

Abstract
Solid-state NMR measurements have been reported for four peptides derived from β-amyloid peptide Aβ(1–42): Aβ(1–40), Aβ(10–35), Aβ(16–22), and Aβ(34–42). Of these, the first two are predicted to be amphiphilic and were reported to form parallel β-sheets, whereas the latter two peptides appear nonamphiphilic and adopt an antiparallel β-sheet organization. These results suggest that amphiphilicity may be significant in determining fibril structure. Here, we demonstrate that acylation of Aβ(16–22) with octanoic acid increases its amphiphilicity and changes the organization of fibrillar β-sheet from antiparallel to parallel. Electron microscopy, Congo Red binding, and one-dimensional 13C NMR measurements demonstrate that octanoyl-Aβ(16–22) forms typical amyloid fibrils. Based on the stability of monolayers at the air-water interface, octanoyl-Aβ(16–22) is more amphiphilic than Aβ(16–22). Measurements of 13C-13C and 15N-13C nuclear magnetic dipole-dipole couplings in isotopically labeled fibril samples, using the constant-time finite-pulse radiofrequency-driven recoupling (fpRFDR-CT) and rotational echo double resonance (REDOR) solid-state NMR techniques, demonstrate that octanoyl-Aβ(16–22) fibrils are composed of parallel β-sheets, whereas Aβ(16–22) fibrils are composed of antiparallel β-sheets. These data demonstrate that amphiphilicity is critical in determining the structural organization of β-sheets in the amyloid fibril. This work also shows that all amyloid fibrils do not share a common supramolecular structure, and suggests a method for controlling the structure of amyloid fibrils.
INTRODUCTION
Amyloid fibrils formed from different peptides and proteins share a remarkable number of common features, including protease resistance, similar fiber diffraction patterns, and a high β-sheet content (Maggio and Mantyh, 1996; Teplow, 1998). However, recent solid-state NMR studies have shown that different amyloidogenic peptides derived from the 42-amino acid Alzheimer's β-amyloid peptide (Aβ) can assemble to form fibrils composed of either parallel or antiparallel β-sheets. The Aβ(1–40) (Antzutkin et al., 2000; Balbach et al., 2002) and Aβ(10–35) (Benzinger et al., 1998, 2000; Gregory et al., 1998) peptides have been found to form fibrils with parallel β-sheets, whereas Aβ(16–22) (Balbach et al., 2000) and Aβ(34–42) (Lansbury et al., 1995) have been found to form fibrils with antiparallel β-sheets. A comparison of the primary structures of these amyloidogenic peptides suggests that amphiphilicity may be critical in determining fibril organization. Both Aβ(1–40) and Aβ(10–35) have clusters of hydrophobic residues at their C-termini and are predicted to be amphiphilic. In the case of Aβ(1–40), the amphiphilicity has been demonstrated experimentally (Soreghan et al., 1994). Assembly of Aβ(1–40) or Aβ(10–35) into parallel β-sheets would juxtapose the C-terminal hydrophobic residues and shield them from the aqueous solution. Conversely, an antiparallel β-sheet organization in Aβ(1–40) and Aβ(10–35) fibrils would result in the unfavorable juxtaposition of hydrophobic and nonhydrophobic regions. In contrast, neither of the peptides that form amyloid fibrils with antiparallel β-sheets, Aβ(16–22) and Aβ(34–42), are predicted to be amphiphilic. The latter peptides have neither a terminal cluster of hydrophobic residues nor an alternation of hydrophobic and hydrophilic residues. These peptides, instead, have a centrally located hydrophobic segment. Thus, maximal clustering of hydrophobic residues could be optimized in either a parallel or antiparallel orientation. In the absence of an amphiphilic structure, the antiparallel orientation may then be more stable because of the hydrogen bond geometry or other factors. In Aβ(16–22) and Aβ(34–42), the antiparallel β-sheet organization could be favored by electrostatic interactions between oppositely charged side chains or termini (Balbach et al., 2000). These data raise the intriguing possibility that the structure of amyloid fibrils is influenced by the degree of amphiphilicity of the peptides.
In this article, we investigate the role of amphiphilicity in determining the organization of β-sheets in amyloid fibrils. We report experiments in which the Aβ(16–22) peptide has been modified to increase its amphiphilicity by acylating the N-terminus with octanoic acid (Takahashi et al., 1999). The modified peptide, octanoyl-Aβ(16–22), forms amyloid fibrils that are indistinguishable from fibrils formed by the unmodified peptide in electron microscopy, Congo Red binding, and one-dimensional (1D) 13C NMR spectra. We demonstrate that octanoyl-Aβ(16–22) is indeed more amphiphilic than Aβ(16–22) from the properties of peptide monolayers formed at the air-water interface. Most significantly, solid-state NMR measurements of nuclear magnetic dipole-dipole couplings in specifically 13C- and 15N-labeled Aβ(16–22) and octanoyl-Aβ(16–22) fibrils clearly demonstrate that octanoyl-Aβ(16–22) fibrils contain parallel β-sheets, whereas Aβ(16–22) fibrils contain antiparallel β-sheets. This work presents direct evidence for the control of β-sheet organization in amyloid fibrils by amphiphilicity.
