

Abstract
The pH-dependence of redox properties and of CO binding to bovine lactoperoxidase has been investigated over the range between 2 and 11. The pH-dependence of redox potentials shows a biphasic behavior, suggesting the existence of (at least) two redox-linked groups, which change their pKa values upon reduction. These values are in close agreement with those observed to play a relevant role in the modulation of CO binding to ferrous bovine lactoperoxidase. They have been tentatively attributed to Arg-372 and His-226, which are located on the distal side of the heme pocket of lactoperoxidase. A complete and unequivocal description of the proton-linked behavior of bovine lactoperoxidase requires, however, three residues, which are redox linked and relevant for the modulation of CO binding. The rate constant for CO binding to bovine lactoperoxidase is slower than what is reported for most hemoproteins, suggesting that these two residues, Arg-372 and His-226, are representing a severe barrier for the access of exogenous ligands to the heme. This aspect has been further investigated by fast kinetics following laser photolysis, trying to obtain information on the ligand binding pathway and on the energy barriers.
INTRODUCTION
Lactoperoxidase (LPO) (donor: hydrogen peroxide oxidoreductase EC.1.11.1.7) is a Fe(III)-heme enzyme, belonging to the mammalian peroxidase family. It is found in milk and other biological fluids (like tears, saliva, etc.) and is a component of an antimicrobial defense system that catalyzes the bielectronic oxidation of SCN− to hypothiocyanite (Wolfson and Sumner, 1993). LPO may also substitute thyroid peroxidase in the biosynthetic pathway of thyroid hormones (Courtin et al., 1982). It is a glycoprotein, characterized by a single polypeptide chain of 78 kDa, whose carbohydrate content approaches 10% of the protein total weight and is mainly represented by high mannose and complex structures (Wolf et al., 2000).
In mammalian peroxidases, the prosthetic group is deeply buried inside the bulk of the protein and is covalently linked to the apoprotein through ester bonds (Rae and Goff, 1996; De Pillis et al., 1997, Kooter et al. 1997a; Suriano et al., 2001). This linkage is responsible for the high stability of the heme core and for the peculiar values of redox potential found in almost all members of this family, that accounts for the ability of these enzymes to oxidize inorganic ions like SCN−, Br− and, in the case of myeloperoxidase, Cl− (Kooter et al., 1997b; Furtmüller et al., 1998).
LPO also catalyzes the bielectronic oxidation (by two 1-electron steps) of a number of physiologically relevant organic substrates, such as phenols (Zhang and Dunford, 1993; Monzani et al., 1997), catecholamines, and catechols (Metodiewa et al., 1989a,b; Ferrari et al., 1993) and in addition, as experimental models, aromatic amines (Doerge and Decker, 1994), polychlorinated biphenyls (Oakley et al., 1996), steroid hormones (Sipe et al., 1994; Cavalieri et al., 1997; Ghibaudi et al., 2000), and polycyclic aromatic hydrocarbons (Ramakrishna et al., 1993), most of which are regarded as important risk factors for breast cancer (Oakley et al., 1996; Josephy, 1996; Ghibaudi et al., 2000).
Notwithstanding the great number of efforts made to get LPO crystals, its three-dimensional structure has never been determined. Therefore, the only structural reference available up to now for LPO is represented by a three-dimensional model built on the scaffold of myeloperoxidase, exploiting the high sequence homology existing between these two enzymes (De Gioia et al., 1996).
Faced with this lack of structural information, all experimental data able to provide indirect confirmation to this model become crucial. Several experimental observations witnessing the reliability of the structural model have already been reported (Ferrari et al, 1997, 1999; Ghibaudi et al., 2003).
