

Abstract
Single particle fluorescence imaging (SPFI) uses the high sensitivity of fluorescence to visualize individual molecules that have been selectively labeled with small fluorescent particles. The positions of particles are determined by fitting the intensity profile of their images to a 2-D Gaussian function. We have exploited the positional information obtained from SPFI to develop a method for detecting colocalization of cell surface molecules. This involves labeling two different molecules with different colored fluorophores and determining their positions separately by dual wavelength imaging. The images are analyzed to quantify the overlap of the particle images and hence determine the extent of colocalization of the labeled molecules. Simulated images and experiments with a model system are used to investigate the extent to which colocalization occurs from chance proximity of randomly distributed molecules. A method of correcting for positional shifts that result from chromatic aberration is presented. The technique provides quantification of the extent of colocalization and can detect whether colocalized molecules occur singly or in clusters. We have obtained preliminary data for colocalization of molecules on intact cells. Cells often exhibit particulate autofluorescence that can interfere with the measurements; a method for overcoming this problem by triple wavelength imaging is described.
INTRODUCTION
Many functions of cell membranes depend on stable or transient associations between membrane proteins. Binding of ligands to cell surface receptors, for example, often promotes molecular associations that initiate complex signaling pathways. Molecular interactions may be facilitated by confining receptors within domains. Such domains may consist of lipid rafts (Simons and Ikonen, 1997) or be formed by cytoskeletal barriers that restrict free diffusion (Fujiwara et al., 2002). Determining the spatial organization of cell surface molecules is a key element in understanding a variety of membrane functions.
Fluorescence techniques are currently at the forefront of methods for observing events in living cells. In particular, fluorescence resonance energy transfer (FRET) is the most widely used method for detecting molecular associations in cells (Tron et al., 1984; Jovin and Arndt-Jovin, 1989; Matko and Edidin, 1997; Damjanovich et al., 1999; Angers et al., 2000; Kenworthy et al., 2000; Cohen-Kashi et al., 2002). FRET occurs between donor and acceptor probe molecules when their separation is of the order of a few nanometers (Stryer, 1978). The occurrence of FRET between donor and acceptors located on different molecules is thus evidence for their close proximity and is usually regarded as indicative of their molecular association. FRET may, however, fail to detect associations if relatively few molecules are associated and/or the donor-acceptor geometry is unfavorable. Most FRET experiments have not provided information on the fraction of molecules participating in an association or their stoichiometry although methods for overcoming this limitation have recently been proposed (Hoppe et al., 2002).
There has of late been much interest in techniques for detecting events at the level of single molecules (Basche et al., 2001). Such an approach can provide much more detailed information than that obtained from more traditional methods. Single particle fluorescence imaging (SPFI) is a method that exploits the high sensitivity of fluorescence to visualize individual receptors in the plasma membrane of living cells. Receptors are selectively labeled with a small fluorescent particle and the particles imaged using a sensitive camera attached to a fluorescence microscope (Gross and Webb, 1988; Anderson et al., 1992; Ghosh and Webb, 1994; Wilson et al., 1996; Cherry et al., 1998; Smith et al., 1999). Suitable probes are made by attaching a monoclonal antibody (or its Fab fragment) to either a phycobiliprotein or a fluorescent latex microsphere (Smith et al., 1998). Phycobiliproteins are particularly advantageous because of their small size (R-phycoerythrin is 11 × 8 nm) and absence of nonspecific binding. The images of the small particles used for SPFI are diffraction patterns that approximate to a two-dimensional (2D) Gaussian function. By fitting the intensity distribution in an individual fluorescent spot to a 2-D Gaussian function, the position of the particle can be determined to an accuracy much greater than the limit of resolution of the microscope (Anderson et al., 1992). This is the basis for the method known as single particle tracking in which the movement of receptors is followed through a sequence of images.
Here we describe the further development of SPFI for studying colocalization at the single molecule level. The method involves labeling two different receptors with different colored (e.g., green and red) fluorescent particles. The positions of the green and red particles are determined separately by dual wavelength imaging and the images analyzed for colocalization of the receptors. Where green and red particles are in close proximity, the overlap of their Gaussian profiles (termed the overlap integral) is determined. The overlap integral, Φ, has a value of 1 when images of the red and green particles exactly coincide and zero when there is no overlap. Simulation of a random distribution of receptors shows that high values of Φ that occur by chance proximity are relatively rare at typical receptor densities. The presence of significant numbers of particles with high values of Φ thus indicates that they are constrained to be in close proximity. In this way it is possible not only to detect colocalization of single molecules but also to quantify the proportion of molecules that are colocalized. Analysis of the intensities of colocalized spots (Morrison et al., 1994) can also give an indication of how many molecules are involved.
Before this approach can successfully be applied to cells, a number of problems have to be addressed. Dual wavelength imaging incurs a positional shift of the images due to chromatic aberration and a prism effect if the glass surfaces of the microscope slide are not perfectly parallel. This is not normally a problem in conventional microscopy but becomes significant when high positional accuracy is required. A further problem is that cells often exhibit particulate autofluorescence. The spots arising from autofluorescence can have similar widths and intensities to the images of the fluorescent particles used for SPFI. Here we present methods for overcoming these problems and demonstrate the applicability of the approach to cellular systems.
MATERIALS AND METHODS
Cell culture
Chinese hamster ovary (CHO) cells transfected with hCD14-DAF cDNA via the vector pRc/RSV (CHO-CD14) were provided by S. Viriyakosol and T. Kirkland (University of California; Viriyakosol and Kirkland, 1995); control cells (CHO-RSV) were transfected with the empty vector. Both types were cultured in 50% Dulbecco's modified Eagles medium, 50% Ham's F12 (DMEM/F12; Gibco BRL, Paisley, UK) supplemented with 2 mM Glutamax (Gibco), 10% fetal calf serum (Gibco), 500 μg/ml Geneticin and 50 μg × ml−1 gentamycin (Sigma, Poole, UK).
The human transfectant fibroblast cell line, M1DR1/Ii/DM, was provided by Professor R. Lechler (Imperial College, London). These cells express the MHC class II molecules, HLA-DR and HLA-DM as well as the invariant chain Ii (Dodi et al., 1994; Lightstone et al., 1995). The cells were cultured in DMEM (Gibco) supplemented with 10% fetal calf serum.
