(6R)-2,4-diamino-5,6,7,8-tetrahydro-6-(L-erythro-1′,2′-dihydroxypropyl)pterin, ABH4] and 4-amino-7,8-dihydro-L-biopterin (ABH2) were purchased from Schircks Laboratories (Jona, Switzerland); L-arginine, CHAPS (3-[(cholamidopropyl)-dimethylammonio]-1-propanesulfonate), and sodium dithionite were from Sigma-Aldrich (Saint Quentin Fallavier, France). Oxygen and CO (99.995% pure) were from Aga (Toulouse, France). All other materials were reagent-grade or better, and were used without further purification.
Experimental procedure
Single turnover experiments under high pressure were performed using a high-pressure stopped-flow apparatus built in our laboratory (Balny et al., 1984). Kinetics were determined by mixing equal volumes of the enzyme and ligand solutions in a thermostated high-pressure stopped-flow cell placed in an Aminco DW2 spectrophotometer (SLM Instruments, Urbana, IL) operating in dual wavelength mode. The buffers were: 50 mM KPi pH 7.5, 1 mM CHAPS, 0.5 mM EDTA, 1 mM 2-mercaptoethanol (NOS), and 50 mM MOPS at pH 7.4 (BM3). Enzyme and gas-saturated solutions were prepared as already described (Gorren et al., 2000 ; Lange et al., 2001). Briefly, enzyme solutions were deoxygenated under argon atmosphere and then reduced by sodium dithionite (1 mM final concentration). During this time, O2- and CO-saturated solutions were prepared by bubbling the buffers with O2 or CO for 45 min. The concentration of these stock solutions was estimated to be 1 mM CO or O2 at 25°C.
The experiments were set up with one syringe filled with reduced enzyme in the presence of substrate and cofactor and the other one containing the O2- or CO-saturated solution. Kinetics were recorded in the dual wavelength mode (absorbance difference between two simultaneously recorded wavelengths). For the formation of the ferrous oxygen complex we used the couples 431/412, 423/412, and 435/405 nm for NOS in the presence of L-Arg or NHA, and for BM3, respectively. The kinetics of the formation of the FeIII-NO complex were recorded at 437/413 nm. For the CO-binding kinetics, we used the wavelength couples 447/413 and 449/409 nm for eNOS and BM3, respectively.
Kinetic data analysis and determination of ΔV‡
First-order rate constants were obtained by nonlinear least-square analysis using a fitting program developed in the laboratory (Balny et al., 1984). In our conditions, the kinetic traces were well-fitted by a mono-exponential equation. An average from four kinetic traces was recorded at each pressure. From the pressure-dependent rate constant, kobs, the activation volume ΔV‡ was calculated according to Eq. 1,
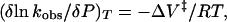 |
(1) |
where P is the hydrostatic pressure, T the absolute temperature, and R the gas constant (8.2 ml MPa mol−1 K−1).
RESULTS
Oxygen- and CO-binding to BM3
Binding of oxygen as well as of CO to dithionite-reduced BM3 produced monoexponential kinetics. The thermodynamic effects on the heme iron in the F393H mutant resulted in stabilization of the ferrous-oxy complex without any significant changes in the structure of the protein. Indeed, the relevant mutation is located on the proximal side of the heme and does not affect the oxygen-binding site, or access to the distal face of the heme. The logarithm of the observed binding-rate constant, kobs, decreased linearly as a function of pressure, with very comparable activation volumes of ΔV‡ = 9–10 ml/mol (See Fig. 1 and Table 1). Previous kinetic work as a function of CO concentration (Lange et al., 1994) indicates that our conditions reflect pseudo-first-order kinetics. High pressure increased to some extent the stability of the oxygen complex: the decomposition rate constant decreased exponentially as a function of pressure with an activation volume of 3.1 ml/mol.
FIGURE 1.

Pressure effect on oxygen and CO complex formation of BM3. Reduced BM3 (2 μM) was mixed in the presence of 60 μM arachidonate with CO or oxygen (0.5 mM) in 50 mM MOPS buffer at pH 7.4. (Upper graph) CO-binding rate (○) and O2-binding rate (•). (Lower graph) Decomposition of the oxygen complex. Data points were the average of three independent experiments. SE on each average was <5%.
TABLE 1.