To obtain structural and functional information on LPO, several investigations have been carried out on this enzyme, both in the ferric and ferrous state, employing substrates analogs or competitive inhibitors, such as CO, CN−, NO, SCN− (Dolman et al., 1968; Lukat et al., 1987; Hu et al., 1993; Sievers et al., 1984; Abu-Soud and Hazen, 2001; Manthey et al., 1986; Sakurada et al., 1987; Modi et al., 1989a ; Lukat et al., 1993; Crull and Goff, 1993). In this study we have carried out a detailed analysis of the pH-dependence of the redox properties of LPO, which has been merged with a deep investigation on the CO binding kinetic properties, allowing to obtain an overall comprehensive view of different proton-linked phenomena. Thus, CO is a heme ligand and its reactivity is modulated by different energetic barriers of the protein moiety, which makes it a very useful probe of structural changes and determinants in the reactivity of a protein. As a matter of fact, our approach allows to better characterize the structural-functional interrelationships, which modulate these events, giving support to the idea that Arg-372 and His-226 play a key role in ligand binding, envisaging as well some relevance also in substrate binding and in the catalytic process.
EXPERIMENTAL PROCEDURE
The lactoperoxidase major cationic isoenzyme was obtained from raw cow's milk, as previously described (Ferrari et al., 1995). The final sample (RZ = 0.80) was stored in 0.1 M phosphate buffer pH 6.8. The concentration of the stock enzyme solution (43 mg/ml) was optically determined by using ɛ412 = 114,000 mM−1 cm−1. The enzyme purity and activity were monitored by UV-visible and electron paramagnetic resonance spectroscopy and by SDS-PAGE analysis with Coomassie Blue staining. For CO binding experiments only the reduced Fe(II) form was obtained by addition of a minimum volume of fresh sodium dithionite to a deoxygenated buffered solution.
Cyclic voltammetry measurements have been carried out using a multipolarograph Amel 433 (AMEL, Milan, Italy). All redox potentials reported in the text refer to the normal hydrogen electrode (NHE). Direct current cyclic voltammograms have been run in different buffers (see below), at a scan rate of 50 mV/s. A pyrolytic graphite electrode (AMEL) was the working electrode, a saturated calomel electrode (+244 mV vs. NHE at 25°C; AMEL) was the reference electrode, and a platinum ring was the counter electrode. The pyrolytic graphite electrode was modified as follows: a suitable amount of TBMPC was dissolved in dimethylsulphoxide, to a final 0.035% w/v concentration. This solution (8 μL) was then mixed with 12 μL of a lactoperoxidase solution and deposited on the surface of a pyrolytic graphite electrode (2-mm diameter) previously polished using an alumina (0.3 μm particle size) water slurry followed by sonication in deionized water (Ferri et al., 1998). The modified electrode was let to dry overnight under vacuum before measurements.
CO binding kinetics has been measured employing a rapid-mixing stopped-flow apparatus SX.18 MV (Applied Photophysics Co., Salisbury, UK) with a 1-ms dead time. The protein was kept in one syringe in a deoxygenated solution of sodium phosphate buffer at pH 7.0 at a very low ionic strength (1.0 mM) and it was mixed with a higher ionic strength buffer (final I = 0.15) at the desired pH value and concentration of CO. Progress curves were recorded at several wavelengths to follow both the formation of the CO adduct (displaying a peak absorbance at 423 nm) and the disappearance of the unliganded form (which is better followed at 440 nm).