Fluorescent probes
Anti-CD14 IgG (26ic) was purified from hybridoma supernatant (ATCC HB246) using affinity chromatography on immobilized protein-A (Pharmacia, St Albans, UK). BU45, an antibody against CD74, was purified from hybridoma supernatant (BU45 clone). DA6.231, an antibody specific for monomorphic HLA-DR, was purified from ascites fluid as previously described (Smith et al., 1998). The pyridyldisulfide derivatives of the phycobiliproteins R-phycoerythrin (PhyE) and allophycocyanin (APhyC) were obtained from Molecular Probes Inc. (Eugene, OR). They were conjugated to antibodies using established methods (Wilson et al., 1996) and the 1:1 complex purified by HPLC. Cell binding was examined by flow cytometry (FACSCalibur, Becton Dickinson, Oxford, UK) to demonstrate that binding specificity was retained after conjugation and to determine saturation levels. Binding of APhyC-IgG(BU45) was not affected by the presence of PhyE-IgG(DA6.231), demonstrating that there was no interference to binding by steric hindrance.
Lipopolysaccharide (LPS) from Escherichia coli 0111:B4 (Sigma) was labeled with Alexa 488 (Molecular Probes Inc., Eugene, OR) after partial oxidation using periodate (Jansson et al., 1981). LPS (1 mg) was incubated with periodate (40 mM) in 200 μl water for 6 h at 4°C, after which the reaction was terminated with 0.1 M glycerol. Low molecular weight components were removed by gel filtration using Sephadex G25. High molecular weight fractions were mixed with Alexa 488 hydrazide (1 mg × ml−1) and the solution incubated at 4°C for 24 h. The hydrazide was reduced using sodium borohydride (2 mM) and low molecular weight components were again removed by gel filtration, eluting in phosphate buffered saline (PBS) containing sodium azide (0.02%). The labeling ratio was estimated to be ∼1:1 from the absorbance at 493 nm, using ɛ493 = 71,000 cm−1 for Alexa 488, and assuming no loss of LPS.
Cell preparation for imaging
Cells from passaging were cultured in eight-well microchamber slides (Labtek, Fisher Scientific, Loughborough, UK) for 48–72 h. The CHO cells were washed with medium, and incubated with Alexa-LPS (∼1 μg, presonicated for 15 min at 4°C) for 30 min in medium containing 10% pooled human serum (Sigma) at 37°C. After two more washes in serum-free medium at ambient temperature, the PhyE-IgG antibody was added in serum-free medium and incubated for 20 min at ambient temperature, before washing in serum-free medium and finally in PBS with 0.02% azide. M1DR1/Ii/DM cells were washed three times in PBS before incubation firstly with PhyE-IgG (DA6.231) and secondly with APhyC-IgG(BU45). All ligands were added in saturating concentrations as determined by flow cytometry. The cells were finally fixed in CellFIX (Becton Dickinson), and a coverslip applied over the minimum amount of PBS.
Fluorescence microscopy
SPFI was performed using an Olympus IX70 microscope and usually with a 40×/NA0.85 UPlanApo objective. Fluorescent specimens were excited by a 100 W mercury lamp and one of four filter assemblies (Omega Optical Inc., Brattleboro VT). For Alexa 488 (blue), an XF71 set with 495 nm dichroic, 450DF55 excitation, and 535DF35 emission bandpass filters was used. PhyE (green) was detected using a modified XF101 set, having a 557 nm dichroic, 525DF35 excitation and 595AF60 emission filters. APhyC (red) was imaged using an XF44 set of 630 nm dichroic, 590DF45 excitation, and 660DF32 emission filters. A filter block designed for Oregon Green 514 was used for some observations; this contained a 525 nm dichroic, 500DF25 excitation, and 545DF35 emission filters.
Images were obtained using a Peltier-cooled slow-scan CCD camera (Wright Instruments, Enfield, UK) with a back-illuminated EEV CCD02-06-1-206 detector, which was attached to the Keller port of the microscope. The camera and its transfer lens were positioned so that the image scale was 4.8 pixels per micron, close to the Nyquist criterion (Inoué and Spring, 1997) for the NA0.85 objective. Spots at optimum focus were found to have 1.6 pixel mean Gaussian width (0.33 μm) using the green filter. Images were obtained with exposure times that gave signal to noise ratios of at least 4:1 for the fluorescent particles in that wavelength region, typically 1–5 s; the exception was Alexa-488 in dual wavelength experiments, for which long exposures caused excessive photobleaching to the other probe.
Polylysine-coated slides
Commercially available coated slides are unsuitable for SPFI as the coating is nonuniform and shows patchy background signals. Low background fluorescence glass slides (BDH, Poole, UK) were cleaned in concentrated nitric acid for 10 min and rigorously washed in double distilled water. The slides were immersed in poly-l-lysine (0.01%) for 10 min at ambient temperature, after which they were shaken free of excess liquid and dried in a slide holder with the lid ajar at 37°C for 2 h. Slides were used within 24 h as the coating degrades in air.
Labeled antibody was diluted to ∼250 ng × ml−1 in low-salt buffer, and 25 μl was applied to the slide center for a few minutes. After removal of the liquid and gentle washing of the area with buffer, the area was sealed with ∼30 μl buffer under a coverslip with a ring of silicone grease. Alexa 488-LPS, 20 ng.ml−1 in PBS, was applied in the same manner. For imaging, the approximate focal plane was located by minute dust particles using differential interference contrast optics, and the final adjustment made by inspection of fluorescent images.
Transfluosphere slides
TransFluoSpheres (T8868, Molecular Probes, Eugene, OR) having diameter 40 nm and designed to emit at 685 nm after excitation at 488 nm, were diluted 20,000 times in PBS and 40 μl was applied to a polylysine-coated slide. After gentle washing, the area was sealed under a coverslip with silicone grease.
Image analysis
Computational methods used in SPFI have been described previously (Anderson et al., 1992). Pixel data in the horizontal rows x and vertical columns y from a small (∼4 μm2) area around the nth spot were fitted to a two-dimensional Gaussian peak of height Zn and width Wn centered at {Xn,Yn}, with local background Z0
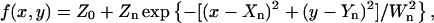 |
(1) |
using a nonlinear least-squares method (Marquardt, 1963) that gives values and standard deviations of the variable parameters when the data noise statistics are known. Closely separated spots (<1 μm apart) were fitted simultaneously by a combination of Gaussians on the same background, including more pixels in the data set. This procedure determined the position of particles to a fraction of a pixel—typically 0.19 with blue and green, and 0.21 with red, filters in the conditions reported here, which convert to 39 and 43 nm, respectively. The statistics of the Gaussian peak width were used to discriminate against unreliable spots; those found to be more than two standard deviations from the center of the distribution can be ignored. Also, those spots whose widths are poorly defined, seen as large standard deviations of the width, can be ignored; these often represent poorly resolved multiple particles. The peak intensity reflects the number of fluorophores under the spot.
