

Abstract
We examined whether the N-terminus of Kv4.2 A-type channels (4.2NT) possesses an autoinhibitory N-terminal peptide domain, which, similar to the one of Shaker, mediates inactivation of the open state. We found that chimeric Kv2.1(4.2NT) channels, where the cytoplasmic Kv2.1 N-terminus had been replaced by corresponding Kv4.2 domains, inactivated relatively fast, with a mean time constant of 120 ms as compared to 3.4 s in Kv2.1 wild-type. Notably, Kv2.1(4.2NT) showed features typically observed for Shaker N-type inactivation: fast inactivation of Kv2.1(4.2NT) channels was slowed by intracellular tetraethylammonium and removed by N-terminal truncation (Δ40). Kv2.1(4.2NT) channels reopened during recovery from inactivation, and recovery was accelerated in high external K+. Moreover, the application of synthetic N-terminal Kv4.2 and ShB peptides to inside-out patches containing slowly inactivating Kv2.1 channels mimicked N-type inactivation. Kv4.2 channels, after fractional inactivation, mediated tail currents with biphasic decay, indicative of passage through the open state during recovery from inactivation. Biphasic tail current kinetics were less prominent in Kv4.2/KChIP2.1 channel complexes and virtually absent in Kv4.2Δ40 channels. N-type inactivation features of Kv4.2 open-state inactivation, which may be suppressed by KChIP association, were also revealed by the finding that application of Kv4.2 N-terminal peptide accelerated the decay kinetics of both Kv4.2Δ40 and Kv4.2/KChIP2.1 patch currents. However, double mutant cycle analysis of N-terminal inactivating and pore domains indicated differences in the energetics and structural determinants between Kv4.2 and Shaker N-type inactivation.
INTRODUCTION
Voltage-dependent potassium (Kv) channel inactivation kinetics, including the time it takes for recovery from inactivation, defines a period of refractoriness which critically determines neuronal excitability and discharge behavior (Hille, 2001). Rapidly inactivating (A-type) Kv channels, like the well-studied archetypal Shaker channel of Drosophila, often utilize an N-terminal inactivation domain, a mechanism referred to as “N-type” inactivation. Shaker N-type inactivation follows a “ball-and-chain” mechanism (Hoshi et al., 1990), where a tethered N-terminal inactivation domain (Zagotta et al., 1990) blocks the pore after channel opening. N-type inactivation can be eliminated by removing the N-terminal autoinhibitory sequence (Hoshi et al., 1990) and be restored by application of the N-terminal peptide to the channel from the cytoplasmic side (Zagotta et al., 1990). The N-terminal inactivation domain interacts with a binding site in the pore cavity (Zhou et al., 2001), which overlaps with the binding site for quaternary amine blockers. Therefore, intracellular tetraethylammonium (TEA) interferes with N-type inactivation (Choi et al., 1991). Furthermore, K+ influx expels the inactivation particle from its binding site, which results in an acceleration of recovery from inactivation in high external K+ (Demo and Yellen, 1991). N-type inactivation also traps the channel in an open-inactivated state and prevents it from closing. Thus, inactivated Shaker channels typically mediate prominent tail currents in high external K+ when the membrane is repolarized, indicative of recovery from inactivation via the open state (Demo and Yellen, 1991; Ruppersberg et al., 1991). Rapid N-type inactivation of Shaker channels is followed by a slower C-type inactivation (Hoshi et al., 1991), which involves a constriction of the outer mouth of the channel pore (Yellen et al., 1994). C-type inactivation is enhanced by N-type inactivation (Hoshi et al., 1991), because the latter leads to ion deprivation of the channel pore and also prolongs the channel open-state from which C-type inactivation occurs (Baukrowitz and Yellen, 1995).
Kv4 channels, which represent the molecular substrate of the cardiac transient outward current Ito (Dixon et al., 1996) and the neuronal somatodendritic A-type current in mammals (Serodio et al., 1994), apparently do not inactivate like Shaker channels. Instead of being trapped in an open-inactivated state, Kv4 channels accumulate in a closed-inactivated state (IC) during prolonged depolarization, after having only transiently occupied an open-inactivated state (IO; Fig. 1; Jerng et al., 1999; Bähring et al., 2001a). Therefore, recovery of Kv4 channels from inactivation does not occur via the open state (Bähring et al., 2001a). This finding, in combination with other experimental evidence, including the resistance of inactivation kinetics to internal TEA and the slowing of recovery from inactivation in high external K+ (Jerng and Covarrubias, 1997), led to the conclusion that occlusion of the inner Kv4 channel vestibule (Jerng et al., 1999; Beck and Covarrubias, 2001; Beck et al., 2002) must differ from the classical Shaker N-type inactivation mechanism. Here we studied the mechanism underlying the initial rapid inactivation of Kv4.2 channels. For this purpose we transferred, by N-terminal chimeric replacement, the fast Kv4.2 inactivation properties onto Kv2.1. Kv2.1(4.2NT) chimeric channels exhibit an open-inactivated state with N-type inactivation features. Our finding that the application of soluble Kv4.2 N-terminal peptide apparently causes channel block suggests that the Kv4.2 N-terminus is involved in a mechanism with features similar to the Shaker N-type inactivation. However, our double mutant cycle analysis indicates differences between Kv4 and Shaker N-type inactivation.
FIGURE 1.

Kv4 inactivation gating. Simplified Kv4 state diagram showing closed (C) and inactivated states (I) near the open state (O). The open-state inactivation pathway may include at least two inactivated states, all represented by [IO]. After opening, Kv4 channels are thought to transiently occupy an open-inactivated state ([IO]), but then they close and finally accumulate in closed-inactivated state(s) (IC; Jerng et al., 1999; Bähring et al., 2001a). State transitions indicated by pairs of arrows are defined by respective on- and off-rates. Transitions leading to closed and closed-inactivated states farther away from the open state, which were of minor interest in the present study, are represented by dashed arrows.
MATERIALS AND METHODS
Channel constructs
In this study, we used the human clones of Kv4.2 (both wild-type and a Δ40 deletion mutant, lacking the proximal N-terminus and starting with the T1-domain; Zhu et al., 1999a), Kv2.1, and Kv1.5 α-subunits and the human KChIP2.1 splice variant, which corresponds to the KChIP2 sequence published by An et al. (2000). In addition, chimeric channel constructs were made: in Kv2.1(4.2NT) the whole cytoplasmic N-terminus (including the proximal N-terminus, the T1-domain, and the T1-S1 linker domain) of Kv2.1 (amino acids 1–176) was replaced by corresponding domains of Kv4.2 (amino acids 1–180); Kv2.1(4.2NTΔ40) lacked the proximal N-terminus. In Kv1.5(+4.2N40) the first 40 amino acids of the Kv4.2 sequence where attached to the N-terminus of Kv1.5. In a Kv4.2R13E point mutant, the only basic residue within the proximal N-terminus was replaced by a negatively charged one. In addition, we created several Kv4.2 double and triple mutants: In a Kv4.2C320S background (Kv4.2CS), point mutations were introduced at the N-terminal end, within the transmembrane region S6, or within both domains. The double mutants were Kv4.2CS(A3V), Kv4.2CS(V5A), Kv4.2CS(V402A), Kv4.2CS(V406A), and Kv4.2CS(Y413A). The triple mutants were Kv4.2CS(A3V,V402A), Kv4.2CS(V5A,V402A), Kv4.2CS(A3V,V406A), Kv4.2CS(V5A,V406A), Kv4.2CS(A3V,Y413A), and Kv4.2CS(V5A,Y413A). The deletion mutant Kv4.2CSΔ40 lacked the proximal N-terminus. All constructs were made using standard PCR methods, cloned in the eukaryotic expression vector pcDNA3 for cell transfection or in the pGEM vector for RNA synthesis, and verified by sequencing.
Heterologous channel expression
Wild-type and mutant Kvα-subunits, in the absence and presence of KChIP2.1, were transiently expressed in human embryonic kidney (HEK) 293 (Kv4.2, Kv2.1, Kv4.2Δ40, and Kv2.1(4.2NT)) or in Chinese hamster ovary (CHO) cells (Kv1.5 and Kv1.5(+4.2N40)). Cells were grown according to standard tissue-culture protocols and plated on 35-mm plastic dishes at densities between 1 and 3 × 104 cells per dish one day before transfection. Both HEK 293 and CHO cells were transfected using LipofectAMINE reagent (Invitrogen, Karlsruhe, Germany) according to the manufacturer's protocol. We used 0.1–1 μg Kvα cDNA per dish, depending on the channel construct. KChIP2.1 cDNA was used at a 5- to 10-fold excess relative to the α-subunit cDNA coexpressed, and 0.5 μg per dish of enhanced green fluorescent protein cDNA (Clontech, Heidelberg, Germany) was used to identify cells for electrophysiological recordings one day after transfection. The Kv4.2CS mutants were expressed in Xenopus oocytes. After surgical removal of the frog's ovarian lobes under 3-aminobenzoic acid ethyl ester (Sigma, Deisenhofen, Germany) anesthesia and removal of follicular tissue by collagenase A (Roche, Basel, Switzerland), stage V–VI oocytes were selected and kept in oocyte ringer (in mM: NaCl 82.5, KCl 2, MgCl2 1, HEPES 5, pH 7.5 with NaOH) at 18°C. Injection of cRNA (1.5 ng per oocyte) was performed with a Nanoliter 2000 microinjector (World Precision Instruments, Berlin, Germany) 1–5 days before recording.
Solutions and recording techniques
For whole-cell patch-clamp recordings cells were superfused with an external solution containing (in mM): NaCl 135, KCl 5, CaCl2 2, MgCl2 2, HEPES 5 and sucrose 10, with 0.01 mg ml−1 phenol red; pH 7.4 (NaOH). Recording pipettes, pulled from thin-walled borosilicate glass using a DMZ puller (Zeitz Instruments, Augsburg, Germany) and heat polished, had bath resistances between 2 and 3 MΩ when filled with internal solution containing (in mM): KCl 125, CaCl2 1, MgCl2 1, EGTA 11, HEPES 10, glutathione 2 and K2-ATP 2; pH 7.2 (KOH). For some experiments external Na+ was replaced by K+ (130 mM, symmetrical K+), and for some we added TEA to the intracellular solution at concentrations of 2, 5, or 10 mM. For experiments with excised inside-out membrane patches, recording pipettes were filled with external solution. Synthetic N-terminal peptides (Jerini, Berlin, Germany) were C-terminally amidated and HPLC purified. The Kv4.2 peptide consisted of the first 20 amino acids of the protein (see Fig. 6 A). In addition, we used a modified 20-amino-acid N-terminal peptide of a Shaker splice variant (ShB E12K, D13K; Murrell-Lagnado and Aldrich, 1993a; Holmgren et al., 1996; see Fig. 6 A) and a 20-amino-acid scrambled peptide with the same amino acid composition as the Kv4.2 N-terminal peptide but in a random order (see Fig. 11). Internal solution with or without N-terminal peptide at a concentration of 100 μM was applied to the cytoplasmic face of inside-out patches using a valve-controlled local superfusion system driven by a peristaltic pump. For two-electrode voltage-clamp experiments, Xenopus oocytes were bathed in a solution containing (in mM): NaCl 96, KCl 2, CaCl2 1.8, MgCl2 1 and HEPES 5; pH 7.4 (NaOH). Recording electrodes plugged with Agar/KCl and filled with 3 M KCl had tip resistances < 1 MΩ. All experiments were conducted at room temperature (20°–22°C).
FIGURE 6.

