

Abstract
The living cell is inherently crowded with proteins and macromolecules. To avoid aggregation of denatured proteins in the living cell, molecular chaperones play important roles. Here we introduce a simple model to describe crowded protein solutions with chaperone-like species based on a dynamic density functional theory. As predicted by others, our simulations show that macromolecular crowding enhances the association of proteins and chaperones. However, when the intrinsic folding rate of the protein is slow, it is possible that crowding also enhances aggregation of proteins. The results of simulation suggest that, when the concentration of the crowding agent is as high as that in the cell, the association of the protein and unbound chaperone becomes correlated with the aggregation process, and that the protein-bound chaperones efficiently destroy the potential nuclei of aggregates and thus prevent the aggregation.
INTRODUCTION
The living cell is inherently crowded with various kinds of macromolecules (Fulton, 1982). It is suspected that such a crowded cell environment imposes significant effects on the properties of proteins compared to the in vitro environment (Minton, 2000, 2001; Ellis, 2001a,b). Some studies have revealed that macromolecular crowding increases the stability of G-actin (Tellam et al., 1983), enhances self-assembly of FtsZ (Rivas et al., 2001), and accelerates amyloid formation (Hatters et al., 2002; Uversky et al., 2002).
Van den Berg et al. (2000) have studied the oxidative folding of reduced hen lysozyme in crowded media and found that the fast track of folding pathways becomes even faster and the slow track even slower under crowded conditions. They have also compared the folding yields of oxidized and reduced lysozyme under various crowded conditions, and examined crowding effects on the folding of reduced lysozyme with or without protein disulfide isomerase (PDI), a folding catalyst (van den Berg et al., 1999). It was found that crowded conditions enhance aggregation of reduced (slow-folding) lysozyme and abolish its folding yield, whereas the folding yield of oxidized (fast-folding) lysozyme was hardly affected (van den Berg et al., 1999). Also found was that the chaperone activity of PDI on the reduced lysozyme folding is enhanced by crowding, which van den Berg et al. (1999) attributed to the increased binding affinity of PDI to non-native lysozyme. Martin and Hartl (1997) also observed the increased affinity of the chaperonin GroEL to non-native proteins under crowded conditions, so that the release of structurally immature substrate proteins from GroEL is prevented in the crowded media. These studies exemplify the interplay among macromolecular crowding, protein folding, and chaperone activity in the cellular context.
Interactions between biological molecules in crowded solutions are, of course, complex. However, as Minton (1997) emphasizes, “although other types of interactions may or may not be present, volume exclusion interactions will always be present” in crowded solutions. Therefore, one of the fundamental aspects of crowded solutions is the excluded volume effect—that caused by the volume excluded by “inert” macromolecules (or crowders) to the target species (the protein of interest). According to this interpretation of crowding effects, Minton have developed theories for a wide spectrum of macromolecular crowding effects (e.g., Minton, 1998). Although thorough and, in some cases, even quantitative (Zimmerman and Minton, 1993; Minton, 2001), most of these theories are based on theories of uniform fluid, so that it cannot directly treat inhomogeneous system such as one containing aggregates of proteins.
To complement Minton's theory, we have recently developed a theoretical framework (Kinjo and Takada, 2002a,b) based on a density functional theory (e.g., Hansen and McDonald, 1986) that can be directly applied to inhomogeneous systems (here, “density” is a synonym of “concentration”). This density functional formulation made it possible to investigate protein stability, folding, and aggregation in a unified framework. By solving the equation of state derived from this theory, we have obtained theoretical phase diagrams with respect to various physical parameters (Kinjo and Takada, 2002a). Fig. 1 shows simplified phase diagrams with bulk crowder concentration (number density, ρC) and bulk protein concentration (number density, ρP) as variables. Fig. 1 A represents a mildly aggregating condition with relatively weak attraction between denatured proteins, and Fig. 1 B, a strongly aggregating condition with strong attraction between denatured proteins. Both phase diagrams exhibit essential features of the crowding effect on protein stability and aggregation tendency. Our assumptions are that the native protein is more compact than the denatured one, and that intermolecular attraction acts only between the denatured proteins and all other interactions are purely repulsive, excluded, volume interactions. There are three phases observed: the first phase is the aggregation phase (AD) in which one spherical aggregate of denatured proteins is formed; the second is a uniform phase in which the native protein is dominant (UN); and the third is another uniform phase in which the denatured protein is dominant (UD). Fig. 1 shows that, as the bulk crowder concentration increases, the aggregation is enhanced, but that the native protein is more stabilized as long as the aggregation does not take place.
FIGURE 1.

Phase diagram with the bulk concentrations of the crowder (ρC) and protein (ρP) as variables. No chaperone is present. The thick solid line indicates the phase boundary between aggregation and uniform phases. The dashed line corresponds to denaturation midpoint where the native and denatured proteins are of equal amount. UN and UD stand for the uniform phase in which proteins are dissolved in the solvent and the native or denatured protein is dominant, respectively, and in the AD phase there is one spherical aggregate of the denatured protein. (A) Mildly aggregating condition with weak attraction between denatured proteins (ɛD,D = −0.058); (B) strongly aggregating condition with strong attraction between denatured proteins (ɛD,D = −0.11).
