

Abstract
Little is known about the heterogenous organization of lipids in biological membranes. Sphingomyelin (SM) is a major plasma membrane lipid that forms lipid domains together with cholesterol and glycolipids. Using SM-specific toxin, lysenin, we showed that in cultured epithelial cells the accessibility of the toxin to SM is different between apical and basolateral membranes. Apical membranes are highly enriched with glycolipids. The inhibitory role of glycolipids in the binding of lysenin to SM was confirmed by comparing the glycolipid-deficient mutant melanoma cell line with its parent cell. Model membrane experiments indicated that glycolipid altered the local density of SM so that the affinity of the lipid for lysenin was decreased. Our results indicate that lysenin recognizes the heterogenous organization of SM in biomembranes and that the organization of SM differs between different cell types and between different membrane domains within the same cell. Isothermal titration calorimetry suggests that lysenin binding to SM is presumably the result of a SM-lysenin complex formation of specific stoichiometry, thus supporting the idea of the existence of small condensed lipid complexes consisting of just a few lipid molecules in living cells.
INTRODUCTION
Sphingomyelin (SM) is a major sphingolipid species of animal cell and is a major lipid constituent of plasma membranes (Barenholz and Thompson, 1999; Gatt, 1999; Ramstedt and Slotte, 2002). Recent reports established the important roles of SM and its metabolites as second messengers in signal transduction events during development and differentiation (Hannun et al., 2001). SM is also a major component of sphingolipid, cholesterol-rich plasma membrane microdomains, called lipid rafts (Brown, 1998; Ostermeyer et al., 1999; Rietveld and Simons, 1998). Lipid rafts are believed to play important roles in cellular functions such as signaling, adhesion, motility, and membrane trafficking (Brown and London, 1998; Simons and Toomre, 2000). Reduction of cellular SM results in disintegration of these domains (Chatterjee et al., 2001). However, little is known about the organization of SM-containing membranes. When cells are treated in the cold with nonionic detergents such as Triton X-100, both SM and glycolipids are recovered in low-density detergent-resistant membranes (DRMs) together with a specific set of proteins (Brown and Rose, 1992; Fiedler et al., 1993). The lack of appropriate probes makes it hard to further analyze these membranes. Therefore, fundamental questions such as the heterogeneity of SM-containing membranes have yet to be answered.
Lysenin is a novel protein derived from coelomic fluid of the earthworm Eisenia foetida. It specifically recognizes SM and induces cytolysis. (Sekizawa et al., 1997; Yamaji et al., 1998; Yamaji-Hasegawa et al., 2003). The specific binding of lysenin to SM makes it possible to use this protein as a unique tool to examine the distribution of cell surface and intracellular SM (Nakai et al., 2000; Yamaji et al., 1998). In the present study, we showed that apical and basolateral membranes of cultured Madin-Darby canine kidney (MDCK) epithelial cell line had altered sensitivity to lysenin. The involvement of glycolipids in lysenin sensitivity was demonstrated by using a glycolipid-deficient mutant cell line. Model membrane experiments indicated that glycolipid altered the local density of SM so that the affinity of the lipid for lysenin was decreased. Our results indicate that lysenin recognizes heterogenous organization of SM in biomembranes and that the organization of SM differs between different cell types and between different membrane domains within the same cell.
EXPERIMENTAL PROCEDURES
Materials
Egg sphingomyelin (Egg SM), dioleoylphosphatidylcholine (diC18:1 PC), dipalmitoylphosphatidylcholine (diC16:0 PC), dilauroylphosphatidylglycerol (diC12:0 PG), dipalmitoylphosphatidylglycerol (diC16:0 PG), bovine cerebrosides (galactosylceramide, GalCer) and bovine phosphatidylserine (PS) were purchased from Avanti Polar Lipids (Alabaster, AL). More than 80% of amide-linked fatty acid in Egg SM is palmitic acid, according to the manufacturer. Palmitoylsphingomyelin (C16:0 SM), ISP-1 (myriocin) and fumonisin B1 were from Sigma (St. Louis, MO). 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI C18), 2-(4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-dodecanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine (BODIPY-C12-PC), N-(1-pyrenedecanoyl)-sphingosylphosphorylcholine (py-SM) and N-(1-pyrenedecanoyl)-sphingosine (py-Cer), Rhodamine Red-X 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (Rho-DHPE) were purchased from Molecular Probes (Eugene, OR). E-RDF medium was purchased from Kyokuto Pharmaceutical Corp. (Tokyo, Japan). The medium was a complete serum-free medium that contained insulin and transferrin. Lysenin and anti-lysenin antiserum were purchased from Peptide Institute (Osaka, Japan).
Cells and cell culture
MDCK strain II cells were provided by Dr. K. Simons of Max-Planck Institute (Dresden, Germany) through Dr. M. Murata (National Institute for Physiological Sciences, Okazaki, Japan). Cells were grown either on glass coverslips or on polycarbonate filters with a pore size of 0.4 μm (Transwell, Costar Corp., Cambridge, MA) in MEM supplemented with 10% fetal calf serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 29.2 μg/ml glutamine (Kobayashi et al., 1992). Mouse melanoma cell line, MEB4, its glycosphingolipid-deficient mutant GM95 (Ichikawa et al., 1994) and the transfectant CG1, which stably express ceramide glucosyltransferase I (CerGlcTI), were cultured in DMEM supplemented with 10% fetal calf serum, 100 units/ml penicillin and 100 μg/ml streptomycin. To decrease SM content of GM95, cells were cultured in E-RDF medium with various concentrations of either ISP-1 or fumonisin B1 for 2 days. To increase SM of MEB4, cells were cultured in E-RDF medium containing fatty acid-free bovine serum albumin-sphingosine complex (Hanada et al., 1992) for 3 days. Final concentration of sphingosine was 2 μM.
Cell staining with lysenin
All manipulations were done at room temperature unless otherwise noted. MDCK cells grown on glass coverslips were incubated with 1 μg/ml lysenin at 4°C for 30 min. After washing with phosphate buffered saline (PBS) containing 0.9 mM CaCl2 and 0.5 mM MgCl2 (PBS+), the cells were fixed with 3% paraformaldehyde in PBS+ for 20 min, quenched with 0.1 M NH4Cl and then blocked with 0.2% gelatin in PBS. The cells were then treated with anti-lysenin antiserum for 30 min followed by the incubation with Alexa 546-conjugated anti-rabbit IgG (Molecular Probes) for 30 min. To label permeabilized MDCK cells, cells were fixed with 3% paraformaldehyde in PBS for 20 min followed by 12 min treatment with 50 μg/ml digitonin in PBS. The permeabilized cells were labeled with lysenin as described above. To label mouse melanoma cells, cells grown on glass coverslips were washed with PBS, fixed with 3% paraformaldehyde and incubated with 0.5 μg/ml lysenin for 1 h at 4°C. Cells were again fixed with 3% paraformaldehyde for 10 min at 4°C. This second fixation was necessary to prevent artificial aggregation of second antibodies. The cells were then incubated with anti-lysenin antiserum for 30 min followed by additional 30-min incubation with Alexa 488-conjugated anti-rabbit IgG (Molecular Probes). The specimens were mounted with Mowiol and examined under Zeiss LSM 510 confocal microscope equipped with C-Apochromat 63XW Korr (1.2 n.a.) objective.
