

Abstract
The interaction of antimicrobial peptides with membranes is a key factor in determining their biological activity. In this study we have synthesized a series of minimized cecropin-mellitin hybrid peptides each containing a single cysteine residue, modified the cysteine with the sulfhydryl-specific methanethiosulfonate spin-label, and used electron paramagnetic resonance spectroscopy to measure membrane-binding affinities and determine the orientation and localization of peptides bound to membranes that mimic the bacterial cytoplasmic membrane. All of the peptides were unstructured in aqueous solution but underwent a significant conformational change upon membrane binding that diminished the rotational mobility of the attached spin-label. Apparent partition coefficients were similar for five of the six constructs examined, indicating that location of the spin-label had little effect on peptide binding as long as the attachment site was in the relatively hydrophobic C-terminal domain. Depth measurements based on accessibility of the spin-labeled sites to oxygen and nickel ethylenediaminediacetate indicated that at high lipid/peptide ratios these peptides form a single α-helix, with the helical axis aligned parallel to the bilayer surface and immersed ∼5 Å below the membrane-aqueous interface. Such a localization would provide exposure of charged/polar residues on the hydrophilic face of the amphipathic helix to the aqueous phase, and allow the nonpolar residues along the opposite face of the helix to remain immersed in the hydrophobic phase of the bilayer. These results are discussed with respect to the mechanism of membrane disruption by antimicrobial peptides.
INTRODUCTION
Over the past two decades a large number of antimicrobial peptides have been identified from a wide range of both vertebrate and invertebrate species (Boman, 1995; Hancock and Scott, 2000; Zasloff, 2002). They include linear peptides such as the magainins (Zasloff, 1987), indolicidin (Selsted et al., 1992), and cecropins (Hultmark et al., 1980), as well as cyclic peptides that contain one or more disulfide bonds such as the defensins (Selsted et al., 1985; Lehrer et al., 1991). These peptides form the “innate immunity” arm of host defense, providing broad-spectrum protection against invading pathogens before activation of classic antibody- and cell-mediated immunity. Although they are of evolutionarily ancient lineage, the occurrence of bacterial resistance to antimicrobial peptides is exceedingly rare (Hancock and Scott, 2000; Zasloff, 2002). This is due to the fact that, in contrast to the vast majority of antibiotics in clinical use, these peptides appear to target general properties of the microbial membrane (Maloy and Kari, 1995; Hancock and Scott, 2000; Zasloff, 2002). Consequently, acquired resistance would require an overall restructuring of membrane composition and/or organization. For this reason, and because of the ever-increasing prevalence of multiple-drug resistant pathogens, there has been considerable interest in the development of these peptides as a new source of antibiotics (Hancock and Scott, 2000; Zasloff, 2002).
Despite their remarkable diversity, antimicrobial peptides share two common features: a net positive charge and the propensity to adopt amphipathic secondary structures in the presence of membranes (Hancock, 2001; Zasloff, 2002). One particularly prominent group of antimicrobial peptides consists of linear peptides that form amphipathic α-helices (Maloy and Kari, 1995; Dathe and Wieprecht, 1999; Giangaspero et al., 2001). Included among these are the cecropins, a family of peptides typically 33–39 amino acids in length first isolated from the silk moth, Hyalophora cecropia (Hultmark et al., 1980; Steiner et al., 1981). Cecropins display broad-spectrum antimicrobial activity (Steiner et al., 1981; Andreu et al., 1985; Fink et al., 1989b) and have a general structural motif consisting of a basic N-terminal amphipathic helical domain connected to a relatively hydrophobic C-terminal domain by a flexible hinge (Holak et al., 1988).
In an effort to reduce the size of the cecropins to facilitate their solid-phase synthesis, Boman, Merrifield, and co-workers identified a group of significantly shortened hybrids of cecropin A and the bee venom peptide, mellitin, that displayed antibacterial activity comparable to the full-length cecropins and yet lacked the hemolytic properties associated with mellitin (Boman et al., 1989; Fink et al., 1989a; Wade et al., 1992; Andreu et al., 1992). A hybrid consisting of the first seven residues of cecropin A and residues two through nine of mellitin, C(1–7)M(2–9) (which we designate CM15), was identified as the minimal sequence with strong antimicrobial efficacy (Andreu et al., 1992). CM15, like the native cecropins, has a highly basic N-terminal domain and a relatively hydrophobic C-terminal domain (Table 1). As with cecropin A (Wade et al., 1990), the all-D enantiomer of CM15 retains biological activity against a broad panel of bacterial species (Merrifield et al., 1995), indicating that interaction with cellular targets occurs in a nonstereospecific manner.
TABLE 1.
Cecropin-mellitin hybrid peptides
| Peptide | Sequence |
|---|---|
| CM15 | Ac-KWKLFKKIGAVLKVL-NH2 |
| CM15-C4 | Ac-KWKCFKKIGAVLKVL-NH2 |
| CM15-C8 | Ac-KWKLFKKCGAVLKVL-NH2 |
| CM15-C10 | Ac-KWKLFKKIGCVLKVL-NH2 |
| CM15-C11 | Ac-KWKLFKKIGACLKVL-NH2 |
| CM15-C12 | Ac-KWKLFKKIGAVCKVL-NH2 |
| CM15-C14 | Ac-KWKLFKKIGAVLKCL-NH2 |
Primary sequences of peptides showing positions of the cysteine labeling sites.