Blocking of Kv2.1 channels by N-terminal peptide application. N-terminal peptides from Kv4.2 and ShB E12K, D13K, respectively (A), were applied to the internal face of inside-out membrane patches containing Kv2.1 channels. (B) Effect of Kv4.2 N-terminal peptide application. Currents were activated by pulses from −100 to +50 mV. Scale bars: 100 pA, 30 ms. (C) Effect of ShB E12K, D13K N-terminal peptide application. In this experiment currents were activated by pulses from −100 to +100 mV, to obtain larger amplitudes. Scale bars: 200 pA, 30 ms. Dashed lines represent zero current.
FIGURE 11.

Application of N-terminal peptides to inactivation-impaired Kv4.2 channels. Kv4.2 N-terminal peptide was applied to the cytoplasmic face of Kv4.2Δ40 (A) and Kv4.2/KChIP2.1 (B) containing inside-out patches. (C) Specificity was tested by applying a scrambled peptide with the sequence H-MGAVPAWAWLAFARAMGAAI-NH2. (D) Application of ShB E12K, D13K N-terminal peptide to Kv4.2Δ40. Currents were activated by pulses from −100 to +50 mV. Horizontal scale bars in all panels: 30 ms; vertical scale bars: 50 pA in A, 10 pA in B, 50 pA in C, and 150 pA in D. Dashed lines represent zero current.
Data acquisition and analysis
HEK 293 and CHO cell currents were recorded with an EPC9 patch-clamp amplifier (HEKA, Lambrecht, Germany), and the program PULSE (HEKA) was used for data acquisition. Signals were digitized at sample intervals between 50 μs and 5 ms depending on the voltage protocol and filtered online at 10 kHz for analysis and digitally offline at 1 kHz for presentation. Leak currents were subtracted using the P/n method. Cells were held at −80 mV and test pulses to activate Kv4.2 channels were preceded by prepulses to −100 mV to remove steady-state inactivation. Recovery from inactivation was measured using a double-pulse protocol with interpulse intervals of variable duration at −80 mV. Oocyte currents were recorded with a TURBO TEC 01C amplifier (npi electronic instruments, Tamm, Germany) and PULSE acquisition software. All current traces were analyzed with PULSEFIT (HEKA), and the obtained data were further processed using Kaleidagraph (Synergy Software, Reading, PA) and Igor Pro (Wavemetrics, Lake Oswego, OR). Statistic analysis was based on unpaired Student's t-tests. Pooled data are presented as mean ± SEM.
Modeling and simulations
For tail current simulations we used a previously developed allosteric model for Kv4.2 channel gating (Bähring et al., 2001a), which is illustrated in a reduced form and briefly explained in Fig. 1. The Markov-model parameters and rates (Bähring et al., 2001a) were implemented in a simulation routine run under PULSESIM (HEKA). Rates were modified to simulate inward tail currents at hyperpolarized voltages for Kv4.2Δ40 as well as Kv4.2/KChIP2.1, such that experimentally obtained outward current kinetics at positive potentials could be reproduced equally well. To simulate Kv4.2Δ40 gating, the open-inactivated state IO was removed from the model, and the closing rate koc was reduced 5-fold. KChIP2.1 remodeling was performed by modifying our existing Kv4.2 model, in principle according to Beck et al. (2002); however, Kv4.2/KChIP2.1 gating required different values, and we had to modify the deactivation rate constant β0, whereby β = β0exp[−zeV/(kT)], to reproduce experimentally observed tail current kinetics. In particular, both the forward-rate kci and the backward-rate kic for the closed-state inactivation were increased twofold. Also, the closing rate koc was increased 2-fold and β0 2.5-fold. Kv4.2/KChIP2.1 open-state inactivation was modeled by reducing the on-rate koi sixfold. Simulated tail currents were fitted with appropriate terms of exponential functions using PULSEFIT, and the results are illustrated together with experimental data (e.g., Fig. 8, D and E; Fig. 9 B, Fig. 10, C and D).
FIGURE 8.

Kinetic analysis of tail currents mediated by Kv4.2 wild-type channels. (A) Representative Kv4.2-mediated currents recorded with a similar protocol as in Fig. 4 A with 130 mM external K+. Reopening becomes visible as a slow component of tail current decay. (B) Fast decay of tail currents after a brief depolarization (4 ms, arrow 1 in A). (C) Tail currents after a longer-lasting depolarization (64 ms, arrow 2 in A) show a pronounced slow component of decay. Dashed lines represent zero current, and dotted lines in B and C represent the best fits of a sum of two exponentials. (D, E) Summary of the results from double-exponential fitting: fast (solid circles) and slow (solid squares) time constants of tail current decay (D), as well as their relative amplitudes (corresponding open symbols in E) were plotted against the duration of the depolarizing prepulse. Note that the relative amplitude of the slower time constant increases with longer prepulse durations. Broken lines in D and E represent predictions of a Kv4.2 Markov model published previously (Bähring et al., 2001a).
FIGURE 9.

Deletion of proximal N-terminus (Δ40) abolishes reopening features of Kv4.2. Tail currents were measured with the same protocol as shown in Fig. 8 A. (A) Superimposed and normalized Kv4.2Δ40-mediated tail currents after depolarizing prepulses lasting 4 and 64 ms, respectively. Single-exponential fits to the current traces are represented by dotted lines, and zero current by a dashed line. (B) Obtained time constants were plotted against the depolarizing prepulse duration, and the prediction of a model, which lacks the open-inactivated state IO and has a slower closing-rate koc, is illustrated as a broken line. (C) Deactivation time constants measured at different test potentials after 4 ms of depolarization for Kv4.2 wild-type (solid circles) and Kv4.2Δ40 (solid triangles). Lines represent monoexponential fits.
FIGURE 10.