METHODS
Peptide synthesis
Peptides were synthesized using standard FMOC chemistry on an Applied Biosystems (Foster City, CA) model 431A instrument. The 13C and 15N amino acids were purchased from Cambridge Isotopes (Andover, MA). Isotopic purity was greater than 97% for all of the amino acids. Peptides were purified by RP-HPLC on a preparative column at 70°C. Peptide purity was greater than 98% by analytical HPLC. The molecular masses of the peptides were verified with electrospray mass spectrometry.
Fibrillization
Peptides were initially dissolved in 1,1,1,3,3,3-hexafluro-2-propanol (HFIP) at 50 mg ml−1. Aliquots of the HFIP stock solutions were then diluted into 9 M urea to final peptide concentrations of ∼200 μM. The urea solutions containing the different peptides were then mixed and dialyzed (Spectra/Por CE Membranes, molecular weight cutoff = 500 Da) extensively against 5 mM phosphate buffer, pH 7.4. After dialysis, the samples were swirled on rotary shaker for 14 days and then flash-frozen, lyophilized, and stored at −20°C. Ratios of 15N-labeled to 13C-labeled peptides in the fibrils (Table 1) were determined from analytical HPLC measurements on the stock solutions.
TABLE 1.
Summary of solid state NMR results
| Peptide mixture in the fibrils | Ratio of 15N- to 13C-labeled peptides | Carbonyl 13C chemical shift (ppm relative to TMS) | Carbonyl 13C MAS NMR linewidth (ppm) | REDOR(15N-13C distances < 6 Å) | fpRFDR-CT(13C-13C distances ≈ 4.8 Å) | β-sheet structure |
|---|---|---|---|---|---|---|
| Aβ(16–22)-15N-Ala21 +Aβ(16–22)-13C-Leu17 | 1.60:1.00 | 171.2 | 2.5 | + | − | Antiparallel |
| Aβ(16–22)-15N-Ala21 +Aβ(16–22)-13C-Phe20 | 1.48:1.00 | 171.6 | 2.1 | − | − | Antiparallel |
| Octanoyl-Aβ(16–22)-15N-Ala21 + octanoyl-Aβ(16–22)-13C-Leu17 | 1.64:1.00 | 171.5 | 1.5 | − | + | Parallel |
| Octanoyl-Aβ(16–22)-15N-Ala21 + octanoyl-Aβ(16–22)-13C-Phe20 | 1.71:1.00 | 171.9 | 2.5 | + | + | Parallel |
Electron microscopy
Aliquots of 200-μM solutions of octanoyl-Aβ(16–22) and Aβ(16–22) that were fibrillized for 14 days were applied to a glow-discharged, 400-mesh, carbon-coated support film and stained with 1% uranyl acetate. Micrographs were recorded using a Philips EM300 (Andover, MA) at magnifications of 17,000×, 45,000×, and 100,000×.
Surface isotherms of peptide monolayers at the air-water interface
Surface isotherms were measured using a Nima surface balance (Coventry, UK) equipped with a Pt-Wilhelmy plate. Peptide dissolved in HFIP was added dropwise through a microsyringe onto the surface of the subphase. After evaporation of the organic solvent the film was compressed at 20 cm2/min. The composition of the subphase was 10 mM phosphate buffer, pH 7.4, 150 mM NaCl, 5% glycerol. For surface pressures of <1 mN/m, the isotherms were analyzed using the equation of a two-dimensional gas:
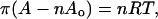 |
in which π is the surface pressure (millinewtons per meter), A is the area (square centimeters), n is the number of moles of peptide at the interface, Ao is the molar exclusion area, R is the gas constant, and T is the absolute temperature. At π > 1 mN/m, the data were fit to equation:
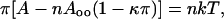 |
where Aoo is the molar exclusion area of the peptide extrapolated to π = 0, κ is a compressibility factor (meters per millinewton), and k is a constant with the same units as the gas constant (Shen et al., 1973; Fukushima et al., 1979; Minakata et al., 1989).
Solid-state NMR
All measurements were performed at a 13C NMR frequency of 100.4 MHz, using a Varian/Chemagnetics Infinity-400 NMR spectrometer and a Varian/Chemagnetics magic-angle spinning (MAS) probe with 3.2-mm diameter rotors. Except as noted below, all data were taken at room temperature on lyophilized fibril samples of ∼8 mg. One-dimensional 13C NMR spectra were recorded at MAS frequencies νR = 10.00 kHz or 5.00 kHz, with standard cross-polarization and proton decoupling methods. Decoupling fields were 100 kHz. Chemical shifts are relative to tetramethylsilane, using the methylene carbon signal of adamantane at 38.56 ppm as an external reference. Rotational echo double resonance (REDOR) data were acquired with the pulse sequence of Anderson et al. (1995) with 180° pulse lengths of 10.0 μs on both 13C and 15N and with νR = 5.00 kHz. Proton decoupling fields during τNC were 100–120 kHz. The constant-time finite-pulse radiofrequency-driven recoupling (fpRFDR-CT) data were acquired with the pulse sequence of Ishii et al. (2001) with 180° pulse lengths of 15.0 μs and 90° pulse lengths of 5.0 μs on 13C and with νR = 20.00 kHz. Proton decoupling fields during τCC were 120 kHz. In both REDOR and fpRFDR-CT measurements, 180° pulses on 13C were actively synchronized with a MAS tachometer signal. The sensitivity of both measurements was enhanced by pulsed spin lock (PSL) detection (Petkova and Tycko, 2002).