Viability of cells exposed to lysenin
Mouse melanoma cells grown on 24-well plates were washed and incubated with 0.3 ml of various concentrations of lysenin in serum-free medium for 30 min at 37°C. MTT (5 mg/ml in DMEM) solution (0.3 ml) was then added followed by a 1-h incubation at 37°C. After removal of MTT-containing medium, formazan produced by the living cells was dissolved in 0.3 ml of DMSO and the absorbance at 595 nm was measured (Mosmann, 1983). As a background control, cells were incubated with 0.2% Tween 20 before the addition of MTT solution. For the measurement of the viability of filter-grown MDCK II cells, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-(phenylamino-carbonyl)2H-tetrazolium hydroxide (XTT) assay was employed. Lysenin was added to the filter-grown cells from either the apical or basolateral side. After 30 min of incubation at 37°C, XTT solution (1 mg/ml) was added from the apical side and the cells were incubated for 3 h at 37°C. 2% Tween 20 was used to prepare 100% lysis control. The absorbance at 490 nm was then measured.
Lipid analysis
Cells grown in a 15-cm culture dish were washed with cold PBS and harvested by scraping. Lipids were extracted according to Bligh and Dyer (1959). After separation of phospholipids by thin layer chromatography, the phosphorus content of each phospholipid was determined (Bartlett, 1958). To measure SM content on the cell surface, cells grown in a 6-cm dish were labeled with 1 μCi/ml [14C]-serine (165 mCi/mmol, PerkinElmer, Boston, MA) in DMEM containing 10% fetal calf serum for 2 days. Cells were then treated with or without 1.25 units/ml Bacillus cereus sphingomyelinase (Sigma) for 30 min at 37°C. After extraction and separation of lipids, SM content was determined by 14C radioactivity. Distribution of radioactivity to each lipid was measured by BAS 2000 (FUJIFILM, Tokyo, Japan).
Preparation of His-Venus-lysenin
The plasmid pRSET-Venus (Nagai et al., 2002), which encodes six residues of histidine followed by Venus, was kindly provided by Drs. A. Miyawaki and T. Nagai of Brain Science Institute, RIKEN. According to the previous study on yellow fluorescent protein (YFP) (Zacharias et al., 2002), the alanine at position 206 was substituted with lysine to prevent dimerization of Venus protein. Lysenin cDNA (generous gift of Y. Sekizawa and H. Kobayashi of Zenyaku Kogyo, Tokyo, Japan) was inserted into downstream of His-Venus. Details of the preparation of His-Venus-lysenin construct is described elsewhere (E. Kiyokawa et al., manuscript in preparation). Recombinant protein, His-Venus-lysenin, was expressed in E. coli and purified by affinity chromatography using a nickel column (Amersham BioSciences, Uppsala, Sweden) and gel filtration choromatography (Superose 6, Amersham BioSciences).
Monolayer measurements
The surface pressure was measured with a fully automated micrometer (DeltaPi, Kibron, Helsinki, Finland). All experiments were carried out at 26 ± 1°C. Monomolecular films of lipids were spread on PBS (pH 7.5) subphase (volume of 500 μl) from hexane:chloroform:ethanol (11:5:4, v/v/v). After spreading of the film, 10 min was allowed for solvent evaporation. To measure the interaction of lysenin with lipid monolayers, 5 μl of 60 μM lysenin solution was injected in the subphase with a 10-μl Hamilton syringe, and the pressure increase was recorded until reaching the equilibrium (maximum surface pressure increase Δπ was usually obtained within 60–120 min of interaction). The data were analyzed with the DeltaGraph 2.15 program (Kibron). To visualize SM domains in lipid monolayers, His-Venus-lysenin instead of native lysenin was introduced into the subphase beneath the lipid monolayer. Fluorescence image was recorded after the binding reached an equilibrium (60–120 min) by using an Olympus Power BX fluorescence microscope equipped with LM Plan Fl 50× objective and Toshiba 3CCD camera.
Preparation of giant unilamellar vesicles (GUVs)
Ten mol % diC16:0 PG was added to the stock chloroform solution of C16:0 SM and diC16:0 PC whereas 10 mol % diC12:0 PG was added to diC18:1 PC. These stock solutions were mixed to prepare GUVs of different lipid composition. PG was added because charged phospholipids were necessary to obtain GUVs. For visualization by confocal fluorescence microscope, fluorescent probes (DiI C18 and BODIPY-C12-PC) were added to the lipid mixture at a concentration of ∼0.1 mol %. GUVs were prepared as described (Akashi et al., 1996; Feigenson and Buboltz, 2001) with minor modifications. One hundred microliters of 10 mg/ml lipid solution in a glass test tube was dried with a rotary evaporator to form a thin lipid film. The tube was placed in vacuo for >6 h. The completely dried lipid film was then prehydrated at 45°C with water-saturated nitrogen for 15–25 min. A total of 5 ml of 5 mM piperazine-1,4-bis(2-ethanesulfonic acid) (PIPES) buffer (pH 7.0) containing 50 mM KCl and 1 mM EDTA was added gently to the test tube. The tube was incubated at 65°C overnight. During incubation, the whole lipid film was gradually stripped off the glass surface and formed lipid balls, which contained GUVs. The samples were slowly cooled to room temperature. Harvested GUVs were placed on a coverslip and were enclosed by a slide glass within a ring of silicone high-vacuum grease. The specimen was then allowed to settle for 10 min. Fluorescence images were obtained with Zeiss LSM 510 confocal microscope equipped with Plan-Apochromat 100 X oil DIC (1.4 n.a.) objective. For three-dimensional image projection of a vesicle, z-scans in 0.2-μm increments were taken through the upper half of a GUV. The scans were then combined using LSM 510 software. We also measured the binding of His-Venus-lysenin to C16:0 SM-containing GUVs. In that experiment, GUVs containing ∼0.1 mol % Rho-DHPE were incubated with His-Venus-lysenin for 10 min at room temperature and the fluorescence image was obtained as described above.
Binding of lysenin to liposomes
Two μl of 1 mg/ml lysenin and 10 μl of 1 mM multilamellar vesicles in PBS were incubated at room temperature for 30 min. The solution was applied to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (6–10% gel) under denatured conditions and lysenin oligomers (Yamaji-Hasegawa et al., 2003) were quantified by silver staining followed by Image Gauge (FUJIFILM). Our control experiment indicated that all membrane-associated lysenin was oligomerized under the experimental conditions.
Fluorescence resonance energy transfer (FRET)
Large unilamellar vesicles (LUVs) with or without 1 mol % py-SM were prepared by extrusion through polycarbonate filters with 0.1-μm pore size (Nuclepore, Maidstone, UK) for 30 times using a two-syringe extruder (MacDonald et al., 1991; Makino et al., 2003). For steady-state fluorescence measurement, 0.3 μM lysenin was incubated with various LUVs (6.25 μM total lipids) in PBS for 10 min. The emission spectra from 300 to 550 nm were recorded with excitation at 280 nm. To measure kinetics of lysenin binding to SM, py-SM fluorescence at 420 nm was followed continuously with excitation at 280 nm after the addition of lysenin (final concentration; 0.06 μM) into LUVs (1.25 μM total lipids). Fluorescence measurements were performed using a FP-6500 spectrofluorometer (Jasco, Tokyo, Japan) at 25°C.