The density functional theory was further extended to treat dynamical phenomena such as aggregation process under crowded conditions based on generalized diffusion-reaction equations (Kinjo and Takada, 2002b). It was shown that, for intrinsically fast-folding proteins (i.e., fast-folding in dilute solutions), crowding can kinetically inhibit aggregation of denatured proteins by accelerating the folding rate (Fig. 3 A), whereas, for intrinsically slow-folding proteins, the aggregation of denatured proteins is always enhanced and accelerated (Fig. 3 B) (Kinjo and Takada, 2002b).
FIGURE 3.

Time course of the fraction of the native protein fN with or without chaperones at various concentration of the crowder ρC = 0.05, 0.2, 0.4, and 0.8. (A) Fast-folding protein (ηTS = 1) without chaperones; (B) slow-folding (ηTS = 5) without chaperones; (C) fast-folding protein with chaperone (ρH = 0.01); and (D) slow-folding protein with chaperones.
In the living cell, however, there exist slow-folding proteins and they do fold to their native structures under highly crowded cellular environments in which amorphous aggregates of denatured proteins are not found. This is possible, at least in part, due to the presence of molecular chaperones which assist folding and/or prevent aggregation. In the present article, we introduce molecular species that mimic chaperone's activity to bind proteins and release them in either native or denatured states (Fig. 2), and show that this model chaperone can inhibit aggregation of slow-folding proteins. Among various mechanisms suggested for chaperones' activity (Thirumalai and Lorimer, 2001; Hartl and Hayer-Hartl, 2002), the present model is related to the kinetic proofreading scheme proposed by Gulukota and Wolynes (1994), which was later refined as the iterative annealing scheme (Thirumalai and Lorimer, 2001). However, our main interest here is the excluded volume or macromolecular crowding effects on chaperone activity, protein folding, and aggregation as well as their correlations. In addition to the chaperone's activity itself, the importance of the large-scale spatial correlations between proteins and chaperones is emphasized. The results of the simulations suggest that crowding enhances not only the association of the chaperone and protein, but also the correlation between the association and the aggregation of the denatured proteins so that the chaperones can efficiently prevent the aggregation.
FIGURE 2.

A schematic diagram of the network of reactions in the system including self-folding/unfolding, chaperone binding/unbinding, and aggregation. The symbols N, D, X, and Y indicate the native protein, the denatured protein, the unbound chaperone, and the bound chaperone species, respectively.
THEORY AND MODELING
Density functional theory
The details of the density functional formulation are described in our previous articles (Kinjo and Takada, 2002a,b), which we briefly summarize here with necessary extension to include chaperone-like species.
In the system considered, there are three chemical species: one protein species (denoted P), one crowder species (C), and one molecular chaperone species (H). The crowders impose only excluded volume interactions on themselves and on other species. The chemical species are further decomposed into physical species depending on their state. That is, the protein can be either in the native state (N) or in the denatured state (D), and the chaperone can be either in its unbound form (X) without bound substrate protein or bound form (Y) with bound substrate protein. Each physical species is characterized by its intrinsic free energy, ηα (α = N, D, C, X, and Y), in dilute solution. All the interactions between physical species, uα,β(r) where α,β = N, D, C, X, and Y, and r is intermolecular distance, are assumed to be isotropic and of hard-core square-well potential,
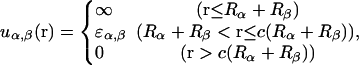 |
(1) |
where Rα is the effective radius of the physical species α, and c is a constant (which we arbitrarily set as c = 3). For simplicity, we set ɛα,β = 0 except for ɛD,D and ɛD,X (=ɛX,D). Since denatured proteins tend to expose their hydrophobic residues and hydrogen-bonding groups on their surface, which cause aggregation, it is natural to assume attractive interaction between them, hence ɛD,D < 0. To be consistent with the previous work (Kinjo and Takada, 2002b), we set ɛD,D = −0.11 (the unit is described in the Numerics subsection), which corresponds to Fig. 1 B, a strongly aggregating condition. Unbound chaperone (X) preferentially binds non-native proteins, thus ɛD,X < 0 should be reasonable.
The system is described by the number density (concentration) of physical species at each point r in space, φα(r). The free energy functional (density functional) of the system, F[{φα}], consists of the ideal part and non-ideal part: F = Fi + Fn. The ideal part, Fi, consists of the intrinsic free energy and mixing entropy terms,
 |
(2) |
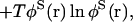 |
where φs(r) is the effective density of solvent defined by φS(r) = ρ0 − Σαφα(r) with the constant ρ0 being the bulk density of the system, and the Boltzmann constant kB is unity in the current unit (see below). The non-ideal part of the free energy originates from the intermolecular interactions. If we take into account up to two-body correlation and if thus obtained effective interaction is short-ranged, we obtain
 |
(3) |
where 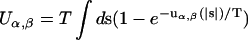 and
and  can be regarded as excluded volume parameters and surface tension parameters, respectively (Kinjo and Takada, 2002a). The local chemical potential, μα(r), of the species α at r can be obtained from the relation μα(r) = δF/δφα(r), the functional derivative of the free energy F with respect to the density field φα(r):
can be regarded as excluded volume parameters and surface tension parameters, respectively (Kinjo and Takada, 2002a). The local chemical potential, μα(r), of the species α at r can be obtained from the relation μα(r) = δF/δφα(r), the functional derivative of the free energy F with respect to the density field φα(r):
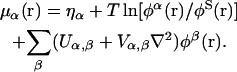 |
(4) |
Accordingly, the local (absolute) activity is defined by aα(r) = eμα(r)/T within a constant factor.
