

Abstract
Spontaneous transient depolarizations in mitochondrial membrane potential (ΔΨm), mitochondrial flickers, have been observed in isolated mitochondria and intact cells using the fluorescent probe, tetramethylrhodamine ethyl ester (TMRE). In theory, the ratio of [TMRE] in cytosol and mitochondrion allows ΔΨm to be calculated with the Nernst equation, but this has proven difficult in practice due to fluorescence quenching and binding of dye to mitochondrial membranes. We developed a new method to determine the amplitude of flickers in terms of millivolts of depolarization. TMRE fluorescence was monitored using high-speed, high-sensitivity three-dimensional imaging to track individual mitochondria in freshly dissociated smooth muscle cells. Resting mitochondrial fluorescence, an exponential function of resting ΔΨm, varied among mitochondria and was approximately normally distributed. Spontaneous changes in mitochondrial fluorescence, indicating depolarizations and repolarizations in ΔΨm, were observed. The depolarizations were reversible and did not result in permanent depolarization of the mitochondria. The magnitude of the flickers ranged from <10 mV to >100 mV with a mean of 17.6 ± 1.0 mV (n = 360) and a distribution skewed to smaller values. Nearly all mitochondria flickered, and they did so independently of one another, indicating that mitochondria function as independent units in the myocytes employed here.
INTRODUCTION
The mitochondrial membrane potential (ΔΨm), established by the proton pumps of the electron transport chain, not only provides the energy for ATP generation but is also required for a variety of other mitochondrial functions such as Ca2+ uptake and apoptosis (Duchen, 1999). Spontaneous transient depolarizations of ΔΨm, sometimes called mitochondrial flickers (Duchen et al., 1998), were first observed in living cells by Loew et al. (1993), using tetramethylrhodamine ethyl ester (TMRE), a fluorescent lipophilic cation. TMRE, and its methyl ester analog TMRM, accumulate rapidly and reversibly in the mitochondria of living cells due to the negative ΔΨm of the mitochondria with respect to the cytosol. Thus, they can serve as “Nernstian” indicators of ΔΨm, with mitochondrial depolarization resulting in a loss of dye from the mitochondrion and a decrease in mitochondrial fluorescence intensity (FI).
The mitochondrial inner membrane contains several ion transporters, activation of which can cause transient mitochondrial flickers. Most frequently, flickers have been attributed to transient activation of the mitochondrial permeability transition pore (PTP) (Collins et al., 2002; De Giorgi et al., 2000; Diaz et al., 2000; Hüser and Blatter, 1999; Jacobson and Duchen, 2002; Zorov et al., 2000; Hüser et al., 1998). However, at least three types of PTP-independent flickers have also been identified. First, flickers have been linked to entry of H+ into the mitochondrial matrix via the F1F0ATPase during ATP generation in neurons (Buckman and Reynolds, 2001). Second, flickers have been attributed to depolarization resulting from Ca2+ influx into a mitochondrion through the Ca2+ uniporter after release from sarcoplasmic reticulum in cardiomyocytes (Duchen et al., 1998). Third, in substrate-deprived cardiomyocytes, flickers are not blocked by inhibition of either mitochondrial Ca2+ uptake or PTP activation, but they can be blocked by inhibitors of inner mitochondrial membrane anion channels (O'Rourke, 2000).
Since the ratio of the free mitochondrial TMRE concentration ([TMRE]m) to free cytosolic TMRE concentration ([TMRE]c) is an exponential function of ΔΨm, then measurement of TMRE fluorescence in the mitochondria and cytosol allows a simple calculation of ΔΨm with the familiar Nernst equation (Loew et al., 1993; Diaz et al., 2000). However, in practice two difficulties have thwarted this straightforward approach. First, TMRE is often used at high concentrations in self-quenching mode so that an increase in mitochondrial fluorescence corresponds to depolarization. This masks the direct Nernstian relationship between mitochondrial fluorescence and ΔΨm, making precise quantitation of changes in ΔΨm impossible although approximations can be made (Duchen et al., 1998). Second, Scaduto and Grotyohann (1999) have shown that TMRE and similar dyes exhibit significant binding to the inner mitochondrial membranes so that most fluorescence comes not from free TMRE but from bound TMRE. In addition to these problems, small movements of mitochondria into or out of the plane of focus in confocal imaging could possibly be mistaken for changes in mitochondrial fluorescence.