Despite extensive study, the precise mechanisms of peptide-membrane interaction and cell killing have not been firmly established for many antimicrobial peptides. It has been suggested that, after translocation across the outer membrane or cell wall, disruption of the cytoplasmic membrane is the lethal event leading to bacterial cell death (Westerhoff et al., 1989; Hancock and Chapple, 1999; Silvestro et al., 2000; Yang et al., 2000). This may occur through a detergentlike “carpet” mechanism (Steiner et al., 1988; Gazit et al., 1995; Shai, 2002), or the formation of discrete channels that dissipate ion gradients (Christensen et al., 1988; Kagan et al., 1990; Juvvadi et al., 1996). Although recent studies have shown that some antimicrobial peptides interact with intracellular targets (Otvos, Jr. et al., 2000) and bind DNA (Zhang et al., 1999), these peptides still must traverse the cell membrane to reach their site of action. Full-length cecropins have been proposed to act through both the carpet mechanism (Steiner et al., 1988) and by formation of ion channels (Christensen et al., 1988; Silvestro et al., 1997). CM15 has also been shown to form ion channels in planar bilayers (Juvvadi et al., 1996); however, there have been no detailed studies on the membrane interactions of these minimized peptides.
In the present study, we have utilized site-directed spin-labeling electron paramagnetic resonance (EPR) spectroscopy to measure partition coefficients for a series of spin-labeled, single-cysteine analogs of CM15, and to examine the structure and localization of CM15 when bound to membranes that mimic the lipid composition of the bacterial cytoplasmic membrane. We find that CM15 is unstructured in aqueous solution, but upon membrane binding forms an α-helix that intercalates just below the surface of the aqueous-membrane interface, aligned parallel to the bilayer surface. Changes in the free energy of binding for different spin-labeling sites did not correlate with standard side-chain free energies, and were apparently dependent on sequence context.
MATERIALS AND METHODS
Escherichia coli bacterial polar lipids (BPL), POPE (1-palmitoyl-2-oleoylphosphatidylethanolamine), POPG (1-palmitoyl-2-oleoylphosphatidylglycerol), and n-PCSL (n-doxyl-phosphatidylcholine; n = 5, 7, 10, 12) spin-labels were obtained from Avanti Polar Lipids (Alabaster, AL). The composition of BPL is PE/PG/cardiolipin (CL) 67:23.2:9.8 (% × wt), corresponding to a molar ratio of ∼68:26:6. This is in good agreement with the composition of E. coli inner membrane lipids (Gennis, 1989). Rink amide MBHA resin and n-Fmoc L-amino acids were purchased from Novabiochem (La Jolla, CA). Acetic anhydride, DIC (diisopropylcarbodiimide), DCM (dichloromethane), HOBt (1-hydroxybenzotriazole), TIS (triisopropylsilane), piperidine, and NMP (n-methylpyrrolidinone) were purchased from Fisher Scientific (Hampton, NH). NiEDDA (nickel ethylenediaminediacetate) was synthesized by a protocol kindly provided by Dr. Christian Altenbach (Jules Stein Eye Institute, UCLA School of Medicine, Los Angeles, CA). The methanethiosulfonate spin-label, MTSL (1-oxy-2,2,5,5-tetramethylpyrroline-3-methyl), was obtained from Toronto Research Chemicals (North York, ON, Canada).
Liposome preparation
Lipids in desired molar ratios were dried down from chloroform stock solutions under nitrogen gas and further dried at least 1 h under vacuum. The resulting lipid film was hydrated by addition of 20 mM MOPS and 100 mM KCl (pH 7.0) to give a concentration of ∼100 mM phospholipid. Large unilamellar vesicles (LUVs) were prepared by freeze-thawing this lipid suspension 5×, followed by extrusion through 100-nm polycarbonate membrane filters (Osmonics, Minnetonka, MN) using a miniextruder syringe device (Lipex Biomembranes, Vancouver, BC, Canada). Final lipid concentration was measured by the method of Stewart (1980).
Peptide synthesis and spin-labeling
Peptides were synthesized on rink amide p-methylbenzhydrylamine resin with acetylated N-termini and amidated C-termini by solid-phase synthesis methods using standard n-(9-fluororenyl)methoxycarbonyl (Fmoc) chemistry. Coupling of Fmoc amino acids was performed with equal volumes of 0.5 M HOBt and 0.5 M DIC in NMP. The Fmoc-protecting group was removed with 25% piperidine in NMP, followed by washing 3× each in NMP and DCM. Amino acid side chains were protected as trityl (Cys) and Boc (Lys and Trp). N-terminal acetylation was carried out with excess acetic anhydride in the presence of coupling reagents HOBt and DIC at room temperature for 4 h. Deprotection and cleavage of peptide from the resin were carried out using TFA, TIS, and double-distilled water (98:1:1, v/v) for 3 h at room temperature. The peptides were precipitated and washed 3× with cold diethyl ether and dried under vacuum. Crude peptides were purified by reverse-phase semipreparative HPLC on a 10-μm, 1.0 × 25-cm C8 column (Vydac, Hesperia, CA) using a linear gradient of 10 to 80% ACN (acetonitrile) in water/0.1% TFA over 40 min. Upon elution the peptides were lyophilized, resuspended in a small volume of ACN:20 mM MOPS, pH7 (1:1), reacted with a fivefold molar excess of MTSL for 3 h at room temperature, and then rechromatographed as described above to remove excess spin-label. Peptide purity was checked by analytical HPLC and molecular mass verified by MALDI-TOF mass spectrometry in the Protein-Nucleic Acid Shared Facility of the Medical College of Wisconsin (Milwaukee, WI).
Listed in Table 1 are the amino acid sequences of the lead peptide, CM15, and six analogs, each of which contains a single cysteine residue to serve as an attachment site for the sulfhydryl-specific nitroxide spin-label, MTSL. Nonpolar residues were selected for substitution with cysteine and spin-labeling inasmuch as the side chain of MTSL is relatively hydrophobic (Yu et al., 1994). In addition, aromatic residues in the amphipathic N-terminal domain were left unchanged as they have been shown to be essential for antibacterial activity in full-length cecropins (Andreu et al., 1985).