Correlating spots from different images
Correlations were established using an algorithm, similar to that employed for tracking moving spots (Anderson et al., 1992), that tests all pairs of spots to find the highest probabilities by a “nearest spot with similar intensity” algorithm; the intensity factor was not included when applied to dual fluorophore correlation. If artifacts or off-cell particles that can be detected at both wavelengths are present, they act as a guide for any offset that occurs between images; in their absence, an initial estimate from the algorithm was chosen by plotting offsets on target diagrams as in Fig. 1 a. A cluster of points on such a diagram showed the best correlations, and the offsets for this cluster could then be used in a second application of the algorithm. All correlations were inspected to reduce the number of poorly correlated particles.
FIGURE 1.

(a) Target plot of the chromatic shifts found for test images of TransfluoSpheres taken under green and red excitation; the mean shifts in the {x,y} values are used as initial offsets before correlation. (b) Chromatic aberration correction for test images of TransfluoSpheres taken under green and red excitation. The spots in the images were fitted to obtain the 2D-Gaussian center, intensity and width, and then correlated. X and Y components of the vector shift between the positions were plotted against the distance from an optical center (𝒳,𝒴); the chi-squared of a straight line with gradient 𝓀 through the points was minimized as 𝒳,𝒴, and 𝓀 varied. The points were weighted by the product of the error values for the spot widths. (c) Overlap integrals for the correlated spots, before correction for chromatic aberration, and (d) overlap integrals after the red positions have been corrected by applying the parameters of the straight line fits in b. A few poorly correlated spots remain below Φ = 0.9, which can be seen as outliers in the aberration correction graphs. (e) Variation of the spot widths with focus position, with blue (•), green (▪), and red (♦) filter sets. (f) Variation of the aberration radial correction parameter with focus position, for blue versus green (•) and green versus red (♦) filter sets. The optical center parameters show similar trends (data not shown).
Chromatic aberration correction
Images of beads that emit at more than one wavelength show the effect of an imperfect optical path; the spot positions do not coincide. This effect is a combination of several sources of aberration; the optical path between the face of the objective lens and the detector is not perfectly achromatic, and the sample slide and coverslip may not be parallel. When colocalized spots are found in the different wavelength images, then a correction can be made by assuming that there is a radial shift from an optical center {𝒳,𝒴} that is linearly dependent on the distance of the spot from this center:
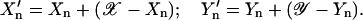 |
(2) |
The values of the radial shift 𝓀 and the coordinates of the optical center are variable parameters for linear regression of the radial shift upon the distance from the chromatic center. Data points were weighted by the product of the errors in the 2D-Gaussian spot widths, σ(Wn,a) × σ(Wn,b). When images are obtained at three wavelengths, the values of 𝓀, 𝒳, and 𝒴 need to be obtained for each pair of images.
Colocalization
The degree of colocalization for correlated spots in two images was quantified by the overlap integral Φ, which represents the volume shared by two 2D-Gaussian shapes.
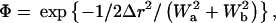 |
(3) |
where Δr is the distance between the computed positions in the two images, and Wa and Wb are the widths of the two Gaussians.
RESULTS AND ANALYSIS
TransFluoSpheres
Initial studies were performed using TransFluospheres bound to polylysine-coated slides. These carboxylate-modified latex beads contain combinations of fluorophores that undergo FRET when illuminated at the excitation wavelength of the donor fluorophore, and thus emit at the acceptor emission wavelength. The beads were imaged using the blue filter set XF71 with the emission filter replaced by a 695AF55 bandpass (“trans” filter set), and showed very intense, submicroscopic spots. Because of their broad excitation and emission spectra, the beads can also be imaged using green and red illumination (filter sets XF44 and XF101), although longer exposure times are required (5 s). They thus provide an ideal system for assessing positional shifts that arise from chromatic aberration.
Images obtained using the three filter sets were analyzed as described under Materials and Methods, and the chromatic aberration investigated; shifts between the red and green filter images are seen in the target diagram in Fig. 1 a, and mean offsets {−1,1} for {x,y} used to assist the correlation. Typical correction graphs are shown in Fig. 1 b. The optical center parameters {𝒳,𝒴} were found to alter considerably on changing the slide, suggesting that nonparallel glass faces dominate this factor; but the radial correction term 𝓀 was fairly constant for each pair of filters. When the optical center was well outside the CCD area, due to slide and coverslip being seriously nonparallel, then convergence of the fitting procedure tended to be poor; however a solution could always be found by varying the initial estimates for the parameters.
The overlap integrals were calculated from the fitted parameters for the correlated spots in the images obtained with the red and the green filters, as described under Material and Methods (Eq. 3). The histogram of these values before and after correction for chromatic aberration is shown in Fig. 1, c and d. As expected, the majority of spots are highly correlated. After correction for chromatic aberration, values of the overlap integral were mostly >0.95. The few outliers can be attributed to beads that have moved in the interval between the images, or where the signal-to-noise in one image is too low for the parameters of the 2D-Gaussian to be determined accurately.
This test slide was also used to investigate whether the chromatic shifts were focus dependent. Sets of three images, using the Oregon Green, green, and red filter sets, were obtained at 0.5 μm focus steps and analyzed as before. Results in Fig. 1 e show the spot width as a function of focus position. There would appear to be a small difference between the trend lines, especially in the case of the green and red filter sets where the wavelength difference was larger. When the chromatic aberration correction algorithm was applied to the data sets at each focus position, then the variable parameters gave values that are plotted in Fig. 1 f; outside this range of focus the spots were too ill-defined to permit analysis. The change of 𝓀-value in particular shows a variation with focus position.
Modeling the overlap integral
To investigate the dependence of the overlap integral upon spot separation, pairs of model images were created with spots randomly positioned at a density approximately the same as that found in images of cells. Spot envelopes were calculated using a Bessel function with width set to that found in optimally focused images, and were randomly positioned in a two-dimensional array of pixels. The intensities were uniform in each image and were representative of those found under the imaging conditions used for PhyE and APhyC probes. Random noise generated by a subtractive algorithm (Press et al., 1986) was added to each pixel to simulate the shot and readout noise found for the CCD camera employed; different seed values for the random noise ensured that noise was not correlated in the two images. Each image was then analyzed by the 2D-Gaussian fitting procedure, correlated, and overlap integrals calculated as before.