We have addressed these difficulties to obtain quantitative measurements of mitochondrial flickers. First, the sensitivity of the imaging system used here allowed the use of very low concentrations of TMRE; thus the shortcomings of using the dye in quenching mode were avoided. Second, in agreement with Scaduto and Grotyohann (1999) our calibrations indicated a substantial level of TMRE binding to the inner mitochondrial membrane, so that absolute ΔΨm cannot be calculated. However, provided [TMRE]c remains constant, binding of TMRE to the membrane does not affect the calculation of flicker amplitudes (changes in ΔΨm) as we outline in Methods. By patching every cell imaged in the whole-cell configuration and holding the plasma membrane potential at 0 mV, we ensured that [TMRE]c was equal to the fixed level in the bath (2.5 nM). We note that this method of quantitation also enables flicker amplitudes to be calculated in the absence of TMRE binding to the mitochondrial membrane. Finally, we have used three-dimensional imaging with wide-field microscopy to track each individual mitochondrion in time and space, ensuring that movement was not mistaken for fluorescence change.
The quantitative approach described here provides opportunity to gain new insights into the nature of mitochondrial flickers; for example, quantitative differences in flickers might be hallmarks of the different mechanisms outlined above. Finally, we have used a preparation of freshly dissociated smooth muscle cells for these studies, which marks the first time that mitochondrial flickers have been observed in smooth muscle.
MATERIALS AND METHODS
Cell preparation
Adult toads (Bufo marinus) were killed by decapitation as approved by the University of Massachusetts Medical Center Animal Care Committee according to the guidelines of the United States Department of Agriculture and Health and Human Services. Single stomach smooth muscle cells were isolated by enzymatic dissociation with trypsin and collagenase as described previously (Fay et al., 1982).
TMRE loading protocol
Freshly isolated smooth muscle cells were incubated with TMRE (25 nM) for 10 min at room temperature. Cells were transferred to a tissue bath where TMRE was diluted to a final concentration of 2.5 nM with bath solution. Cells were allowed to equilibrate for 10 min before establishing whole-cell patch clamp recording. To the best of our knowledge, this is the lowest concentration of TMRE used to monitor ΔΨm in living cells.
Electrophysiology
All cells were patch-clamped in the whole-cell configuration at room temperature (∼21°C) using thin-walled borosilicate patch electrodes (∼5 MΩ) (World Precision Instruments, Sarasota, FL). The membrane potential was voltage-clamped to 0 mV and cells were allowed to equilibrate for 5–10 min before commencement of the experiment. First, this ensured that the only potential gradient for TMRE accumulation was across the mitochondrial membrane. Second, that there would be no variations in cell membrane potential to alter [TMRE]c and hence change [TMRE]m as TMRE re-equilibrated between the two compartments. Thus maintaining cells at a holding potential of 0 mV ensured that the equilibrium [TMRE]c was constant and equal to that in the bathing solution, i.e., 2.5 nM.
Imaging
TMRE-labeled mitochondria were imaged using a high-speed, wide-field digital imaging microscope and charge-coupled device camera (Massachusetts Institute of Technology, Lincoln Laboratory, Lexington, MA) which have been described in detail elsewhere (ZhuGe et al., 1999) and a Nikon (Melville, NY) 60×, NA 1.4, oil-immersion objective. The pixel size was 333 nm × 333 nm, and the total area imaged was 42.6 μm × 42.6 μm. The 514-nm line of an argon-krypton laser was used for TMRE excitation, providing a wide-field illumination flux of ∼4 × 1019 photons cm−2 s−1. A shutter was used to limit the total light exposure of the cells. A 550-nm long-pass filter was used for TMRE emission. A piezoelectric translator was used to focus the objective rapidly. A 10-plane through-focus series of images was taken at 0.5-μm Z-spacing to create each three-dimensional image stack. Each stack was acquired in 140 ms using 5-ms exposures with 10-ms intervals to allow movement of the objective. 20 image stacks were acquired at 5-s intervals to generate each 95-s image sequence. Three-dimensional imaging made it possible to resolve the fluorescence of individual mitochondria reliably without contamination of the signal by fluorescence from mitochondria in other planes. It also ensured that movement into and out of the plane of focus (due to movement of the entire cell or contraction in a portion of the cell) was not mistaken for changes in mitochondrial FI.