The instrument used for photolysis experiments was described elsewhere (Arcovito et al., 2001). Briefly, the 5-ns pulse (λ = 532 nm, E = 80 mJ) of a Nd-YAG solid-state laser (HIL 101, Quanta System, Milano, Italy) was focused onto an optical Thunberg tube containing the desired solution and the transmittance of the sample was monitored using either of two separate recording devices, differently oriented with respect to the laser light pulse: a photomultiplier tube and a CCD solid-state camera. The photomultiplier monitoring line is orthogonal to the laser beam; it consists of a 100-W lamp (Oriel, Stratford, CT), a Spex 1681 monochromator, a Hamamatsu H6780 (Hamamtsu Photonics, Shimokanzo, Japan) photomultiplier tube, and a digital Tektronix TDS 360 (Tektronix, Beaverton, OR) oscilloscope equipped with either a fast amplifier (160 Mhz Comlinear Corporation model E203; later replaced by a 350-MHz Analog Modules model 353A) or the preamplifier Tektronix ADA400. The CCD optical line is arranged at 15 cm from the laser beam; it consists of a 300-W lamp (ILC Technology, Sunnyvale, CA), an Acton-Princeton 320 PI spectrometer (Princeton Instruments, Trenton, NJ) and a pulsed CCD camera (Princeton Instruments) capable of time resolution down to 3.5 ns. The delay between the laser pulse and the action of recording a transmittance spectrum is controlled either by the Princeton FG100 pulse generator necessary to operate the CCD camera, or for longer time delays, by a Tektronix AFG 310 function generator. The experimental data thus collected are either time courses at single wavelength or series of spectra; they are stored as MS-DOS files on an Intel Pentium-based PC and converted to absorbances by means of the package Matlab (Math Works Inc., Natick, MA). Single wavelength time courses are fitted to the desired kinetic model using the nonlinear least-squares routines provided by Matlab.
All experiments were performed at T = 25°C in 1.0 mM CaCl2, employing the following buffer systems: 0.15 M sodium acetate (pH 4.0–5.5), 0.15 M sodium phosphate (pH 3.0–4.0; 5.0–7.5), 0.15 M Tris/HCl (pH 7.5–9.0), 0.15 M sodium borate (pH 8.5–9.5), 0.15 M sodium bicarbonate (pH 9.0–10.5), 0.15 M sodium carbonate (pH 10.0–13.0); no buffer-dependent effects were observed at overlapping pH values.
RESULTS AND DISCUSSION
Fig. 1 shows the influence of pH on the redox potential of bovine LPO; the experimental points were obtained from cyclic voltammograms run at different pH values (see the “Experimental Procedure” section for details). It comes out clear that the redox-proton linkage is regulated by (at least) two residues, which change their pKa values upon change of the heme-iron oxidation state. However, data obtained at acid pH may suggest the existence of a third redox-linked residue characterized by a very low pKa value; the low protein stability observed under low pH conditions did not allow to extend the pH range of the investigation. The observed pH-dependence of redox potential has been analyzed according to the following equation
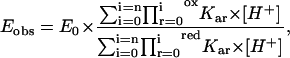 |
(1) |
where Eobs is the observed redox potential at a given pH value, E0 is the redox potential of the form present at very alkaline pH values, redKa and oxKa are the proton binding constants in the reduced and oxidized forms, respectively (such as Ka = 10pKa), outlining that in Eq. 1 Ka0 × [H+] = 1, and n is the total number of protonating redox-linked groups. It must be pointed out that in Eq. 1 we are using the formalism Ka referring to the groups, which are sequentially protonated as pH is lowered, as also depicted in Scheme 1; therefore, the numbering r follows this sequence and r = 1 relates to the residue displaying the highest pKa value. The values of pKa (considering n = 2, see dashed line in Fig. 1) indicate that the proton binding properties of these two residues decrease upon Fe(III)-Fe(II) reduction (oxpKa1 = 11.13 ± 0.14 shifting to redpKa1 = 9.53 ± 0.15 and oxpKa2 = 6.51 ± 0.15 shifting to redpKa2 = 5.18 ± 0.17). Therefore the very negative redox potential shown by lactoperoxidase at pH > 11 (E = −0.342 V vs. NHE) becomes less negative as pH is lowered, and changes to positive at pH < 6 (see Fig. 1). This behavior suggests that LPO resting state displays an increasing difficulty to become oxidized when pH is lowered under anaerobic conditions.
FIGURE 1.

pH-dependence of redox potentials of bovine lactoperoxidase at 25°C. Dashed line corresponds to the nonlinear least-squares fitting of data according to Eq. 1 with n = 2. Continuous line corresponds to the nonlinear least-squares fitting of data according to Eq. 1 with n = 3 by global analysis. For further details, see text.