Thermodynamic activation parameters of O2- and CO-binding to P450 BM3 and NO synthase
| O2-binding
|
CO-binding
|
||||
|---|---|---|---|---|---|
| Substrate |
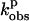 = 0.1 MPa (s−1) = 0.1 MPa (s−1) |
ΔV‡ (ml/mol−1) |
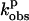 = 0.1 MPa (s−1) = 0.1 MPa (s−1) |
ΔV‡ (ml/mol−1) | |
| P450 BM3 | Arachidonate | 10.0 ± 0.1 | +8.9 ± 0.1 | 10.6 ± 0.2 | +10.8 ± 0.2 |
| NO synthase | |||||
| +amino BH4 | L-Arg | 6.8 ± 0.1 | −5.1 ± 0.1 (0.1–55 MPa) | 1.1 ± 0.1 | |
| +6.5 ± 0.1 (55–90 MPa) | |||||
| + amino BH2 | L-Arg | 9.1 ± 0.1 | +12.3 ± 0.1 | 0.9 ± 0.1 | −16.7 ± 0.2 |
| +amino BH4 | NHA | 2.2 ± 0.1 | −42.1 ± 0.2 (0.1–48 MPa) | 1.0 ± 0.1 | |
| +9.0 ± 0.1 (48–90 MPa) | |||||
| + amino BH2 | NHA | 6.7 ± 0.1 | −6.9 ± 0.1 | 0.9 ± 0.1 | |
Oxygen-binding to eNOS in the presence of ABH2
To compare oxygen-binding of BM3 and eNOS, care has to be taken that the oxygen complex which is formed with eNOS does not evolve further to an activated complex via reduction by a second electron. BH4 can donate this second electron. However, ABH2 cannot deliver an electron. Therefore, we measured oxygen-binding to eNOS in the presence of ABH2. As shown in Figs. 2 and 3, the oxygen-binding rate varies linearly as a function of pressure. It decreases with pressure in the first reaction cycle (in the presence of L-Arg), and it increases in the second reaction cycle (in the presence of NOHLA). The rate of the oxygen complex decay is nearly pressure-independent in the first reaction cycle, and it decreases strongly as a function of pressure in the second reaction cycle. The thermodynamic parameters are listed in Table 1.
FIGURE 2.

Pressure effect on formation and decomposition of the oxygen complex of eNOS in the first reaction cycle. (Upper graph) Formation of the oxygen complex. (Lower graph) Decomposition of the oxygen complex in the presence of ABH4 (•) and in the presence of ABH2 (Δ). Exponential bursts of oxycomplex production and breakdown were followed by rapidly mixing a solution containing 7 μM ferrous enzyme, 500 μM arginine, and 50 μM ABH4 or ABH2 with an oxygen-saturated buffer solution at 4°C. Three independent experiments were performed to determine the rate constant at each pressure. SE on each experimental point was <2%.
FIGURE 3.

Pressure effect on formation and decomposition of the oxygen complex of eNOS in the second reaction cycle. (Upper graph) Formation of oxygen complex. (Lower graph) Decomposition of oxygen complex in the presence of ABH4 (•) and in the presence of ABH2 (Δ). The experimental conditions were those of Fig. 2 except with NOHLA, 1 mM, as substrate, in the place of L-Arg.
CO-binding to eNOS
As we have already shown, at atmospheric pressure, the CO-binding rate was considerably slower (by a factor of 10) than oxygen-binding (Lange et al., 2001). However, the CO-binding rate increased strongly as a function of pressure, attaining a threefold higher value at 140 MPa (see Fig. 4 and Table 1). This pressure-induced increase of the CO-binding rate is reflected by an activation volume of ΔV‡ = −16.7 ml/mol. The CO-binding rate and its activation volume were not significantly affected by the nature of the pterin cofactor (ABH4 or ABH2). Furthermore, the kinetics did not depend on the nature of the substrate (L-Arg or NOHLA).
FIGURE 4.

Kinetics of CO complex formation of eNOS. Effect of ABH4 in the presence of L-Arg (•) and NOHLA (○); effect of ABH2 in the presence of L-Arg (▾) and NOHLA (▿). Conditions before mixing: 50 μM ABH4 or ABH2, 500 μM L-Arg or 1 mM NOHLA and 7 μM enzyme. Three independent experiments were performed to determine the rate constant at each pressure.