Image processing and data analysis
Image processing and data analysis were performed using custom software on a Silicon Graphics workstation. Since the images were taken with wide-field illumination, blurred fluorescence from out-of-focus objects contributes to the sum of the fluorescence at any given point. Image restoration is essential to allow accurate data analysis. To reassign out-of-focus light to its point of origin, the data was processed using an iterative constrained deconvolution algorithm (Carrington et al., 1990, 1995). Mitochondria were bright and easily distinguished from background in the restored three-dimensional images (see Results). The central z-plane of each mitochondrion was identified in each three-dimensional image stack. Voxels were selected at several points along the length of the mitochondrion in the central plane; each voxel was centered in the midpoint of the mitochondrial width. The number of voxels used depended upon the length of the mitochondrion. The average fluorescence of these voxels was designated as the FI. Mitochondria that drifted out of focus due to small movements of the cell were not included in the analysis, avoiding false identification of changes in ΔΨm. Root mean square noise value was computed for 115 mitochondria and divided by the mean FI × 100. This gave the percent noise in the signal (average, 5.9%; range, 2.5–10.8%). A change in FI that is >20% (3.4-fold the mean percent noise) over 2–3 consecutive images (i.e., within 5–10 s) was considered a change in mitochondrial potential and designated a flicker, i.e., the limit of resolution was a change of 5.8 mV (see below).
The percentage of mitochondria that flickered in each cell was calculated as
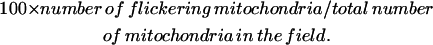 |
Flicker frequency in flickering mitochondria was
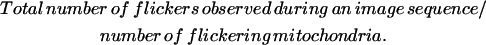 |
Calibration of TMRE fluorescence
To relate [TMRE] to measured FI, a calibration curve was constructed using the same microscope and optical setup employed for the experiments. Glass capillaries (Vitro Dynamics, Rockaway, NJ), 200-μm wide and 20-μm deep, loaded with known concentrations of TMRE were imaged, and the total FI (TFI) at each concentration was measured by summing the light over an area of 450.65 μm2 (127 × 32 pixels). The TFI data were plotted against the total number of molecules in the imaged volume, 9013 μm3 (Fig. 1, inner axes), and the data fit with a straight line (R = 0.999) with a slope of 0.93 FI/molecule of TMRE. Thus, each FI unit measured in 5 ms corresponds to ∼1 molecule of TMRE. There was no evidence of quenching up to 100 μM.
FIGURE 1.

TMRE calibration curve. Inner axes show the TFI plotted against total number of TMRE molecules. The data were fit with a straight line (R = 0.999) having a slope of 0.93 FI/molecule of TMRE. Outer axes show mitochondrial FI plotted against [TMRE] in μM. There was no evidence of self-quenching. The dashed line shows the extrapolation of [TMRE] from the mean FI.
To compare the mitochondrial FI to [TMRE]m, TFI data were normalized to the typical mitochondrial volume imaged by one pixel. Assuming a 333-nm pixel is used to image a rod-shaped mitochondrion of 220-nm diameter (Loew et al., 1993), the volume of a mitochondrion imaged by a pixel would be at most ∼0.013 μm3. I.e., normalized TFI data = TFI data/9013 μm3 × 0.013 μm3. The normalized TFI data were then multiplied by 10 to account for intensity scaling in the deconvolution software, and plotted against their corresponding TMRE concentrations (Fig. 1, outer axes). The factor of 10 results from the fact that, as this is wide-field illumination, each of the 10 optical sections through a cell redundantly detects all the light from all the mitochondria (and cytosol), whether in or out of focus, within the field of view. In the image deconvolution algorithm, the point spread function integrates over all 10 optical sections (Femino et al., 1998). Thus, in a restored three-dimensional image of a cell, every photon is counted 10 times rather than just once. In contrast, only one optical section is taken through a microcapillary tube and each photon is counted only once. This scaling factor could be corrected in the deconvolution process (divide the image intensities by 10), but here we have applied it to the calibration value for numerical reasons in the computations and image data storage.