EPR measurements of accessibility and immersion depth
The accessibility of the spin-label to the diffusible relaxation agents O2 and NiEDDA was determined by continuous-wave power saturation (Altenbach et al., 1989). Spin-labeled peptides were mixed with LUVs to give final concentrations of 40 mM lipid and 0.2 mM peptide in a final volume of 5 to 10 μl, and placed into gas-permeable TPX capillaries (Molecular Specialties, Milwaukee, WI). Samples containing NiEDDA were incubated at 37°C for at least 2 h before analysis to allow equilibration across the membrane bilayer. No differences were observed between samples incubated 2 h at 37°C and samples allowed to equilibrate at room temperature overnight. As shown in Fig. 2, at these high lipid/peptide ratios the spin-labeled peptides were fully membrane-bound. EPR spectra for power saturation studies were obtained on a Varian E-102 Century series spectrometer equipped with an X-band two-loop one-gap resonator (Molecular Specialties, Milwaukee, WI). Values for the saturation parameter P1/2 were determined for each sample under three conditions: saturated with N2, saturated with air (20% O2), and under N2 in the presence of 20 mM NiEDDA. The change in P1/2, ΔP1/2 is a direct measure of the bimolecular collision rate between the spin-label and the relaxation agent and, hence, the accessibility of the spin-label to a given paramagnetic probe (Altenbach et al., 1989). To account for differences in spin-label mobility and resonator performance, ΔP1/2 values are multiplied by the inverse width of the center line (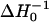 ) and normalized according to ΔH0 and P1/2 for a DPPH (diphenylpicrylhydrazine) standard giving the accessibility parameter, Π (Farahbakhsh et al., 1992),
) and normalized according to ΔH0 and P1/2 for a DPPH (diphenylpicrylhydrazine) standard giving the accessibility parameter, Π (Farahbakhsh et al., 1992),
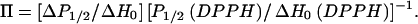 |
(1) |
FIGURE 2.

Room temperature EPR spectra of single-cysteine CM15 analogs labeled with MTSL (left) in aqueous solution, and (right) bound to LUVs composed of bacterial inner membrane lipids (PE/PG/CL, 68:26:6) at a lipid/peptide molar ratio of 200:1. Scan widths are 100 G.
The concentrations of oxygen and NiEDDA have been shown to vary along the bilayer normal in inverse fashion, providing the basis of a method to measure the transmembrane location of a spin-label exposed to the lipid phase (Altenbach et al., 1994). The depth parameter, Φ, is defined as
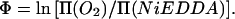 |
(2) |
The dependence of Φ on bilayer depth is calibrated with n-doxyl-phosphatidylcholine spin-labels (n-PCSL) in the same membrane system (including unlabeled peptide) that is employed for protein studies (Altenbach et al., 1994; Klug et al., 1997). Liposomes containing 0.5 mol % of 5-, 7-, 10-, or 12-PCSL were prepared in BPL and extruded to prepare LUVs as described above. The parent peptide, CM15 (Table 1), was added to give a lipid/peptide ratio of 200:1 before analysis. A linear fit to the known depths of the PCSL standards (Dalton et al., 1987) yielded the relationship of depth (Å) = 4.62 Φ + 3.89.
EPR measurement of partition coefficients
For peptide-binding assays a constant amount of spin-labeled peptide was mixed with various concentrations of LUVs to give a final sample volume of 40 μl and a final peptide concentration of 35 to 50 μM. All stock solutions and dilutions were made with 20 mM MOPS and 100 mM KCl, pH 7.0. Peptide-lipid mixtures were incubated at room temperature for 30 min, loaded into 50-μl glass capillaries, and the EPR spectrum recorded on a Varian E-102 Century series spectrometer equipped with a TE102 cavity at a microwave power at 10 mW using a 100-kHz, 1.0-G field modulation. Signal averaging and measurement of signal amplitudes were accomplished with software written in LabView by Dr. C. Altenbach.
Methodology for quantitating the binding of spin-labeled peptides to membranes is well-established (Archer et al., 1991; Mchaourab et al., 1994; Thorgeirsson et al., 1996; Victor and Cafiso, 2001; reviewed by Feix and Klug, 1998). Shown in Fig. 1 are EPR spectra for MTSL-labeled CM15-C12 free in solution and in equilibrium with LUVs where ∼90% of the peptide is membrane-bound. The fraction of bound peptide, fb, is calculated according to the relation
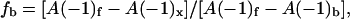 |
(3) |
where A(−1)f and A(−1)x are the peak-peak amplitudes of the high-field (MI = −1) line for the peptide free in solution and in the given experimental sample, respectively (Fig. 1). A(−1)b. The amplitude of the high-field line when the peptide is fully bound was measured at high lipid/peptide concentrations, and was typically <3% of A(−1)f, thus representing a small correction. The concentrations of membrane-bound peptide, Cb, and peptide remaining free in solution, Cf, were calculated from fb and the known total concentration of peptide. To determine the apparent partition coefficient, Kp, the molar ratio of bound peptide to lipid, Xb, was plotted against Cf (e.g., see Fig. 5). For ideal partitioning behavior a plot of Xb against Cf will yield a straight line of slope, Kp, i.e., Xb = Kp Cf where Kp is in units of M−1 (Spuhler et al., 1994; Russell et al., 1996; Han and Tamm, 2000). It was assumed that the peptides bound only to the outer surface of the LUVs, so that the accessible lipid concentration was taken to be one-half the total lipid concentration. It was observed that addition of 50 mM chromium oxalate, a nonpermeant relaxation agent, to vesicle-peptide suspensions completely broadened the EPR signal of the spin-labeled peptide. This indicates that these peptides remain on the exterior of the vesicles, at least in the time frame (<1 h) of these experiments. Partition coefficients were determined under conditions where bound peptide was a small fraction of the accessible membrane lipid, so as to not perturb the membrane surface charge density.