Frictional heating at the high MAS speed required for fpRFDR-CT measurements leads to sample temperatures of ∼40°C at ambient conditions. To rule out possible effects of thermally activated molecular motions or structural changes on the fpRFDR-CT measurements, data for one octanoyl-Aβ(16–22) sample were also recorded at a sample temperature of −70°C. The low-temperature data were not significantly different from the data reported in Fig. 4 b, below.
FIGURE 4.

(a) 13C-detected 15N-13C REDOR measurements on Aβ(16–22) and octanoyl-Aβ(16–22) fibrils. Data are shown for Aβ(16–22)-15N-Ala21:Aβ(16–22)-13C-Leu17 (▵), Aβ(16–22)-15N-Ala21:Aβ(16–22)-13C-Phe20 (○), octanoyl-Aβ(16–22)-15N-Ala21:octanoyl-Aβ(16–22)-13C-Leu17 (□), and octanoyl-Aβ(16–22)-15N-Ala21:octanoyl-Aβ(16–22)-13C-Phe20 (⋄) fibrils. Error bars represent uncertainty from the root mean-square (rms) noise in the NMR spectra. Simulated REDOR curves assume an in-register antiparallel β-sheet structure in Aβ(16–22) (dashed line) and an in-register parallel β-sheet structure in octanoyl-Aβ(16–22) (solid line). (b) 13C-13C fpRFDR-CT measurements on Aβ(16–22) and octanoyl-Aβ(16–22) fibrils (same symbols). Simulated curves assume an in-register antiparallel β-sheet structure in Aβ(16–22) (dashed line) and an in-register parallel β-sheet structure in octanoyl-Aβ(16–22) (solid line).
Numerical simulations of REDOR data included only 15N-13C dipole-dipole couplings. Because of the random cofibrillization of 13C-labeled and 15N-labeled peptides in the experiments, a given carbonyl 13C label may have zero, one, or two 15N neighbors within 6 Å in the β-sheet structure, with probabilities (1 + ρ)−2, ρ (1 + ρ)−2, and ρ2(1 + ρ)−2 where ρ is the ratio of 15N-labeled to 13C-labeled molecules in Table 1. The REDOR simulations in Fig. 4 a were calculated as the sum of probability-weighted contributions from carbonyl 13C nuclei coupled to no 15N nuclei, to a single 15N nucleus at 4.2 Å (d1 in Fig. 3, below), to a single 15N nucleus at 5.7 Å (d2 in Fig. 3), and to 15N nuclei at both 4.8 Å and 5.7 Å, with an angle ξ between the two 15N-13C internuclear vectors. Based on examination of model β-sheets, ξ was taken to be 15° and 30° for antiparallel and parallel β-sheets, respectively. To fit the experimental REDOR data on Aβ(16–22)-15N-Ala:Aβ(16–22)-13C-Leu and octanoyl-Aβ(16–22)-15N-Ala:octanoyl-Aβ(16–22)-13C-Phe fibril samples in Fig. 4 a (below), overall scaling factors of 0.60 and 0.65 were applied to the corresponding REDOR simulations. These scaling factors suggest that 40% and 35% of the total 13C NMR signals are not attributable to carbonyl 13C labels in the β-sheets in the two samples, respectively. Given that ∼18% of the total 13C NMR signal in these experiments is expected to arise from natural-abundance 13C at carbonyl and carboxyl sites, and that an additional 20% may arise from unfibrillized peptides and from other 13C background signals that are not resolved under pulsed spin lock detection, these scaling factors are reasonable.
FIGURE 3.


Labeling scheme for REDOR and fpRFDR-CT solid-state NMR measurements designed to distinguish in-register parallel (a) from in-register antiparallel (b) β-sheet structures. Carbonyl 13C labels at Leu17 and Phe20 and amide 15N labels at Ala21 are indicated by brown square, cyan hexagon, and green circle outlines, respectively. The figure shows only the carbonyl and the β-carbon atom of the N-terminal acyl group, which is an acetyl group in Aβ(16–22) and an octanoyl group in octanoyl-Aβ(16–22). The figure shows the hydrogen atom on the α-carbon atom and only the β-carbon of the side chains.
Numerical simulations of fpRFDR-CT data were performed at the average Hamiltonian level (Ishii et al., 2001), considering only 13C-13C dipole-dipole couplings. To approximate the random cofibrillization of 13C-labeled and 15N-labeled peptides and the experimental labeling ratios, simulations were carried out for systems of six 13C spins randomly distributed on 16 possible sites, with averaging over 200 random configurations. For in-register parallel β-sheet simulations, the 16 sites were positioned in a linear chain with 4.8-Å spacings. For in-register antiparallel β-sheet simulations, the 16 sites were positioned in a plane with coordinates (Å units) given by (x, y) = (0 + 19.2k, 0), (9.6 + 19.2k, 0), (3.7 + 19.2k, 9.5), and (13.3 + 19.2k, 9.5), with k = 0, 1, 2, 3. These coordinates were derived by examination of model β-sheet structures. To account for the estimated 35% contributions of natural-abundance 13C and other background signals to the experimental fpRFDR-CT data (estimated from the REDOR data as described above), simulated curves in Fig. 4 b are 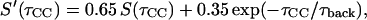 where S(τCC) is the curve from the numerical six-spin simulation and τback is an empirical decay time for background signals. In Fig. 4 b, τback = 200 ms.
where S(τCC) is the curve from the numerical six-spin simulation and τback is an empirical decay time for background signals. In Fig. 4 b, τback = 200 ms.