Suppressed N-type features of Kv4.2/KChIP2.1-mediated tail currents. Tail current analysis with protocols as in Fig. 8 A. (A) Superimposed and normalized Kv4.2/KChIP2.1-mediated tail currents after depolarizing prepulses lasting 4 and 64 ms, respectively. Tail current decay was fitted with a double-exponential function (dotted lines). Dashed line represents zero current. (B) Time constants of deactivation measured after 4 ms of depolarization at different test potentials for Kv4.2 wild-type (solid circles) and Kv4.2/KChIP2.1 channel complexes (solid diamonds). Lines represent monoexponential fits. (C, D) Fast (solid diamonds) and slow (solid squares) time constants of tail current decay, obtained from double-exponential fitting, as well as their relative amplitudes (corresponding open symbols) were plotted against the prepulse duration. Broken lines show the predictions of a model with modified rates to simulate Kv4.2/KChIP2.1 gating.
Double mutant cycle analysis
We determined the affinity for the fast inactivation gate of Kv4.2CS channels by calculating the equilibrium constant Kd as koff/kon, similar to the kinetic analysis of ball peptide blockade of Shaker channels done previously by Holmgren et al. (1996): for all Kv4.2CS mutants the current decay was fitted with a double-exponential function, and the time constant τ1 and its relative amplitude A1 were used to estimate kon and koff for the fast inactivation reaction. The fraction of current remained fb = 1 − A1 was used as an estimate for the steady state (Holmgren et al., 1996; see also Fig. 13 B), with koff = fb/τ1 and kon =  − koff. Regarding Kv4.2CS as the wild-type reference (wt), we refer to the double mutants as (wt,mut) or (mut,wt) and to the triple mutants as (mut,mut). Similar to the investigations on Shaker-related channel N-type inactivation (Zhou et al., 2001), the coupling coefficient Ω obtained with double mutant cycle analysis was used to quantify the degree of coupling of point mutation-induced effects on fast Kv4.2CS inactivation, with Ω = (
− koff. Regarding Kv4.2CS as the wild-type reference (wt), we refer to the double mutants as (wt,mut) or (mut,wt) and to the triple mutants as (mut,mut). Similar to the investigations on Shaker-related channel N-type inactivation (Zhou et al., 2001), the coupling coefficient Ω obtained with double mutant cycle analysis was used to quantify the degree of coupling of point mutation-induced effects on fast Kv4.2CS inactivation, with Ω = (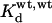

 ). For ease of comparison, if Ω was smaller than unity, its reciprocal was used. Thus, coupling coefficients higher than unity (ln Ω > 0) indicate that the effects of two mutations are coupled, and it can be concluded that the respective mutated amino acids normally interact due to their close spatial proximity. We used the mean and SEM (n = 5 to 7) of Kd to obtain the range of uncertainty on Ω, assuming linear propagation of independent errors through the above equation.
). For ease of comparison, if Ω was smaller than unity, its reciprocal was used. Thus, coupling coefficients higher than unity (ln Ω > 0) indicate that the effects of two mutations are coupled, and it can be concluded that the respective mutated amino acids normally interact due to their close spatial proximity. We used the mean and SEM (n = 5 to 7) of Kd to obtain the range of uncertainty on Ω, assuming linear propagation of independent errors through the above equation.
FIGURE 13.

Double mutant cycle analysis of Kv4.2CS N-type inactivation. (A) Incomplete sequence alignment showing only the residues of interest for N-type inactivation: Main interacting residues found by Zhou et al. (2001) in Kvβ1.1 N-terminal and Kv1.4 S6 domains, respectively, as well as the corresponding residues in Kv4.2, which were mutated in the present study, are highlighted. (B) Analysis of fast inactivation, estimation of steady state (dotted line), and calculation of fb illustrated for Kv4.2CS. (C) Example recordings for the mutant channels Kv4.2CSA3V and Kv4.2CSV406A (for comparison Kv4.2CS inactivation kinetics are indicated by a dotted line). Dashed lines represent zero current. (D) Changes in Kd for the fast inactivation gate caused by individual point mutations on a Kv4.2CS background relative to Kv4.2CS (Kd,wt). (E) Double mutant cycle analysis of Kv4.2CS N-terminal mutations and pore-lining S6 mutations. Mutated residues are homologous to the ones shown previously by Zhou et al. (2001) to interact during Shaker-related channel N-type inactivation. The coupling coefficient Ω is shown for three pore residues and two N-terminal residues. The presented data are based on 5 to 7 experiments per mutant.
RESULTS
Chimeric Kv2.1(4.2NT) channels show typical Kv4.2 inactivation properties
K+ channel inactivation mediated by the Kv4.2 cytoplasmic N-terminus (NT) was studied in transiently transfected HEK 293 cells. In agreement with previous results (Bähring et al., 2001a), rapidly inactivating outward currents were recorded in whole-cell patch-clamp experiments at depolarized test potentials (Fig. 2 A) from cells expressing Kv4.2 wild-type channels. The inactivation time course of Kv4.2 currents at +50 mV was well described by the sum of three exponential functions with τ1 = 15 ± 1 ms (72 ± 4%), τ2 = 62 ± 6 ms (23 ± 3%), and τ3 = 776 ± 103 ms (5 ± 1%; n = 8). The multiexponential decay kinetics of Kv4.2 currents is thought to be a consequence of the various ways by which inactivated states can be reached during strong depolarization (Jerng et al., 1999; Bähring et al., 2001a). The gating scheme in Fig. 1 defines these transitions: Some channels directly inactivate from closed states (C → IC) before they open; since the opening step is reverse-biased, open channels may quickly close again and then enter closed-inactivated state(s) (O → C → IC); open channels may also enter an open-inactivated state (O ↔ IO). The key kinetic features of such Kv4 gating models (Jerng et al., 1999; Bähring et al., 2001a) are a transient occupancy of this open-inactivated state and the final accumulation of channels in a closed-inactivated state (IO → O → C → IC). So far, an open-inactivated state IO in Kv4.2 channels has not been directly observed in electrophysiological experiments. The existence of an IO-state may have escaped experimental observation, because closed-state inactivation of Kv4.2 channels is relatively fast and the open-inactivated state IO is short-lived (Jerng et al., 1999; Bähring et al., 2001a). Alternatively, inactivation of an open-state, which is the primary cause for the rapid inactivation of Shaker channels, may not occur in Kv4.2 channels.
FIGURE 2.

Kv4.2 N-terminus confers Kv4 inactivation properties to Kv2.1. Currents recorded at +50 mV from transiently transfected HEK 293 cells expressing wild-type Kv4.2 (A), wild-type Kv2.1 (B), and Kv2.1(4.2NT) chimeric channels with the Kv4.2 proximal N-terminus intact (C) or deleted (F). Respective channel constructs are illustrated by cartoons (Kv4.2 parts in black and Kv2.1 parts in gray). KChIP2.1 coexpression causes slowing of the initial Kv4.2 inactivation component (D) and removal of fast inactivation in Kv2.1(4.2NT) chimeric channels (E). Dotted lines in D and E represent normalized currents mediated by Kv4.2 and Kv2.1(4.2NT), respectively, in the absence of KChIP2.1. Kv4.2 expressing cells were depolarized from −100, Kv2.1 (wild-type or chimera) expressing cells from −80 mV. Dashed lines represent zero current.
We tested experimentally whether the Kv4.2 N-terminus possesses the ability to function as an autoinhibitory inactivating domain, like the N-terminus of Shaker channels. For this purpose, we constructed the chimera Kv2.1(4.2NT) where the whole cytoplasmic N-terminus of Kv2.1 was replaced by the corresponding part of Kv4.2 (see Materials and Methods). Kv2.1 channels are normally resistant to inactivation in their open state; rather, all Kv2.1 inactivation is mediated by transitions from pre-open closed to closed-inactivated states (Klemic et al., 1998). In comparison to Kv4.2, this reaction apparently occurs on a much slower timescale in Kv2.1 (Fig. 2 B; τinact = 3.4 ± 0.3 s; n = 14; Zhu et al., 1999b). Kv2.1 channels, therefore, were regarded as a suitable channel template, where cumulative closed-state inactivation did not confound investigations of open-state inactivation mediated by a Kv4.2 inactivating domain.
Fig. 2 C shows that during a 1-s depolarizing test pulse to +50 mV, Kv2.1(4.2NT) chimeric channels mediated outward currents, which in contrast to Kv2.1 displayed a prominent component of fast inactivation. The Kv2.1(4.2NT) inactivation kinetics were well described by the sum of two exponential functions (τ1 = 120 ± 13 ms, 82 ± 3%; τ2 = 5.9 ± 1.1 s; n = 6). The results indicated that the Kv4.2 N-terminus conferred a fast inactivation to Kv2.1 channels.
In Kv4 channels the association of Kv channel interacting proteins (KChIPs) modifies several aspects of gating. One aspect relates to the initial inactivation component of Kv4-mediated currents, which is markedly slowed in the presence of KChIP (Bähring et al., 2001b; Beck et al., 2002). Accordingly, Kv4.2/KChIP2.1 channel complexes inactivated with τ1 = 33 ± 5 ms (41 ± 4%), τ2 = 74 ± 7 ms (58 ± 2%), and τ3 = 500 ± 129 (1 ± 1%; n = 6; Fig. 2 D). When we coexpressed Kv2.1(4.2NT) channels with KChIP2.1, we also observed an effect on inactivation (Fig. 2 E). Kv2.1(4.2NT)/KChIP2.1 currents decayed as slowly (τ = 4.9 ± 0.4 s; n = 10) as Kv2.1 wild-type currents. Apparently, KChIP2.1 attenuated the inactivating activity of the Kv4.2 N-terminus in the Kv2.1(4.2NT) chimera.
The fast inactivation of Kv2.1(4.2NT) currents depended on the presence of the first 40 amino acid residues of the Kv4.2 N-terminal sequence. N-terminal truncation caused a dramatic effect in Kv2.1(4.2NT). Kv2.1(4.2NTΔ40) channels mediated outward currents, which inactivated even slower (τ = 11.1 ± 1.6 s ; n = 6) than Kv2.1 (Fig. 2 F). In summary, our experimental results were compatible with the hypothesis that a KChIP-sensitive inactivating domain resides in the Kv4.2 proximal N-terminus. In the following, we show experimental evidence that the fast Kv2.1(4.2NT) inactivation bears features similar to the ones described for the Shaker N-type inactivation.
Kv2.1(4.2NT) inactivation affected by intracellular channel blocker
Intracellular channel blockers like TEA slow the decay kinetics of Shaker-mediated currents because of a competition reaction between the pore-blocking reagent and the N-terminal inactivation domain for binding within the pore cavity (Choi et al., 1991; Zhou et al., 2001). Here, we explored, due to low current amplitudes in inside-out patches, the effect of TEA added to the internal solution on the inactivation kinetics of whole-cell currents mediated by Kv2.1(4.2NT). At the highest concentration tested (10 mM), internal TEA induced a dramatic slowing of inactivation (Fig. 3 A), and the fast component of decay obtained with double-exponential fitting was 280 ± 37 ms (n = 7). Analysis of the linear concentration dependence of this effect suggested a twofold slowing of the fast inactivation component at ∼5 mM TEA (Fig. 3 B).
FIGURE 3.