Dynamics equations
The dynamics of the system consists of diffusion of physical species, folding-unfolding reactions of proteins (N ↔ D), and binding and unbinding between proteins and molecular chaperones (D + X ↔ Y ↔ N + X). These reactions are depicted in Fig. 2. Aggregation of the denatured proteins—D(monomers) ↔ D(aggregate)—occurs by diffusion. Note that the apparent reversibility does not necessarily mean that the reaction D(aggregate) → D(monomers) can actually occur in biologically relevant timescale: depending on free energies of D(aggregate) relative to D(monomer), the aggregation can be practically irreversible. Hence, we assume the following generalized diffusion-reaction equations (Kinjo and Takada, 2002b):
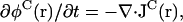 |
(5) |
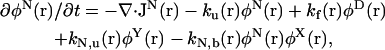 |
(6) |
 |
(7) |
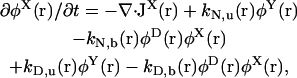 |
(8) |
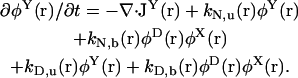 |
(9) |
The flux Jα(r) in the diffusion term is given as the spatial gradient of the local activity as a consequence of the non-ideality of crowded solutions: Jα(r) = −D0α(φS)2∇aα(r) where D0α is a diffusion constant. This is a generalization of Fick's law for diffusion which states that Jα(r) = −D0α∇φα(r).
The various reaction rate constants are site-dependent reflecting the non-ideality and inhomogeneity of the crowded system. The unfolding and folding reaction rates are given previously (Kinjo and Takada, 2002b) by ku(r) = k0 exp[−{ηTS + WTS(r) − ηN − WN(r)}] and 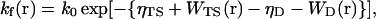 respectively, where ηTS parameterizes the intrinsic free energy of the transition state in dilute solution, and WTS, WN, and WD are the potentials of mean force of the folding-unfolding transition state, the native state, and the denatured state, respectively. The potential of mean force Wα(r) for α = N, D, C, X, and Y are defined by the third term on the RHS of Eq. 4, i.e., Wα(r) = Σβ(Uα,β + Vα,β∇2)φβ(r). The potential of mean force for the transition state is defined by WTS(r) = ωWN(r) + (1 − ω)WD(r) where ω is a constant which we set ω = 0.8 in the following (see Kinjo and Takada, 2002b).
respectively, where ηTS parameterizes the intrinsic free energy of the transition state in dilute solution, and WTS, WN, and WD are the potentials of mean force of the folding-unfolding transition state, the native state, and the denatured state, respectively. The potential of mean force Wα(r) for α = N, D, C, X, and Y are defined by the third term on the RHS of Eq. 4, i.e., Wα(r) = Σβ(Uα,β + Vα,β∇2)φβ(r). The potential of mean force for the transition state is defined by WTS(r) = ωWN(r) + (1 − ω)WD(r) where ω is a constant which we set ω = 0.8 in the following (see Kinjo and Takada, 2002b).
The chaperone-related reaction rates are modeled in the following way. For the reaction X + α → Y (α = N,D), the rate constant is defined by 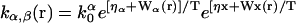 where k0α is a constant factor. For the reverse reaction Y → X + α, the rate constant is defined by
where k0α is a constant factor. For the reverse reaction Y → X + α, the rate constant is defined by 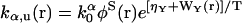 . Note that with this choice of rate constants, the equations
. Note that with this choice of rate constants, the equations 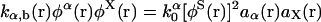 and
and 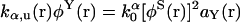 hold. In a similar manner as the previous article without the chaperone species (Kinjo and Takada, 2002b), it can be shown that the free energy is a decreasing function of time: dF/dt ≤ 0.
hold. In a similar manner as the previous article without the chaperone species (Kinjo and Takada, 2002b), it can be shown that the free energy is a decreasing function of time: dF/dt ≤ 0.
Numerics
The total bulk density of the system ρ0 (see Eq. 2) is set to unity. The radius of the native protein is set RN = 0.4 in an arbitrary unit of length. The denatured protein is expected to be more expanded by 50–100% in its radius than the native protein (e.g., Fersht, 1999), hence we set RD = 0.6. For example, let the unit of length be 5 nm, then the radius of the native protein is 2 nm, and that of the denatured protein is 3 nm. Throughout this study, we consider the crowder that is of the same size as the native protein (RC = 0.4). With this choice of RC, the bulk crowder concentration of ρC = 0.8 corresponds to the volume fraction of ≈22% which is a biologically relevant value (Ellis, 2001a). The bound chaperone species, Y, is expected to be larger than the unbound counterpart, X, hence RY > RX should hold. We set RX = 0.4 and RY = 0.6. The temperature is defined to be the unit of energy, hence T = 1. Furthermore, this temperature is assumed to correspond to the folding temperature of the protein in dilute solution, hence ηN = ηD. The intrinsic free energy of the crowder species does not play any role in the theory so we set ηC = 0 for convenience. The relation between ηX and ηY is not trivial. For simplicity, we set ηX = ηY. After all, we have set ηα = 0 for all the physical species α = N, D, C, X, and Y. All the diffusion constants D0α are arbitrarily set to 0.1, and reaction constants are set to k0 = 0.1 as in the previous article (Kinjo and Takada, 2002b). In most cases, the chaperone-related reaction rates are set such that k0N = K0D = 0.1, but we also use the values K0N = 0.01 and 0.001 to examine the significance of chaperone's ability to assist protein folding.