Interaction of eNOS with oxygen in the presence of BH4
In the course of the second reaction cycle of NOS, i.e., in the presence of NOHLA, interaction of reduced eNOS with oxygen results in the transient formation of a ferric NO complex. Low temperature studies have shown that this reaction requires the presence of its natural cofactor BH4. It does not require the presence of the reductase domain (Gorren et al., 2000). The FeIII-NO complex is characterized by an absorbance maximum near 440 nm. In our present study we studied the formation and decomposition of this complex under high pressure. As shown in Fig. 5, its formation was very rapid even at 5°C. It was completed within 100 ms. The decomposition was ∼10× slower. The logarithmic decomposition rate decreased linearly as a function of pressure with an activation volume of ΔV‡ = 7 ml/mol.
FIGURE 5.

Pressure effect on formation and decomposition of the FeIII-NO complex of eNOS. The complex was observed after mixing of reduced eNOS (3.5 μM final) in the presence of NOHLA (500 μM) and BH4 (25 μM) with oxygen (0.5 mM final) at 4°C. The complex formation and decomposition were followed at 437 nm. Three independent determinations were performed to determine the rate constants at each pressure. SE of each experimental point was <2%. (Inset) Time course of the O2-binding to eNOS after stopped-flow mixing at 50 MPa. The solid line shows the mono-exponential fit for the decomposition kinetics of the ferric NO complex.
Interaction of eNOS with oxygen in the presence of ABH4
ABH4 is a strong inhibitor of NOS activity (Werner et al., 1996; Pfeiffer et al., 1997). Nevertheless, an electron transfer from ABH4 to the oxygen complex cannot be excluded. It was therefore interesting to study the pressure dependence of this reaction. Our previous low temperature work (Gorren et al., 2000) had revealed that oxygen-binding to reduced eNOS in the presence of ABH4 results in optically detectable oxygen complexes within the first reaction cycle (in the presence of L-Arg) as well as in the second reaction cycle (in the presence of NOHLA). Figs. 2 and 3 show the pressure dependence of the complex formation and decomposition kinetics. For both reaction cycles, the pressure dependence of the oxy-complex formation was biphasic: up to ∼50 MPa, the rate increased. Above this pressure, the rate decreased. This effect was especially important within the second reaction cycle: the activation volume changed from ΔV‡ = −42 ml/mol (<50 MPa) to ΔV‡ = +9 ml/mol (>50 MPa). In contrast, the pressure dependence of the oxygen complex decomposition rate did not show a break. In the first reaction cycle, the activation volume was nearly pressure-independent (ΔV‡ = −3.3 ml/mol). However, in the second reaction cycle, the decomposition rate decreased strongly as a function of pressure with an activation volume of ΔV‡ = 25.4 ml/mol.
DISCUSSION
Although the interaction of reduced heme-thiolate proteins with molecular oxygen plays a central role in the reaction mechanism, it is always difficult to study this reaction. This is due to the transient nature of the ferrous oxygen complex: in the absence of a specific electron donor it decomposes quickly to ferric enzyme and superoxide, and in the presence of an electron donor it transforms immediately to an activated complex leading to product release. Apart from the relatively stable oxygen complex of cytochrome P450cam, which can be observed for a couple of minutes (Bangcharoenpaurpong et al., 1986), the detection of oxygen complexes with other P450 forms requires the use of low-temperature spectroscopic techniques (Bonfils et al., 1979; Larroque and van Lier, 1980). But even for P450cam, a closer structural inspection needs cryogenic techniques (Vidakovic et al., 1998; Schlichting et al., 2000; Denisov et al., 2002). Similarly, for nitric oxide synthase, oxygen complexes have been observed by low-temperature spectroscopy (Bec et al., 1998; Ledbetter et al., 1999). In this article, stopped-flow kinetics of oxygen-binding under high pressure are reported for the first time. In the past, we had used high pressure stopped-flow (HPSF) to study the mechanism of CO-binding to heme-thiolate enzymes (Lange et al., 1994; Jung et al., 2002). The work with oxygen became feasible now after an instrumental adaptation (use of a ruby sphere acting as an inner valve), and by a very careful design of anaerobic experimental conditions: the concentration of dithionite must be high enough to ensure enzyme reduction, and sufficiently low to prevent reduction of oxygen.