Internal TEA affects Kv2.1(4.2NT) inactivation kinetics. (A) Normalized Kv2.1(4.2NT) current traces recorded from two different cells in the absence (control) and in the presence of 10 mM internal TEA, respectively. (B) Mean values of τ1 obtained from double-exponential fits to the current decay in the presence of 0, 2, 5, and 10 mM TEA in the pipette solution. Concentration dependence of τ1 is characterized by a linear regression line with y = 100 + 18 x.
Recovery of Kv2.1(4.2NT) channels from N-type inactivation
Recovery of Shaker channels from inactivation displays several characteristic features due to the unblocking reaction of the Shaker N-terminal inactivation domain (Demo and Yellen, 1991; Ruppersberg et al., 1991). In particular, Shaker channels reopen during recovery from N-type inactivation, giving rise to a significant temporary increase in inward current at negative membrane potentials under suitable experimental conditions. To test directly reopening of Kv2.1(4.2NT) channels during recovery from inactivation, we examined the kinetics of tail currents in the presence of 130 mM external K+. The kinetics critically depended on the length of the preceding depolarizing pulse (Fig. 4 A). After brief depolarizing pulses (e.g., 40 ms; arrow 1 in Fig. 4 A) tail currents decayed without delay (Fig. 4 B). After longer-lasting depolarizations (e.g., > 1 s), a significant fraction of Kv2.1(4.2NT) channels had already inactivated (Fig. 4 A), and repolarization resulted in tail currents with altered kinetics (arrow 2 in Fig. 4 A) and a temporary increase in amplitude (Fig. 4 C). The temporary increase in tail current amplitude indicated that inactivated Kv2.1(4.2NT) channels reopened during recovery from inactivation.
FIGURE 4.

Kv2.1(4.2NT) channels reopen during recovery from inactivation. (A) Currents mediated by Kv2.1(4.2NT) were activated by voltage steps to +50 mV of variable length in 130 mM external K+, before tail current recording at −50 mV. Tail currents obtained after a brief (40 ms; arrow 1 in A) and after a longer depolarization (1.3 s; arrow 2 in A) are shown at a higher magnification and normalized to the largest negative current deflexion in B and C, respectively. Note the initial increase (indicative of channel reopening) followed by slow decay of current in C. Dashed lines represent zero current.
High external K+ concentrations, as were used in the tail current experiments shown in Fig. 4, lower the apparent affinity of the Shaker inactivating particle for its receptor site within the channel pore. This leads to an acceleration of recovery from inactivation (Demo and Yellen, 1991). We investigated whether this characteristic feature also applies to the recovery of Kv2.1(4.2NT) channels from inactivation. Therefore, we compared the time courses of recovery of Kv2.1(4.2NT) channels from inactivation in 5 and 130 mM external K+, respectively. Recovery from inactivation in 5 mM K+ was relatively slow (Fig. 5 A), with no apparent recovery within the first 40 ms (Fig. 5 C). The recovery kinetics (Fig. 5 D) were well described by a monoexponential function with a recovery time constant τrec = 1.7 ± 0.2 s (n = 6). By contrast, the recovery from inactivation in 130 mM K+ (Fig. 5 B) was very fast (apparent τrec = 27 ± 7 ms; n = 5), such that a considerable fraction of Kv2.1(4.2NT) channels recovered from inactivation within 40 ms (Fig. 5 C). The results showed that high external K+ concentrations dramatically accelerated the recovery of Kv2.1(4.2NT) channels from inactivation. Under these conditions Kv2.1(4.2NT)-mediated currents showed an unusual property: the fast recovery of Kv2.1(4.2NT) channels from inactivation in 130 mM K+ was followed by a current overshoot at longer interpulse intervals (Fig. 5 B). Amplitudes of the second peak increased to values higher than control and then gradually leveled off (Fig. 5, B and D). Apparently, at 130 mM K+ Kv2.1(4.2NT) channels recover from N-type inactivation much faster than they subsequently enter closed states. Thus, channels may temporarily accumulate in the open state(s), yielding more peak current flow than during the initial control pulse, where activation and inactivation kinetics overlap.
FIGURE 5.

High external K+ accelerates recovery of Kv2.1(4.2NT) channels from inactivation. A and B show example recordings performed to study recovery of Kv2.1(4.2NT) channels from inactivation in the presence of different external K+ concentrations. Currents were activated at +50 mV using a double-pulse protocol with variable interpulse durations at −80 mV. (C) Whereas no recovery can be observed after 40 ms in 5 mM [K+]o, currents reach control during this interval in 130 mM [K+]o. Zero current is represented by a dashed line. (D) Peak currents of the second pulse were normalized to peak currents of the first pulse and plotted against the interpulse duration. Kinetics of recovery from inactivation in 5 (open symbols) and 130 mM [K+]o (solid symbols) were analyzed by fitting a monoexponential function to the data. Note that after 40-ms intervals currents in high external K+ exceeded the peak control amplitudes and gradually leveled off within intervals longer than 10 s (B); therefore, only a fraction of the data in 130 mM external K+ (solid circles) was analyzed. Data contaminated by overshoot are presented as solid diamonds (D).
Kv4.2 N-terminus behaves like an inactivating “ball peptide”
The activity of the N-terminal Shaker inactivation domain may also be observed if it is added as a soluble peptide to the cytoplasmic face of inside-out patches containing noninactivating Shaker channels (Zagotta et al., 1990). We wanted to see if the Kv4.2 N-terminus exhibited a similar activity when applied to inside-out patches containing noninactivating channels. For such experiments, we used a Kv4.2 peptide corresponding to the first 20 amino acid residues of the Kv4.2 N-terminus and the previously described 20-amino-acid-residues-long ShB E12K, D13K peptide (Murrell-Lagnado and Aldrich, 1993a; Fig. 6 A). Application of Kv4.2 peptide to the cytoplasmic face of inside-out patches containing Kv2.1 channels induced a rapid inactivation of currents (Fig. 6 B, trace 2). In the presence of 100 μM peptide the Kv2.1 current decay could be described by a monoexponential function with τ = 65 ± 11 ms (n = 5). Upon washout of the Kv4.2 peptide, Kv2.1 currents fully regained their slow decay kinetics (Fig. 6 B). Application of 100 μM ShB E12K, D13K peptide induced a similarly rapid inactivation of Kv2.1 currents (τ = 51 ± 11 ms; n = 5; Fig. 6 C, trace 2), and inactivation was also reversed upon washout of the peptide (Fig. 6 C). The results indicated that the Kv4.2 N-terminal peptide acted as a Kv2.1 channel blocker, like the ShB E12K, D13K peptide.
Rapid inactivation conferred to Kv1.5 channels by proximal Kv4.2 N-terminus
The results described so far showed that the fast inactivation of Kv2.1(4.2NT) channels displayed key features of N-type inactivation very similar to the ones described for Shaker channels. This strongly suggested that the proximal Kv4.2 N-terminus is a Shaker-like N-type inactivating domain. Therefore, it should be possible to confer inactivation to Shaker-related Kv channels via the proximal Kv4.2 N-terminus. To test this proposition, we chose Kv1.5 as a representative member of the Kv1 family. When expressed in CHO cells, wild-type Kv1.5 channels exhibited only a slow C-type inactivation during test pulses to +60 mV (Fig. 7 A). We attached the proximal 40 amino acid residues of the Kv4.2 N-terminus to Kv1.5 (see Materials and Methods and Fig. 7 A) to generate Kv1.5(+4.2N40) channels. In contrast to Kv1.5, Kv1.5(+4.2N40) channels yielded rapidly inactivating outward currents (Fig. 7 A). The results demonstrated that the Kv4.2 inactivating domain conferred a rapid inactivation to Kv1.5 channels. Kv1.5(+4.2N40) current decay was well described by the sum of two exponential functions with τ1 = 14 ± 1 ms (87 ± 2%) and τ2 = 104 ± 11 ms (n = 12), probably representing the coupled reactions of N- and C-type inactivation. The recovery of Shaker channels from inactivation shows two components which reflect recovery from N- and C-type inactivation, respectively. Both inactivation mechanisms can be influenced by changes in the external K+ concentration; high external K+ concentrations may prevent C-type inactivation of Shaker channels and, furthermore, lead to an accelerated recovery from N-type inactivation (Demo and Yellen, 1991; Baukrowitz and Yellen, 1995). We compared the recovery of Kv1.5(+4.2N40) channels from inactivation in 5 and 130 mM external K+, respectively (Fig. 7 B). In 5 mM K+ the recovery of Kv1.5(+4.2N40) channels from inactivation showed two components, a relatively fast one (τrec1 = 185 ± 5 ms, 83 ± 5%; n = 5) and a slow one (τrec2 = 1.1 ± 0.1 s). Most likely, the fast component reflected recovery from N-type inactivation and the slow component recovery from C-type inactivation, as in Shaker channels. In 130 mM external K+ the recovery of Kv1.5(+4.2N40) channels from inactivation was accelerated and showed a monoexponential time course (Fig. 7 B) with τrec = 100 ± 10 ms (n = 3). These experiments demonstrated for the inactivation of Kv1.5(+4.2N40) channels a characteristic K+ dependence, as observed for the Shaker N- and C-type inactivation.
FIGURE 7.