SCHEME 1.

Values of oxpKa and redpKa values, of different Ei and ki values for redox properties and CO binding kinetics of lactoperoxidase.
To quantitatively describe the proton-linked equilibria operating in lactoperoxidase according to Scheme 1 (see above), we have investigated the influence of pH on the CO association kinetics (Fig. 2). The data have been analyzed according to the following equation:
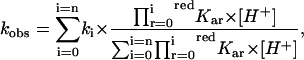 |
(2) |
where kobs is the observed CO binding second-order rate constant at a given pH value, ki is the CO binding second-order rate constant of the ith-protonated species, whereas the other symbols are as in Eq. 1. As from Scheme 1, the pH-dependence of CO binding kinetics allows determination of redpKa (but not of oxpKa) values because CO binds only to the Fe(II) species.
FIGURE 2.

pH-dependence of CO binding to bovine lactoperoxidase at 25°C. Dashed line corresponds to the nonlinear least-squares fitting of data according to Eq. 2 with n = 3 (independent fitting). Continuous line corresponds to the nonlinear least-squares fitting of data according to Eq. 2 with n = 3 by global analysis. For further details, see text.
The pH-dependence reported in Fig. 2 allows a detailed description of the proton linkages regulating CO binding by LPO. Thus, at a very alkaline pH (i.e., pH > 10) lactoperoxidase displays a fairly fast CO binding second-order kinetic constant, even faster than Mb (Coletta et al., 1985). As pH is lowered (pH < 10), the CO association rate constant dramatically decreases (by almost two orders of magnitude), attaining a plateau between pH 7 and 6 (see Fig. 2), with values similar to those observed for other peroxidases (Coletta et al., 1986) and much lower than those observed in Mb (Coletta et al., 1985). Between pH 6.0 and pH 3.5, the CO binding rate constant further decreases (by about one order of magnitude), becoming very slow. Below pH 3.5 an inverted tendency is observed with an increase of the second-order rate constant (see Fig. 2).
A previous paper dealing with CO binding kinetics to lactoperoxidase only at pH 7.0 and 10°C (Abu-Soud and Hazen, 2001) reported a biphasic time course not observed in our measurements. This may be ascribed to the fact that in the previous work the enzyme was from commercial source. The biphasicity observed previously may therefore be related to the fact that commercial LPO displays a value of RZ ≪ 0.8, consistent with the presence of impurities such as lactoferrin and met-myoglobin (Ferrari et al., 1995; Ferrari and Traversa, 2000) or some heterogeneity in the N-terminal region (R. P. Ferrari, unpublished data). Thanks to the high degree of purification achieved for the sample used in this work (Ferrari et al., 1995), our samples did not display impurities or microheterogeneity, showing a single rate constant.