An example of a simulated image is shown in Fig. 2 a, with a histogram of overlap integrals for perfectly correlated images in Fig. 2 b; as expected, all values are >0.95. When the positions of the spots in the second image were independently random, and correlations were taken with the nearest spot, then the distribution of Φ-values shown in Fig. 2 c was found. Clearly mostly low values of overlap occur in this case; from five simulations 59 ± 3% of particles have Φ < 0.2, and only 7 ± 3% above Φ = 0.8. When the density of receptors is increased threefold (Fig. 2 d), the fraction having overlap integrals >0.2 increases due to chance proximity, until only ∼30% have Φ < 0.2. Eventually images cannot be analyzed as the spots overlap excessively, but there is no sign of a peak for Φ > 0.8 as was observed in Fig. 2 b and in on-cell experiments presented below.
FIGURE 2.

(a) An example of an image created to simulate a PhyE probe, with 200 spots having Bessel function profiles that approximate to the width at optimum focus obtained with the camera/microscope combination used in these studies. The spots have equal intensities, are randomly positioned on a uniform background, and shot and readout noise are applied. Those spots that appear brighter than the majority are chance superpositions. (b) The histogram of the overlap integrals found from correlating the image that exactly corresponds to a but simulated for APhyC. Only one spot falls below Φ = 0.95. (c) A histogram of Φ-values found when attempting to correlate a with an image simulating APhyC probes randomly placed in the image. (d) The distribution of Φ-values found for simulated images at three times the density of spots employed for a–c. (e) Dependence of the peak value of Φ on the shift in positions of spots in the two simulated images. The image a was compared with simulations where all the red image spots were shifted by 0.1–1.6 pixels; the peak value was obtained from histograms of log(1 − Φ) fitted by a normal distribution.
Simulated images were also created with uniform small shifts added to the positions of spots in the second image. Overlap integrals were obtained for a range of values of these shifts; in each case a representative value for the peak in the histogram was found by fitting log(1 − Φ) as a normal distribution. The dependence of this peak value upon the shift is shown in Fig. 2 e.
Antibodies on polylysine slides
As an experimental test of noncorrelated images, PhyE-IgG and APhyC-IgG were both bound to a polylysine-coated slide at densities similar to that of the simulation in Fig. 2 a. The two probes were imaged using the green and red filter systems and the images analyzed to determine overlap integrals. Fig. 3 a shows that the overlap integrals are qualitatively similar to those of the simulation in Fig. 2 c, and the target diagram Fig. 3 b indicates that this is not due to poor registration of the images. To ensure that the lack of correlation was not due to chromatic aberration, we determined the values of the adjustable parameters in Eq. 2 that maximized the overlap integrals by adjusting the offsets using the means of shifts plotted in Fig. 3 b. Fig. 3 a shows that even after applying this correction, there are few spots that exhibit a high degree of overlap.
FIGURE 3.

(a) Histogram of overlap integrals for PhyE-IgG and APhyC-IgG codeposited onto a polylysine slide. (b) Target plot of the chromatic shifts from the attempt to correlate spots; the points are scattered through the diagram, with no “hot-spot” to indicate preferred offsets.
LPS and CD14 on CHO cells
Images of CHO-CD14 cells labeled with Alexa 488-LPS and PhyE-IgG(26ic) are shown in Fig. 4 a; Alexa 488 is the blue channel, the PhyE is green, and a third image with red filters is merged with these two. It is immediately apparent that many submicroscopic spots are visible in all three images. Because Alexa 488 and PhyE do not emit in the red, these spots are assigned to autofluorescence and are hereafter referred to as artifacts. Control images of unlabeled CHO-CD14 cells showed only these artifacts, as did the images of empty vector CHO-RSV cells (results not shown). Their presence is fortuitously very useful, as they allow the chromatic aberration correction to be determined without making assumptions about colocalization of LPS and CD14.
FIGURE 4.


Color-merged images of (a) CHO-CD14 cells labeled with Alexa 488-LPS (blue) and PhyE-IgG(CD14) (green) and imaged with suitable filters and exposure times; the red channel detects artifacts that appear as white spots, and (b) M1DR1/Ii/DM cells labeled with PhyE-IgG(DA6.231) (green) and APhyC-IgG(BU45) (red); artifacts are detected using the blue channel. Colocalized spots and artifacts show imperfect registration due to chromatic aberration before correction. Insets are the separate images (shown by the edge colors) from a small area; the final inset is a map of the chromatically corrected fitted spot positions: an artifact (triple correlated) is shown as a black triangle, isolated spots appear as circles, correlated blue/green (Alexa488-LPS and PhyE-antiCD14) spots in a as cyan diamonds, and a correlated green/red (PhyE-antiHLA-DR and APhyC-antiCD74) spot in b, where it is marked by a short arrow, as a yellow diamond. Correlated spots are plotted at the coordinates of the green spot.
The green image showed many uncorrelated spots that could be analyzed. Fig. 5 a shows a histogram of intensities of such spots, with outliers removed as described in Image Analysis. The histogram is fitted to a normal distribution and its convolution representing double particles. Fig. 5 d contains a similar histogram and fit for PhyE-IgG(26ic) on polylysine slides. Although there are some differences between the histograms in Fig. 5, a and d, the data suggest that most of the CD14 is monomeric.
FIGURE 5.

Histograms of spot intensities of Alexa488-LPS and PhyE-IgG(26ic) on CHO-CD14 cells and on polylysine-coated slides. (a–d) Intensities of spots seen in the green channel in Fig. 4 a; the solid line is a best-fit normal distribution with a second contribution from the convolution of this distribution with itself. (a) Uncorrelated spots; (b) correlated with blue channel; (c) artifacts; (d) for PhyE-IgG(26ic) on polylysine using the same imaging conditions as in a. (e–h) Intensities of spots seen in the blue channel in Fig. 4 a; (e) uncorrelated spots; (f) correlated with green channel; (g) artifacts; (h) for Alexa488-LPS on a polylysine-coated glass surface, using exposure times 5× those used for cell images; note the increased intensity scale. (i–k) Intensity ratios, with normal distribution fitted on the log axis. The parameters of the centroid with upper and lower standard deviations are: (i) correlated (green/blue) 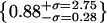 (j) artifacts (green/blue),
(j) artifacts (green/blue), 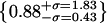 , (k) artifacts (green/red),
, (k) artifacts (green/red), 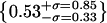 . (l) The overlap integrals found for all possible blue∼green spots in Fig. 4 a.
. (l) The overlap integrals found for all possible blue∼green spots in Fig. 4 a.