FIGURE 1.

EPR spectra of MTSL-labeled CM15-C12 (top) in aqueous solution and (bottom) in the presence of POPE/POPG (80:20) LUVs with ∼90% bound. At bottom, the spectrum of fully-bound peptide (dashed line) is superimposed. The peak-peak amplitudes A(−1)f and A(−1)x are used to calculate the fraction of peptide bound (Eq. 3).
FIGURE 5.

Binding isotherms obtained with POPE/POPG (80:20) vesicles for CM15 analogs spin-labeled at (Δ) C4, (•) C8, (×) C10, and (○) C12. Xb is the molar ratio of bound peptide/accessible lipid, where accessible lipid is one-half the total lipid concentration and Cf is the concentration of peptide remaining free in solution. Straight lines indicate regions of maximum slope used for calculation of Kp. All experiments were done at room temperature in the presence of 20 mM MOPS and 100 mM KCl at pH 7.
RESULTS
Structure of the membrane-bound peptide
Shown in Fig. 2 are the EPR spectra for a series of MTSL-labeled single-cysteine analogs of CM15 in aqueous solution and following the addition of LUVs that mimic the composition of the bacterial cytoplasmic membrane. In aqueous solution the spectra all consist of three narrow lines indicative of rapid rotational motion of the spin-label with subnanosecond rotational correlation times. This is consistent with what is expected for small, unstructured peptides in solution. Upon binding of the peptides to membranes their EPR spectra are all significantly broadened, indicating a decrease in mobility with rotational correlation times in the range of 3 to 5 ns. Line widths and rotational correlation times were independent of the concentration of membrane-bound peptide for lipid/peptide ratios >120:1 (60:1 if one considers only the outer leaflet of the bilayer), and there was no evidence in the spectra of strong spin-spin interaction, indicating that these peptides are monomeric when bound at high lipid/peptide ratios. The ability to prepare samples in which peptides are fully bound and in the monomeric aggregation state facilitated the mapping of secondary structure and immersion depth for the membrane-bound peptides, as described below.
To characterize the structure of the membrane-bound state of the peptide, we examined the accessibility of the spin-label side chain to the paramagnetic relaxation agents, O2 and NiEDDA. NiEDDA is a polar, neutral solute that partitions primarily into the aqueous phase, whereas oxygen is nonpolar and partitions favorably into membranes with a gradient of increasing concentration that reaches a maximum near the center of the bilayer. The inverse concentration gradients of O2 and NiEDDA provide a basis for determining the depth of a spin-label in the lipid bilayer (Altenbach et al., 1994). The EPR accessibility parameters for O2 and NiEDDA are given in Table 2 and shown as a function of spin-label position in Fig. 3. Notably, the residue/residue variation in accessibilities to O2 and NiEDDA are out-of-phase (Fig. 3), which is a classic indication of interaction with a lipid bilayer (Feix and Klug, 1998; Hubbell et al., 1998).
TABLE 2.
EPR accessibility and depth parameters
| Labeling site | Π(O2)* | Π(NiEDDA)† | Φ‡ | Depth (Å)‡ |
|---|---|---|---|---|
| C4 | 0.48 ± 0.02 | 0.14 ± 0.02 | 1.23 ± 0.20 | 9.5 ± 1.0 |
| C8 | 0.60 ± 0.02 | 0.10 ± 0.03 | 1.79 ± 0.39 | 12.2 ± 1.8 |
| C10 | 0.42 ± 0.02 | 1.48 ± 0.02 | −1.26 ± 0.06 | −1.9 ± 1.0 |
| C11 | 0.51 ± 0.03 | 0.48 ± 0.02 | 0.06 ± 0.10 | 4.2 ± 0.4 |
| C12 | 0.64 ± 0.01 | 0.10 ± 0.02 | 1.86 ± 0.22 | 12.5 ± 0.8 |
| C14 | 0.45 ± 0.02 | 0.84 ± 0.02 | −0.62 ± 0.06 | 1.0 ± 0.4 |
Accessibility parameter for air (20% O2).
Accessibility parameter for 20 mM NiEDDA. Uncertainties in Π values are standard errors based on replicate experiments (n = 2–3 for each site).
Uncertainties in Φ and depth are calculated from the maximum uncertainties in Π values.
FIGURE 3.

Accessibility parameters for oxygen, Π(O2) (•), and 20 mM NiEDDA, Π(NiEDDA) (○), as a function of labeling position. Horizontal error bars indicate the standard error of the mean from replicate experiments. Lines between points are shown to emphasize the out-of-phase behavior of the accessibilities.
Depth parameters based on differential accessibility to O2 and NiEDDA (Eq. 2) are given in Table 2, and immersion depths of the spin-label side chain are shown as a function of labeling position in Fig. 4. For the sites examined in this study, all of which correspond to nonpolar residues in the native peptide (Table 1), the spin-label attached at C10 had the greatest accessibility to NiEDDA, and C12 was the most deeply buried site (i.e., least accessible to NiEDDA and most accessible to O2). As seen in Fig. 4, the experimental data maps very well to a periodicity of 3.6 residues/turn, consistent with a well-defined α-helix. Furthermore, the similarity in depths for sites C4, C8, and C12, as well as for C10 and C14, indicates that the axis of the helix is oriented parallel to the bilayer surface. Immersion depths varied from ∼12 Å deep in the membrane to 2 Å above the bilayer surface (Fig. 4). This 14 Å span is in good agreement with the dimensions expected for a helical cylinder aligned parallel to the bilayer surface, given that the length of the MTSL side chain is ∼6–7 Å (Rabenstein and Shin, 1995). This suggests that the central axis of the helix is positioned ∼5 Å below the aqueous-membrane interface, or at about the level of the phospholipid glycerol backbone. Such localization would allow lysine residues along one face of the helix to extend into the aqueous phase and potentially interact with lipid phosphates, and the nonpolar residues along the other face of the helix to be buried in the hydrophobic phase of the lipid alkyl chains.