RESULTS
Electron microscopy
Fig. 1 a shows electron micrographs of negatively stained Aβ(16–22) and octanoyl-Aβ(16–22) amyloid fibrils formed over a 14-day incubation at pH 7.4. The fibrils formed by the two peptides are indistinguishable by electron microscopy. The lengths of the fibrils vary from ∼800 Å to greater than 8000 Å. The diameters of the fibrils range from 80 Å to 250 Å. Additional properties characteristic of amyloid fibrils include an unbranched morphology and a periodic twist (Kirschner et al., 1987). Both the Aβ(16–22) and octanoyl-Aβ(16–22) fibrils bind Congo Red, and the stained fibrils exhibit the characteristic green birefringence (Klunk et al., 1989) of amyloid under polarized light (data not shown).
FIGURE 1.

(a) Electron micrographs of negatively stained fibrils formed by the Aβ(16–22) and octanoyl-Aβ(16–22) peptides over a 14-day incubation. Magnification, 45,000×. Fibrils formed by both peptides exhibit the long, unbranched, and twisting morphology that is typical of amyloid fibrils. This demonstrates that the addition of the octanoyl group to Aβ(16–22) does not significantly alter the structure of the amyloid fibril on the macromolecular level. (b) Carbonyl regions of 1D 13C MAS NMR spectra of Aβ(16–22) and octanoyl-Aβ(16–22) fibrils with the indicated isotopic labels. Dashed lines indicate random coil chemical shift values for the carbonyl 13C labels. Experimental chemical shifts and linewidths in all spectra are consistent with well-structured β-strand conformations.
One-dimensional 13C NMR
The secondary structure and structural order in amyloid fibrils formed by the two peptides were characterized by 1D 13C NMR spectroscopy with magic-angle spinning. Fig. 1 b shows 13C MAS NMR spectra of Aβ(16–22) and octanoyl-Aβ(16–22) fibrils with 13C labels at carbonyl sites of Phe20 and Leu17 (and 15N labels at Ala21, see below). Chemical shifts and linewidths are summarized in Table 1. Carbonyl 13C chemical shifts are sensitive to secondary structure (Saito et al., 1983; Wishart et al., 1991; de Dios and Oldfield, 1994). Experimental chemical shifts in all samples in Fig. 1 b are reduced relative to random coil values (Wishart et al., 1995), consistent with β-strand conformations at Phe20 and Leu17 in both Aβ(16–22) and octanoyl-Aβ(16–22). Carbonyl 13C MAS NMR linewidths are sensitive to structural disorder, with 6–8-ppm linewidths observed in fully unstructured peptides (Long and Tycko, 1998; Weliky et al., 1999). The experimental linewidths in Fig. 1 b (full width at half maximum, FWHM) are in the 1.5–2.5-ppm range. Similar linewidths have been observed for amyloid fibrils formed by other Aβ peptides (Long and Tycko, 1998; Weliky et al., 1999) as well as for other well-structured peptides in rigid, noncrystalline environments (Balbach et al., 2000, 2002).
Peptide monolayers at the air-water interface
To assess the amphiphilicity of Aβ(16–22) and octanoyl-Aβ(16–22), we measured surface isotherms of peptide monolayers at the air-water interface (Fig. 2 and Table 2). Aβ(16–22) and octanoyl-Aβ(16–22) exhibit collapse pressures of 6 mN/M and 20 mN/M, respectively. This demonstrates that the octanoyl chain significantly increases the amphiphilicity of the octanoyl-Aβ(16–22) peptide relative to Aβ(16–22). In fact, the collapse pressure of octanoyl-Aβ(16–22) is comparable to the collapse pressure of Aβ(1–40) (26 mN/M). The excluded area for all of these peptides is ∼30–35 Å2 per amino acid. Random coil peptides typically have excluded areas of 50 Å2 per amino acid, which indicates that these peptides are ordered at the interface (Shen and Scanu, 1980).
FIGURE 2.

Surface isotherms of Aβ(16–22) (•), octanoyl-Aβ(16–22) (▪), and Aβ(1–40) (▴) peptide monolayers spread on a subphase of 25 mM phosphate buffer, 150 mM NaCl, 5% glycerol, pH 7.4. Data were fit to the equations described in Methods. Octanoyl-Aβ(16–22) and Aβ(1–40) form more stable monolayers than Aβ(16–22) based on the collapse pressures of the peptide monolayers. This demonstrates that the octanoyl group increases the amphiphilicity of octanoyl-Aβ(16–22) relative to Aβ(16–22).
TABLE 2.