High-sensitivity titration calorimetry
Isothermal titration calorimetry (ITC) was performed using a MicroCal VP-ITC high sensitivity titration calorimeter (MicroCal, Northampton, MA). Solutions were degassed under vacuum before use. The calorimeter was calibrated electrically. The heats of dilution were determined in control experiments by injecting lipid suspension into buffer. The heats of dilution were subtracted from the heats determined in the corresponding lysenin-lipid binding experiments.
Other methods
Protein concentration was measured by amino acid analysis.
RESULTS
Apical and basolateral membranes of cultured epithelial cells showed altered sensitivity to lysenin
Epithelial cells contain two distinct plasma membranes: apical domains confront the external lumen whereas basolateral membranes face the underlying cell layer (Hubbard, 1991; Simons et al., 1992). Each plasma membrane domain has a specialized function and contains a different set of lipids and proteins. Of particular interest are glycolipids that are highly enriched in the apical domain (Simons and van Meer, 1988). Apical and basolateral polarity of MDCK II cells was observed both in filter-grown cells and, to a lesser degree, in cells grown on coverslips (Simons and Virta, 1987). Development of polarity is dependent on cell density. With low density, cells are not well polarized whereas with high density, cells contact each other, form tight junctions, and polarize with the apical membrane facing to the top. When MDCK II cells were grown on coverslips at low density, plasma membranes were stained with lysenin (Fig. 1 A). Fluorescence was not uniformly distributed on the cell surface; rather, it showed a punctate pattern throughout the cell. Some cells were completely devoid of lysenin labeling. In contrast, lysenin did not significantly stain cell surface when cells were grown at high density (Fig. 1 B). However, when these cells were permeabilized with digitonin, the periphery of each cell became labeled (Fig. 1 C). These results suggest that lysenin recognizes SM in the basolateral but not in the apical membrane of MDCK II cells. This was confirmed by measuring the lysenin sensitivity of filter-grown cells. In Fig. 1 G, we added lysenin to filter-grown MDCK II cells from either the apical or the basolateral side. Cells were highly sensitive to lysenin when the toxin was added from the basolateral side. In contrast, the cells showed resistance to apically added lysenin. Previously, it was shown that apical membranes contain 19.0% of SM in total phospholipids whereas basolateral membranes had 26.4% (van Meer and Simons, 1982). Model membrane study has indicated that the specific binding of lysenin to the membrane was observed when SM content was as low as 5% (see below), indicating that the difference of SM content between apical and basolateral membranes does not explain the different sensitivity of these membranes to lysenin. Since apical membranes are highly enriched with glycolipids with which SM is known to interact (Brown and London, 2000; Johnston and Chapman, 1988), we then asked whether glycolipids affect lysenin sensitivity.
FIGURE 1.

Apical and basolateral membranes of cultured epithelial cells showed altered sensitivity to lysenin. (A–F) Binding of lysenin to MDCK II cells grown on coverslips. MDCK II cells at low density (A and D) or at high density (B, C, E and F) were stained with lysenin as described under Experimental Procedures. Lysenin staining (A–C) and DIC micrographs (D–F) of the same specimens are shown. Bar, 10 μm. (G) Lysenin was added to filter-grown MDCK II cells from apical side (solid column) or basolateral side (hatched column). The cells were incubated with lysenin for 30 min at 37°C. Viability was measured as described in Experimental Procedures.
Glycolipid content affects lysenin sensitivity in mouse melanoma cells
To examine the effect of glycolipids on the recognition of SM by lysenin, we compared lysenin binding between the mouse melanoma cell line MEB4 and its glycolipid-deficient mutant GM95 (Fig. 2, A and B). GM95 has a defect in ceramide glucosyltransferase I (CerGlcTI), that catalyzes the first step of glycosphingolipid synthesis (Ichikawa et al., 1994). Although lysenin brightly stained the cell surface of GM95 (Fig. 2 B), parent MEB4 was almost devoid of fluorescence (Fig. 2 A). Consistent with cell labeling, GM95 was sensitive to lysenin-induced killing whereas MEB4 was resistant to lysenin under our experimental conditions (Fig. 2 E). The involvement of CerGlcTI in lysenin sensitivity was further confirmed by the observation that CG1, the stable CerGlcTI transfectant of GM95, was resistant to lysenin (Fig. 2 E).
FIGURE 2.

Glycolipid content affects lysenin sensitivity in mouse melanoma cells. (A–D) Immnunofluorescence staining of mouse melanoma cells with lysenin. MEB4 (A and C) and GM95 (B and D) were stained with lysenin as described under Experimental Procedures. Lysenin staining (A and B) and DIC micrographs (C and D) of the same specimens are shown. Bar, 10 μm. (E) Cytolytic sensitivity of MEB4 (filled bar), GM95 (hatched bar), or CG1 (open bar) to lysenin. Cells were incubated with various concentrations of lysenin for 30 min at 37°C. Viability was measured as described in Experimental Procedures.
Lipid analysis revealed that, consistent with the published data (Hidari et al., 1996), the mole percentage of SM in total phospholipids of GM95 cells was 8–9%, which was about twice that of MEB4 cells (4–5%) (Table 1). We also measured SM on cell surfaces using sphingomyelinase treatment. SM content on the cell surface of MEB4 (61% of total SM) was similar to that of GM95 (66%). Since the difference in SM content between two cells might affect the sensitivity to lysenin, we tried to adjust SM content of these cells to the same level using various reagents. The results are summarized in Table 1. The content of SM of GM95 cells was decreased by the treatment with fumonisin B1 or ISP-1. When SM of GM95 was decreased to the level of MEB4 (20 μM fumonisin B1 or 10 nM ISP-1 treatment), the viability of GM95 was increased. However, cells were still sensitive to lysenin. In contrast, the increase of SM content in MEB4 cells to 6% by the addition of sphingosine did not affect the viability of the cells. Our results indicate that the content of SM was not the cause of lysenin resistance of MEB4 cells. These results, together with the results of MDCK cells, suggest that the glycolipid contents are crucial in the sensitivity of the cells to lysenin.
TABLE 1.
Mouse melanoma MEB4 and its glycolipid deficient mutant GM95 showed altered lysenin sensitivity
| Cell | Treatment* | SM (%) | Viability† (%) |
|---|---|---|---|
| MEB4 | None | 4.5 | 100 |
| Sphingosine (2 μM) | 6.0 | 100 | |
| GM95 | None | 8.9 | 10 |
| Fumonisin B1 (10 μM) | 7.1 | 19 | |
| Fumonisin B1 (20 μM) | 5.5 | 30 | |
| ISP-1 (5 nM) | 6.6 | 16 | |
| ISP-1 (10 nM) | 4.1 | 60 |
Cells were treated with various reagents as described in Experimental Procedures.
Cells were treated with 250 ng/ml lysenin for 30 min at 37°C. Viability of cells was measured as described in Experimental Procedures.