Inactivation properties of Kv1.5(+4.2N40) chimeric channels. (A) Normalized currents mediated by Kv1.5 and Kv1.5(+4.2N40), respectively. Currents were elicited by voltage steps from −80 to +60 mV; [K+]o = 5 mM. Dashed line represents zero current. (B) Effects of high external K+ (130 mM) on the recovery kinetics of Kv1.5(+4.2N40). A double-exponential function describes the recovery kinetics in 5 mM [K+]o (open symbols) and a monoexponential function describes the recovery kinetics in 130 mM [K+]o (solid symbols). Dotted line represents a hypothetical fit, where the fast component of recovery in low external K+ accounts for 100%.
N-type inactivation of Kv4.2 channels
Our experimental results with Kv2.1(4.2NT) chimeric channels demonstrated transitions between open and open-inactivated state(s). Our data support an occlusion mechanism mediated by the Kv4.2 proximal N-terminus. Consequently, the recovery of Kv2.1(4.2NT) channels from inactivation caused typical reopening features of tail currents.
Next, we revisited the inactivation of Kv4.2 wild-type channels. The decay kinetics of tail currents elicited by repolarization to −50 mV after depolarizing prepulses to +50 mV of variable length (Fig. 8 A) was taken as an indirect measure of Kv4.2 N-type inactivation. Kv4.2 tail currents decayed with double-exponential kinetics. After brief depolarizing prepulses, e.g., 4 ms (arrow 1 in Fig. 8 A), a fast tail current component (τ1 = 3.9 ± 0.7 ms; n = 4) accounted for 95 ± 1% of the total decay and was followed by a slower component (τ2 = 180 ± 55 ms; Fig. 8 B). After longer depolarizations, e.g., 64 ms (arrow 2 in Fig. 8 A), the fast tail current component was slowed (τ1 = 10.0 ± 2.0 ms; n = 4) and the relative amplitude was reduced to 63 ± 7%. Now the slow component (τ2 = 199 ± 30 ms; n = 4) accounted for a considerable fraction of the total decay (Fig. 8 C). The results of our kinetic tail current analyses are shown in Figs. 8 D and 8 E. Prolongation of the depolarizing prepulse from a few to more than 100 ms led to a reduction of the relative amplitude of the fast component τ1 in favor of the slower component τ2 (Fig. 8 E). Possibly, the slow component of tail current decay was caused by channels, which opened during recovery from inactivation before they closed.
Because Kv4.2Δ40 channels lack the N-terminal inactivating domain, transitions to open-inactivated state(s) should not occur during activation, and the decay kinetics of Kv4.2Δ40-mediated tail currents should simply reflect channel closure. To test this prediction, we analyzed the decay kinetics of Kv4.2Δ40 tail currents after depolarizing test pulses of variable length, according to the protocol shown in Fig. 8 A. In contrast to Kv4.2 wild-type, Kv4.2Δ40 tail currents were adequately fitted by a single-exponential function, with τ = 14.3 ± 3.5 ms (n = 3) when measured after a 4-ms depolarizing prepulse, and τ = 20.3 ± 2.3 ms (n = 3) when measured after 64 ms of depolarization (Fig. 9 A). The data further indicated that the time constant of Kv4.2Δ40 tail current decay did not markedly depend on the duration of the depolarizing prepulse (Fig. 9, A and B). It should be noted at this point that Kv4.2 channel deactivation kinetics are apparently slowed in a voltage-independent manner by the Δ40 N-terminal deletion (Fig. 9 C; Bähring et al., 2001a).
Assuming that KChIP2.1 attenuates the Kv4.2 N-type inactivation, one would predict, first, that tail currents have mainly a fast component, and second, that the kinetics of tail current decay is not markedly affected by the duration of the depolarizing prepulse. Double-exponential fitting of the Kv4.2/KChIP2.1 tail currents, obtained with protocols like the one shown in Fig. 8 A, yielded fast time constants τ1 between 1.4 ± 0.4 ms after 4 ms and 2.3 ± 0.4 ms (n = 5) after 64 ms of depolarization (Fig. 10, A and C). Within this time interval the fast time constant accounted for more than 90% of the total tail current amplitude, independent of the duration of the depolarizing prepulse (Fig. 10 D). In addition, Kv4.2 channel deactivation was faster in the presence of KChIP2.1 at all test potentials (Fig. 10 B), similar to previous observations made with other Kv4/KChIP complexes (Beck et al., 2002).
Kv4.2 channels blocked by N-terminal peptide
The results described so far indicated that N-type inactivation, i.e., the open-state inactivation transition, was completely absent in Kv4.2Δ40 channels, and largely impaired in Kv4.2/KChIP2.1 channel complexes, as was suggested previously by KChIP remodeling of Kv4 gating (Beck et al., 2002). We considered the possibility that KChIP2.1 hinders the Kv4.2 proximal N-terminus from reaching to the intracellular Kv4.2 channel opening, where it can block the conduction pathway. We intended to restore Kv4.2 N-type inactivation by adding the Kv4.2 N-terminal peptide to the cytoplasmic face of inside-out patches containing inactivation-impaired channels like Kv4.2Δ40 or Kv4.2/KChIP2.1 (Fig. 11). Under control conditions Kv4.2Δ40 patch currents exhibited a double-exponential decay with τ1 = 10 ± 1 ms (63 ± 3%) and τ2 = 124 ± 21 ms (n = 18). During the application of 100 μM Kv4.2 N-terminal peptide, the Kv4.2Δ40 current decay was markedly accelerated (τ1 = 4 ± 1 ms, 71 ± 5% and τ2 = 34 ± 6 ms; n = 8; p < 0.001; Fig. 11 A, trace 2). Upon washout of the peptide, the acceleration was diminished and the original Kv4.2Δ40 current decay was seen (Fig. 11 A). Kv4.2/KChIP2.1 channels also mediated patch currents with double-exponential decay (τ1 = 28 ± 7 ms, 63 ± 8% and τ2 = 100 ± 24 ms; n = 4). Similar to Kv4.2Δ40, the application of 100 μM Kv4.2 N-terminal peptide caused a noticeable acceleration (p = 0.078) of Kv4.2/KChIP2.1 current decay with τ1 = 11 ± 3 ms (58 ± 16%) and τ2 = 69 ± 17 ms (n = 4; Fig. 11 B, trace 2), which could be reversed upon washout (Fig. 11 B). We tested the specificity of the effect caused by the Kv4.2 N-terminal peptide application with a 20-amino-acid scrambled sequence peptide (see Materials and Methods and legend to Fig. 11). Application of 100 μM of the scrambled sequence peptide did not significantly influence the decay kinetics of Kv4.2Δ40-mediated patch currents (τ1 = 8 ± 1 ms, 80 ± 2% and τ2 = 104 ± 33 ms; n = 7; p = 0.1889; Fig. 11 C), suggesting that the observed effect with Kv4.2 N-terminal peptide was not due to unspecific hydrophobic interactions. Notably, however, the application of 100 μM ShB E12K, D13K peptide (Fig. 11 D) did influence the Kv4.2Δ40 current decay kinetics (τ1 = 4 ± 1 ms, 73 ± 9% and τ2 = 58 ± 34 ms; n = 3; p = 0.02). These results showed that Kv4.2 N-type inactivation, lost by N-terminal truncation, can be mimicked by the 20-amino-acid N-terminal peptides, and they imply that KChIPs suppress Kv4.2 N-type inactivation.
Double mutant cycle-based kinetic and structural analysis of Kv4.2 N-type inactivation
The interaction between an N-type inactivation domain and its receptor in Shaker-related channels has been studied in great detail with double mutant cycle analysis, and the interacting amino acid residues in the proximal N-terminus and in S6, respectively, have been identified (Zhou et al., 2001). We asked if homologous amino acid residues in Kv4.2 (Fig. 13 A) participate in an interaction between the Kv4.2 N-terminus and S6. Accordingly, we used double mutant cycle analysis of Kv4.2 N-terminal and S6 mutations to assess the proximity of amino acids in the open-inactivated state. Double mutant cycle analysis was performed on a Kv4.2C320S background (Kv4.2CS). Since this point mutation slowed Kv4.2 deactivation (data not shown), similar to previous results obtained with a corresponding C322S point mutation in Kv4.1 (Jerng et al., 1999), it should prevent rapid accumulation of Kv4.2 channels in closed-inactivated states and favor open-state inactivation.
The inactivation kinetics of Kv4.2CS as well as Kv4.2CS double and triple mutants (see Materials and Methods) were studied with two-electrode voltage-clamp in Xenopus oocytes. Whereas no significant current was measured in water-injected oocytes (not shown), Kv4.2CS expression yielded A-type currents (Fig. 12 A) with inactivation kinetics that could be described by the sum of two exponential functions, with τ1 = 48 ± 5 ms and a relative amplitude A1 of 0.59 ± 0.01 (n = 7). It is important to note that the fast inactivation component was completely abolished by deleting the proximal N-terminus (Kv4.2CSΔ40; Fig. 12 A). Furthermore, a close inspection of tail current kinetics (Fig. 12 B) revealed reopening features for Kv4.2CS but not for Kv4.2CSΔ40.
FIGURE 12.