Given a [TMRE]c of 2.5 nM, and a Nernstian relationship, the free [TMRE]m is calculated to be 1 μM for a ΔΨm of –150 mV. However, the mean mitochondrial FI per pixel, measured experimentally from 243 mitochondria, was 4407 ± 170 FI, which when calibrated using Fig. 1, corresponds to a [TMRE]m of ∼63 μM, greater than expected if mean ΔΨm is −150 mV. Hence, we obtain a ratio of 63:1, bound:free.
Calculation of flicker amplitude
TMRE, even at low concentrations, exhibits significant binding to the mitochondrial membrane as found by Scaduto and Grotyohann (1999). Hence the absolute value for ΔΨm cannot be determined by a simple Nernstian relationship between [TMRE]m and [TMRE]c, but changes in ΔΨm (flicker amplitude) can be determined from changes in FI as follows. The FI in a mitochondrion is a measure of its total TMRE content, as given by
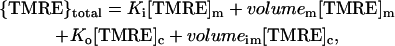 |
(1) |
where {TMRE}total = total mitochondrial TMRE in units of mass, and [TMRE]c was always 2.5 nM and includes [TMRE] in the mitochondrial intermembrane space.
The following are given in units of volume:
volumem = mitochondrial matrix volume
volumeim = volume of the intermembrane space
Ki = partition coefficient for [TMRE]m binding to the inner mitochondrial membrane
Ko = partition coefficient for [TMRE]c binding to the inner mitochondrial membrane
[TMRE]c is >300-fold less than [TMRE]m in a mitochondrion with ΔΨm = −150 mV; volumem and volumeim are small compared to Ko or Ki; the value of Ko is 129, and that of Ki is 60 at 28°C (Scaduto and Grotyohann, 1999). (This value for Ki is in precise agreement with the bound:free ratio of 63:1 which we obtained as outlined in the previous paragraph.) Hence, the above expression simplifies to
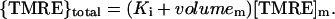 |
(2) |
Therefore, flicker amplitude
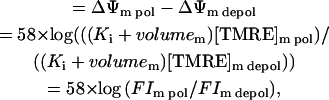 |
(3) |
where pol and depol are the state of mitochondrial polarization before and during a flicker, respectively. Since volumem is much smaller than Ki by a factor of 60/1 (Ki/volumem) (Scaduto and Grotyohann, 1999), possible changes in mitochondrial volume would affect our determination of flicker amplitude by a negligible amount. Thus flicker magnitude can be calculated directly from the FI, and since [TMRE]c need not be detected, it can be kept at a very low concentration. This methodology is independent of TMRE binding to the mitochondrial membrane; that is, even if Ki is equal to zero the determination of flicker amplitude remains valid.
Image preparation
Custom software was used to prepare the images shown throughout the article. In general a single plane from the three-dimensional image stack is displayed. In some cases the 10 planes of a three-dimensional image stack were projected onto a single plane to form a composite image, as stated in figure legends. Polarized mitochondria appear brighter than depolarized mitochondria. The slight pixelization of the images is caused by the imaging resolution (the pixel size is slightly larger than the optical limit of resolution) and the enhanced contrast produced by image restoration.