Some uncorrelated spots can also be seen in the blue image. The analysis of these spots gives the intensity histogram displayed in Fig. 5 e. Applying Alexa 488-LPS to polylysine slides, it was found that images only showed spots reliably by using long exposures, five times that used for cells; analysis of such images provided the histogram of Fig. 5 h. To ensure that the system was detecting all fluorescent particles, even longer exposures were obtained for the same sample, and the mean spot intensity was found to scale with the exposure time by 7 counts s−1. These data suggest that the Alexa 488 spots seen in Fig. 4 a with 2 s exposure time arise from small aggregates of LPS. Photobleaching of some particles is expected during the long exposures, so a linear response may not extend down to normal exposure times; also it is possible that aggregation of Alexa 488-LPS could result in intermolecular quenching of the fluorescence, so accurate quantitation of the LPS aggregates is difficult. It is unlikely that any single LPS particles would be visible on the cell due to the increased noise resulting from the diffuse autofluorescent background at these wavelengths.
The intensity distributions of the correlated spots in the green and blue images are shown in Fig. 5, b and f, and are not significantly different from those in Fig. 5, a and e; those of the artifacts are given in Fig. 5, c and g. Intensity ratios are shown on a log scale in Fig. 5, i–k, with the best-fit normal distributions; the parameters are given in the figure legend. Those spots that remain after removing the artifacts were subjected to the colocalization analysis. Positions were corrected after determining the chromatic aberration correction from the artifacts, and the overlap integrals calculated. The histogram of these values is shown in Fig. 5 l.
HLA-DR and CD74 on M1DR1/Ii/DM cells
A merged image of an M1DR1/Ii/DM cell labeled with PhyE-IgG(DA6.231) and APhyC-IgG(BU45) is shown in Fig. 4 b. In this case, artifacts were identified using the blue filter set and used to determine the correction for chromatic aberration for the red and green images. The intensity distributions for uncorrelated and correlated spots are shown in Fig. 6, a and b, and Fig. 6, e and f, for PhyE-IgG(DA6.231) and APhyC-IgG(BU45), respectively. The corresponding intensities for the probes bound to polylysine-coated slides are shown in Fig. 6, d and h. After aberration correction, the overlap integrals were calculated as before. The histogram of these values is presented in Fig. 6 l, which shows all possible correlations. Fig. 6, i–k, shows the intensity ratios of correlated spots in the green and red channels, and of the artifacts in the green/red and green/blue channels.
FIGURE 6.

Histograms of spot intensities on M1DR1/Ii/DM cells labeled by PhyE-IgG(DA6.231) and APhyC-IgG(BU45) cells and on polylysine-coated slides. (a–d) Intensities of spots seen in the green channel in Fig. 4 b; the solid line is a best fit normal distribution with a second contribution from the convolution of this distribution with itself; (a) uncorrelated spots; (b) correlated with red channel; (c) artifacts; (d) on polylysine using the same imaging conditions as in a. (e–h) Intensities of spots seen in the red channel in Fig. 4 b; the solid line is a best-fit normal distribution and two-particle convolution. (e) Uncorrelated spots; (f) correlated with green channel; (g) artifacts; (h) on polylysine using the same imaging conditions as e. (i–k) Intensity ratios, with normal distribution fitted on the log axis. The parameters of the centroid with upper and lower standard deviations are: (i) correlated (green/red) 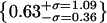 (j) artifacts (green/red),
(j) artifacts (green/red), 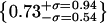 , (k) artifacts (green/blue),
, (k) artifacts (green/blue), 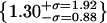 . The outlying artifacts with high green/blue ratios are off-cell dust particles. (l) The overlap integrals found for all possible green∼red spots in Fig. 4 b.
. The outlying artifacts with high green/blue ratios are off-cell dust particles. (l) The overlap integrals found for all possible green∼red spots in Fig. 4 b.
Some cells have very few artifacts, in which case the chromatic aberration correction depends on finding colocalized spots. These can be identified as having similar vector shifts between the green and red images as described in Materials and Methods and displayed in Fig. 1 a. This procedure produced results indistinguishable from those obtained using artifacts to assist the correction.
DISCUSSION
SPFI utilizes small fluorescent particles to visualize single molecules in intact cells. The approach offers two methods of detecting two or more molecules that are in too close proximity to be resolved by conventional optical microscopy. The first method depends on analyzing the intensities of the particle images and has been used to determine clustering of low density lipoprotein receptors and dimerization of MHC class II molecules (Gross and Webb, 1988; Morrison et al., 1994; Cherry et al., 1998). Here we describe the development of a second method in which molecules are colocalized with high spatial accuracy using dual wavelength imaging.
Quantification of colocalization in conventional confocal microscopy relies on Pearson coefficients (Manders et al., 1993). Such calculations rely on a good signal to noise ratio, and a well-defined background signal. Wide-field fluorescence images generally show a variable background signal due to cell autofluorescence; the signals in SPFI can be lower than this background variation. It might in principle be possible to “flatfield” the images, but the chromatic aberration is large enough to destroy the correlation. An alternative approach would be to use deconvolution techniques (Agard, 1984) to reconstruct the second image of a pair, thus removing the aberrations and also correcting for focus imperfections. However, this would require collections of images at each wavelength at different focal positions to establish the point spread functions; the phycobiliproteins used in this study are only marginally photostable, and the bleaching effects of imaging at different wavelengths are severe.
Trabesinger et al. (2001) presented results for single molecule dual-color imaging of DNA adhering to glass. This work is directed at evaluation of colocalization assays by statistical analysis, using a probability density function of the spatial separation of two probes; this includes a term for the standard deviation of molecular positions established for the microscope system. This may be appropriate for glass slides, but on cell membranes the small variation in focal position causes this parameter to vary from particle to particle. Thus we have chosen to compute an overlap integral for each correlated pair of spots using the individual spot parameters.
Previously, Betzig (1995) suggested methods for studying colocalization by near-field scanning optical microscopy. Enderle et al. (1997) used this method to investigate colocalization of malarial parasite proteins with host skeletal proteins in infected erythrocytes. A pixel-by-pixel comparison of the images of fluorescent antibodies permitted detection of colocalization with a resolution of ∼100 nm. Schütz et al. (1998) imaged single fluorophores to detect ligand-receptor recognition between biotin and streptavidin in a model system. Fluorophores bound to biotin and streptavidin were colocalized within ∼40 nm when biotin was bound to streptavidin immobilized on supported phospholipid membranes. More recently, Michalet et al. (2001) described methodology for high-resolution distance measurements using confocal microscopy. The sample consisted of 40 nm TransFluospheres or 10 nm nanocrystals that were deposited onto glass coverslips in suspension and then dried.
In the present work, we have addressed problems that arise when attempting colocalization studies with cells at high spatial resolution. The phycobiliproteins used here are less bright than TransfluoSpheres and cannot be excited with a single excitation wavelength to minimize chromatic aberration. Their small size is, however, a distinct advantage because it reduces the risk that steric hindrance will prevent saturation binding to closely associated receptors.