Lysenin specifically bound to SM at the air-water interface
The above experiments suggest that the binding of lysenin to SM is affected by the presence of glycolipid. To address this question, we employed a monolayer system in which lipid composition is easily manipulated. Specific binding of lysenin to SM has been shown by solid-phase binding analysis, TLC blotting, as well as liposome binding analysis (Yamaji et al., 1998; Yamaji-Hasegawa et al., 2003). We first asked whether lysenin specifically bound to SM at the air-water interface. Lysenin was added in the aqueous subphase underneath a monolayer film of lipid, and the resulting interaction was measured as an increase in the surface pressure of the film. As shown in Fig. 3 A, the surface pressure was increased after the injection of lysenin solution into the subphase, on which the SM monolayer was formed. This result indicates that lysenin interacts with SM and penetrates into the lipid monolayer. After 1–2 h, the surface pressure stopped increasing when saturation was reached. The maximal surface pressure increase at that time was defined as Δπ. In Fig. 3 A, Δπ was 12.5 mN/m. To assess the lipid specificity of the penetration process, monolayer films of SM or other lipids were prepared at various initial pressures (πi), and the Δπ was determined (Fig. 3 B). In the absence of a lipid monolayer, the surface pressure was also increased (Fig. 3 B, white circle), indicating that lysenin has an ability to adsorb at the air-water interface. Below a πi of 20 mN/m, lysenin penetrated into the lipid monolayers of diC18:1 PC, diC16:0 PC, as well as PS. However, Δπ gradually decreased as πi increased. For SM monolayers, the Δπ values were almost the same (10–13 mN/m), between πi value of 10 and 30 mN/m. When πi was above 20 mN/m, lysenin specifically bound to SM at the air-water interface. Above a πi of 30 mN/m, Δπ gradually decreased as πi increased. At a πi of ∼40 mN/m, the surface pressure increase was not observed even with SM. At this pressure, the lipid monolayer was collapsed as monitored by Langmuir-type film balance (data not shown).
FIGURE 3.

Lysenin specifically binds SM at the air-water interface. (A) Time course of lysenin penetration into egg SM monolayer. Lysenin was injected into the subphase at a final concentration of 0.6 μM. Injection performed where indicated. (B) Δπ reached after injection of lysenin under egg SM (closed circle), diC18:1 PC (closed square), diC16:0 PC (closed triangle), or PS (closed diamond) monolayers at various πi values. Spontaneous penetration of lysenin into an air-water interface was observed in the absence of lipid monolayer (open circle).
Glycolipid alters binding of lysenin to SM in a binary mixture at the air-water interface
Having established the condition of specific binding of lysenin to SM, we then asked whether glycolipid and other lipids affect the interaction between SM and the protein. For this purpose we chose diC18:1 PC and diC16:0 PC in addition to a glycolipid, galactosylceramide (GalCer). It has been reported that diC18:1 PC was immiscible with SM (Yuan et al., 2002) whereas diC16:0 and SM were completely miscible (Maulik and Shipley, 1996b). SM and GalCer were miscible (Johnston and Chapman, 1988). Various monolayers were prepared and surface pressure increase by lysenin was determined at a πi of 20 ± 1 mN/m. Fig. 4 A shows the surface pressure increase as a function of SM content. The Δπ values for SM/diC18:1 PC were always higher than those for SM/diC16:0 PC and SM/GalCer. This result indicates that lysenin has a higher affinity for SM in the SM/diC18:1 PC monolayer than for SM in the SM/diC16:0 PC or SM/GalCer monolayers. Fig. 4 B shows the Δπ values for SM/diC18:1 PC (molar ratio 1:9), SM/diC16:0 PC (1:9), or SM/GalCer (1:9) in the absence or presence of equimolar cholesterol to SM at a πi of 20 mN/m. Addition of cholesterol did not significantly alter the binding of lysenin to the SM/diC18:1 PC or SM/GalCer monolayers. In contrast, the binding of lysenin to the SM/diC16:0 PC monolayer was slightly increased by the addition of cholesterol.
FIGURE 4.

Binding of lysenin to SM was dependent upon the local density of SM at the air-water interface. (A) Δπ values reached after injection of lysenin under egg SM/diC18:1 PC (closed circle), egg SM/diC16:0 PC (closed square), or egg SM/GalCer (closed triangle) mixed monolayers with various concentrations of SM at a πi of 21.5 ± 2.0 mN/m. (B) Δπ values reached after injection of lysenin under egg SM/diC18:1 PC (molar ratio1:9), egg SM/diC16:0 PC (1:9), or egg SM/GalCer (1:9) in the absence (solid column) or presence (hatched column) of equimolar cholesterol to SM at πi of 20.9 ± 1 mN/m. Results are the mean of duplicate or triplicate experiments ± difference. (C–F) His-Venus-lysenin was injected into the subphase beneath egg SM/diC18:1 PC (1:9) (C), egg SM/diC16:0 (1:9) (D), egg SM/diC18:1 PC/cholesterol (1:9:1) (E), or egg SM/diC16:0/cholesterol (1:9:1) (F). Fluorescence image was recorded as described in Experimental Procedures. Bar, 20 μm.
For direct visualization of lysenin binding, we employed lysenin tagged with the YFP-homolog, Venus, which has higher quantum yield than green fluorescent protein or YFP (Nagai et al., 2002). In Fig. 4, C and D, His-Venus-lysenin was added to the subphase of the SM/diC18:1 PC (molar ratio 1:9) or SM/diC16:0 PC (1:9) monolayers at a pressure of ∼20 mN/m. The fluorescence image was recorded as described in Experimental Procedures. The distribution of lysenin in the monolayer was not uniform. In SM/diC18:1 PC, lysenin accumulated as big (30–50 μm diameter) aggregates (Fig. 4 C). In contrast, lysenin faintly stained as smaller (2–5 μm) dots in the SM/diC16:0 monolayer (Fig. 4 D). The addition of cholesterol to SM/diC18:1 did not affect the fluorescent pattern of lysenin (Fig. 4 E), whereas in the SM/diC16:0 monolayer, lysenin accumulated in bigger aggregates in the presence of cholesterol (Fig. 4 F). Lysenin staining was not observed in the SM/GalCer (molar ratio 1:9) monolayer even after the addition of cholesterol (data not shown). Without lysenin, Venus protein alone was not concentrated at the air-water interface (data not shown). These results suggest that lysenin efficiently recognizes SM only when the lipid forms aggregates or domains.
Lysenin recognizes local density of SM in lipid bilayers
We then asked whether the additional lipids affect lysenin binding to SM in bilayer membranes. We first examined SM distribution in GUVs of SM/diC18:1 PC (molar ratio 7:3) and SM/diC16:0 PC (5:5). Both GUVs contained 10 mol % PG as described in Experimental Procedures. At room temperature the SM employed (C16:0 SM) and diC16:0 PC were in gel state whereas diC18:1 PC was a liquid crystalline. To identify different phases in GUVs, we used two dyes that partition differently between the coexisting phases: DiI C18, which favors solid phase, and BODIPY-C12-PC, which favors the fluid phase (Feigenson and Buboltz, 2001). Although lipid distribution in GUVs are heterogenous as reported (Veatch and Keller, 2003) and relative green/red ratios are not exactly the same among different liposomes, these complementary probes produced clear visualizations of coexisting phases in SM/diC18:1 PC vesicles (Fig. 5, A and B). The results suggest that red fluorescence from DiI C18 identified the SM-rich ordered phase, and green fluorescence from BODIPY-C12-PC identified the diC18:1 PC-rich fluid phase. In contrast, in GUVs of SM/diC16:0 PC, uniform fluorescence of DiI C18 was observed (Fig. 5 C). These results suggest that SM forms clusters in SM/diC18:1 PC bilayers whereas it is uniformly distributed in the presence of diC16:0 PC. The binding of lysenin to SM/diC18:1 PC (molar ratio 3:7), SM/diC16:0 PC (3:7), as well as SM/GalCer (3:7) was quantified in Fig. 5 D. Similar to the monolayer experiment, lysenin had higher affinity for SM/diC18:1 PC than for SM/diC16:0 PC or SM/GalCer. Fig. 5, E and F, show the binding of His-Venus-lysenin to SM/diC18:1 PC (molar ratio 3:7) (Fig. 5 E) and to SM/diC16:0 PC (3:7) (Fig. 5 F). His-Venus-lysenin bound SM/diC18:1 PC liposomes and formed aggregates on the membrane as observed in the monolayer experiment. In contrast, the protein did not bind SM/diC16:0 PC liposomes.