The physical size of the system is set to 64 × 64 × 64 in the given unit length, which is discretized into 32 × 32 × 32 lattice sites. Therefore, if the native protein has RN = 2 nm, then the linear length of the system corresponds to 0.32 μm. The periodic boundary condition is imposed on all directions. The differential operation with respect to the spatial coordinates is calculated using the 27-point isotropic stencil of van Vlimmeren and Fraaije (1996). For the details of the discretization scheme, see Kinjo and Takada (2002b). The numerical integration of the diffusion-reaction equations (Eqs. 5–9) are carried out with the second-order, variable time step Runge-Kutta-Chebyshev method of Abdulle and Medovikov (2001).
RESULTS AND DISCUSSION
In the following, the average concentration (density) of the protein, ρP, is always set to ρP = 0.1, and that of the chaperone to ρH = 0.01. The protein concentration ρP = 0.1 does not correspond to a dilute solution even in absence of other molecular species because this amount of protein can trigger aggregation (see Fig. 1 and below). Initially, most of the chaperones are set to be unbound to proteins: ρY = 0.001 ρH, and thus ρX = 0.999 ρH. Proteins are set initially mostly denatured so that the concentration of the native protein is set ρN = 0.001 ρP, and that of the denatured protein is set ρD = ρP − ρN − ρY. Initial concentrations are set weakly fluctuated in space such that φα(r) = ρα[1 + ξα(r)] where −0.1 < ξα(r) < 0.1 is a random field. We repeated simulations with different random fields but the results are hardly affected. As noted above, the temperature is set to the folding temperature of the protein in dilute solution in all the following simulations so that the intrinsic free energies of the native and denatured proteins are the same: ηN = ηD. This temperature condition can be regarded as a mild heat-shock condition.
In most of the cases, the time course of simulations is monitored in terms of the fraction of the native protein, fN = 〈φN〉 / ρP where 〈·〉 is the spatial average (i.e., 〈·〉 = (1/V) f × dr with V being the volume of the system). Although not shown explicitly, aggregation in the system is monitored in terms of relative spatial deviation of a density field: sα = [〈(φα − 〈φα〉)2 〉]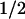 /〈φα〉. If the value of sα (e.g., sN) is very small (i.e., sN < 10−8), the system is considered as uniform, otherwise the system is non-uniform and contains aggregates.
/〈φα〉. If the value of sα (e.g., sN) is very small (i.e., sN < 10−8), the system is considered as uniform, otherwise the system is non-uniform and contains aggregates.
Effects of molecular chaperones on fast- and slow-folding proteins
First, the simulation results without molecular chaperones (i.e., ρH = 0) are shown as a reference (Fig. 3, A and B). Folding (initially denatured) simulations of fast- and slow-folding proteins can be performed by adjusting the parameter for the intrinsic free energy of the folding transition state, ηTS. As in our previous study, we set ηTS = 1 for the fast-folding protein, and ηTS = 5 for the slow-folding one (Kinjo and Takada, 2002b). As was shown previously, these two cases show qualitatively different behavior regarding folding and aggregation (Fig. 3, A and B). That is, the fast-folding protein forms aggregates at low bulk concentrations of the crowder (ρC = 0.05, 0.2), but becomes dissolved in more crowded solutions ρC = 0.4, 0.8 (Fig. 3 A), whereas the slow-folding protein exhibits the aggregation of denatured proteins at all the tested crowder concentrations (Fig. 3 B). Note that the drops in the fraction of the native protein, fN, toward zero means aggregation in these cases (Kinjo and Takada, 2002b). It should also be noted that thermodynamically, as Fig. 1 indicates, the aggregation of the denatured proteins is enhanced by crowding (Kinjo and Takada, 2002a) regardless of the folding rate of the protein. Therefore, the inhibition of aggregation by crowding for the fast-folding protein is a kinetic one.
Next the molecular chaperone species was added with the bulk concentration ρH = 0.01. The attraction parameter between the denatured protein and unbound form of the chaperone was set to a small negative value, ɛD,X = −0.01, compared to the attraction between the denatured proteins, ɛD,D = −0.11. In Fig. 3, C and D, we see that the fast- and slow-folding proteins show qualitatively the same behavior regarding folding and aggregation. In both the fast- and slow-folding cases, aggregation occurs at low concentrations of the crowder, ρC = 0.05, 0.2, but not at higher concentrations ρC = 0.4, 0.8. Closer examination showed that aggregation takes place for the crowder concentration ρC ≤ 0.34 for both the fast- and slow-folding cases with chaperone. However, this value depends on the choice of the rate constants k0N and K0D (see below). It is not trivial that the present model for the chaperone can prevent the aggregation of the denatured proteins, because not only the reaction D + X → Y but also the reactions Y → D + X and N + X → Y are included in the dynamics. The denatured proteins released from the chaperones can, in principle, again attract each other and form aggregates.