Solutions and reagents
Experiments were carried out using the following solutions (in mM). Pipette: KCl 137, MgCl2 3, HEPES 20, and Na2ATP 3, at pH 7.2 with KOH. Bath: NaCl 130, KCl 3, CaCl2 1.8, MgCl2 1, and HEPES 10, at pH 7.4 with NaOH. TMRE was purchased from Molecular Probes (Eugene, OR). All other reagents were from Sigma-Aldrich (St. Louis, MO). Stock solutions of TMRE (10 mM) and FCCP (10 mM) were made up in DMSO. Stocks were stored at −20°C.
Statistical analysis
Data are presented as mean ± SE. Statistical significance was assessed using Instat (3.05) (Graphpad Software, San Diego, CA). In general, nonparametric tests were utilized to assess the data, which did not follow a normal distribution. P ≤ 0.05 was considered statistically significant.
RESULTS
Three-dimensional imaging of mitochondria labeled with TMRE
Fig. 2 shows a stereo-pair of images of a freshly isolated smooth muscle cell loaded with TMRE. TMRE was localized to rod-shaped organelles of varying length arranged in alignment with the long axis of the cell, consistent with mitochondrial accumulation of dye (Drummond et al., 2000). The FI differed among the mitochondria; some were clearly brighter than others. The variations imply that subpopulations of mitochondria may have different resting ΔΨm (RΔΨm) but they may also be indicative of mitochondrial flickering. 1 μM FCCP was applied to the cells to dissipate ΔΨm (Fig. 3 A). After FCCP exposure, the FI of 32 of the 34 labeled organelles decayed, demonstrating that the dye had accumulated in the mitochondria (Fig. 3 B). Only 2 of the 34 mitochondria shown in Fig. 3 A exhibited an initial increase in FI after FCCP application. Interestingly, both of these mitochondria had very low FI at the start of the recording period, suggesting that the two mitochondria were already depolarized due to flickering. The increase in FI is attributable to repolarization; the FI subsequently decreased, undoubtedly due to the effects of FCCP. The decrease in mitochondrial FI elicited by FCCP confirms that the loading protocol employed in this study resulted in the utilization of TMRE in nonquenching mode.
FIGURE 2.

Mitochondrial accumulation of TMRE. A high resolution stereo-pair fluorescence image of a toad stomach smooth muscle cell loaded with TMRE. Each image is a composite consisting of a projection of a three-dimensional image stack onto a single plane. The discrete localization of the dye to the mitochondria and the arrangement of the mitochondria in alignment with the long axis of the cell are apparent. Differences in mitochondrial FI, and therefore mitochondrial ΔΨm, are clear. The scale bars are 5 μm.
FIGURE 3.

Effect of FCCP upon TMRE signal. (A) Images of a myocyte before (first image) and after exposure to 1 μM FCCP. The images are at 10-s intervals. FCCP elicited an immediate decrease in FI, characteristic of mitochondrial depolarization. Scale bar is 5 μm. (B) The trace represents the FI (mean ± SE on a log scale) of 34 mitochondria before and after the application of 1 μM FCCP to the cell shown in A. The arrows above the trace indicate the time points for which images are shown in A. The last point on the trace, labeled as BG, represents the mean background FI outside the cell.
Transient depolarizations of ΔΨm: mitochondrial flickers
Two successive image sequences were obtained from each cell. Examples of spontaneous, brief fluctuations in mitochondrial FI are shown in the images of Fig. 4, A and C, and quantified in Fig. 4, B and D. These fluctuations indicate transient changes in ΔΨm. These examples provide an indication of the range in magnitude, frequency, and time-course of the mitochondrial flickers. Results are presented on a log scale as the change in ΔΨm is proportional to the log of the ratio of the free [TMRE]m before and during depolarization. The traces show the changes in ΔΨm during a flicker rather than absolute ΔΨm. It can be seen from the images that the mitochondria flickered independently of their neighbors. Synchronous flickering among neighboring mitochondria was observed rarely, as expected from a chance event. This suggests that mitochondria are not functionally coupled in these cells.
FIGURE 4.