FIGURE 5.

Local density of SM influenced the binding of lysenin to SM in lipid bilayers. (A–C) GUVs composed of C16:0 SM/diC18:1 PC (molar ratio 7:3) containing 7 mol % diC16:0 PG and 3 mol % diC12:0 PG (A and B, two representative fields were shown) and C16:0 SM/diC16:0 PC (molar ratio 1:1) containing 10 mol % diC 16:0 PG (C) labeled with 0.1% DiI C18 (red) and 0.1% BODIPY-C12-PC (green) were prepared as described under Experimental Procedures. Color merged images were shown. Bar, 2 μm. (D) Binding of lysenin to egg SM/diC18:1 PC (molar ratio 3:7), egg SM/diC16:0 PC (3:7), or egg SM/GalCer (3:7) liposomes. The binding was measured as described in Experimental Procedures. The data are representative of two independent experiments yielding similar results. (E and F) GUVs of C16:0 SM/diC18:1 PC (3:7) (E) or C16:0 SM/diC16:0 PC (3:7) (F) were incubated with His-Venus-lysenin as described in Experimental Procedures. His-Venus-lysenin fluorescence images were obtained with confocal microscope. Bar, 5 μm.
Energy transfer between pyrene-labeled SM and lysenin reveals organization-dependent interaction of SM with lysenin
The binding of lysenin to SM-containing liposomes was further analyzed by measuring FRET between tryptophan residues of lysenin and pyrene-labeled SM (py-SM). When lysenin was incubated with SM/diC18:1 PC (molar ratio1:4), tryptophan fluorescence of lysenin was increased and blue shifted as described previously (Yamaji-Hasegawa et al., 2003) (Fig. 6 A). The presence of py-SM containing SM/diC18:1 PC (1:4) liposomes decreased the tryptophan fluorescence, indicating energy transfer between lysenin and py-SM. Accordingly, the fluorescence of py-SM was increased. SM/GalCer/diC18:1 PC (1:1:3) also induced blue shift of tryptophan fluorescence of lysenin and energy transfer was observed in the presence of py-SM (Fig. 6 B). However, SM/GalCer/diC18:1 PC (1:1:3) was less effective than SM/diC18:1 PC (1:4) liposomes. SM/diC16:0 PC (1:4) did not significantly alter tryptophan fluorescence nor increase py-SM fluorescence (Fig. 6 C). In the absence of SM, lysenin did not affect pyrene fluorescence (Fig. 6 D). Fig. 6 E shows the effect of different liposomes on time-dependent increase of py-SM fluorescence in the presence of lysenin. When py-SM was incorporated into SM/diC18:1 PC (1:4), fluorescence intensity increased rapidly and reached plateau within 100 s. Incorporation of GalCer to the membrane slowed the increase and decreased the plateau level of fluorescence. Replacing diC18:1 PC with diC16:0 PC abolished fluorescence increase.
FIGURE 6.

Energy transfer between pyrene-labeled SM and lysenin reveals organization dependent interaction of SM with lysenin. Lysenin (0.3 μM) was incubated with 6.25 μM (total lipids) LUVs composed of SM/diC18:1 PC (1:4) (A), SM/GalCer/diC18:1 PC (1:1:3) (B), SM/diC16:0 PC (1:4) (C) with (green) or without (red) 1 mol % py-SM. In D, lysenin was incubated with diC18:1 PC LUVs with (green) or without (red) py-Cer. Fluorescence of lysenin alone (blue) and pyrene-containing LUVs in the absence of lysenin (light blue) were also recorded. Fluorescence spectra were obtained with the excitation wavelength at 280 nm at 25°C. (E) Fluorescence of py-SM was measured continuously after the addition of 0.06 μM (final concentration) lysenin to 1.25 μM LUVs of SM/diC18:1 PC (1:4) (blue), SM/GalCer/diC18:1 PC (1:1:3) (green), SM/diC16:0 PC (1:4) (red) in the presence of 1 mol % py-SM. Excitation wavelength, 280 nm; emission wavelength; 420 nm; temperature, 25°C.
Glycolipid alters stoichiometry and thermodynamic parameters of SM-lysenin complex formation
ITC is useful to study the thermodynamic parameters of protein-lipid interactions (Wieprecht and Seelig, 2002). Fig. 7 A illustrates the titration of lysenin solution with SM/diC18:1 PC (molar ratio 1:4) LUVs. For the first six injections, each addition of lipid to lysenin solution caused a distinct exothermic reaction. The reaction enthalpy suddenly dropped at the seventh injection. This titration curve may be interpreted as follows (Machaidze et al., 2002): Initially, the peptide is much in excess over the added lipid. Upon injection of lipid vesicles the peptide molecules react immediately with the added SM, and the concentration of free peptide in solution is reduced accordingly. After six injections, a sufficient amount of SM has been added to bind all available lysenin and the addition of further lipid has no effect. The reaction enthalpy ΔH0 can be calculated from the titration profile. The total heat measured was 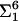 hi = −120 μcal, the total amount of protein in the sample cell was
hi = −120 μcal, the total amount of protein in the sample cell was 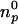 = 6.664 nmol, and the reaction enthalpy ΔH0 =
= 6.664 nmol, and the reaction enthalpy ΔH0 = 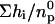 = −18.0 kcal/mol protein. The amount of SM added in the first six steps was 33.6 nmol. Thus the SM/lysenin ratio was 33.6:6.664 = 5.04. This calculation indicates that one lysenin molecule binds 5 SM molecules. The reaction enthalpy per mol of SM can be calculated by dividing the enthalpy per mol of lysenin by the stoichiometry (lipid/protein) of the interaction. This gives −3.6 kcal/mol SM. Addition of GalCer to the vesicles (SM/GalCer/diC18:1 PC (1:1:3)) altered the shape of titration curve (Fig. 7 B). The reaction enthalpy decreased gradually and ΔH0 was calculated to be −13.0 kcal/mol protein and −1.4 kcal/mol SM. SM/lysenin ratio was increased to 9.08 by the addition of GalCer (Table 2). These results indicate that GalCer alters the stoichiometry and thermodynamic parameters of SM-lysenin complex formation.
= −18.0 kcal/mol protein. The amount of SM added in the first six steps was 33.6 nmol. Thus the SM/lysenin ratio was 33.6:6.664 = 5.04. This calculation indicates that one lysenin molecule binds 5 SM molecules. The reaction enthalpy per mol of SM can be calculated by dividing the enthalpy per mol of lysenin by the stoichiometry (lipid/protein) of the interaction. This gives −3.6 kcal/mol SM. Addition of GalCer to the vesicles (SM/GalCer/diC18:1 PC (1:1:3)) altered the shape of titration curve (Fig. 7 B). The reaction enthalpy decreased gradually and ΔH0 was calculated to be −13.0 kcal/mol protein and −1.4 kcal/mol SM. SM/lysenin ratio was increased to 9.08 by the addition of GalCer (Table 2). These results indicate that GalCer alters the stoichiometry and thermodynamic parameters of SM-lysenin complex formation.