Oxygen-binding to BM3 and eNOS
To study oxygen-binding, we chose conditions under which the oxygen complexes could not evolve further in the reaction cycles. That is, for BM3 we worked with the oxygenase domain (the absence of the reductase domain preventing a reduction by a second electron). For eNOS, we replaced BH4 by ABH2, a cofactor which cannot deliver a second electron. As one may expect, oxygen-binding to BM3 appears to occur via the same mechanism as CO-binding with similar rate constants and the same activation volumes. The oxygen-binding mechanism of eNOS appears not to be very different from that of BM3: the observed rate constants are comparable, and in the first reaction cycle, i.e., with L-Arg as substrate, the activation volume of the complex formation is similar. However, within the second reaction cycle, the activation volume is negative (it is positive in the first cycle). This means that the oxygen complex formation is accelerated by pressure within the second cycle. This points to a different binding mechanism in the two reaction cycles.
In contrast to BM3, in eNOS the mechanism of oxygen-binding is very different from that of CO-binding. Indeed, whatever the cofactor or the substrate, the CO-binding rate was ∼7× lower at atmospheric pressure. However, it increased strongly under high pressure. In our previous work, we had explained the low reactivity of eNOS with CO at atmospheric pressure in terms of inaccessibility of CO to the heme iron when substrate was present. Indeed, in the absence of substrate, the CO-binding rate is considerably higher (Lange et al., 2001). The effect of pressure was then attributed to an expulsion of the substrate of the active center, facilitated by the CO-binding. However, if the presence of substrate obstructs the active site for CO-binding, one may expect a similar effect on oxygen-binding, and high pressure should accelerate oxygen-binding. Since this is not the case, we must conclude that 1), oxygen- and CO-binding mechanisms in eNOS are different; 2), the CO-binding mechanisms of BM3 and eNOS are different; and 3), care must be taken in using CO-binding as a model for oxygen-binding. In the latter case, using CO-binding as a model for O2-binding in eNOS appears highly inappropriate. The fact that O2 binds up to 10-fold faster than does CO at atmospheric pressure suggests the existence of a specific pathway of oxygen entry into the eNOS active site.
Formation and decay of FeIII-NO
In the presence of BH4 and NOHLA, the ferrous oxygen complex of eNOS was not detected. Instead, as previously reported, the first detectable intermediate was the ferric NO complex (Bec et al., 2000). Thus, our stopped-flow experiments reflected formation and decay of this intermediate state as a function of pressure. The rather small activation volume of its decay rate, as well as its linear logarithmic pressure dependence, indicate that no further intermediate in the reaction cycle may be expected, and that the transformation from FeIII-NO to FeIII is not accompanied by a major protein conformational change.
The case of ABH4
It is more difficult to interpret the results obtained in the presence of ABH4. In its presence, product is not formed, and consequently we did not observe a ferric NO complex. However, the pressure dependence of the oxygen-binding rate constant shows a break, both in the first and in the second reaction cycle. This break can be interpreted in two ways: 1), high pressure induces a transition between two protein conformers of different oxygen-binding properties; and 2), in the presence of ABH4 oxygen-binding is followed by another reaction, and at high pressure a change of the rate-limiting step takes place.
Support for the first hypothesis comes from nonlinear pressure dependence of ligand-binding rates of myoglobin (mb) which have been attributed to a pressure-induced transition between two protein conformers (Uchida et al., 2000). Although mb has a different heme ligation (imidazole) than P450 and NOS (thiolate), and different ligand-binding mechanisms can be expected, recent progress in the understanding of ligand-binding to mb may be relevant also for P450 and NOS. Indeed, CO-binding to mb has been shown to proceed via different paths, comprising different CO “docking sites” (Šrajer et al., 2001; Lamb et al., 2002). These docking sites are related to protein cavities or packing defects (Chu et al., 2000; Brunori and Gibson, 2001) that may conceivably change size under pressure. Further support for the conformational explanation comes from flush-photolysis experiments of geminate CO rebinding to P450 and NOS, which gave evidence of doublet substate conformers of different CO-binding characteristics (Tétreau et al., 1997, 1999). Moreover, a high pressure stopped-flow study of CO-binding to P450cam also revealed the coexistence of conformers of different activation volumes ΔV‡ (Jung et al., 2002).