Although not used for on-cell studies, TransfluoSpheres are very useful for investigating the corrections required for positional shifts due to chromatic aberration. Fig. 1 shows the results of analyzing dual wavelength images obtained with TransfluoSpheres bound to polylysine-coated slides. Fig. 1 a shows that the shifts between images obtained at different wavelengths can be satisfactorily fitted by Eq. 2, which assumes that there is a radial shift from an optical center that is linearly dependent on distance from this center. There are a few outliers that probably arise from the bead movement that could occur if a bead detaches and rapidly rebinds to the slide. The overlap integrals calculated according to Eq. 3 are shown with and without the correction for chromatic aberration in Fig. 1 b. The correction clearly results in a substantial improvement in the overlap of the two images.
The methodology was tested by simulating images with similar particle densities and signal-to-noise as are obtained using phycobiliproteins as probes (Fig. 2 a). Two simulated images were generated with the particles in identical positions but with different random noise added to each image. This resulted in a high degree of colocalization, as expected (Fig. 2 b). Next, two images, in which the particles occupied random positions, were generated; the histogram of the overlap integrals in this case is shown in Fig. 2 c. Clearly at this particle density, the probability of random coincidences is low. Images were also simulated with the spot density increased threefold; the fraction of correlations near Φ = 0 decreases as displayed in Fig. 2 d but there is no peak at high values of Φ. Lastly, pairs of images were generated with a fixed positional shift between images. Fig. 2 e shows how the overlap integral varies with the shift. The overlap integral falls from a value of 1 for zero shift to 0.95 for a shift of 0.7 pixel and to 0.9 for a shift of 1 pixel (205 nm).
A model experimental system was created by depositing PhyE-IgG and APhyC-IgG together on the same polylysine-coated slide. This sample was then imaged using the green and red filter sets. As expected there was little or no correlation between the two images. We thus conclude that high values of the overlap integrals rarely arise from random coincidence at these particle densities. The experiment also demonstrates that the PhyE-IgG and APhyC-IgG do not mutually associate to any appreciable extent.
A feature of dual wavelength SPFI measurements is that they permit the extent of any colocalization to be quantified. This requires deciding upon a cut-off value for the histogram of overlap integrals. Although the cutoff is somewhat arbitrary, the data in Figs. 1–3 suggest that 0.8 is a reasonable value. Molecules are thus assigned as colocalized or noncolocalized according to whether their overlap integral is greater or less than this value. As can be seen from Figs. 1 d, 2, b and c, and 3 a, a value of 0.8 is sufficiently low to include virtually all colocalized spots but sufficiently high to eliminate nearly all random colocalization. The cut-off value might need to be adjusted in different experiments, however, if the receptor density were very high or if the signal to noise of the images were much different from that obtained here.
To test the methodology on cells, we investigated the colocalization of LPS with CD14 on CHO-CD14 cells. It is well established that CD14 is the primary receptor for LPS (Wright et al., 1990). It is thus probable that LPS bound to cells is colocalized with CD14. We conjugated PhyE to the monoclonal antibody 26ic, which binds to CD14 without inhibiting LPS binding (Wright et al., 1990). LPS was labeled with Alexa 488. We bound both these probes to the cells and imaged the PhyE and Alexa 488 with green and blue filter sets, respectively.
In control experiments we imaged CHO cells that do not express CD14 and also CHO-CD14 cells in the absence of any probes. In both cases, there were significant numbers of fluorescent spots in both images. We have observed a similar phenomenon in other cell lines. The spots presumably arise from organelles that are weakly autofluorescent. Many of these spots are indistinguishable in intensity and spot width from those arising from the fluorescent probes used in these experiments.
To discriminate the artifactual (autofluorescent) spots from those arising from PhyE and Alexa 488 on CHO-CD14 cells, a third image was obtained with the red filter set. The artifacts could then be eliminated from further analysis because they appear in all three images whereas the PhyE and Alexa 488 spots only appear in the green and blue images, respectively (Fig. 4 a).
While analyzing data from cells, a tendency was observed for some artifacts to show poor correlation, particularly in the vicinity of the nucleus. This may be due to distortions to the light path caused by the different refractive index of the organelle, but might also be a result of slight variations of spot position from the plane of best focus. This was investigated using the TransfluoSphere system, by taking images at the three wavelengths at focus positions away from that considered to be optimum. A dependence of the radial compensation factor 𝓀 on the focus position was observed for the green and red images (Fig. 1 d); these have the best signal to noise, and also the larger difference in the emission wavelengths: 595–660 nm, compared with 595–545 nm. The effect can be explained by considering the shapes of the combined point-spread functions (PSF) of the lens and slide system; the aberrations are due to incongruence of the PSFs at the different wavelengths, and the magnitude will depend on the design of the achromatic lens system. Initial results with an older microscope (Nikon Diaphot) but the same camera gave much larger values of 𝓀. In cell imaging, spots that are not at best focus are easily recognized because they are too wide (out of the optimum plane on one side) or too narrow (out of focus in the other direction; the Airy rings are usually too faint for detection), and probably would be omitted from further analysis.
The presence of the artifacts does have one advantage, namely that they can be used to determine the parameters in Eq. 2 required to correct for chromatic aberration. The corrected overlap integrals so obtained for PhyE and Alexa 488 (Fig. 5 l) show that as expected, there is a strong correlation between the positions of many CD14 and LPS molecules. Some 41% of the Alexa 488 spots and 23% of the PhyE spots have overlap integrals >0.8. This plot is less clearly defined than that for the M1DR1/Ii/DM cells, which may reflect the lower precision of the aberration correction on these rather rounded cells. The colocalized CD14 and LPS molecules may correspond to LPS bound to CD14 or to LPS that has been transferred to a nearby coreceptor. It is well-established that receptors other than CD14 are required for signal transduction (Schmitz and Orso, 2002) and there is evidence that LPS may be released from CD14 soon after binding (Triantafilou et al., 2001).
The intensities of the fluorescent spots contain information on the self-association or clustering of the labeled molecules (Gross and Webb, 1988; Morrison et al., 1994; Cherry et al., 1998). Essentially, images are analyzed by comparing the histogram of the on-cell spot intensity with that obtained when the probe is bound to a polylysine-coated slide. Fig. 5, e and h, demonstrates that the on-cell LPS-Alexa 488 images correspond to small clusters. The possibility that there are also monomers present cannot be ruled out, however, as they may be too weakly fluorescent to detect against the background noise. The CD14 molecules appear to be largely monomeric irrespective of whether or not they are colocalized with LPS. Conceivably steric hindrance could prevent two or more PhyE-IgG molecules binding to dimers or higher oligomers of CD14 but there is no evidence that we are aware of that CD14 has a propensity to self-associate.