Localized transient depolarizations of ΔΨm. A and C show a selection of images taken of two cells. Each image shows one plane of a three-dimensional image stack. The arrows point to mitochondria in which changes in ΔΨm were observed during the image sequence. A decrease in FI denotes mitochondrial depolarization. Scale bars are 5 μm. B and D are a graphical representation of the FI (on a log scale) of the mitochondria indicated in A and C. The arrows above the traces indicate the time points for which images are shown. The scale bar on the right shows the change in ΔΨm (mV). Flickers vary in amplitude, frequency, time to depolarize, and duration of depolarization.
Experiments were conducted in the absence of Kreb's cycle substrates as done in previous studies on the cell type employed here (Drummond et al., 2000; ZhuGe et al., 1999). To determine whether substrate deprivation caused mitochondrial flickers, sodium pyruvate was added to the bath solution. Flickers were observed in the presence of pyruvate, indicating that flickers do not result from substrate deprivation (data not shown).
A quantitative comparison of the flickers observed in the two consecutive image sequences was performed. The flicker parameters investigated were amplitude, the percentage of flickering mitochondria, and the frequency of flickers within flickering mitochondria. There was no significant difference in any of the parameters studied over the two image sequences, each lasting 95 s (Mann-Whitney U test). Therefore the data was grouped together. 83% of mitochondria (n = 150) flickered at least once during the two sequences. Over the entire mitochondrial population the flicker rate was 0.01 flickers per mitochondrion per second. 64.3 ± 5.9% of the mitochondria were observed to flicker during individual image sequences. 1.9 ± 0.1 flickers per sequence were observed in each flickering mitochondrion, i.e., the rate of flickering was 0.02 flickers per flickering mitochondrion per second.
Fig. 5 A demonstrates the frequency with which depolarizations of different amplitudes occurred. The mean amplitude was 17.6 ± 1.0 mV (n = 360); and ∼75% of the depolarizations were <20 mV in amplitude. The histogram in Fig. 5 B illustrates the distribution of resting mitochondrial FI. Interestingly, resting mitochondrial FI varies over a wide range (see Fig. 2). This suggests that mitochondria may exist at quite different RΔΨm values since more negative potentials should result in brighter fluorescence. The amplitudes of mitochondrial flickers are plotted as a function of the resting mitochondrial FI in Fig. 5 C. Larger flicker amplitudes are associated with brighter mitochondrial FI (more negative RΔΨm). The correlation, though low (r2 = 0.03), is highly significant (P ≤ 0.0005).
FIGURE 5.

Distribution of flicker amplitudes and resting FI. (A) The frequency with which depolarizations of different amplitudes occur. The distribution is skewed to smaller values. (B) The distribution of resting FI (FI before the onset of a flicker). Resting FI varies over a wide range, suggesting that mitochondria exist at different RΔΨm. (C) Amplitudes of mitochondrial flickers plotted as a function of the resting FI. Larger flicker amplitudes are associated with brighter resting FI. The correlation, though low, r2 = 0.03, is highly significant (P ≤ 0.0005). Solid circles are those points used for determination of maximum flicker amplitude (ΔVmax) at each RΔΨm·ΔVmax are plotted against resting FI before the flicker. The slope of the line is 58.1 mV per 10-fold change in FI m pol (r2 = 0.94).
In some cases flicker amplitudes may be underestimated. With a 5-s sampling frequency it is possible that, during shorter flickers, mitochondria might reach maximum depolarization between images. In addition, we have no information regarding the rate of dye disassociation from the inner mitochondrial membrane or the rate of dye diffusion across this membrane. If the rate at which depolarization proceeds exceeds the rate at which dye exits a mitochondrion (i.e., if the dye movement is rate-limiting), short-lived depolarizations may occur and start to reverse before dye equilibration has taken place. However, for ∼30% of flickers, the FI is stable for several time points. In these cases ΔΨm remains depolarized and the maximum depolarization can be detected. Analysis of this “plateau” data yields an average flicker amplitude of 31.7 ± 2.4 mV (n = 109), significantly greater (P < 0.0001) than the flicker amplitude calculated using all acquired data (i.e., 17.6 ± 1.0 mV). In either case the distributions appear quite similar but with fewer instances of the smallest depolarizations in the case of the distribution of flickers that have clearly reached a plateau.