FIGURE 4.

Immersion depth as a function of labeling position. Depths relative to the membrane-aqueous interface were calculated from the depth parameter, Φ (Eq. 2), and comparison to a series of n-doxylphosphatidylcholine spin-labels. Error bars are based on the maximum propagation of the uncertainties in Π values. Positive values indicate sites embedded in the hydrophobic phase and negative values exposure to the aqueous phase. The solid line is a sine curve with a periodicity of 3.6.
Membrane-binding affinity
Binding isotherms were determined by titrating spin-labeled peptides with LUVs (see Materials and Methods) composed of either PE/PG/CL (68:26:6 molar ratio) or POPE/POPG (80:20 mol/mol). Representative binding curves are shown for several CM15 analogs in Fig. 5, and molar partition coefficients are given in Table 3. All of the peptides showed an upward curvature in their binding isotherms at relatively low mole fractions of bound peptide, suggesting a positive cooperativity in the early stages of membrane binding (Spuhler et al., 1994; Han and Tamm, 2000). This was followed by an essentially linear phase of maximal binding affinity, and finally a decrease in apparent binding affinity at higher concentrations of bound peptide due to partial neutralization of the negative surface charge of the liposomes by bound peptide. Binding appeared to saturate at 3–4 mol % of the outer leaflet lipid concentration with POPE/POPG bilayers (Fig. 5) and at slightly higher concentrations (4–6 mol %) with BPL liposomes, consistent with the higher surface potential in the latter system.
TABLE 3.
Partition coefficients for the binding of MTSL-labeled CM15 analogs with PE/PG and bacterial polar lipid vesicles
| Kp (M−1) | ||
|---|---|---|
| Labeling site | POPE/POPG (8:2) | BPL |
| C4 | 0.04 ± 0.02 × 104 | 0.46 ± 0.15 × 104 |
| C8 | 0.39 ± 0.04 × 104 | 1.78 ± 0.35 × 104 |
| C10 | 0.31 ± 0.04 × 104 | 0.95 ± 0.31 × 104 |
| C11 | 0.30 ± 0.03 × 104 | 0.83 ± 0.26 × 104 |
| C12 | 0.40 ± 0.04 × 104 | 1.81 ± 0.32 × 104 |
| C14 | 0.33 ± 0.03 × 104 | 1.53 ± 0.33 × 104 |
Kp was determined from the maximum slope of binding isotherms as shown in Fig. 5. Error estimates are based on the uncertainty in the linear regression analysis. Replicate experiments were in good agreement with the values shown (i.e., within the given error limits). The composition of BPL is PE/PG/cardiolipin (68:26:6 mol %).
Apparent partition coefficients, Kp, were calculated based on the linear regions of the binding isotherms where binding affinity is maximal (Fig. 5). Kp values were relatively similar among the various peptides, with the exception of CM15-C4 for which binding was significantly weaker (Table 3). Given that PG and CL carry one and two negative charges per molecule, respectively, membranes composed of BPL have a very high negative surface charge density (i.e., ∼38 mol %). Consequently, it is expected that electrostatic interactions will promote strong binding of CM15 analogs to BPL liposomes. Kp values were significantly lower for PE/PG (80:20) LUVs than for liposomes composed of BPL, confirming a strong electrostatic component/membrane association even in the presence of 100 mM KCl.
Based on apparent partition coefficients it is possible to evaluate differences in the free energies of binding for the various peptides. Considering only those analogs labeled in the C-terminal domain, the free energy of transfer from the aqueous to the membrane phase, ΔGt, for the peptide with the greatest binding affinity (CM15-C12), was −0.4 kcal/mol greater than that for the weakest binding peptide, CM15-C10 (Table 3). For CM15-C4, ΔGt was less negative than that of C10 by an additional 0.35 kcal/mol. Differences in ΔGt between BPL liposomes and LUVs composed of POPE/POPG (80:20) ranged from ∼0.4 kcal/mol for CM15-C10 to 1.4 kcal/mol for CM15-C4.
DISCUSSION
The interaction of small, basic antimicrobial peptides with membranes is a key factor in determining their biological activity. For many of these peptides, membrane disruption is considered to be the primary mechanism of cell killing (Maloy and Kari, 1995; Silvestro et al., 2000; Zasloff, 2002; Shai, 2002). With this in mind we have investigated a series of small, cecropin-mellitin (CM) hybrid peptides with the goals of determining their structure and orientation upon initial membrane binding, their localization in the lipid bilayer, and their binding affinity for bilayers having a lipid composition similar to that of the bacterial cytoplasmic membrane.
All of the peptides bound to liposomes composed of either PE/PG/CL (68:26:6) or POPE/POPG (80:20) with high affinity, and with positive cooperativity at relatively low concentrations of bound peptide. Self-promoted uptake (Hancock and Chapple, 1999; Sawyer et al., 2003) and sigmoidal binding curves (Hancock and Scott, 2000; Chen et al., 2002) are a common characteristic of antimicrobial peptides, and we have previously observed a similar positive cooperativity in the binding isotherms of a full-length cecropin (Mchaourab et al., 1994). This is usually taken as an indication of peptide-peptide interaction (Spuhler et al., 1994; Han and Tamm, 2000), although it could also reflect changes in bilayer structure that enhance subsequent peptide binding (Chen et al., 2002). We observed no evidence for peptide aggregation in the initial stages of peptide binding (at lipid/peptide ratios of ≅120:1 or greater). There was also no obvious indication of peptide aggregation at higher concentrations of bound peptide. Maintaining membrane-bound peptides in a monomeric aggregation state was important in facilitating the mapping of secondary structure that was the focus of this study; however, further studies are in progress to determine if aggregation occurs in the membrane as the concentration of bound peptide is increased.