As a further application, we investigated the colocalization of HLA-DR with CD74, the surface isoform of the invariant chain Ii. It is well-established that cytoplasmic Ii is required for the correct assembly of MHC class II molecules in the endoplasmic reticulum and their targeting to endocytic compartments (Sant and Miller, 1994; Stumptner-Cuvelette and Benaroch, 2002). The role of CD74 is, however, less certain but immunoprecipitation experiments suggest that CD74 is associated with HLA-DR at the cell surface (Triantafilou et al., 1999).
For these experiments, we labeled HLA-DR with PhyE-IgG(DA6.231) and CD74 with APhyC-IgG(BU45). The cells (M1DR1/Ii/DM) were then imaged using the green and red filter sets (Fig. 4 b). Artifacts in this case were detected by imaging with the blue filter set and used as before to determine the correction for chromatic aberration. Fig. 6 e shows the overlap integrals calculated for the green and red images. It was found that 26% of the PhyE spots and 28% of the APhyC spots had overlap integrals >0.8. We thus conclude that HLA-DR and CD74 are in part in close proximity, probably as a result of a molecular association.
We analyzed the intensities of the fluorescent spots (Fig. 6, a–d) and found that HLA-DR is present as a mixture of monomers and dimers, in agreement with previous studies (Cherry et al., 1998). As the earlier experiments were performed on unfixed cells, this suggests that associations are not produced by fixation. This was confirmed in a control experiment in which MHC class I molecules were found not to be colocalized with CD74 in fixed cells (data not shown). There was no significant difference in the intensity distributions of the colocalized and noncolocalized spots for probes bound to either HLA-DR or CD74; the latter also showed evidence of self-association.
It is noteworthy that the artifact spots showed intensity ratios that fell into a rather narrow band (Fig. 6 j) rather similar to those of the colocalized particles (Fig. 6 i); this might suggest that those spots considered to be colocalized were in fact artifacts that had not been recognized in the third wavelength image. This is unlikely because artifacts in unlabeled control cells all showed the same (blue:green:red) intensity ratios; however the ratios on the CHO and M1DR1/Ii/DM cells were slightly different. The intensity ratios of the PhyE-APhyC spots also fell into a narrow band, whereas those of the Alexa-488 with PhyE cells were much wider, reflecting the heterogeneity of the binding in this system.
The above experiments establish that colocalization of single molecules on cells can be determined by dual wavelength SPFI. The distance scale for these measurements is intermediate between that of FRET and conventional microscopy. Observation of a FRET signal requires molecules to be in close proximity, typically 0.5–5 nm, and is often regarded as evidence of a molecular association. In dual wavelength SPFI, high values of the overlap integral are detected for separations up to ∼300 nm. Because of the noise in individual spot images, a given value of overlap integral cannot be assigned to a precise molecular separation, hence a high overlap integral could result from either a molecular association or confinement of molecules within a small domain. The larger distance scale of SPFI could be advantageous for investigating domains in cell membranes of a few hundred nanometers diameter, where FRET experiments have given conflicting results (Varma and Maya, 1998; Kenworthy et al., 2000). Probably the most informative approach would be to combine SPFI with FRET measurements. Together, they have the potential to elucidate in considerable detail the organization of molecules in cell membranes.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council. The advice of Professor John Dowden (Department of Mathematical Sciences, University of Essex, Colchester, UK) is gratefully acknowledged. We thank S. Viriyakosol and T. Kirkland (University of California, San Diego, CA) for supplying transfected CHO cells and R. Lechler (Imperial College of Science, Technology and Medicine, London, UK) for supplying M1DR1/Ii/DM cells.
References
- Agard, D. A. 1984. Optical sectioning microscopy: cellular architecture in three dimensions. Annu. Rev. Biophys. Bioeng. 13:191–219. [DOI] [PubMed] [Google Scholar]
- Anderson, C. M., G. N. Georgiou, I. E. G. Morrison, G. V. W. Stevenson, and R. J. Cherry. 1992. Tracking of cell surface receptors by fluorescent digital imaging microscopy using a charge-coupled device camera: low-density lipoprotein and influenza virus receptor mobility at 4°C. J. Cell Sci. 101:415–425. [DOI] [PubMed] [Google Scholar]
- Angers, A., A. Salahpour, E. Joly, S. Hilairat, D. Chelsky, M. Dennis, and M. Bouvier. 2000. Detection of β2- adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. USA. 97:3684–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basche, T., S. Nie, and J. M. Fernandez. 2001. Single molecules. Proc. Natl. Acad. Sci. USA. 98:10527–10528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig, E. 1995. Proposed method for molecular optical imaging. Opt. Lett. 20:237–239. [DOI] [PubMed] [Google Scholar]
- Cherry, R. J., K. M. Wilson, K. Triantafilou, P. J. O'Toole, I. E. G. Morrison, and N. Fernández. 1998. Detection of dimers of dimers of HLA-DR on the surface of living cells by single particle fluorescence imaging. J. Cell Biol. 140:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Kashi, M., S. Moshkov, N. Zurgil, and M. Deutsch. 2002. Fluorescence resonance energy transfers measurements on cell surfaces via fluorescence polarization. Biophys. J. 83:1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanovich, S., L. Bene, J. Matko, L. Matyus, Z. Krasznai, G. Szabo, C. Pieri, R. Gaspar, and J. Szollosi. 1999. Two-dimensional receptor patterns in the plasma membrane of cells. A critical evaluation of their identification, origin and information content. Biophys. Chem. 82:99–108. [DOI] [PubMed] [Google Scholar]
- Dodi, A. I., S. Brett, T. Nordeng, S. Sidhu, R. J. Batchelor, G. Lombardi, O. Bakke, and R. I. Lechler. 1994. Invariant chain inhibits presentation of endogenous antigens by a human fibroblast cell line. Eur. J. Immunol. 24:1632–1639. [DOI] [PubMed] [Google Scholar]
- Enderle, T., T. Ha, D. F. Ogletree, D. S. Chemla, C. Magowan, and S. Weiss. 1997. Membrane specific mapping and colocalization of malarial and host skeletal proteins in the Plasmodium falciparum infected erythrocyte by dual-color near-field scanning optical microscopy. Proc. Natl. Acad. Sci. USA. 94:520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara, T., K. Ritchie, H. Murakoshi, K. Jacobson, and A. Kusumi. 2002. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 157:1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh, R. N., and W. W. Webb. 1994. Automated detection and tracking of individual and clustered cell surface low density lipoprotein receptor molecules. Biophys. J. 66:1301–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross, D. J., and W. W. Webb. 1988. Cell surface clustering and mobility of the liganded LDL receptor measured by digital video fluorescence microscopy. In Spectroscopic Membrane Probes, Vol.II. L. M. Loew, editor. CRC Press, Boca Raton, FL. 9–45.