Summary of monolayer results
| Peptide | Collapse pressure (mN/M) | Compressibility (M/mN) | Apparent molecular weight (actual MW) | Exclusion area (Å2/aa) |
|---|---|---|---|---|
| OA-Aβ(16–22) | 21 | 0.019 | 705 (895.1) | 30.5 |
| Ac-Aβ(16–22) | 6 | 0.028 | 910 (979.1) | 35.3 |
| Aβ(1–40) | 26 | 0.029 | 3851 (4330) | 32.5 |
REDOR
Rotational echo double resonance experiments were designed to determine the structural organization of β-sheets in Aβ(16–22) and octanoyl-Aβ(16–22) fibrils. REDOR is a solid state MAS NMR method that measures heteronuclear dipole-dipole couplings (15N-13C couplings in the present experiments) (Anderson et al., 1995; Gullion and Schaefer, 1989; Pan et al., 1990). The dipole-dipole coupling strengths are proportional to 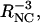 where RNC is the distance between 15N and 13C labels. In our 13C-detected REDOR measurements, the 13C signal is measured after a dephasing period τNC during which a train of rotation-synchronized 180° pulses is applied at either the 13C frequency alone (S0) or both the 13C and 15N frequencies (S1). The dependence of the normalized difference signal ΔS/S0 on τNC, where ΔS = S0 − S1, is a direct measure of the 13C-15N dipole-dipole couplings. 15N-13C distances up to roughly 6 Å can be measured with REDOR.
where RNC is the distance between 15N and 13C labels. In our 13C-detected REDOR measurements, the 13C signal is measured after a dephasing period τNC during which a train of rotation-synchronized 180° pulses is applied at either the 13C frequency alone (S0) or both the 13C and 15N frequencies (S1). The dependence of the normalized difference signal ΔS/S0 on τNC, where ΔS = S0 − S1, is a direct measure of the 13C-15N dipole-dipole couplings. 15N-13C distances up to roughly 6 Å can be measured with REDOR.
To probe the β-sheet organization, fibrils were prepared from a mixture of 13C-labeled and 15N-labeled peptides according to the labeling strategy depicted in Fig. 3. 15N labels were placed at the amide nitrogen of Ala21. 13C labels were placed at the carbonyl carbon of either Leu17 or Phe20. In an in-register parallel β-sheet (intermolecular hydrogen bonds between residue i of one chain and residues i − 1 and i + 1 of a neighboring chain), the shortest intermolecular distance between 15N-Ala2l and 13C-Phe20 is ∼4.2 Å (the hydrogen-bonded distance, d1 in Fig. 4), whereas the distance between 15N-Ala2l and 13C-Leu17 is greater than 8 Å. Conversely, in an in-register antiparallel β-sheet (intermolecular hydrogen bonds between residue 19 + i of one chain and residue 19 − i of a neighboring chain), the shortest intermolecular distance between 15N-Ala2l and 13C-Leu17 is ∼4.2 Å, whereas the distance between 15N-Ala2l and 13C-Phe20 is greater than 8 Å. Four different peptide mixtures were prepared and allowed to fibrillize over 14 days (see Table 1 and Methods).
Fig. 4 a shows REDOR data for the four fibril samples. Experimental ΔS/S0 values for the Aβ(16–22)-15N-Ala:Aβ(16–22)-13C-Leu and octanoyl-Aβ(16–22)-15N-Ala:octanoyl-Aβ(16–22)-13C-Phe samples build up to ∼0.4 at τNC ≈ 35 ms. These data are in good agreement with simulated REDOR curves based on an in-register antiparallel β-sheet structure for Aβ(16–22) and an in-register parallel β-sheet structure for octanoyl-Aβ(16–22) (see Methods for simulation details). Experimental ΔS/S0 values are significantly smaller for the other two fibril samples (Aβ(16–22)-15N-Ala:Aβ(16–22)-13C-Phe and octanoyl-Aβ(16–22)-15N-Ala:octanoyl-Aβ(16–22)-13C-Leu), indicating the absence of 15N-13C distances less than 6 Å, again consistent with an in-register antiparallel β-sheet structure for Aβ(16–22) and an in-register parallel β-sheet structure for octanoyl-Aβ(16–22).
fpRFDR-CT
The β-sheet organization indicated by the REDOR data was confirmed by measurements of 13C-13C dipole-dipole couplings in the same samples, using the constant-time finite-pulse radiofrequency-driven recoupling solid-state NMR technique (Ishii et al., 2001). In fpRFDR-CT measurements, the 13C signal intensity is recorded as a function of an effective dephasing period τCC during which the dipole-dipole couplings are switched on by the recoupling pulse sequence. The fpRFDR-CT signals decrease with increasing τCC, on a timescale inversely proportional to the 13C-13C coupling strengths. These couplings are proportional to 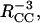 where RCC is the distance between 13C labels. As in REDOR measurements, the upper limit on distance measurements by fpRFDR-CT is roughly 6 Å. In an in-register parallel β-sheet, the 13C labels form a linear chain with spacings of ∼4.8 Å (d3 in Fig. 3). In an antiparallel arrangement, the 13C labels form a zigzag pattern with significantly longer internuclear distances.
where RCC is the distance between 13C labels. As in REDOR measurements, the upper limit on distance measurements by fpRFDR-CT is roughly 6 Å. In an in-register parallel β-sheet, the 13C labels form a linear chain with spacings of ∼4.8 Å (d3 in Fig. 3). In an antiparallel arrangement, the 13C labels form a zigzag pattern with significantly longer internuclear distances.