Kv4.2CS current kinetics. (A) Currents recorded during depolarizing pulses from −100 to +50 mV under two-electrode voltage-clamp in Xenopus oocytes expressing Kv4.2C320S mutant channels (Kv4.2CS) or the N-terminal deletion construct Kv4.2CSΔ40. Current traces were leak-subtracted and normalized. Note that the fast component of inactivation is absent in Kv4.2CSΔ40. (B) The tail currents recorded after repolarization are shown at a higher magnification and without leak-subtraction (truncated capacitive transients are visible). Note that reopening features are seen only with Kv4.2CS-mediated tail currents. Dashed lines represent zero current.
Calculation of kon and koff via τ1 and fb (Fig. 13 B) eventually yielded Kd = 0.699 and ln(Kd/Kd,wt) = 0 for Kv4.2CS (see Materials and Methods). Individual mutations introduced on the Kv4.2CS background caused distinct alterations of inactivation kinetics. Two examples are shown in Fig. 13 C: Kv4.2CSA3V showed a very low fb value, and Kv4.2CSV406A showed a relatively slow τ1 and a high fb value. The relative changes in Kd for these two and the other point mutants tested are plotted in Fig. 13 D. Apparently, the A3V mutation caused a decrease, whereas the V406A mutation caused an increase in Kd. The coupling of the effects on inactivation kinetics caused by pairs of these mutations was analyzed in a double mutant cycle (see Materials and Methods), and the respective coupling coefficients are plotted in Fig. 13 E. We obtained the largest coupling coefficient for A3V in combination with V406A (ln Ω = 0.85 kT). On the other hand, a coupling coefficient close to unity (ln Ω = 0.01 kT) was obtained for A3V in combination with Y413A. The coupling coefficients were lower than the corresponding ones reported for the Shaker-related channel N-type inactivation (ln Ω values ≥ 2; Zhou et al., 2001).
DISCUSSION
In the present study we provide strong evidence for the existence of Kv4.2 open-state inactivation. In particular, we show that it is mediated by an N-type inactivation mechanism, similar to the one described for Shaker channels, albeit the structural determinants on the amino acid level and, as a consequence, the reaction kinetics for Kv4.2 and Shaker N-type inactivation may be distinct. In addition, we show that KChIP2.1 largely attenuates Kv4.2 N-type inactivation.
N-type inactivation of chimeric Kv2.1(4.2NT) channels
Kv4 channels only transiently occupy an open-inactivated state, which is the reason why this inactivation reaction is difficult to examine. Therefore, we used Kv2.1(4.2NT) chimeric channels, where the study of open-state inactivation was not contaminated by fast, cumulative closed-state inactivation. We demonstrated the conferral of typical Kv4.2 inactivation properties, including its modulation by KChIP2.1 and its impairment by N-terminal truncation. The slower decay of Kv2.1(4.2NT) in comparison to Kv4.2-mediated currents may be due to the overlap of fast inactivation and slow activation kinetics in the chimeric channels. However, we cannot exclude the possibilities that the inactivating domain receptor sites in Kv2.1 and Kv4.2 may differ, or that other Kv2.1 domains may impede the action of the Kv4.2 N-terminus. Nevertheless, the inactivation of Kv2.1(4.2NT) channels displayed features very similar to the ones described for Shaker N-type inactivation (Hoshi et al., 1990; Zagotta et al., 1990; Choi et al., 1991; Demo and Yellen, 1991); e.g., temporary reopening during recovery from inactivation, slowing of current decay in the presence of internal TEA, acceleration of recovery from inactivation in high external K+, and onset of block when N-terminal peptides were applied to slowly inactivating channels to mimic N-type inactivation during voltage-dependent activation.
In a previous study, the mode of Kv4.1 inactivation has been examined in great detail. It was proposed that Kv4.1 channel inactivation differs from a classical Shaker N-type inactivation mechanism (Jerng and Covarrubias, 1997), because, in contrast to Shaker (Choi et al., 1991; Demo and Yellen, 1991), internal TEA did not modulate the decay kinetics of Kv4.1-mediated currents, and elevated external K+ did not accelerate the recovery of Kv4.1 channels from inactivation.
Kv4 channel gating is well described by models in which both the opening step and open-state inactivation are reverse-biased (Jerng et al., 1999; Bähring et al., 2001a); i.e., open-state inactivation is brief, and the channels tend to immediately recover back to the open state and then close. As the models predict, rising phase, peak amplitude, and a considerable fraction of the macroscopic current decay are dominated by cumulative closed-state inactivation, rather than open-state N-type inactivation. This is corroborated by the finding that macroscopic Kv4 channel inactivation is slowed when the outward currents are carried by Rb+, which stabilizes the open state by delaying channel closure (Bähring et al., 2001a; Shahidullah and Covarrubias, 2003). Kv4.1-mediated currents, in particular, show a very small initial inactivation component (only 15%–20% of the total decay), and N-terminal truncation has only modest effects on Kv4.1-mediated current decay (Pak et al., 1991; Jerng and Covarrubias, 1997), as if open-state N-type inactivation was nearly absent in these channels. This may be the reason why Jerng and Covarrubias (1997) did not observe a slowing of Kv4.1 current kinetics by internally applied TEA. By contrast, internal TEA affected the inactivation kinetics of Kv2.1(4.2NT)-mediated currents in a concentration-dependent manner. Although, based on our experiments, we cannot correlate the amount of slowing with the amount of block, it is likely that the binding of TEA within the pore interferes with the action of the proximal Kv4.2 N-terminus. Noteworthy, whereas Shaker channel N-type inactivation was slowed twofold by 1 mM TEA (Choi et al., 1991), Kv2.1(4.2NT) inactivation was slowed to a similar degree only at 5 mM TEA. Possibly, the energetics of binding for TEA or for the N-terminal inactivating domain or for both differ between Kv2.1(4.2NT) and Shaker channels. Alternatively, the degree of overlap of the binding sites for TEA and inactivating domain, respectively, may be lower in Kv2.1(4.2NT) than in Shaker channels.
The absence of an accelerating effect of high external K+ on the recovery of Kv4.1 channels from inactivation (Jerng and Covarrubias, 1997) may be explained by the finding that, at all relevant voltages, Kv4 channels readily accumulate in closed-inactivated state(s), from which they directly recover in a strongly voltage-dependent manner (Bähring et al., 2001a). Apparently, the recovery from closed-state inactivation cannot be accelerated by high external K+. By contrast, experiments from the present study with Kv2.1(4.2NT) chimeric channels show that, in the absence of a fast, cumulative closed-state inactivation, high external K+ dramatically accelerated the recovery from inactivation.
Kv4.2 N-type inactivation
After having demonstrated that the Kv4.2 proximal N-terminus can behave like an autoinhibitory ball peptide in a foreign channel environment, we revisited Kv4.2 inactivation. We examined hyperpolarization-induced Kv4.2-mediated tail currents after brief depolarizations to positive membrane potentials. If the prepulse duration was increased to produce an increasing fraction of open-inactivated channels, tail current kinetics varied accordingly, showing, in addition to the relatively fast deactivation component, a slower component of decay. The slow component of tail current decay is most likely associated with recovery of Kv4.2 channels from N-type inactivation via the open state, since it was absent in Kv4.2Δ40-mediated tail currents. Using a previously published model with corresponding rate parameters for Kv4.2 gating (Bähring et al., 2001a), we were able to adequately simulate the experimentally observed tail current kinetics (see Fig. 8, D and E). Based on our hypothesis that Kv4.2 open-state inactivation is mechanistically similar to Shaker N-type inactivation, N-terminally truncated Kv4.2Δ40 channels would lack a transition from the open state (O) to an open-inactivated state (IO; see Fig. 1). Consistent with this assumption, we were able to simulate our experimentally obtained Kv4.2Δ40 tail current kinetics with a model lacking the [IO] state(s) and having a decreased closing rate koc (see Fig. 9 B).