FIGURE 7.

ITC suggests that lysenin binding to SM is the result of a SM-lysenin complex formation of specific stoichiometry. Lysenin was titrated with LUVs composed of SM/diC18:1 PC (molar ratio 1:4) (A) or SM/GalCer/diC18:1 PC (molar ratio 1:1:3) (B). The concentration of the protein in the reaction cell was 4.76 μM. The total lipid concentration was ∼5 mM. Concentration of SM was 0.934 mM in A and 1.12 mM in B. Each peak corresponds to the injection of 6 μl of lipid suspension into the reaction cell (Vcell = 1.4 ml). ITC was performed as described in Experimental Procedures. Buffer, 20 mM Hepes-NaOH, pH 7.2; temperature, 25°C.
TABLE 2.
GalCer alters stoichiometry and thermodynamic parameters of SM-lysenin complex formation
| N (SM/lysenin) | ΔH0 (kcal/mol lysenin) | ΔH0 (kcal/mol SM) | |
|---|---|---|---|
| SM/diC18:1 PC(1:4) | 5 | −18.0 | −3.6 |
| SM/GalCer/diC18:1 PC (1:1:3) | 9 | −13.0 | −1.4 |
DISCUSSION
It has been reported that 16:0 SM was immiscible with diC18:1 PC by π-A isotherms of the SM/diC18:1 PC monolayer (Yuan et al., 2002). Atomic force microscope images of SM/diC18:1 PC monolayers also showed that SM-rich domains and C18:1 PC-rich domains were segregated at surface pressure of 10–30 mN/m (Yuan et al., 2002) . In contrast, SM and diC16:0 PC have been shown to be completely miscible by differential scanning calorimetry analysis of multilamellar vesicles composed of C16:0 SM/diC16:0 PC (Maulik and Shipley, 1996b) and stearoyl (C18:0) SM/diC16:0 PC (Maulik and Shipley, 1996a). Differential scanning calorimetry analysis also showed SM and GalCer were miscible (Johnston and Chapman, 1988). These results indicate that physical property of the added lipids affects the distribution of SM in the binary mixture. When SM is mixed with liquid crystalline (disordered) lipids like diC18:1 PC, SM forms clusters. In other words, local density of SM is high. In contrast, the presence of solid (ordered) lipids such as diC16:0 PC or GalCer decreases the local density of SM. In most of our experiments, we have used egg SM, of which >80% of amide-linked fatty acids was palmitic acid. Our results, together with the published data, suggest that the binding of lysenin to SM is dependent upon the local density of the lipid. Cholesterol is known to facilitate phase separation of sphingolipid/phospholipid binary mixture (Ohvo-Rekila et al., 2002; Ramstedt and Slotte, 2002). Increased lysenin binding to the SM/diC16:0 PC monolayer in the presence of cholesterol could be explained by the segregation of SM and diC16:0 PC and the formation of SM-rich domains by the addition of cholesterol. Since diC18:1 PC was disordered and diC16:0 PC and GalCer were solid under our experimental conditions, one can speculate that the observed difference of binding of lysenin in vitro might be because of the different physical states of the membranes. We think this is unlikely because lysenin bound equally to C18:1 SM and C16:0 SM under our experimental conditions (data not shown). Since diC18:1 PC and diC16:0 PC have the same headgroup and GalCer contains only one sugar at the headgroup, it is also unlikely that the observed differences were due to steric hindrance.
Unlike model membranes, the estimated size of SM-rich lipid domains in biomembranes is much smaller (Anderson and Jacobson, 2002). Therefore, the conclusions based on model membrane experiments in this study may be qualitative, rather than quantitative. Since most cells tested are sensitive to lysenin (Hanada et al., 1998; Kobayashi et al., 2000; Ohta et al., 2003; Yamaji et al., 1998), it is speculated that the size of SM-rich domains in cell membranes is big enough for efficient binding of lysenin. Our ITC results suggest that domains containing five molecules of SM may be the smallest units for efficient binding of lysenin. Apical membrane of MDCK and plasma membrane of MEB4 are rare examples of lysenin-resistant membranes. Considering that the content of glycolipids in melanoma cells is comparatively higher than that of many other cells (Deng et al., 2000) and apical membranes are highly enriched with glycolipids, perhaps high concentrations of glycolipids are required to alter the distribution of SM. It was shown that MEB4 and glycolipid-deficient GM95 had similarly detergent-resistant membranes (Ostermeyer et al., 1999). Our results indicate that the organization of SM is different between these two cell types. Our results also indicate that the apical and basolateral membranes of MDCK cells display altered SM organization. Cell labeling with lysenin suggests that local density of SM in apical membranes reorganizes during the establishment of polarization. The heterogenous distribution of lysenin labeling in low density cells suggests the heterogeneity of reorganization of lipids during polarized cell growth on coverslips.
Sphingomyelin has attracted attention as a reservoir of ceramide in sphingomyelinase-dependent signal transduction (Kolesnick and Hannun, 1999; Hoffman and Dixit, 1999). The big difference in physical properties of SM and ceramide affects the local structural reorganization of the membrane during hydrolysis (Fanani et al., 2002). Recently it has been proposed that this structural change is critical for ceramide-dependent transmembrane signal transduction (Cremesti et al., 2002). Ceramide-dependent membrane structural change is dependent on the local concentration of ceramide, which is directly affected by the local density of SM. Our results suggest that the sphingomyelinase-dependent signal transduction is affected by glycolipids.
Recently we showed that lysenin bound membranes and assembled to SDS-resistant oligomers in a SM-dependent manner, leading to the formation of pores with a hydrodynamic diameter of ∼3 nm (Yamaji-Hasegawa et al., 2003). Immunoelectron microscopy revealed that lysenin was not uniformly distributed on SM-containing membranes; rather, it accumulated in limited regions of the membrane. Altered distribution of His-Venus-lysenin and fluorescent lipid probe in GUVs is likely to be the result of aggregation of lysenin on SM/diC18:1 PC membranes. Lysenin contains six tryptophan residues. The tryptophan fluorescence increased and the wavelength of maximum emission underwent a blue shift after incubation with SM/cholesterol (Yamaji-Hasegawa et al., 2003) or SM/diC18:1 PC (this study) liposomes. This suggests that the conformation of lysenin is altered during oligomerization. ITC suggests the formation of a 1:5 complex of lysenin and SM in SM/diC18:1 PC. This ratio is increased to 1:9 in the presence of GalCer. Since the addition of GalCer diminishes SM-induced alteration of the tryptophan spectrum, it is suggested that changing the lysenin/SM ratio in the complex affects the conformation change of lysenin.