With the exception of CM15-C4, apparent partition coefficients were similar for all of the peptides studied, indicating that the position of the spin-label had little effect on peptide binding as long as the attachment site was in the relatively hydrophobic C-terminal domain. The diminished binding affinity of CM15-C4 affirms that the energetics of membrane association depends on more than just the average physical properties of a given peptide, such as net charge or mean residue hydrophobicity. For example, CM15-C4 and CM15-C12 are both leucine-to-cysteine substitutions and have identical amino acid compositions, yet differ significantly in their binding affinities for both membrane systems examined. Thus, sequence context also plays a role in the effects of a given amino acid substitution on the membrane interactions of these small heterogeneous peptides. This is in agreement with the conclusions reached in comparative studies based on antimicrobial and hemolytic activities—that, although general trends in the relationship between physical properties and biological activity can be identified for groups of peptides, the effects of a given amino acid substitution on any individual peptide are difficult to predict (Dathe and Wieprecht, 1999; Giangaspero et al., 2001).
Our studies indicate that at low concentrations of bound peptide, small cecropin-mellitin hybrid peptides intercalate into the membrane just below the surface of the bilayer, and adopt an α-helical conformation with the helix axis parallel to the membrane surface. Peptide localization is such that side chains along the hydrophobic face of the helix, including those of Leu-4, Ile-10, and Leu-12, are buried in the hydrophobic phase of the bilayer whereas the hydrophilic residues, notably the four lysines in the N-terminal domain and Lys-13, are within reach of the membrane-aqueous interface with the potential for ion pairing with lipid phosphates. Our results are consistent with formation of a single α-helix that encompasses the full length of the peptide. Evidence for helical structure is very strong for the C-terminal half of the peptide, i.e., from residues 8–14, where five of the seven sites were labeled (Fig. 4). Although the depth for C4 mapped closely to that expected if the helix extends through the entire length of the peptide, evidence for helical structure in the N-terminal half of CM15 is less rigorous given that only one of the first seven sites was labeled, and flexibility about the glycine residue at position 9 cannot be ruled out. Nonetheless, our results are consistent with previous CD studies indicating that CM15 forms ∼100% α-helix in 16–20% HFIP (Andreu et al., 1992; Juvvadi et al., 1996).
Previous studies using internal reflectance Fourier-transform infrared spectroscopy (Silvestro and Axelsen, 2000), oriented CD (Chen et al., 2001), and solid-state NMR (Marassi et al., 1999) have all concluded that, in the initial stages of membrane binding, full-length (35–37 residue) cecropins adopt helical secondary structures that are aligned parallel the bilayer surface. Similarly, based on oriented CD studies Huang and co-workers have shown that a number of amphipathic antimicrobial peptides are initially embedded in the headgroup region of the bilayer, parallel to the membrane surface (Ludtke et al., 1996; Heller et al., 1998; Huang, 2000). They have suggested that this initial interaction leads to a thinning of the lipid bilayer that progresses with increasing concentrations of bound peptide until a critical threshold is reached, at which point a structural transition occurs resulting in reorientation of membrane-bound peptide (Huang, 2000). The localization that we have observed for CM15 is consistent with the initial phase of this mechanism, in that immersion of the peptide near the phospholipid glycerol backbone would necessitate surface expansion and a concomitant thinning of the hydrophobic phase. Whether this leads to membrane disruption by a detergentlike mechanism, ion-channel formation, or translocation across the membrane and interaction with cytoplasmic targets for this particular group of minimized antimicrobial peptides, remains to be determined.
Acknowledgments
We thank Dr. Candice Klug for a reading and critique of the manuscript.
This work was supported by an Institutional Research Grant from the Medical College of Wisconsin.
References
- Altenbach, C., S. L. Flitsch, H. G. Khorana, and W. L. Hubbell. 1989. Structural studies on transmembrane proteins. 2. Spin-labeling of bacteriorhodopsin mutants at unique cysteines. Biochemistry. 28:7806–7812. [DOI] [PubMed] [Google Scholar]
- Altenbach, C., D. A. Greenhalgh, H. G. Khorana, and W. L. Hubbell. 1994. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 91:1667–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu, D., R. B. Merrifield, H. Steiner, and H. G. Boman. 1985. N-terminal analogs of cecropin A: synthesis, antibacterial activity, and conformational properties. Biochemistry. 24:1683–1688. [DOI] [PubMed] [Google Scholar]
- Andreu, D., J. Ubach, A. Boman, B. Wahlin, D. Wade, R. B. Merrifield, and H. G. Boman. 1992. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. 296:190–194. [DOI] [PubMed] [Google Scholar]
- Archer, S. J., J. F. Ellena, and D. S. Cafiso. 1991. Dynamics and aggregation of the peptide ion channel alamethicin. Measurements using spin-labeled peptides. Biophys. J. 60:389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boman, H. G. 1995. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 13:61–92. [DOI] [PubMed] [Google Scholar]
- Boman, H. G., D. Wade, I. A. Boman, B. Wahlin, and R. B. Merrifield. 1989. Antibacterial and antimalarial properties of peptides that are cecropin-melittin hybrids. FEBS Lett. 259:103–106. [DOI] [PubMed] [Google Scholar]
- Chen, F.-Y., M.-T. Lee, and H. W. Huang. 2002. Sigmoidal concentration dependence of antimicrobial peptide activities: a case study on alamethicin. Biophys. J. 82:908–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. M., A. H. A. Clayton, W. Wang, and W. H. Sawyer. 2001. Kinetics of membrane lysis by custom lytic peptides and peptide orientations in membrane. Eur. J. Biochem. 268:1659–1669. [DOI] [PubMed] [Google Scholar]
- Christensen, B., J. Fink, R. B. Merrifield, and D. Mauzerall. 1988. Channel-forming properties of cecropins and related model compounds incorporated into planar lipid membranes. Proc. Natl. Acad. Sci. USA. 85:5072–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton, L. A., J. O. McIntyre, and R. F. Flewelling. 1987. Distance estimate of the active center of D-β-hydroxybutyrate dehydrogenase from the membrane surface. Biochemistry. 26:2117–2130. [DOI] [PubMed] [Google Scholar]
- Dathe, M., and T. Wieprecht. 1999. Structural features of helical antimicrobial peptides. Biochim. Biophys. Acta. 1462:71–87. [DOI] [PubMed] [Google Scholar]
- Farahbakhsh, Z. T., C. Altenbach, and W. L. Hubbell. 1992. Spin-labeled cysteines as sensors for protein-lipid interaction and conformation in rhodopsin. Photochem. Photobiol. 56:1019–1033. [DOI] [PubMed] [Google Scholar]
- Feix, J. B., and C. S. Klug. 1998. Site-directed spin-labeling of membrane proteins and peptide-membrane interactions. In Biological Magnetic Resonance, Vol. 14: Spin-Labeling: The Next Millennium. L. J. Berliner, editor. Plenum Press, New York. 252–281.