However, despite many of the conformational arguments given above, the second hypothesis, which assumes an additional reaction step in the presence of ABH4, appears to be more plausible. Indeed, we found a linear pressure dependence of the oxygen-binding rate for BM3 as well as for NOS. A pressure break was only observed with NOS when ABH2 was replaced by ABH4. Since ABH2 and ABH4 have the same structure, they differ only in their oxidation state, so a differential effect on oxygen-binding rates under pressure would be difficult to explain. However, unlike ABH2, ABH4 could play an electron donor role, like authentic BH4. A possible interpretation of the additional reaction step would therefore be that the formation of the ferrous oxygen complex is followed by its reduction via ABH4. If this hypothesis is correct, then we do not know exactly which step we are following; it can be the oxygen complex formation as well as its reduction. However, the break in the pressure profile would then indicate a change of the rate-limiting step. Interestingly, since we do not observe an FeIII-NO complex, the reduction of the oxygen complex by ABH4 may lead only to a peroxy complex which does not evolve further to the higher valence iron-oxo complex considered to be the catalytically active species. The possibility of ABH4 acting as a mechanism-based inhibitor is intriguing, but needs to be proven. One possibility to decide between the conformational and the mechanistic explanations would be to carry out HPSF experiments with spectral resolution. The instrumental setup for such experiments is currently being built in our laboratory.
CONCLUSION
The above results show the potential of the HPSF technique to study enzyme elementary steps as a complement to conventional stopped-flow technique. With respect to high temperature, high pressure offers the possibility to study enzyme reactions without denaturing the protein. Instead, pressure may be used to switch from one elementary step to another. This possibility appears to be particularly important to study complex reactions such as reaction cycles occurring in hemoproteins.
Acknowledgments
We thank Christian Valentin for continuous development and assistance in the high pressure stopped-flow technique.
This work was supported by the Human Frontier Science Program (grant RGP0026/2001-M).
Abbreviations used: BM3, the F393H mutant of cytochrome P450 BM3 (CYP102); eNOS, the oxygenase domain of endothelial nitric oxide synthase (EC 1-14-13-39); BH4, (6R)-5,6,7,8-tetrahydro-6-(L-erythro-1′,2′-dihydroxypropyl)pterin; NHA, NG-hydroxy-l-arginine.
References
- Abu-Soud, H. M., R. Gachhui, F. M. Raushel, and D. J. Stuehr. 1997. The ferrous-dioxy complex of neuronal nitric oxide synthase. Divergent effects of L-arginine and tetrahydrobiopterin on its stability. J. Biol. Chem. 272:17349–17353. [DOI] [PubMed] [Google Scholar]
- Abu-Soud, H. M., K. Ichimori, A. Presta, and D. J. Stuehr. 2000. Electron transfer, oxygen binding, and nitric oxide feedback inhibition in endothelial nitric-oxide synthase. Biochemistry. 39:2332–2339. [DOI] [PubMed] [Google Scholar]
- Balny, C., J. L. Saldana, and N. Dahan. 1984. High pressure stopped flow spectrometry at low temperature. Anal. Biochem. 139:178–189. [DOI] [PubMed] [Google Scholar]
- Bancel, F., G. Hui Bon Hoa, P. Anzenbacher, C. Balny, and R. Lange. 2002. High pressure: a new tool to study P450 structure and function. Methods Enzymol. 357:145–157. [DOI] [PubMed] [Google Scholar]
- Bangcharoenpaurpong, O., A. K. Rizos, P. M. Champion, D. Jollie, and S. G. Sligar. 1986. Resonance Raman detection of bound dioxygen in cytochrome P-450 cam. J. Biol. Chem. 261:8089–8092. [PubMed] [Google Scholar]