DISCUSSION
Characterization of mitochondrial flickers
The mitochondrial flickers observed in the present study were transient events of varying duration with amplitudes ranging from 6 to 130 mV and a mean of 17.6 mV. Almost all of the mitochondria (83%) were observed to flicker at least once with an average frequency of 0.02 flickers per flickering mitochondrion per second. Thus flickers appear to be characteristic of virtually all mitochondria, at least in the smooth muscle cell type used here.
In most previous studies, analysis of flicker amplitude was limited to qualitative assessment and not translated into millivolt changes in ΔΨm with some notable exceptions. Loew et al. (1993) did not observe binding of TMRE to neuroblastoma cell membranes, and thus used the Nernst equation to calculate ΔΨm and flicker amplitude, and a similar approach was taken by Diaz et al. (2000). Duchen et al. (1998) used proportional changes in FI unquenching to assess flicker amplitudes indirectly. Despite the different methodologies, the mean value of 17.6 mV found here is in broad agreement with these earlier reports in all of which transient mitochondrial depolarizations were assessed as falling in the range of ∼5–20 mV. Furthermore, as in the study by Jacobson and Duchen (2002), flicker amplitudes in the present work do not exhibit a Gaussian distribution but instead are skewed toward smaller values (Jacobson and Duchen, 2002).
As a rule mitochondria flickered independently of one another and there was a large variation in RΔΨm. Thus, it appears that in these smooth muscle cells, mitochondria operate individually and not as a network. Similarly, transient mitochondrial depolarizations occur asynchronously in neuroblastoma, endothelial cells, and astrocytes (Fall and Bennett, 1999; Hüser and Blatter, 1999; Jacobson and Duchen, 2002). However, simultaneous flickers have been observed in cardiac myocytes, astrocytes, hepatocytes, and cos-7 cells (Collins et al., 2002; De Giorgi et al., 2000; Diaz et al., 2000; Zorov et al., 2000). It appears that in some cell types, mitochondria are electrically isolated from one another and in others, part of a continuous syncitium.
Quantitative characteristics of flickers and mechanisms for flicker generation
At high resting FI (more negative RΔΨm), flickers are larger in amplitude (Fig. 5 C). This has not been previously reported, and it suggests that an ion channel or electrogenic ion exchanger must cause flickers. When a channel or exchanger in the mitochondrial membrane is activated, it drives ΔΨm toward a defined reversal potential (Vrev). If maximal flicker amplitude at a given RΔΨm (ΔVmax) (caused by the longest period of channel or carrier activation) drives the membrane potential close to Vrev, then at any given potential
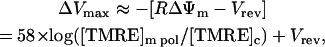 |
with pol being the resting or polarized state for a mitochondrion. Therefore a line drawn to connect the maximum flicker amplitudes at each level of resting TMRE fluorescence should have a slope of 58 mV per 10-fold change in resting mitochondrial FI. If such a line is fit to the solid points in Fig. 5 C, the slope is 58.1 mV per 10-fold change in FI, in agreement with this hypothesis. Thus, either an ion channel or an electrogenic carrier could be responsible for mitochondrial flickers.
To the best of our knowledge, this is the first study of mitochondrial flickers which quantitates flicker amplitude over a large population of flickering mitochondria. The data suggests that an ion channel or electrogenic exchanger in the mitochondrial inner membrane underlies these transient depolarizations in ΔΨm. The quantitative analysis of flickers should be useful in the further elucidation of the mechanism and role of mitochondrial flickers in smooth muscle and other cell types.
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance of the late Rebecca McKinney. Thanks to Larry Lifshitz and Les Loew for comments and discussions.
This research was funded by National Institutes of Health grant HL61297 to J.V.W.
Robert M. Drummond's present address is Department of Physiology and Pharmacology, University of Strathclyde, Glasgow, U.K.