Fig. 4 b shows experimental fpRFDR-CT data for the four fibril samples. The fpRFDR-CT signals for both octanoyl-Aβ(16–22) samples decay significantly more rapidly than for both Aβ(16–22) samples, indicating shorter intermolecular 13C-13C distances in the octanoyl-Aβ(16–22) fibrils. Data for octanoyl-Aβ(16–22) samples are in good agreement with fpRFDR-CT simulations for an in-register parallel β-sheet structure. Data for Aβ(16–22) samples decay more rapidly than predicted by simulations for an in-register antiparallel β-sheet structure, but this discrepancy may be attributed to long-range 13C-13C couplings not included in the simulations (see Methods).
DISCUSSION
Four peptides derived from Aβ(1–42) have been investigated by solid-state NMR (Lansbury et al., 1995; Benzinger et al., 1998, 2000; Gregory et al., 1998; Antzutkin et al., 2000; Balbach et al., 2000, 2002). Balbach et al. (2002) recently demonstrated that Aβ(16–22), unlike Aβ(1–40) (Antzutkin et al., 2000) and Aβ(10–35) (Benzinger et al., 1998, 2000), forms amyloid fibrils composed of antiparallel β-sheets. An antiparallel organization was also observed for Aβ(34–42) by Lansbury et al. (1995). In this work, we have shown that modification of Aβ(16–22) with an octanoyl chain increases its amphiphilicity, and that this is sufficient to change the organization of the β-sheets in the amyloid fibrils from antiparallel to parallel.
The octanoyl-Aβ(16–22) peptide forms amyloid fibrils that are indistinguishable from Aβ(16–22) fibrils in electron microscopy (Fig. 1 a), Congo Red binding, and 1D 13C MAS NMR spectra (Fig. 1 b). However, a significant difference is observed between the collapse pressures of Aβ(16–22) and octanoyl-Aβ(16–22) peptide monolayers formed at the air-water interface (Fig. 2). Octanoyl-Aβ(16–22) is more amphiphilic than Aβ(16–22) and exhibits a collapse pressure similar to Aβ(1–40). Soreghan et al. (1994) previously described the amphiphilic properties of a number of Aβ peptides. The hydrophobic C-terminus of Aβ was noted to be essential for the amphiphilicity of the peptides.
The carbonyl 13C chemical shifts and linewidths in the 1D MAS NMR measurements indicate that octanoyl-Aβ(16–22) and Aβ(16–22) adopt well-structured β-strand conformations in the fibrils. The structural organization of the β-sheets in the fibril samples was determined using a combination of 15N-13C REDOR and 13C-13C fpRFDR-CT experiments (Fig. 4). As summarized in Table 1, all solid-state NMR data are consistent with an in-register parallel β-sheet organization in octanoyl-Aβ(16–22) fibrils and an in-register antiparallel organization in Aβ(16–22) fibrils. These data indicate that amphiphilicity is a significant factor in determining the organization of β-sheets in amyloid fibrils. Other factors, such as charge interactions, electric dipoles, and hydrogen bond geometry, may also be critical in determining fibril structure.
Parallel β-sheets in the absence of neighboring α-helices are uncommon, suggesting that such structures may be inherently less stable than antiparallel β-sheets (Yoder et al., 2003; Cohen, 1993). The questions then arise why and how the addition of an octanoyl group to Aβ(16–22) reverses the orientation of the β-sheets. Several authors have proposed that β-amyloid peptides organize into micelle-like structures, in which hydrophobic regions are shielded from the aqueous environment (Soreghan et al., 1994; Torok et al., 2002). Pre- or protofibrillar structures may have this feature in common with micelles, though some micelle-like structures may not be on the pathway toward fibril formation (Pallitto and Murphy, 2001). The formation of β-sheet fibrillar structures from protofibrillar structures may be thermodynamically controlled, in which case the parallel structure would represent the lowest free energy state when the octanoyl chain is present but not when it is absent. Alternatively, formation of β-sheet fibrillar structures may be kinetically controlled, in which case the parallel structure of octanoyl-Aβ(16–22) would arise because the octanoyl chains produces a parallel alignment in a prefibrillar aggregated state of the peptide. In such a scenario, the parallel β-sheet would represent a kinetically trapped intermediate, rather than the thermodynamically preferred state. In the parallel orientation, the aggregation of octanoyl groups of octanoyl-Aβ(16–22) may be sufficient to overcome not only intrinsically greater stability of antiparallel over parallel β-sheets but also unfavorable charge repulsions between Lys16 and Glu22 residues. In contrast, these charge interactions may favor the antiparallel orientation of acetyl-Aβ(16–22).
Our results demonstrate that there is not a strict structural requirement for β-sheet organization, parallel or antiparallel, in amyloid fibrils, contrary to the assumption that all amyloid fibrils share a specific structure (Egnaczyk et al., 2001). The ability to control fibril structure by modulation of the amphiphilicity of amyloidogenic peptides may be useful in the development of self-assembling, nanoscale materials (Aggeli et al., 1997; Zhang and Rich, 1997). The amphiphilic nature of peptide secondary structures, for example, has often been utilized in the design of tertiary and supramolecular structures (Kaiser and Kézdy, 1984).