- Fink, J., A. Boman, H. G. Boman, and R. B. Merrifield. 1989a. Design, synthesis and antibacterial activity of cecropin-like model peptides. Int. J. Pep. Prot. Res. 33:412–421. [DOI] [PubMed] [Google Scholar]
- Fink, J., R. B. Merrifield, A. Boman, and H. G. Boman. 1989b. The chemical synthesis of cecropin-D and an analog with enhanced antibacterial activity. J. Biol. Chem. 264:6260–6267. [PubMed] [Google Scholar]
- Gazit, E., A. Boman, H. G. Boman, and Y. Shai. 1995. Interaction of the mammalian antibacterial peptide cecropin P1 with phospholipid vesicles. Biochemistry. 34:11479–11488. [DOI] [PubMed] [Google Scholar]
- Gennis, R. B. 1989. Biomembranes: Molecular Structure and Function. Springer-Verlag, New York.
- Giangaspero, A., L. Sandri, and A. Tossi. 2001. Amphipathic α-helical antimicrobial peptides. Eur. J. Biochem. 268:5589–5600. [DOI] [PubMed] [Google Scholar]
- Han, X., and L. K. Tamm. 2000. pH-dependent self-association of influenza hemagglutinin fusion peptides in lipid bilayers. J. Mol. Biol. 304:953–965. [DOI] [PubMed] [Google Scholar]
- Hancock, R. E. 2001. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1:156–164. [DOI] [PubMed] [Google Scholar]
- Hancock, R. E., and D. S. Chapple. 1999. Peptide antibiotics. Antimicrob. Agents Chemother. 43:1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, R. E., and M. G. Scott. 2000. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA. 97:8856–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller, W. T., A. J. Waring, R. I. Lehrer, and H. W. Huang. 1998. Multiple states of β-sheet peptide protegrin in lipid bilayers. Biochemistry. 37:17331–17338. [DOI] [PubMed] [Google Scholar]
- Holak, T. A., A. Engstrom, P. J. Kraulis, G. Lindeberg, H. Bennich, T. A. Jones, A. M. Gronenborn, and G. M. Clore. 1988. The solution conformation of the antibacterial peptide cecropin A: a nuclear magnetic resonance and dynamical simulated annealing study. Biochemistry. 27:7620–7629. [DOI] [PubMed] [Google Scholar]
- Huang, H. W. 2000. Action of antimicrobial peptides: two-state model. Biochemistry. 39:8347–8352. [DOI] [PubMed] [Google Scholar]
- Hubbell, W. L., A. Gross, R. Langen, and M. A. Lietzow. 1998. Recent advances in site-directed spin-labeling of proteins. Curr. Opin. Struct. Biol. 8:649–656. [DOI] [PubMed] [Google Scholar]
- Hultmark, D., H. Steiner, T. Rasmuson, and H. G. Boman. 1980. Insect immunity. Purification and properties of three inducible bactericidal proteins from hemolymph of immunized pupae of Hyalophora cecropia. Eur. J. Biochem. 106:7–16. [DOI] [PubMed] [Google Scholar]
- Juvvadi, P., S. Vunnam, E. L. Merrifield, H. G. Boman, and R. B. Merrifield. 1996. Hydrophobic effects on antibacterial and channel-forming properties of cecropin A-melittin hybrids. J. Pep. Sci. 2:223–232. [DOI] [PubMed] [Google Scholar]
- Kagan, B. L., M. E. Selsted, T. Ganz, and R. I. Lehrer. 1990. Antimicrobial defensin peptides form voltage-dependent ion-permeable channels in planar lipid bilayer membranes. Proc. Natl. Acad. Sci. USA. 87:210–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug, C. S., W. Su, and J. B. Feix. 1997. Mapping of the residues involved in a proposed β-strand located in the ferric enterobactin receptor FepA using site-directed spin-labeling. Biochemistry. 36:13027–13033. [DOI] [PubMed] [Google Scholar]
- Lehrer, R. I., T. Ganz, and M. E. Selsted. 1991. Defensins: endogenous antibiotic peptides of animal cells. Cell. 64:229–230. [DOI] [PubMed] [Google Scholar]
- Ludtke, S. J., K. He, W. T. Heller, T. A. Harroun, L. Yang, and H. W. Huang. 1996. Membrane pores induced by magainin. Biochemistry. 35:13723–13728. [DOI] [PubMed] [Google Scholar]
- Maloy, W. L., and U. P. Kari. 1995. Structure-activity studies on magainins and other host defense peptides. Biopolymers. 37:105–122. [DOI] [PubMed] [Google Scholar]
- Marassi, F. M., S. J. Opella, P. Juvvadi, and R. B. Merrifield. 1999. Orientation of cecropin-A helices in phospholipid bilayers determined by solid-state NMR spectroscopy. Biophys. J. 77:3152–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mchaourab, H. S., J. S. Hyde, and J. B. Feix. 1994. Binding and state of aggregation of spin-labeled cecropin AD in phospholipid bilayers: effects of surface charge and fatty acyl chain length. Biochemistry. 33:6691–6699. [DOI] [PubMed] [Google Scholar]
- Merrifield, R. B., P. Juvvadi, D. Andreu, J. Ubach, A. Boman, and H. G. Boman. 1995. Retro and retroenantio analogs of cecropin-melittin hybrids. Proc. Natl. Acad. Sci. USA. 92:3449–3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos, L., Jr., M. E. Rogers, P. J. Consolvo, B. A. Condie, S. Lovas, P. Bulet, and M. Blaszczyk-Thurin. 2000. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 39:14150–14159. [DOI] [PubMed] [Google Scholar]