The results of our peptide application experiments with Kv4.2Δ40 suggested that also in Kv4.2 channels the proximal N-terminus is capable of acting as an autoinhibitory channel-blocking domain. In particular, the similarity of the results obtained with the application of soluble N-terminal peptide fragments from Kv4.2 and ShB, respectively, shows that rapid open-state inactivation mediated by the proximal Kv4.2 N-terminus and by the Shaker “ball”-domain are based on similar mechanisms. This was not unexpected, since ShB and Kv4.2 proximal N-termini show some similarity (see Fig. 6 A). Both contain a number of hydrophobic or uncharged residues with two consecutive Ala residues right at the beginning. The hydrophobicity of this initial part of the N-terminus seems to be essential for binding within the channel, with no absolute requirement for sequence similarities (Murrell-Lagnado and Aldrich, 1993a). In accordance with previous results obtained with ShB peptide applied to Kv2.1 (Isacoff et al., 1991) and large conductance Ca2+-activated K+ channels (Foster et al., 1992; Toro et al., 1992), our peptide application experiments showed that this reaction is not subfamily-specific.
Positively charged residues in a region next to the proximal hydrophobic inactivation domain may influence the blocking reaction by long-range electrostatic interactions in Shaker channels (Murrell-Lagnado and Aldrich, 1993a,b) or may directly interact with parts of the channel protein outside the pore (Gulbis et al., 2000). The absence of a large number of positive charges in Kv4 N-termini is conspicuous. In experiments with Kv4.2R13E (data not shown), in which the only basic residue within the proximal Kv4.2 N-terminus was replaced by a negatively charged one, we observed triple-exponential inactivation kinetics with τ1 = 13 ± 1 ms (72 ± 1%), τ2 = 59 ± 4 ms (22 ± 3%), and τ3 = 820 ± 51 ms (6 ± 1%; n = 8), none of these values being significantly different from wild-type. This result is in sharp contrast to the slowing of inactivation seen when negative charges were introduced in the proximal Kv3.4 N-terminus (Beck et al., 1998). Apparently, electrostatic interactions mediated by charged residues play a minor role for the Kv4.2 channel N-type inactivation. This notion is also supported by our finding that the application of the poorly charged Kv4.2 N-terminal peptide and the application of ShB E12K, D13K peptide, which bears a large number of positive charges, respectively, caused Kv4.2Δ40 patch current decay with identical kinetics.
The above results indicate differences in the structural determinants that contribute to Kv4.2 and Shaker N-type inactivation, respectively. Some of the differences may be directly connected to the instability of the open-inactivated state(s) in Kv4 channels and to the observation that macroscopic Kv4 current decay is not dominated by open-state N-type inactivation. This notion is consistent with our mutational analysis of possible interactions between N-terminal and pore-lining residues in Kv4.2CS. The mutational analysis was based on previous work by Zhou et al. (2001), who studied Kvβ1.1-mediated N-type inactivation of N-terminally truncated Kv1.4 channels. The results of Zhou et al. (2001) showed that amino acids at positions 3 and 5 in the Shaker-related Kvβ1.1 N-terminus represent major pore-interacting residues. Therefore we chose to mutate the corresponding amino acid residues in Kv4.2CS. In Shaker channels the N-terminus contains a valine at the third and an alanine at the fifth position. For comparison, we mutated Ala-3 in Kv4.2CS to valine and Val-5 in Kv4.2CS to alanine. Corresponding to the main interacting pore-lining residues in the Kv1.4 S6 domain we mutated Val-402, Val-406, and Tyr-413 in Kv4.2CS to alanine (see Fig. 13 A). Comparison of our results with the ones obtained by Zhou et al. (2001) showed the following: the decrease in Kd caused by our Kv4.2CS A3V mutation corresponds to an increase in Kd observed for the reverse mutation (V3A) in the Shaker Kvβ inactivation domain (Zhou et al., 2001). Mutation to Ala of both Kv4.2CS Val-5 and Kvβ Ile-5 causes an increase in Kd. Similar to the Kv1.4/Kvβ mutation pair Y569A/V3A, a sharp decline for the coupling coefficient was observed when we analyzed the corresponding mutation pair Y413A/A3V in Kv4.2CS. However, Zhou et al. (2001) obtained up to 80-fold larger changes in Kd when they mutated the main interacting residues individually. Moreover, they obtained 4- to 5-fold larger Ω-values with double mutant cycle analysis. By contrast, the Ω-values we obtained for Kv4.2CS were close to the noise range for such analyses, albeit the mutation pair Y413A/A3V with a coupling coefficient close to unity significantly differed from the rest of the examined putative interaction pairs. Our results suggest that the larger coupling coefficients obtained for the Shaker-related channel N-type inactivation indicate a stronger interaction between the N-terminal inactivation domain and the pore as compared to Kv4.2 N-type inactivation. Taken together, our results suggest that in Kv4.2 channels N-terminal residues may come close to pore-lining S6 residues during open-state N-type inactivation; however, such an interaction cannot be of high affinity. This is in agreement with the proposed short-lived nature of the open-inactivated state(s) [IO] in Kv4 channels (Fig. 1).
The apparent differences in the energetics and structural determinants for Kv4.2 and Shaker N-type inactivation may reflect different functional roles of this inactivation reaction. In Shaker channels, N-type inactivation is tightly coupled to C-type inactivation (Baukrowitz and Yellen, 1995). In fact, our results obtained with rapidly inactivating Kv1.5(+4.2N40) chimeric channels showed an N-type inactivation mediated by the Kv4.2 proximal N-terminus coupled to the intrinsic Kv1.5 C-type inactivation. In particular, the effects of high external K+ on the kinetics of recovery from inactivation suggested Shaker-like N-C–coupling in Kv1.5(+4.2N40). In a recent study (Eghbali et al., 2002), a pronounced decline of Kv4.3-mediated currents during repetitive stimulation, reminiscent of C-type inactivation, was shown with all external Na+ and K+ replaced by N-methyl-d-glucamine. When we performed experiments under similar nonphysiological conditions with Kv4.2, we observed a faster decline of current magnitude for wild-type than for N-terminally truncated Kv4.2Δ40 channels (R.B., unpublished observation). Possibly, the inactivation mediated by the Kv4.2 proximal N-terminus has features comparable, albeit not necessarily structurally related, to the positive N-C–coupling seen in Shaker channels (Baukrowitz and Yellen, 1995). However, it seems unlikely that N-C–coupling plays any role for Kv4.2 channel function under physiological conditions.
Modulation of Kv4 N-type inactivation by KChIPs
Native Kv4 channels associate with KChIPs (An et al., 2000). Heterologous coexpression of KChIPs causes a characteristic modulation of Kv4 channel-mediated current kinetics (An et al., 2000; Bähring et al., 2001b; Beck et al., 2002), including a slowing of the initial rapid component and an acceleration of the cumulative slow component of current decay, as well as a speeding-up of recovery from inactivation. In addition, Kv4 channel deactivation kinetics and the onset of low-voltage closed-state inactivation are enhanced by KChIPs. The experimentally obtained kinetic alterations of depolarization-induced Kv4 current kinetics in the presence of KChIPs can be reproduced by a gating model, in which the on-rate for open-state inactivation (koi; see Fig. 1) is decreased. Furthermore, the closing rate (koc) and both on- and off-rate for closed-state inactivation (kci and kic) need to be increased (Beck et al., 2002).