- Bec, N., A. C. Gorren, C. Voelker, B. Mayer, and R. Lange. 1998. Reaction of neuronal nitric-oxide synthase with oxygen at low temperature. Evidence for reductive activation of the oxy-ferrous complex by tetrahydrobiopterin. J. Biol. Chem. 273:13502–13508. [DOI] [PubMed] [Google Scholar]
- Bec, N., A. C. F. Gorren, B. Mayer, P. P. Schmidt, K. K. A. Andersson, and R. Lange. 2000. The role of tetrahydrobiopterin in the activation of oxygen by nitric-oxide synthase. J. Inorg. Biochem. 81:207–211. [DOI] [PubMed] [Google Scholar]
- Boggs, S., L. Huang, and D. J. Stuehr. 2000. Formation and reactions of the heme-dioxygen intermediate in the first and second steps of nitric oxide synthesis as studied by stopped-flow spectroscopy under single-turnover conditions. J. Biol. Chem. 275:17349–17357. [DOI] [PubMed] [Google Scholar]
- Bonfils, C., P. Debey, and P. Maurel. 1979. Highly purified microsomal P-450: the oxyferro intermediate stabilized at low temperature. Biochem. Biophys. Res. Commun. 88:1301–1307. [DOI] [PubMed] [Google Scholar]
- Brunori, M., and Q. H. Gibson. 2001. Cavities and packing defects in the structural dynamics of myoglobin. EMBO Rep. 2:674–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, K., J. Vojtchovský, B. H. McMahon, R. M. Sweet, J. Berendzen, and I. Schlichting. 2000. Structure of a ligand-binding intermediate in wild-type carbonmonoxy myoglobin. Nature. 403:921–923. [DOI] [PubMed] [Google Scholar]
- Davis, K. L., E. Martin, I. V. Turko, and F. Murad. 2001. Novel effects of nitric oxide. Annu. Rev. Pharmacol. Toxicol. 41:203–236. [DOI] [PubMed] [Google Scholar]
- Denisov, I. G., T. M. Makris, and S. G. Sligar. 2002. Cryoradiolysis for the study of P450 reaction intermediates. Methods Enzymol. 357:103–115. [DOI] [PubMed] [Google Scholar]
- Gorren, A. C. F., N. Bec, A. Schrammel, E. R. Werner, R. Lange, and B. Mayer. 2000. Low-temperature optical absorption spectra suggest a redox role for tetrahydrobiopterin in both steps of nitric oxide synthase catalysis. Biochemistry. 39:11763–11770. [DOI] [PubMed] [Google Scholar]
- Grisham, M. B., D. Jourd'heuil, and D. A. Wink. 1999. Nitric oxide. I. Physiological chemistry of nitric oxide and its metabolites: implications in inflammation. Am. J. Physiol. 276:G315–G321. [DOI] [PubMed] [Google Scholar]
- Jung, C., N. Bec, and R. Lange. 2002. Substrates modulate the rate-determining step for CO binding in cytochrome P450cam (CYP101). A high-pressure stopped-flow study. Eur. J. Biochem. 269:2989–2996. [DOI] [PubMed] [Google Scholar]
- Lamb, D. C., K. Nienhaus, A. Arcovito, F. Draghi, A. E. Miele, M. Brunori, and G. U. Nienhaus. 2002. J. Biol. Chem. 277:11636–11644. [DOI] [PubMed] [Google Scholar]
- Lange, R., I. Heiber-Langer, C. Bonfils, I. Fabre, M. Negishi, and C. Balny. 1994. Activation volume and energetic properties of the binding of CO to hemoproteins. Biophys. J. 66:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, R., N. Bec, P. Anzenbacher, A. W. Munro, A. C. F. Gorren, and B. Mayer. 2001. Use of high pressure to study elementary steps in P450 and nitric oxide synthase. J. Inorg. Biochem. 87:191–195. [DOI] [PubMed] [Google Scholar]
- Lange, R. 2003. Cellular biology of cytochrome P450 regulation. Preface. Biochim. Biophys. Acta. 1619:221–222. [Google Scholar]
- Larroque, C., and J. E. van Lier. 1980. The subzero temperature stabilized oxyferro complex of purified cytochrome P450scc. FEBS Lett. 115:175–177. [DOI] [PubMed] [Google Scholar]
- Ledbetter, A. P., K. McMillan, L. J. Roman, B. S. Masters, J. H. Dawson, and M. Sono. 1999. Low-temperature stabilization and spectroscopic characterization of the dioxygen complex of the ferrous neuronal nitric oxide synthase oxygenase domain. Biochemistry. 38:8014–8021. [DOI] [PubMed] [Google Scholar]
- Lewis, D. F. V. 2001. Guide to Cytochromes P450. Structure and Function. Taylor and Francis, London and New York.