References
- Buckman, J. F., and I. J. Reynolds. 2001. Spontaneous changes in mitochondrial membrane potential in cultured neurons. J. Neurosci. 21:5054–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington, W. A., K. E. Fogarty, and F. S. Fay. 1990. 3D fluorescence imaging of single cells using image restoration. In Non-Invasive Techniques in Cell Biology. K. Foster, editor. Wiley-Liss, New York. 53–72.
- Carrington, W. A., R. M. Lynch, E. D. Moore, G. Isenberg, K. E. Fogarty, and F. S. Fay. 1995. Superresolution three-dimensional images of fluorescence in cells with minimal light exposure. Science. 268:1483–1487. [DOI] [PubMed] [Google Scholar]
- Collins, T. J., M. J. Berridge, P. Lipp, and M. D. Bootman. 2002. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 21:1616–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giorgi, F., L. Lartigue, and F. Ichas. 2000. Electrical coupling and plasticity of the mitochondrial network. Cell Calcium. 28:365–370. [DOI] [PubMed] [Google Scholar]
- Diaz, G., A. M. Falchi, F. Gremo, R. Isola, and A. Diana. 2000. Homogeneous longitudinal profiles and synchronous fluctuations of mitochondrial transmembrane potential. FEBS Lett. 475:218–224. [DOI] [PubMed] [Google Scholar]
- Drummond, R. M., T. C. H. Mix, R. A. Tuft, J. V. Walsh, Jr., and F. S. Fay. 2000. Mitochondrial Ca2+ homeostasis during Ca2+ influx and Ca2+ release in gastric myocytes from Bufo marinus. J. Physiol. 522:375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen, M. R. 1999. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. J. Physiol. 516:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen, M. R., A. Leyssens, and M. Crompton. 1998. Transient mitochondrial depolarizations reflect focal sarcoplasmic reticular calcium release in single rat cardiomyocytes. J. Cell Biol. 142:975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fall, C. P., and J. P. Bennett, Jr. 1999. Visualization of cyclosporin A and Ca2+-sensitive cyclical mitochondrial depolarizations in cell culture. Biochim. Biophys. Acta. 1410:77–84. [DOI] [PubMed] [Google Scholar]
- Fay, F. S., R. Hoffmann, S. Leclair, and P. Merriam. 1982. Preparation of individual smooth muscle cells from the stomach of Bufo marinus. Methods Enzymol. 85:284–292. [DOI] [PubMed] [Google Scholar]
- Femino, A. M., F. S. Fay, K. Fogarty, and R. H. Singer. 1998. Visualization of single RNA transcripts in situ. Science. 280:585–590. [DOI] [PubMed] [Google Scholar]
- Hüser, J., and L. A. Blatter. 1999. Fluctuations in mitochondrial membrane potential caused by repetitive gating of the permeability transition pore. Biochem. J. 343:311–317. [PMC free article] [PubMed] [Google Scholar]
- Hüser, J., C. E. Rechenmacher, and L. A. Blatter. 1998. Imaging the permeability pore transition in single mitochondria. Biophys. J. 74:2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson, J., and M. R. Duchen. 2002. Mitochondrial oxidative stress and cell death in astrocytes—requirement for stored Ca2+ and sustained opening of the permeability transition pore. J. Cell Sci. 115:1175–1188. [DOI] [PubMed] [Google Scholar]
- Loew, L. M., R. A. Tuft, W. Carrington, and F. S. Fay. 1993. Imaging in five dimensions: time-dependent membrane potentials in individual mitochondria. Biophys. J. 65:2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke, B. 2000. Pathophysiological and protective roles of mitochondrial ion channels. J. Physiol. 529:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaduto, R. C., Jr., and L. W. Grotyohann. 1999. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J. 76:469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhuGe, R., R. A. Tuft, K. E. Fogarty, K. Bellve, F. S. Fay, and J. V. Walsh, Jr. 1999. The influence of sarcoplasmic reticulum Ca2+ concentration on Ca2+ sparks and spontaneous transient outward currents in single smooth muscle cells. J. Gen. Physiol. 113:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov, D. B., C. R. Filburn, L. O. Klotz, J. L. Zweier, and S. J. Sollott. 2000. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 192:1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]