By comparing Fig. 3, B and D, we can see that the acceleration of folding of the slow-folding protein by the chaperone is most pronounced in the case with ρC = 0.8, the most crowded condition. On the other hand, the acceleration of the folding of the fast-folding protein is not so pronounced even at ρC = 0.8.
To see the effect of crowding on the stability of the bound chaperone species Y, the time course of its fraction, fY = 〈φY〉/ρH, is plotted in Fig. 4. It can be seen that crowding enhances the formation of the bound chaperone Y. There is no apparent difference between the cases of the fast-folding and slow-folding proteins. When the crowder concentration ρC is small (ρC = 0.05 and 0.2), there is not a significant amount of the bound chaperone, and fY approaches to zero as aggregation proceeds. When the aggregation of the denatured protein does not occur (ρC = 0.4, 0.8), the value of fY approaches to a finite constant as the system becomes uniform (fY ≈ 0.1 or 0.47 for ρC = 0.4 or 0.8, respectively). The difference between the cases for ρC = 0.4 and ρC = 0.8 is significant in both the rates of increase and final values of fY. In terms of the equilibrium constant fY/fX, the bound form of the chaperone Y is more favored by more than eightfold when the crowder concentration ρC is increased from ρC = 0.4 to ρC = 0.8.
FIGURE 4.

Time course of the fraction of the bound chaperone species. (A) Fast-folding protein; (B) slow-folding protein.
In terms of the native fraction fN and bound chaperone fraction fY, the fast- and slow-folding proteins show qualitatively the same behavior for folding in the presence of the chaperone species (Figs. 3 and 4). The difference becomes apparent if the folding yield is divided into the “self-folding” and “chaperone-assisted folding” parts (Fig. 5). The (instantaneous) self-folding yield, GP, is defined by GP = 〈kf(r)φD(r) − ku(r)φN(r)〉, and the (instantaneous) chaperone-assisted folding yield, GH = 〈ku,N(r)φY(r) − kb,N(r)φN(r)φX(r)〉. It is evident from Fig. 5 that the self-folding yield GP is dominant for the fast-folding protein, but for the slow-folding protein, the chaperone-assisted folding yield GP is dominant and self-folding yield GP is relatively negligible. In both fast- and slow-folding cases, the increase in the crowder concentration significantly increases the chaperone-assisted folding yield. The peaks in GH for the slow-folding protein are slightly higher and significantly broader than those for the fast-folding protein; see Fig. 5, C and D. The fast-folding protein in the present study should correspond to a class of proteins which do not require chaperone's assistance for its folding (Thirumalai and Lorimer, 2001).
FIGURE 5.

Self-folding yield GP and chaperone-assisted folding yield GH for the fast-folding protein (A) and the slow-folding protein (B). Colors in A and B are indicated as follows: red, GP with ρC = 0.4; green, GH with ρC = 0.4; blue, GP with ρC = 0.8; and magenta, GH with ρC = 0.8. C and D compare the chaperone-assisted folding yield GH for fast-folding and slow-folding proteins, ρC = 0.4 in C, and ρC = 0.8 in D. Note that vertical scales are different in C and D.
To examine the significance of chaperone's activity to assist the folding of the slow-folding protein, we also carried out simulations with slower rate constants k0N for the reactions N + X ↔ Y. With the rate k0N = 0.01, aggregation was observed for ρC ≤ 0.4, but not for ρC = 0.8. With k0N = 0.001 and ρC = 0.8, the aggregation did occur, but by increasing the crowder concentration to ρC = 0.85 the aggregation was again inhibited (data not shown). Therefore, although crowding can enhance the chaperone's activity, the chaperone must intrinsically have its ability to assist protein folding. Chaperone's ability to bind denatured proteins is not sufficient to avoid aggregation.
Spatial correlations between the protein and molecular chaperone
Protein folding, chaperone binding, and protein aggregation—all of these processes take place inhomogeneously in space. In particular, induced spatial inhomogeneity plays a crucial role in aggregation. As mentioned above, the current density functional formalism can directly treat this inhomogeneity. Here we look into the above processes more closely for the intrinsically slow-folding case because it is in this case where the molecular chaperone plays a critical role. The parameters are the same as in the previous section (ɛD,X = −0.01, k0N = 0.1). The spatial correlation between the proteins and chaperones can be monitored in terms of the quantity Cα,β, which is defined by Cα,β = 〈(φα − 〈φα〉)(φβ − 〈φβ〉)〉/〈φα〉〈φβ〉 where α = N,D and β = X,Y. The positive value of Cα,β means that the species α and β, on average, tend to be close to each other in space.