- Marletta, M. A., A. R. Hurshman, and K. M. Rusche. Catalysis by nitric oxide synthase. 1998. Curr. Opin. Chem. Biol. 2:656–663. [DOI] [PubMed] [Google Scholar]
- Mozhaev, V. V., K. Heremans, J. Frank, P. Masson, and C. Balny. 1996. High pressure effects on protein structure and function. Proteins. 24:81–91. [DOI] [PubMed] [Google Scholar]
- Noble, M. A., C. S. Miles, S. K. Chapman, D. A. Lysek, A. C. Mackay, G. A. Reid, R. P. Hanzlik, and A. W. Munro. 1999. Roles of key active-site residues in flavocytochrome P450 BM3. Biochem. J. 339:371–379. [PMC free article] [PubMed] [Google Scholar]
- Ortiz de Montellano, P. R. 1995. Cytochrome P450: Structure, Function and Biochemistry. Plenum Press, New York.
- Ost, T. W. B., C. S. Miles, A. W. Munro, J. Murdoch, G. A. Reid, and S. K. Chapman. 2001. Phenylalanine 393 exerts thermodynamic control over the heme of flavocytochrome P450 BM3. Biochemistry. 40:13421–13429. [DOI] [PubMed] [Google Scholar]
- Pfeiffer, S., A. C. F. Gorren, E. Pitters, K. Schmidt, E. R. Werner, and B. Mayer. 1997. Allosteric modulation of rat brain nitric oxide synthase by the pterin-site enzyme inhibitor 4-aminotetrahydrobiopterin. Biochem. J. 328:349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer, S., B. Mayer, and B. Hemmens. 1999. Nitric oxide: chemical puzzles posed by a biological messenger. Angew. Chem. Int. Ed. 38:1714–1731. [DOI] [PubMed] [Google Scholar]
- Schlichting, I., J. Berendzen, K. Chu, A. M. Stock, S. A. Maves, D. E. Benson, R. M. Sweet, D. Ringe, G. A. Petsko, and S. G. Sligar. 2000. The catalytic pathway of cytochrome P450cam at atomic resolution. Science. 287:1615–1622. [DOI] [PubMed] [Google Scholar]
- Sono, M., D. J. Stuehr, M. Ikeda-Saito, and J. H. Dawson. 1995. Identification of nitric oxide synthase as a thiolate-ligated heme protein using magnetic circular dichroism spectroscopy. Comparison with cytochrome P-450-CAM and chloroperoxidase. J. Biol. Chem. 270:19943–19948. [DOI] [PubMed] [Google Scholar]
- Šrajer, V., Z. Ren, T.-Y. Teng, M. Schmidt, T. Ursby, D. Bourgeois, C. Pradervand, W. Schildkamp, M. Wulff, and K. Moffat. 2001. Protein conformational relaxation and ligand migration in myoglobin: a nanosecond to millisecond molecular movie from time-resolved Laue x-ray diffraction. Biochemistry. 40:13802–13815. [DOI] [PubMed] [Google Scholar]
- Stuehr, D. J. 1999. Mammalian nitric oxide synthases. Biochim. Biophys. Acta. 1411:217–230. [DOI] [PubMed] [Google Scholar]
- Tétreau, C., C. Di Primo, R. Lange, H. Tourbez, and D. Lavalette. 1997. Dynamics of carbon monoxide binding with cytochromes P-450. Biochemistry. 36:10262–10275. [DOI] [PubMed] [Google Scholar]
- Tétreau, C., M. Tourbez, A. Gorren, B. Mayer, and D. Lavalette. 1999. Dynamics of carbon monoxide binding with neuronal nitric oxide synthase. Biochemistry. 38:7210–7218. [DOI] [PubMed] [Google Scholar]
- Uchida, T., K. Ishimori, and I. Morishima. 2000. Unusual pressure effects on ligand rebinding to the human myoglobin leucine-29 mutants. J. Biol. Chem. 275:30309–30316. [DOI] [PubMed] [Google Scholar]
- Vidakovic, M., S. G. Sliar, H. Li, and T. L. Poulos. 1998. Understanding the role of the essential Asp251 in cytochrome P450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry. 37:9211–9219. [DOI] [PubMed] [Google Scholar]
- Werner, E. R., E. Pitters, K. Schmidt, H. Wachter, G. Werner-Felmayer, and B. Mayer. 1996. Identification of the 4-amino analogue of tetrahydrobiopterin as a dihydropteridine reductase inhibitor and a potent pteridine antagonist of rat neuronal nitric oxide synthase. Biochem. J. 320:193–196. [DOI] [PMC free article] [PubMed] [Google Scholar]