The results from this work provide an explanation for the different β-sheet organizations described in the previous solid-state NMR work with peptides derived from Aβ(1–42). The two peptides that form parallel fibrils, Aβ(10–35) and Aβ(1–40), are both amphiphilic molecules, with hydrophobic, C-terminal segments. Similar to octanoyl-Aβ(16–22), these amphiphilic peptides may adopt a parallel organization to shield hydrophobic residues from aqueous solvents. In nonamphiphilic peptides, such as Aβ(16–22) and Aβ(34–42), other factors that favor an antiparallel organization, such as charge interactions and hydrogen bond geometry, may dominate. Based on the small sizes of Aβ(16–22) and octanoyl-Aβ(16–22), these peptides may serve as an excellent model system for investigating the role of these other factors in determining β-strand orientation in amyloid fibrils.
Acknowledgments
We thank the Alzheimer's Association (IIRG 98-1344), and the National Institutes of Health (5 T32 GM07281 to D.J.G.; 1 RO1 NS042852 to S.C.M.) for support for this work.
References
- Aggeli, A., M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. McLeish, M. Pitkeathly, and S. E. Radford. 1997. Responsive gels formed by the spontaneous self-assembly of peptides into polymeric β-sheet tapes. Nature. 386:259–262. [DOI] [PubMed] [Google Scholar]
- Anderson, R. C., T. Gullion, J. M. Joers, M. Shapiro, E. B. Villhauer, and H. P. Weber. 1995. Conformation of [1-13C,15N]acetyl-L-carnitine. Rotational-echo double resonance nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 117:10546–10550. [Google Scholar]
- Antzutkin, O. N., J. J. Balbach, R. D. Leapman, N. W. Rizzo, J. Reed, and R. Tycko. 2000. Multiple quantum solid-state NMR indicates a parallel, not antiparallel, organization of β-sheets in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. USA. 97:13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbach, J. J., Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed, and R. Tycko. 2000. Amyloid fibril formation by Aβ 16–22, a seven-residue fragment of the Alzheimer's β-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 39:13748–13759. [DOI] [PubMed] [Google Scholar]
- Balbach, J. J., A. T. Petkova, N. A. Oyler, O. N. Antzutkin, D. J. Gordon, S. C. Meredith, and R. Tycko. 2002. Supramolecular structure in full-length Alzheimer's β-amyloid fibrils: evidence for a parallel β-sheet organization from solid state NMR. Biophys. J. 83:1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger, T. L., D. M. Gregory, T. S. Burkoth, H. Miller-Auer, D. G. Lynn, R. E. Botto, and S. C. Meredith. 1998. Propagating structure of Alzheimer's β-amyloid(10–35) is parallel β-sheet with residues in exact register. Proc. Natl. Acad. Sci. USA. 95:13407–13412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger, T. L., D. M. Gregory, T. S. Burkoth, H. Miller-Auer, D. G. Lynn, R. E. Botto, and S. C. Meredith. 2000. Two-dimensional structure of β-amyloid(10–35) fibrils. Biochemistry. 39:3491–3499. [DOI] [PubMed] [Google Scholar]
- Cohen, F. E. 1993. The parallel β-helix of pectate lyase C: something to sneeze at. Science. 260:1444–1445. [DOI] [PubMed] [Google Scholar]
- de Dios, A. C., and E. Oldfield. 1994. Chemical shifts of carbonyl carbons in peptides and proteins. J. Am. Chem. Soc. 116:11485–11488. [Google Scholar]
- Egnaczyk, G. F., K. D. Greis, E. R. Stimson, and J. E. Maggio. 2001. Photoaffinity cross-linking of Alzheimer's disease amyloid fibrils reveals interstrand contact regions between assembled β-amyloid peptide subunits. Biochemistry. 40:11706–11714. [DOI] [PubMed] [Google Scholar]
- Fukushima, D., J. P. Kupferberg, S. Yokoyama, D. J. Kroon, E. T. Kaiser, and F. J. Kézdy. 1979. Synthetic amphiphilic helical docosapeptide with the surface properties of plasma apolipoprotein A-I. J. Am. Chem. Soc. 101:3703–3704. [Google Scholar]
- Gregory, D. M., T. L. Benzinger, T. S. Burkoth, H. Miller-Auer, D. G. Lynn, S. C. Meredith, and R. E. Botto. 1998. Dipolar recoupling NMR of biomolecular self-assemblies: determining inter- and intrastrand distances in fibrilized Alzheimer's β-amyloid peptide. Solid State Nucl. Magn. Reson. 13:149–166. [DOI] [PubMed] [Google Scholar]
- Gullion, T., and J. Schaefer. 1989. Rotational-echo double-resonance NMR. J. Magn. Reson. 81:196–200. [DOI] [PubMed] [Google Scholar]
- Ishii, Y., J. J. Balbach, and R. Tycko. 2001. Measurement of dipole-coupled lineshapes in a many-spin system by constant-time two-dimensional solid state NMR with high-speed magic-angle spinning. Chem. Phys. 266:231–236. [Google Scholar]
- Kaiser, E. T., and F. J. Kézdy. 1984. Amphiphilic secondary structure: design of peptide hormones. Science. 223:249–255. [DOI] [PubMed] [Google Scholar]