The time evolution of the correlation shows similar tendency for ρC = 0.05, 0.2, and 0.4, except that aggregation does not take place for ρC = 0.4. Fig. 6 A shows the case with ρC = 0.05. The correlation between the denatured protein and the bound chaperone starts to increase at time t ∼ 0.01, which corresponds to the onset of increase in the bound chaperone fraction shown in Fig. 4 B. This positive correlation corresponds to the fact that the bound chaperone Y is likely to be created at the sites where the denatured proteins are relatively abundant. Then the positive correlation between the native protein and bound chaperone starts to appear at t ∼ 0.1, indicating that the native proteins are likely to appear near the bound chaperones, that is, the native proteins are released from the bound chaperones. In fact, the peak in CN,Y roughly corresponds to the onset of increase in the native fraction shown in Fig. 3 D. Until the aggregation of the denatured proteins is triggered, there is no apparent correlation between the native protein and the unbound chaperone (i.e., CN,X ≈ 0). This suggests that the native proteins released from the chaperones quickly diffuses away from them, which in turn explains the negative correlation between the denatured protein and unbound chaperone at t ≈ 1. That is, after the native proteins diffuse away from the chaperones, the proteins unfold and start to aggregate at the sites where the chaperones are less abundant. Due to the fast diffusion of proteins away from the chaperones, proteins are unlikely to stay near the chaperone whether the proteins are native or denatured.
FIGURE 6.

Time course of density field correlations with the bulk concentration ρC = 0.05 in A and ρC = 0.8 in B.
The case with ρC = 0.8 (Fig. 6 B) exhibits quantitatively and qualitatively different behavior from those with ρC ≤ 0.4. The correlations are in general much stronger than those for ρC ≤ 0.4. The correlation between the denatured protein and bound chaperone starts to increase earlier at t ∼ 10−4 and its height is higher than that for ρC ≤ 0.4. However, this positive correlation is not conspicuous when visualized as cross-sections of local concentrations in Fig. 7, A and B, in which the blue spots indicate the sites where both the denatured and chaperone concentrations are higher than their respective average values. On the other hand, the correlation between the native and unbound chaperone is very strong at t ∼ 1 which is clearly seen in Fig. 7, C and D. The correlation between the native protein and bound chaperone now shows a negative peak at t ∼ 0.1 but it soon becomes positive at t ∼ 1. The behavior of the correlation CD,X is also completely different from that for ρC ≤ 0.4. That is, for the case with ρC = 0.8, the correlation is positive whereas it was negative for the cases with ρC ≤ 0.4. This positive correlation indicates that the denatured proteins tend to exist around the unbound chaperones, which facilitates the reaction D + X → Y so that the danger of aggregation is reduced. Time course of these reactions is summarized in Table 1. Although not apparent in the values of Cα,β, a large-scale spatial correlation is observed between the protein and chaperone density fields at t ∼ 1000 (Fig. 8, A and B).
FIGURE 7.

Correlation of density fields in terms of density distribution on a cross-section of the system with ρC = 0.8. Red sites indicate that φα(r) > 〈φα〉 and φβ(r) < 〈φβ〉; green sites, φα(r) < 〈φα〉 and φβ(r) > 〈φβ〉; blue sites, φα(r) > 〈φα〉 and φβ(r) > 〈φβ〉; and black sites, φα(r) < 〈φα〉 and φβ(r) < 〈φβ〉, where α = D and β = Y and in A; α = N in B; and β = X in C and D.
TABLE 1.
Time course of the system with ρC = 0.8 and chaperones (corresponding to Fig. 6 B)
| Time range | Reaction | Correlation |
|---|---|---|
| ∼10−2 | D + X → Y proceeds | CD,Y > 0 |
| ∼10−1 | D → N proceeds far from chaperones | CN,Y > 0 |
| ∼1 | Y → N + X proceeds | CN,X > 0 |
| ∼10 | N → D proceeds near X | CD,X > 0 |
| N + X → Y proceeds | CN,Y > 0 | |
| D + X → Y proceeds | CD,Y > 0 |
FIGURE 8.

Large-scale correlations of density fields at a late stage in terms of density distribution on a cross-section of the system with ρC = 0.8. Color scheme is the same as Fig. 7 with α = N and β = X in A; and α = N and β = Y in B.
It was shown above that crowding enhances the aggregation of the denatured proteins in the absence of the chaperone (Fig. 3 B) as well as the formation of the bound chaperone (Fig. 4 B). Also shown was that, under the presence of the chaperone, crowding prevents the aggregation of the denatured proteins and enhances the correlation between the denatured protein and the bound chaperone (Fig. 6). Can we further expect that crowding enhances the correlation between the aggregation and the formation of the bound chaperone? This expectation is clearly confirmed by Fig. 9, which shows that the denatured proteins and bound chaperones are spatially more correlated when ρC = 0.8 (Fig. 9 B) than when ρC = 0.4 (Fig. 9 A). In other words, the bound chaperones are likely to exist in a potential nucleus of the aggregate of the denatured proteins under more crowded conditions. However, since the bound chaperone imposes repulsion on its surrounding as well as releases native proteins, it acts to disperse and destroy the potential nucleus of the aggregate. Hence, by strongly correlating the precursor of protein aggregates with the formation of the bound chaperones, crowding can help the chaperone to efficiently assist protein folding and to prevent aggregation.
FIGURE 9.

Correlation of the denatured protein and bound chaperone density fields with ρC = 0.4 (A) and ρC = 0.8 (B) at time t = 100. Coloring scheme is the same as Fig. 7.