- Hoppe, A., K. Christenson, and J. A. Swanson. 2002. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83:3652–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoué, S., and K. R. Spring. 1997. Video Microscopy: The Fundamentals, 2nd ed. Plenum Press, New York, NY/London, UK. 312.
- Jansson, P. E., A. A. Lindberg, B. Lindberg, and R. Wollin. 1981. Structural studies on the hexose region of the core in lipopolysaccharides from enterobacteriaceae. Eur. J. Biochem. 115:571–577. [DOI] [PubMed] [Google Scholar]
- Jovin, T. M., and D. J. Arndt-Jovin. 1989. FRET microscopy: digital imaging of fluorescence resonance energy transfer. Application in cell biology. In Cell Structure and Function by Microspectrofluorimetry. E. Kohen, J. S. Ploem, and J. G. Hirschberg, editors. Academic Press, Orlando, FL. 99–117.
- Kenworthy, A. K., N. Petranova, and M. Edidin. 2000. High resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes. Mol. Biol. Cell. 11:1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightstone, L., R. Hargreaves, G. Bobek, G. Lombard, H. Stauss, and R. Lechler. 1995. Absence of invariant chain (Ii) alters the repertoire of peptides displayed by class II. Immunology. 86(Suppl. 1):79.7590886 [Google Scholar]
- Manders, E. M. M., F. J. Verbeek, and J. A. Aten. 1993. Measurement of colocalization of objects in dual-colour confocal images. J. Microsc. 169:375–383. [DOI] [PubMed] [Google Scholar]
- Marquardt, D. 1963. An algorithm for least-squares estimation of non-linear parameters. J. Soc. Indust. Appl. Math. 11:431–441. [Google Scholar]
- Matko, J., and M. Edidin. 1997. Energy transfer methods for detecting molecular clusters on cell surfaces. Methods Enzymol. 278:444–462. [DOI] [PubMed] [Google Scholar]
- Michalet, X., T. D. Lacoste, and S. Weiss. 2001. Ultra-resolution colocalization of separable point-like fluorescent probes. Methods. 25:87–102. [DOI] [PubMed] [Google Scholar]
- Morrison, I. E. G., C. M. Anderson, G. N. Georgiou, G. V. W. Stevenson, and R. J. Cherry. 1994. Analysis of receptor clustering on cell surfaces by imaging fluorescent particles. Biophys. J. 67:1280–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press, W. H., S. A. Teukolsky, W. T. Vetterling, and B. P. Flannery. 1986. Numerical Recipes: The Art of Scientific Computing. Cambridge University Press, London, UK/New York, NY. 198.
- Sant, J. A., and J. Miller. 1994. MHC class II antigen processing: the biology of the invariant chain. Curr Opin. Immunol. 6:57–63. [DOI] [PubMed] [Google Scholar]
- Schmitz, G., and E. Orso. 2002. CD14 signalling in lipid rafts: new ligands and co-receptors. Curr. Opin. Lipidol. 13:513–521. [DOI] [PubMed] [Google Scholar]
- Schütz, G. J., W. Trabesinger, and T. Schmidt. 1998. Direct observation of ligand colocalization on individual receptor molecules. Biophys. J. 74:2223–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 1997. Functional rafts in cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- Smith, P. R., I. E. G. Morrison, K. M. Wilson, N. Fernández, and R. J. Cherry. 1999. Anomalous diffusion of major histocompatibility complex class I molecules on HeLa cells determined by single particle tracking. Biophys. J. 76:3331–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, P. R., K. M. Wilson, I. E. G. Morrison, R. J. Cherry, and N. Fernández. 1998. Imaging of individual cell surface MHC antigens using fluorescent particles. In MHC: Biochemistry and Genetics. N. Fernández, and G. Butcher, editors. Practical Approach Series, Oxford University Press. 131–151.
- Stumptner-Cuvelette, P., and P. Benaroch. 2002. Multiple roles of the invariant chain in MHC class II function. Biochim. Biophys. Acta. 1542:1–13. [DOI] [PubMed] [Google Scholar]
- Stryer, L. 1978. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 47:819–846. [DOI] [PubMed] [Google Scholar]
- Trabesinger, W., B. Hecht, U. P. Wild, G. J. Schütz, H. Schindler, and T. Schmidt. 2001. Statistical analysis of single-molecule colocalization assays. Anal. Chem. 73:1100–1105. [DOI] [PubMed] [Google Scholar]
- Triantafilou, K., M. Triantafilou, K. M. Wilson, R. J. Cherry, and N. Fernández. 1999. Intracellular and cell surface heterotypic associations of human leukocyte antigen-DR and human invariant chain. Hum. Immunol. 60:1101–1112. [DOI] [PubMed] [Google Scholar]
- Triantafilou, K., M. Triantafilou, S. Ladha, A. Mackie, R. L. Dedrick, N. Fernández, and R. J. Cherry. 2001. Fluorescence recovery after photobleaching reveals that LPS rapidly transfers from CD14 to hsp70 and hsp90 on the cell membrane. J. Cell Sci. 114:2535–2545. [DOI] [PubMed] [Google Scholar]
- Tron, L., J. Szöllösi, S. Damjanovich, S. H. Helliwell, D. J. Arndt-Jovin, and T. M. Jovin. 1984. Flow cytometric measurements of fluorescence resonance energy transfer on cell surfaces. Quantitative evaluation of the transfer efficiency on a cell-by-cell basis. Biophys. J. 45:939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma, R., and S. Maya. 1998. GPI-anchored proteins are organised in submicron domains at the cell surface. Nature. 394:798–801. [DOI] [PubMed] [Google Scholar]
- Viriyakosol, S., and T. N. Kirkland. 1995. A region of human CD14 required for lipopolysaccharide binding. J. Biol. Chem. 270:362–368. [DOI] [PubMed] [Google Scholar]
- Wilson, K. M., I. E. G. Morrison, P. R. Smith, N. Fernández, and R. J. Cherry. 1996. Single particle tracking of cell-surface HLA-DR molecules using R-phycoerythrin labelled monoclonal antibodies and fluorescence digital imaging. J. Cell Sci. 109:2101–2109. [DOI] [PubMed] [Google Scholar]
- Wright, S. D., R. A. Ramos, P. S. Tobias, R. J. Ulevitch, and J. C. Mathison. 1990. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding-protein. Science. 249:1431–1433. [DOI] [PubMed] [Google Scholar]