- Kirschner, D. A., H. Inouye, L. K. Duffy, A. Sinclair, M. Lind, and J. J. Selkoe. 1987. Synthetic peptide homologous to β-protein from Alzheimer disease forms amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA. 84:6953–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk, W. E., J. W. Pettegrew, and D. J. Abraham. 1989. Quantitative evaluation of congo red binding to amyloid-like proteins with a β-pleated sheet conformation. J. Histochem. Cytochem. 37:1273–1281. [DOI] [PubMed] [Google Scholar]
- Lansbury, P. T., P. R. Costa, J. M. Griffiths, E. J. Simon, M. Auger, K. J. Halverson, D. A. Kocisko, Z. S. Hendsch, T. T. Ashburn, R. G. Spencer, B. Tidor, and R. G. Griffin. 1995. Structural model for the β-amyloid fibril based on interstrand alignment of an antiparallel-sheet comprising a C-terminal peptide. Nat. Struct. Biol. 2:990–998. [DOI] [PubMed] [Google Scholar]
- Long, H. W., and R. Tycko. 1998. Biopolymer conformational distributions from solid-state NMR: α-helix and 310-helix contents of a helical peptide. J. Am. Chem. Soc. 120:7039–7048. [Google Scholar]
- Maggio, J. E., and P. W. Mantyh. 1996. Brain amyloid—a physicochemical perspective. Brain Pathol. 6:147–162. [DOI] [PubMed] [Google Scholar]
- Minakata, H., J. W. Taylor, M. W. Walker, R. J. Miller, and E. T. Kaiser. 1989. Characterization of amphiphilic secondary structures in neuropeptide Y through the design, synthesis, and study of model peptides. J. Biol. Chem. 264:7907–7913. [PubMed] [Google Scholar]
- Pallitto, M. M., and R. M. Murphy. 2001. A mathematical model of the kinetics of β-amyloid fibril growth from the denatured state. Biophys. J. 81:1805–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Y., T. Gullion, and J. Schaefer. 1990. Determination of C-N internuclear distances by rotational-echo double-resonance NMR of solids. J. Magn. Reson. 90:330–340. [Google Scholar]
- Petkova, A. T., and R. Tycko. 2002. Sensitivity enhancement in structural measurements by solid state NMR through pulsed spin locking. J. Magn. Reson. 155:293–299. [DOI] [PubMed] [Google Scholar]
- Saito, H., R. Tabeta, A. Shoji, T. Ozaki, and I. Ando. 1983. A high-resolution 13C NMR study of poly(β-benzyl-L-aspartate) by the cross-polarization-magic angle spinning method. Distinction of the right-handed α-helix, left-handed α-helix, ω-helix, and β-sheet forms by conformation-dependent 13C chemical shifts. Macromolecules. 16:1050–1057. [Google Scholar]
- Shen, B. W., and A. M. Scanu. 1980. Properties of human apolipoprotein A-I at the air-water interface. Biochemistry. 19:3643–3650. [DOI] [PubMed] [Google Scholar]
- Shen, B. W., A. M. Scanu, and F. J. Kézdy. 1973. Behavior of human apo-high density lipoproteins A-I (Fraction III) and A-II (Fraction IV) at air-water interface. Circulation. 48:218. [Google Scholar]
- Soreghan, B., J. Kosmoski, and C. Glabe. 1994. Surfactant properties of Alzheimer's Aβ peptides and the mechanism of amyloid aggregation. J. Biol. Chem. 269:28551–28554. [PubMed] [Google Scholar]
- Takahashi, Y., A. Ueno, and H. Mihara. 1999. Optimization of hydrophobic domains in peptides that undergo transformation from α-helix to β-fibril. Bioorg. Med. Chem. 7:177–185. [DOI] [PubMed] [Google Scholar]
- Teplow, D. B. 1998. Structural and kinetic features of amyloid β-protein fibrillogenesis. Amyloid. 5:121–142. [DOI] [PubMed] [Google Scholar]
- Torok, M., S. Milton, R. Kayed, P. Wu, T. McIntire, C. G. Glabe, and R. Langen. 2002. Structural and dynamic features of Alzheimer's Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J. Biol. Chem. 277:40810–40815. [DOI] [PubMed] [Google Scholar]
- Weliky, D. P., A. E. Bennett, A. Zvi, J. Anglister, P. J. Steinbach, and R. Tycko. 1999. Solid-state NMR evidence for an antibody-dependent conformation of the V3 loop of HIV-1 gp120. Nat. Struct. Biol. 6:141–145. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., C. G. Bigam, A. Holm, R. S. Hodges, and B. D. Sykes. 1995. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J. Biomol. NMR. 5:67–81. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., B. D. Sykes, and F. M. Richards. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222:311–333. [DOI] [PubMed] [Google Scholar]
- Yoder, M. D., N. T. Keen, and F. Jurnak. 1993. New domain motif: the structure of pectate lyase C, a secreted plant virulence factor. Science. 260:1503–1507. [DOI] [PubMed] [Google Scholar]
- Zhang, S., and A. Rich. 1997. Direct conversion of an oligopeptide from a β-sheet to an α-helix: a model for amyloid formation. Proc. Natl. Acad. Sci. USA. 94:23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]