We have also examined an extreme case in which the attraction between the denatured protein and unbound chaperone is very strong, ɛD,X = −0.1. This value is comparable to the attraction between the denatured proteins (ɛD,D = −0.11) and is so strong that aggregation, when it occurred, involved not only the denatured proteins but also the unbound chaperones. However, at the crowder concentration of ρC = 0.8, the result was essentially the same as above, that is, the aggregation did not take place and the final fraction of the native protein was almost the same as in the case with ɛD,X = −0.01 (data not shown).
Validity of the model molecular chaperone
In the present study, we have introduced a crude model for the molecular chaperone. The internal structure and mechanism of the molecular chaperone are not treated explicitly. We examine the extent to which the present model represents the characteristics of real molecular chaperones. First of all, no external energy source is exerted upon the system in the present simulations. Therefore, those chaperones which require ATP hydrolysis to catalyze protein folding cannot be fully represented by our model. Some molecular chaperones are known not to require ATP. Examples are the HSP110 family of heat shock proteins, and a class of protein-folding catalysts such as peptidyl-prolyl isomerase or protein disulfide isomerase (Ohtsuka, 2001). The role of ATP hydrolysis in molecular chaperone's ability to assist protein folding and to avoid aggregation is left for future study. A straightforward prediction is that exerting external energy via ATP hydrolysis will shift the equilibrium of the protein toward its native state, and thus aggregation may be inhibited at lower crowder concentrations than the present study.
Secondly, due to the limitation of the theoretical framework, only those chaperones whose sizes, in terms of the effective radii, are comparable to those of the target proteins can be treated. This limitation stems from the parameter ρ0 in the present treatment of mixing entropy (see Theory and Modeling). Because of the value of ρ0 = 1, it becomes technically difficult to set Rα > ≈0.6, otherwise the volume fraction can exceed unity. Nevertheless, if the unbound and bound chaperones were larger in their size, the results would be qualitatively the same and quantitatively become more exaggerated, because the reaction D + X → Y will be more enhanced and the repulsion caused by the bound chaperone (Y) on its surrounding will be more stringent due to the larger excluded volume.
Apart from the above limitations, the present model captures essential features of molecular chaperones in that binding of denatured proteins is preferred to that of native proteins, and that after bound complex (Y) is formed the chaperone can release either the denatured or native proteins. The reaction N + X → Y may seem to contradict the reality. This reaction can be interpreted as the reaction between the unbound chaperone and instantaneous non-native protein which exists in equilibrium with the native one (Thirumalai and Lorimer, 2001). In fact, this reaction with the rate kN,b(r) becomes increasingly unfavorable as the native protein becomes intrinsically more stable than the denatured one (see the expression for the site-dependent reaction rate kN,b(r) in Theory and Modeling). This behavior is also consistent with the consideration of Thirumalai and Lorimer (2001).
There are a number of adjustable parameters regarding the intrinsic stability of each molecular species and the strength of interaction between each couple of species as well as the rates of various chemical reactions. We examined various different sets of parameters although it is difficult to cover all the possible combinations of these parameters. In particular, we have tested the dependence of our results on the strength of attraction between the denatured protein and the unbound chaperone (ɛD,X), and the rate of reaction N + X ↔ Y (k0N). The robust observations thus far obtained are that chaperone's activity to assist protein folding (the path Y → N + X) is required to avoid aggregation, and that, for the crowder concentration ρC ≥ 0.8, aggregation is inhibited irrespective of the strength of ɛD,X.
Biological implications
The average concentration of the crowder, ρC, ranges from 0.05 to 0.8 in most of the calculations above. With the given radius of the crowder (RC = 0.4), the crowder concentration ρC = 0.8 corresponds to the volume fraction of ≈22%. On the other hand, the estimated volume fraction of the living cell is ≈20–30% (Ellis, 2001a). Therefore, among the above simulations, those with ρC = 0.8 are the most biologically relevant ones. With the present choice of the parameters for the interaction strength and reaction rates, the aggregation of the denatured proteins and the reaction D + X → Y can take place in the absence of the crowders. When the crowder concentration is low, the aggregation and the reaction D + X → Y can occur independently at different sites in the system. Both of these two reactions are enhanced by crowding. This raises an interesting possibility for the interplay among crowding, chaperone-assisted protein folding, and aggregation. The aggregation and chaperone-binding reactions become more correlated as the crowder concentration increases. In other words, when the system is sufficiently crowded, the D + X → Y reaction is more likely to take place near the nucleus of an aggregate. The bound chaperone Y either imposes strong repulsion on its surrounding, or it releases the native and/or denatured proteins. As a result, the potential nuclei of aggregates become unstable and their surrounding becomes heterogeneous, which hinder the further growth of the nucleus and prevent the aggregation. The present results suggest that the inhibition of aggregation by molecular chaperones becomes pronounced under conditions as crowded as the intracellular environment, and less crowded conditions may only enhance the aggregation when there are a small number of chaperones compared to denatured proteins. This implies that the crowded environment of the living cell is not merely a consequence of the existence of many kinds of macromolecules, but may have an active role in preventing aggregation in the cell.
Acknowledgments
A.R.K. thanks Prof. Hiroyuki Toh for suggesting the possibility of the present work.