- Rabenstein, M. D., and Y. K. Shin. 1995. Determination of the distance between two spin-labels attached to a macromolecule. Proc. Natl. Acad. Sci. USA. 92:8239–8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, C. J., T. E. Thorgeirsson, and Y. K. Shin. 1996. Temperature dependence of polypeptide partitioning between water and phospholipid bilayers. Biochemistry. 35:9526–9532. [DOI] [PubMed] [Google Scholar]
- Sawyer, J. G., N. L. Martin, and R. E. W. Hancock. 2003. Interaction of macrophage cationic proteins with the outer membrane of Pseudomonas aeruginosa. Infect. Immun. 56:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsted, M. E., S. S. Harwig, T. Ganz, J. W. Schilling, and R. I. Lehrer. 1985. Primary structures of three human neutrophil defensins. J. Clin. Invest. 76:1436–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsted, M. E., M. J. Novotny, W. L. Morris, Y. Q. Tang, W. Smith, and J. S. Cullor. 1992. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 267:4292–4295. [PubMed] [Google Scholar]
- Shai, Y. 2002. Mode of action of membrane-active antimicrobial peptides. Biopolymers. 66:236–248. [DOI] [PubMed] [Google Scholar]
- Silvestro, L., and P. H. Axelsen. 2000. Membrane-induced folding of cecropin A. Biophys. J. 79:1465–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvestro, L., K. Gupta, J. N. Weiser, and P. H. Axelsen. 1997. The concentration-dependent membrane activity of cecropin A. Biochemistry. 36:11452–11460. [DOI] [PubMed] [Google Scholar]
- Silvestro, L., J. N. Weiser, and P. H. Axelsen. 2000. Antibacterial and antimembrane activities of cecropin A in Escherichia coli. Antimicrob. Agents Chemother. 44:602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spuhler, P., G. M. Anantharamaiah, J. P. Segrest, and J. Seelig. 1994. Binding of apolipoprotein A-I model peptides to lipid bilayers. J. Biol. Chem. 269:23904–23910. [PubMed] [Google Scholar]
- Steiner, H., D. Andreu, and R. B. Merrifield. 1988. Binding and action of cecropin and cecropin analogues: antibacterial peptides from insects. Biochim. Biophys. Acta. 939:260–266. [DOI] [PubMed] [Google Scholar]
- Steiner, H., D. Hultmark, A. Engstrom, H. Bennich, and H. G. Boman. 1981. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature. 292:246–248.7019715 [Google Scholar]
- Stewart, J. C. M. 1980. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 104:10–14. [DOI] [PubMed] [Google Scholar]
- Thorgeirsson, T. E., C. J. Russell, D. S. King, and Y. K. Shin. 1996. Direct determination of the membrane affinities of individual amino acids. Biochemistry. 35:1803–1809. [DOI] [PubMed] [Google Scholar]
- Victor, K. G., and D. S. Cafiso. 2001. Location and dynamics of basic peptides at the membrane interface: electron paramagnetic resonance spectroscopy of tetramethyl-piperidine-n-oxyl-4-amino-4-carboxylic acid-labeled peptides. Biophys. J. 81:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, D., D. Andreu, S. A. Mitchell, A. M. Silveira, A. Boman, H. G. Boman, and R. B. Merrifield. 1992. Antibacterial peptides designed as analogs or hybrids of cecropins and melittin. Int. J. Pep. Prot. Res. 40:429–436. [DOI] [PubMed] [Google Scholar]
- Wade, D., A. Boman, B. Wahlin, C. M. Drain, D. Andreu, H. G. Boman, and R. B. Merrifield. 1990. All-D amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. USA. 87:4761–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerhoff, H. V., D. Juretic, R. W. Hendler, and M. Zasloff. 1989. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA. 86:6597–6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, L., T. M. Weiss, R. I. Lehrer, and H. W. Huang. 2000. Crystallization of antimicrobial pores in membranes: magainin and protegrin. Biophys. J. 79:2002–2009 [erratum appears in Biophys. J. 80:1029]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, Y. G., T. E. Thorgeirsson, and Y. K. Shin. 1994. Topology of an amphiphilic mitochondrial signal sequence in the membrane-inserted state: a spin-labeling study. Biochemistry. 33:14221–14226. [DOI] [PubMed] [Google Scholar]
- Zasloff, M. 1987. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA. 84:5449–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff, M. 2002. Antimicrobial peptides of multicellular organisms. Nature. 415:389–395. [DOI] [PubMed] [Google Scholar]
- Zhang, L., R. Benz, and R. E. Hancock. 1999. Influence of proline residues on the antibacterial and synergistic activities of α-helical peptides. Biochemistry. 38:8102–8111. [DOI] [PubMed] [Google Scholar]