Previously, Nores et al. showed that the recognition of the glycolipid GM3 by anti-GM3 antibody was dependent on the concentration of the glycolipid in liposomes (Nores et al., 1987). The authors concluded that the antibody recognized GM3 clusters. It was also shown that the lectin alloA recognized the glycolipid lactosylceramide in a density-dependent manner (Hashizuma et al., 1998). In the present study, we showed that lysenin binds SM in a local density-dependent manner and that glycolipids alter both the stoichiometry and thermodynamic parameters of SM-lysenin complex formation. Our titration experiments demonstrate that lysenin readily partitions into membranes even in the presence of glycolipid. However, the measured reaction enthalpy/SM was small (−1.4 kcal/mol). This low H0 is also observed for the partitioning of phosphatidylethanolamine-specific peptide cinnamycin to phosphatidylcholine bilayers (Machaidze et al., 2002). Our results thus indicate that lysenin is not only a lipid-specific protein but also a lipid organization-specific toxin. Recently the heterogeneity of lipid raft has been reported by using altered sensitivity of raft components to cholesterol extraction (Schade and Levine, 2002), different solubility of proteins in a combination of detergents (Drobnik et al., 2002), as well as altered distribution of glycolipids as revealed by specific antibodies (Gomez-Mouton et al., 2001). Our results suggest that lysenin will be an additional tool to study the heterogeneity of lipid rafts.
Acknowledgments
We are grateful to G.W. Feigenson for the suggestion of the preparation of GUVs. We wish to thank E. Kiyokawa, T. Yamaji, Y. Shimizu, and M. Takahashi for critically reading the manuscript, T. Nagai for providing pRSET-Venus cDNA, E. Kiyokawa and N. Otsuka for the preparation of His-Venus-lysenin, and S.B. Sato for valuable discussion.
This work was supported by Grants-in-Aid for Scientific Research 14370753 (to T.K.) and 13771400 (to A.Y.-H.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and RIKEN Presidential Research Grant for Intersystem Collaboration (T.K. and A.Y.-H.). T.K. was supported by the International HDL Award Program. R.I. and A.Y.-H. were special postdoctoral fellows of RIKEN.
References
- Akashi, K., H. Miyata, H. Itoh, and K. Kinosita, Jr. 1996. Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys. J. 71:3242–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, R. G., and K. Jacobson. 2002. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 296:1821–1825. [DOI] [PubMed] [Google Scholar]
- Barenholz, Y., and T. E. Thompson. 1999. Sphingomyelin: biophysical aspects. Chem. Phys. Lipids. 102:29–34. [DOI] [PubMed] [Google Scholar]
- Bartlett, G. R. 1958. Phosphorus assay in column chromatography. J. Biol. Chem. 234:466–468. [PubMed] [Google Scholar]
- Bligh, E. G., and W. J. Dyer. 1959. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37:911–917. [DOI] [PubMed] [Google Scholar]
- Brown, D. A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- Brown, D. A., and E. London. 2000. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221–17224. [DOI] [PubMed] [Google Scholar]
- Brown, D. A., and J. K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- Brown, R. E. 1998. Sphingolipid organization in biomembranes: what physical studies of model membranes reveal. J. Cell Sci. 111:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee, S., E. R. Smith, K. Hanada, V. L. Stevens, and S. Mayor. 2001. GPI anchoring leads to sphingolipid-dependent retention of endocytosed proteins in the recycling endosomal compartment. EMBO J. 20:1583–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremesti, A. E., F. M. Goni, and R. Kolesnick. 2002. Role of sphingomyelinase and ceramide in modulating rafts: do biophysical properties determine biologic outcome? FEBS Lett. 531:47–53. [DOI] [PubMed] [Google Scholar]
- Deng, W., R. Li, and S. Ladisch. 2000. Influence of cellular ganglioside depletion on tumor formation. J. Natl. Cancer Inst. 92:912–917. [DOI] [PubMed] [Google Scholar]
- Drobnik, W., H. Borsukova, A. Bottcher, A. Pfeiffer, G. Liebisch, G. J. Schutz, H. Schindler, and G. Schmitz. 2002. Apo AI/ABCA1-dependent and HDL3-mediated lipid efflux from compositionally distinct cholesterol-based microdomains. Traffic. 3:268–278. [DOI] [PubMed] [Google Scholar]
- Fanani, M. L., S. Hartel, R. G. Oliveira, and B. Maggio. 2002. Bidirectional control of sphingomyelinase activity and surface topography in lipid monolayers. Biophys. J. 83:3416–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenson, G. W., and J. T. Buboltz. 2001. Ternary phase diagram of dipalmitoyl-PC/dilauroyl-PC/cholesterol: nanoscopic domain formation driven by cholesterol. Biophys. J. 80:2775–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler, K., T. Kobayashi, T. V. Kurzchalia, and K. Simons. 1993. Glycosphingolipid-enriched, detergent-insoluble complexes in protein sorting in epithelial cells. Biochemistry. 32:6365–6373. [DOI] [PubMed] [Google Scholar]
- Gatt, S. 1999. Studies of sphingomyelin and sphingomyelinases. Chem. Phys. Lipids. 102:45–53. [DOI] [PubMed] [Google Scholar]
- Gomez-Mouton, C., J. L. Abad, E. Mira, R. A. Lacalle, E. Gallardo, S. Jimenez-Baranda, I. Illa, A. Bernad, S. Manes, and A. C. Martinez. 2001. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. USA. 98:9642–9647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada, K., T. Hara, M. Fukasawa, A. Yamaji, M. Umeda, and M. Nishijima. 1998. Mammalian cell mutants resistant to a sphingomyelin-directed cytolysin. Genetic and biochemical evidence for complex formation of the LCB1 protein with the LCB2 protein for serine palmitoyltransferase. J. Biol. Chem. 273:33787–33794. [DOI] [PubMed] [Google Scholar]
- Hanada, K., M. Nishijima, M. Kiso, A. Hasegawa, S. Fujita, T. Ogawa, and Y. Akamatsu. 1992. Sphingolipids are essential for the growth of Chinese hamster ovary cells. Restoration of the growth of a mutant defective in sphingoid base biosynthesis by exogenous sphingolipids. J. Biol. Chem. 267:23527–23533. [PubMed] [Google Scholar]
- Hannun, Y. A., C. Luberto, and K. M. Argraves. 2001. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry. 40:4893–4903. [DOI] [PubMed] [Google Scholar]
- Hashizume, M., T. Sato, and Y. Okahata. 1998. Selective binding of a lectin for phase-separated glycolipid monolayers. Chem Lett. 27:399–400. [Google Scholar]
- Hidari, K., S. Ichikawa, T. Fujita, H. Sakiyama, and Y. Hirabayashi. 1996. Complete removal of sphingolipids from the plasma membrane disrupts cell to substratum adhesion of mouse melanoma cells. J. Biol. Chem. 271:14636–14641. [DOI] [PubMed] [Google Scholar]
- Hoffman, K., and V. M. Dixit. 1999. Reply to Kolesnick and Hannun, and Perry and Hannun. Trends Biochem Sci. 24:227. [DOI] [PubMed] [Google Scholar]
- Hubbard, A. L. 1991. Targeting of membrane and secretory proteins to the apical domain in epithelial cells. Semin. Cell Biol. 2:365–374. [PubMed] [Google Scholar]