Akira R. Kinjo's present address is the Center for Information Biology and DNA Data Bank of Japan, National Institute of Genetics, Mishima, Shizuoka 411-8540, Japan.
References
- Abdulle, A., and A. A. Medovikov. 2001. Second order Chebyshev methods based on orthogonal polynomials. Numer. Math. 90:1–18. [Google Scholar]
- Ellis, R. J. 2001a. Macromolecular crowding: an important but neglected aspect of the intracellular environment. Curr. Opin. Struct. Biol. 11:114–119. [DOI] [PubMed] [Google Scholar]
- Ellis, R. J. 2001b. Macromolecular crowding: obvious but underappreciated. Trends Biochem. Sci. 26:597–604. [DOI] [PubMed] [Google Scholar]
- Fersht, A. 1999. Structure and Mechanism in Protein Science. W. H. Freeman & Company, New York.
- Fulton, A. B. 1982. How crowded is the cytoplasm? Cell. 30:345–347. [DOI] [PubMed] [Google Scholar]
- Gulukota, K., and P. G. Wolynes. 1994. Statistical mechanics of kinetic proofreading in protein folding in vivo. Proc. Natl. Acad. Sci. USA. 91:9292–9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, J. P., and I. R. McDonald. 1986. Theory of Simple Liquids, 2nd Ed. Academic Press, London.
- Hartl, F. U., and M. Hayer-Hartl. 2002. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 295:1852–1858. [DOI] [PubMed] [Google Scholar]
- Hatters, D. M., A. P. Minton, and G. J. Howlett. 2002. Macromolecular crowding accelerates amyloid formation by human apolipoprotein C-II. J. Biol. Chem. 277:7824–7830. [DOI] [PubMed] [Google Scholar]
- Kinjo, A. R., and S. Takada. 2002a. Effects of macromolecular crowding on protein folding and aggregation studied by density functional theory: statics. Phys. Rev. E. 66:031911. [DOI] [PubMed] [Google Scholar]
- Kinjo, A. R., and S. Takada. 2002b. Effects of macromolecular crowding on protein folding and aggregation studied by density functional theory: dynamics. Phys. Rev. E. 66:051902. [DOI] [PubMed] [Google Scholar]
- Martin, J., and F. U. Hartl. 1997. The effect of macromolecular crowding on chaperonin-mediated protein folding. Proc. Natl. Acad. Sci. USA. 94:1107–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton, A. P. 1997. Influence of excluded volume upon macromolecular structure and associations in “crowded” media. Curr. Opin. Biotechnol. 8:65–69. [DOI] [PubMed] [Google Scholar]
- Minton, A. P. 1998. Molecular crowding: analysis of effects of high concentrations of inert cosolutes on biochemical equilibria and rates in terms of volume exclusion. Methods Enzymol. 295:127–149. [DOI] [PubMed] [Google Scholar]
- Minton, A. P. 2000. Implications of macromolecular crowding for protein assembly. Curr. Opin. Struct. Biol. 10:34–39. [DOI] [PubMed] [Google Scholar]
- Minton, A. P. 2001. The influence of macromolecular crowding and macromolecular confinement on biochemical reactions in physiological media. J. Biol. Chem. 276:10577–10580. [DOI] [PubMed] [Google Scholar]
- Ohtsuka, K. 2001. A list of molecular chaperones. In Regulation of Cellular Functions by Molecular Chaperones. K. Nagata, M. Mori, and M. Yoshida, editors. Springer-Verlag, Tokyo. 205–214.
- Rivas, G., J. A. Fernández, and A. P. Minton. 2001. Direct observation of the enhancement of noncooperative protein self-assembly by macromolecular crowding: indefinite linear self-association of bacterial cell division protein FtsZ. Proc. Natl. Acad. Sci. USA. 98:3150–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellam, R. L., M. J. Sculley, and L. W. Nichol. 1983. The influence of poly(ethylene glycol) 6000 on the properties of skeletal-muscle actin. Biochem. J. 213:651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirumalai, D., and G. H. Lorimer. 2001. Chaperonin-mediated protein folding. Annu. Rev. Biophys. Biomol. Struct. 30:245–269. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N., E. M. Coopera, K. S. Bowera, J. Lia, and A. L. Fink. 2002. Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 515:99–103. [DOI] [PubMed] [Google Scholar]
- van den Berg, B., R. J. Ellis, and C. M. Dobson. 1999. Effects of macromolecular crowding on protein folding and aggregation. EMBO J. 18:6927–6933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg, B., R. Wain, C. M. Dobson, and R. J. Ellis. 2000. Macromolecular crowding perturbs protein refolding kinetics: implications for folding inside the cell. EMBO J. 19:3870–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vlimmeren, B. A. C., and J. G. E. M. Fraaije. 1996. Calculation of noise distribution in mesoscopic dynamics models for phase separation of multicomponent complex fluids. Comput. Phys. Comm. 99:21–28. [Google Scholar]
- Zimmerman, S. B., and A. P. Minton. 1993. Macromolecular crowding: biochemical, biophysical, and physiological consequences. Annu. Rev. Biophys. Biomol. Struct. 22:27–65. [DOI] [PubMed] [Google Scholar]