KChIP remodeling of Kv4 channel inactivation by reducing the on-rate for open-state inactivation implies, in the light of the present findings, that KChIP2.1 may interfere with N-type inactivation of Kv4.2 channels. In fact, the proximal Kv4 N-terminus seems to be essential for the interaction with KChIPs, because truncated Kv4.2Δ40 channels are no longer modulated by these accessory subunits (Bähring et al., 2001b). Thus, in wild-type Kv4 channels, KChIPs may interfere with N-type inactivation by binding to at least part of the proximal N-terminus. Our experimental findings that KChIP2.1 coexpression suppressed the slow tail current component and that peptide application accelerated the decay of Kv4.2/KChIP2.1-mediated currents strongly support the idea that KChIP2.1 binding interferes with Kv4.2 N-type inactivation. This is in accordance with a previously suggested model, in which KChIPs are thought to immobilize the Kv4 proximal N-termini, thereby hindering inactivation from the open state (Beck et al., 2002).
KChIPs apparently modulate both N-type inactivation and closure of Kv4 channels, such that the former is slowed and the latter enhanced (Beck et al., 2002). Simulating Kv4.2/KChIP2.1 tail currents (see Fig. 10, C and D) required a modification of the voltage dependence for the transitions between closed states in the deactivation chain, which either means that our previously published model did not cover all aspects of Kv4.2 channel gating or that KChIP2.1 remodeling of Kv4.2 gating involves more parameters than currently thought (Beck et al., 2002). Notably, N-terminal truncation of Kv4.2 channels (Kv4.2Δ40), which apparently eliminates N-type inactivation, also modulates Kv4.2 deactivation kinetics (Bähring et al., 2001a). In contrast to the modulation by KChIP2.1, however, deactivation of Kv4.2 channels is not accelerated but slowed by the N-terminal truncation (Bähring et al., 2001a). This indicates that deletion of the Kv4.2 proximal N-terminus and interaction of the proximal N-terminus with KChIP2.1 did not produce exactly comparable effects. Future studies have to show whether the Kv4.2 proximal N-terminus, in addition to inactivating the open channel, has other functions, which may be related to channel closure.
Acknowledgments
We thank Andrea Zaisser, Sabine Wehrmann, and Iris Meier for excellent technical assistance and Thomas Baukrowitz for critical comments on the manuscript.
This work was supported by a grant to O.P. from the Deutsche Forschungsgemeinschaft.
References
- An, W. F., M. R. Bowlby, M. Betty, J. Cao, H. P. Ling, G. Mendoza, J. W. Hinson, K. I. Mattsson, B. W. Strassle, J. S. Trimmer, and K. J. Rhodes. 2000. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 403:553–556. [DOI] [PubMed] [Google Scholar]
- Bähring, R., L. M. Boland, A. Varghese, M. Gebauer, and O. Pongs. 2001a. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J. Physiol. 535:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähring, R., J. Dannenberg, H. C. Peters, T. Leicher, O. Pongs, and D. Isbrandt. 2001b. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J. Biol. Chem. 276:23888–23894. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., and G. Yellen. 1995. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 15:951–960. [DOI] [PubMed] [Google Scholar]
- Beck, E. J., M. Bowlby, W. F. An, K. J. Rhodes, and M. Covarrubias. 2002. Remodelling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J. Physiol. 538:691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, E. J., and M. Covarrubias. 2001. Kv4 channels exhibit modulation of closed-state inactivation in inside-out patches. Biophys. J. 81:867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, E. J., R. G. Sorensen, S. J. Slater, and M. Covarrubias. 1998. Interactions between multiple phosphorylation sites in the inactivation particle of a K+ channel. Insights into the molecular mechanism of protein kinase C action. J. Gen. Physiol. 112:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, K. L., R. W. Aldrich, and G. Yellen. 1991. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc. Natl. Acad. Sci. USA. 88:5092–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo, S. D., and G. Yellen. 1991. The inactivation gate of the Shaker K+ channel behaves like an open-channel blocker. Neuron. 7:743–753. [DOI] [PubMed] [Google Scholar]
- Dixon, J. E., W. Shi, H. S. Wang, C. McDonald, H. Yu, R. S. Wymore, I. S. Cohen, and D. McKinnon. 1996. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ. Res. 79:659–668. [DOI] [PubMed] [Google Scholar]
- Eghbali, M., R. Olcese, M. M. Zarei, L. Toro, and E. Stefani. 2002. External pore collapse as an inactivation mechanism for Kv4.3 K+ channels. J. Membr. Biol. 188:73–86. [DOI] [PubMed] [Google Scholar]
- Foster, C. D., S. Chung, W. N. Zagotta, R. W. Aldrich, and I. B. Levitan. 1992. A peptide derived from the Shaker B K+ channel produces short and long blocks of reconstituted Ca(2+)-dependent K+ channels. Neuron. 9:229–236. [DOI] [PubMed] [Google Scholar]
- Gulbis, J. M., M. Zhou, S. Mann, and R. MacKinnon. 2000. Structure of the cytoplasmic β subunit-T1 assembly of voltage-dependent K+ channels. Science. 289:123–127. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes, 3rd ed. Sinauer Associates, Inc., Sunderland, MA.
- Holmgren, M., M. E. Jurman, and G. Yellen. 1996. N-type inactivation and the S4–S5 region of the Shaker K+ channel. J. Gen. Physiol. 108:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1991. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 7:547–556. [DOI] [PubMed] [Google Scholar]
- Isacoff, E. Y., Y. N. Jan, and L. Y. Jan. 1991. Putative receptor for the cytoplasmic inactivation gate in the Shaker K+ channel. Nature. 353:86–90. [DOI] [PubMed] [Google Scholar]
- Jerng, H. H., and M. Covarrubias. 1997. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys. J. 72:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng, H. H., M. Shahidullah, and M. Covarrubias. 1999. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J. Gen. Physiol. 113:641–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemic, K. G., C. C. Shieh, G. E. Kirsch, and S. W. Jones. 1998. Inactivation of Kv2.1 potassium channels. Biophys. J. 74:1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado, R. D., and R. W. Aldrich. 1993a. Interactions of amino terminal domains of Shaker K channels with a pore blocking site studied with synthetic peptides. J. Gen. Physiol. 102:949–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell-Lagnado, R. D., and R. W. Aldrich. 1993b. Energetics of Shaker K channels block by inactivation peptides. J. Gen. Physiol. 102:977–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak, M. D., K. Baker, M. Covarrubias, A. Butler, A. Ratcliffe, and L. Salkoff. 1991. mShal, a subfamily of A-type K+ channel cloned from mammalian brain. Proc. Natl. Acad. Sci. USA. 88:4386–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruppersberg, J. P., R. Frank, O. Pongs, and M. Stocker. 1991. Cloned neuronal IK(A) channels reopen during recovery from inactivation. Nature. 353:657–660. [DOI] [PubMed] [Google Scholar]
- Serodio, P., C. Kentros, and B. Rudy. 1994. Identification of molecular components of A-type channels activating at subthreshold potentials. J. Neurophysiol. 72:1516–1529. [DOI] [PubMed] [Google Scholar]
- Shahidullah, M., and M. Covarrubias. 2003. The link between ion permeation and inactivation gating of Kv4 potassium channels. Biophys. J. 84:928–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro, L., E. Stefani, and R. Latorre. 1992. Internal blockade of a Ca(2+)-activated K+ channel by Shaker B inactivating “ball” peptide. Neuron. 9:237–245. [DOI] [PubMed] [Google Scholar]
- Yellen, G., D. Sodickson, T. Y. Chen, and M. E. Jurman. 1994. An engineered cysteine in the external mouth of a K+ channel allows inactivation to be modulated by metal binding. Biophys. J. 66:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta, W. N., T. Hoshi, and R. W. Aldrich. 1990. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 250:568–571. [DOI] [PubMed] [Google Scholar]
- Zhou, M., J. H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]
- Zhu, X. R., R. Netzer, K. Böhlke, Q. Liu, and O. Pongs. 1999b. Structural and functional characterization of Kv6.2 a new gamma-subunit of voltage-gated potassium channel. Receptors Channels. 6:337–350. [PubMed] [Google Scholar]
- Zhu, X. R., A. Wulf, M. Schwarz, D. Isbrandt, and O. Pongs. 1999a. Characterization of human Kv4.2 mediating a rapidly-inactivating transient voltage-sensitive K+ current. Receptors Channels. 6:387–400. [PubMed] [Google Scholar]