- Ichikawa, S., N. Nakajo, H. Sakiyama, and Y. Hirabayashi. 1994. A mouse B16 melanoma mutant deficient in glycolipids. Proc. Natl. Acad. Sci. USA. 91:2703–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, D. S., and D. Chapman. 1988. A calorimetric study of the thermotropic behaviour of mixtures of brain cerebrosides with other brain lipids. Biochim. Biophys. Acta. 939:603–614. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., S. W. Pimplikar, R. G. Parton, S. Bhakdi, and K. Simons. 1992. Sphingolipid transport from the trans-Golgi network to the apical surface in permeabilized MDCK cells. FEBS Lett. 300:227–231. [DOI] [PubMed] [Google Scholar]
- Kobayashi, H., Y. Sekizawa, M. Aizu, and M. Umeda. 2000. Lethal and non-lethal responses of spermatozoa from a wide variety of vertebrates and invertebrates to lysenin, a protein from the coelomic fluid of the earthworm Eisenia foetida. J. Exp. Zool. 286:538–549. [DOI] [PubMed] [Google Scholar]
- Kolesnick, R., and Y. A. Hannun. 1999. Ceramide and apoptosis (letter to the editor). Trends Biochem Sci. 24:224–225. [DOI] [PubMed] [Google Scholar]
- MacDonald, R. C., R. I. MacDonald, B. P. Menco, K. Takeshita, N. K. Subbarao, and L. R. Hu. 1991. Small-volume extrusion apparatus for preparation of large, unilamellar vesicles. Biochim. Biophys. Acta. 1061:297–303. [DOI] [PubMed] [Google Scholar]
- Machaidze, G., A. Ziegler, and J. Seelig. 2002. Specific binding of Ro 09–0198 (cinnamycin) to phosphatidylethanolamine: a thermodynamic analysis. Biochemistry. 41:1965–1971. [DOI] [PubMed] [Google Scholar]
- Makino, A., T. Baba, K. Fujimoto, K. Iwamoto, Y. Yano, N. Terada, S. Ohno, S. B. Sato, A. Ohta, M. Umeda, K. Matsuzaki, and T. Kobayashi. 2003. Cinnamycin (Ro 09–0198) promotes cell binding and toxicity by inducing transbilayer lipid movement. J. Biol. Chem. 278:3204–3209. [DOI] [PubMed] [Google Scholar]
- Maulik, P. R., and G. G. Shipley. 1996a. Interactions of N-stearoyl sphingomyelin with cholesterol and dipalmitoylphosphatidylcholine in bilayer membranes. Biophys. J. 70:2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulik, P. R., and G. G. Shipley. 1996b. N-palmitoyl sphingomyelin bilayers: structure and interactions with cholesterol and dipalmitoylphosphatidylcholine. Biochemistry. 35:8025–8034. [DOI] [PubMed] [Google Scholar]
- Mosmann, T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 65:55–63. [DOI] [PubMed] [Google Scholar]
- Nagai, T., K. Ibata, E. S. Park, M. Kubota, K. Mikoshiba, and A. Miyawaki. 2002. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 20:87–90. [DOI] [PubMed] [Google Scholar]
- Nakai, Y., Y. Sakurai, A. Yamaji, H. Asou, M. Umeda, K. Uyemura, and K. Itoh. 2000. Lysenin-sphingomyelin binding at the surface of oligodendrocyte lineage cells increases during differentiation in vitro. J. Neurosci. Res. 62:521–529. [DOI] [PubMed] [Google Scholar]
- Nores, G. A., T. Dohi, M. Taniguchi, and S. Hakomori. 1987. Density-dependent recognition of cell surface GM3 by a certain anti-melanoma antibody, and GM3 lactone as a possible immunogen: requirements for tumor-associated antigen and immunogen. J. Immunol. 139:3171–3176. [PubMed] [Google Scholar]
- Ohta, N., M. Aizu, T. Kaneko, T. Sato, and H. Kobayashi. 2003. Damage to the gills, skin and other tissues by lysenin and the coelomic fluid of the earthworm Eisenia foetida in two teleosts, Tanichthys albonubes and Oreochromis mossambicus. J. Exp. Zool. 295A:117–126. [DOI] [PubMed] [Google Scholar]
- Ohvo-Rekila, H., B. Ramstedt, P. Leppimaki, and J. P. Slotte. 2002. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 41:66–97. [DOI] [PubMed] [Google Scholar]
- Ostermeyer, A. G., B. T. Beckrich, K. A. Ivarson, K. E. Grove, and D. A. Brown. 1999. Glycosphingolipids are not essential for formation of detergent-resistant membrane rafts in melanoma cells. methyl-beta-cyclodextrin does not affect cell surface transport of a GPI-anchored protein. J. Biol. Chem. 274:34459–34466. [DOI] [PubMed] [Google Scholar]
- Ramstedt, B., and J. P. Slotte. 2002. Membrane properties of sphingomyelins. FEBS Lett. 531:33–37. [DOI] [PubMed] [Google Scholar]
- Rietveld, A., and K. Simons. 1998. The differential miscibility of lipids as the basis for the formation of functional membrane rafts. Biochim. Biophys. Acta. 1376:467–479. [DOI] [PubMed] [Google Scholar]
- Schade, A. E., and A. D. Levine. 2002. Lipid raft heterogeneity in human peripheral blood T lymphoblasts: a mechanism for regulating the initiation of TCR signal transduction. J. Immunol. 168:2233–2239. [DOI] [PubMed] [Google Scholar]
- Sekizawa, Y., T. Kubo, H. Kobayashi, T. Nakajima, and S. Natori. 1997. Molecular cloning of cDNA for lysenin, a novel protein in the earthworm Eisenia foetida that causes contraction of rat vascular smooth muscle. Gene. 191:97–102. [DOI] [PubMed] [Google Scholar]
- Simons, K., P. Dupree, K. Fiedler, L. A. Huber, T. Kobayashi, T. Kurzchalia, V. Olkkonen, S. Pimplikar, R. Parton, and C. Dotti. 1992. Biogenesis of cell-surface polarity in epithelial cells and neurons. Cold Spring Harb. Symp. Quant. Biol. 57:611–619. [DOI] [PubMed] [Google Scholar]
- Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- Simons, K., and G. van Meer. 1988. Lipid sorting in epithelial cells. Biochemistry. 27:6197–6202. [DOI] [PubMed] [Google Scholar]
- Simons, K., and H. Virta. 1987. Perforated MDCK cells support intracellular transport. EMBO J. 6:2241–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer, G., and K. Simons. 1982. Viruses budding from either the apical or the basolateral plasma membrane domain of MDCK cells have unique phospholipid compositions. EMBO J. 1:847–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch, S. L., and S. L. Keller. 2003. A closed look at the canonical raft mixture in model membrane studies. Biophys. J. 84:725–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieprecht, T., and J. Seelig. 2002. Isothermal Titration Calorimetry for Studying Interactions between Peptides and Lipid Membranes. Academic Press, New York.
- Yamaji, A., Y. Sekizawa, K. Emoto, H. Sakuraba, K. Inoue, H. Kobayashi, and M. Umeda. 1998. Lysenin, a novel sphingomyelin-specific binding protein. J. Biol. Chem. 273:5300–5306. [DOI] [PubMed] [Google Scholar]
- Yamaji-Hasegawa, A., A. Makino, T. Baba, Y. Senoh, H. Kimura-Suda, S. B. Sato, N. Terada, S. Ohno, E. Kiyokawa, M. Umeda, and T. Kobayashi. 2003. Oligomerization and pore formation of a sphingomyelin-specific toxin, lysenin. J. Biol. Chem. 278:22762–22770. [DOI] [PubMed] [Google Scholar]
- Yuan, C., J. Furlong, P. Burgos, and L. J. Johnston. 2002. The size of lipid rafts: an atomic force microscopy study of ganglioside GM1 domains in sphingomyelin/DOPC/cholesterol membranes. Biophys. J. 82:2526–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias, D. A., J. D. Violin, A. C. Newton, and R. Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 296:913–916. [DOI] [PubMed] [Google Scholar]