The proton-linked behavior of CO binding kinetics requires three protonating groups (and a nonlinear least-squares fitting of the pH-dependence of data according to Eq. 2 gives pKa1 = 9.53 ± 0.14, pKa2 = 5.41 ± 0.16, and pKa3 = 0.38 ± 0.18; see dashed line in Fig. 2). However, it must be remarked that the resulting value of pKa3, which is well outside the pH range investigated, renders this value fairly unreliable. Interestingly, the two higher pKa values obtained from CO binding kinetics (see above) practically overlap the pKa values obtained from the redox measurements (redpKa1 = 9.53 ± 0.15 and redpKa2 = 5.18 ± 0.17; see Fig. 1); this result clearly indicates that these two residues are involved in the proton-linked modulation of both redox properties and of CO binding dynamics. To check if also the third residue with the lowest pKa in CO binding may be involved in the modulation of redox data with a value consistent for both sets of data, we have carried out a global simultaneous fitting relative to both redox and CO binding measurements, imposing that the same pKa values must apply to both the redpKa (from redox data) and the pKa (from the CO binding kinetics). The three oxpKa and redpKa values resulting from this procedure (reported in Scheme 1) were utilized to draw the continuous line shown in Figs. 1 and 2. We want to stress that the constraint imposed on the fitting procedure actually improved the fitting of both sets of data (see continuous lines in Figs. 1 and 2), clearly indicating that the overlapping of redpKa (from redox data) and of pKa (from CO binding data) was not simply a mathematical outcome, but it likely reflects a possibility that indeed three residues involved in the modulation of redox properties are also involved in the regulation of the pH-dependence of CO binding kinetics. The redpKa3 obtained from this global procedure (reported in Scheme 1) is now significantly different from the pKa3 obtained from the fitting relative to only the CO binding kinetics, and its value (redpKa3 = 2.11 ± 0.18, which shifts to oxpKa3 = 2.94 ± 0.19 upon oxidation) is perfectly compatible with both the redox and kinetic data. At this point, we want to stress that the proton-linked behavior of LPO seems to be fully described through the three redox- and ligand-linked groups, whose pKa values satisfy at the same time the proton-linked behavior resulting from redox (in the absence of sodium dithionite) and CO binding measurements (in the presence of sodium dithionite).
At present, the x-ray structure of lactoperoxidase is not available; this renders it difficult to identify the residues responsible for the observed proton-linked behavior. However, the structural model (De Gioia et al., 1996) together with binding studies (Ferrari et al., 1999) indeed suggest that the ligand inward pathway (for substrates) is characterized by a hydrophobic channel formed by several Phe residues and ending into a pocket stereochemically hindered by Arg-372 and His-226 (Fig. 3). In this respect, values of pKa1 and pKa2 are compatible with the assignment to these two residues (namely Arg-372 for pKa1 and His-226 for pKa2), even though the lack of site-directed mutants of lactoperoxidase impairs a definite identification. However, the oxpKa2 value of 6.63 (see Scheme 1) is perfectly consistent with previous evidence concerning the binding of inorganic substrates to LPO. Therefore, the dissociation constants of the LPO adducts with SCN− and I− have been shown to depend on the protonation state of a site with pKa ∼ 6.0, which is likely the Nɛ atom of the distal His-226. An analogous pH-dependence was shown for the activity of LPO toward both substrates and attributed to an amino acid residue located in the active site (Ferrari et al., 1997).
FIGURE 3.

View of the LPO active site according to the structural model by De Gioia et al. (1996). The amino acid residues forming the walls of the organic substrate access channel are shown as a dotted cloud.
On the other hand, pKa3 is referable to the protonation of the Nɛ of the imidazole group of the axial proximal His-468; this is supported by the fact that the value of redpKa3 is very similar to that determined for Mb (Coletta et al., 1985) and its protonation brings about an enhancement on the CO binding kinetics.
To have a clearer view of the mechanism of CO binding and on the dynamic role of these residues, we have carried out an investigation of CO binding dynamics through laser photolysis, which allows one to discriminate the role of different steps along the ligand pathway within the protein matrix.
In the absence of additional information, laser photolysis data can be analyzed according to the following minimum scheme (see also Henry et al. (1983)): where state 1 refers to the unliganded protein with the ligand in the bulk solvent, state 2 refers to the unliganded protein with the ligand inside the protein matrix (the so called geminate pair), and state 3 refers to the liganded protein. In the case of the stopped-flow experiments we follow the kinetics from state 1 to state 3, whereas in the laser photolysis experiment starting from state 3 we induce the formation of state 2 by a very fast laser pulse and we follow the temporal evolution of state 2 toward either state 3 (i.e., the geminate rebinding) or to state 1, from which we have the same bimolecular process observed by the rapid-mixing method (i.e., kbim from state 1 to state 3). Therefore, under the most straightforward conditions, two parameters can be determined: i), kgem, corresponding to the observed rate constant of the geminate process (in LPO it is almost but not perfectly exponential), and ii), Agem, corresponding to the absorbance change referable to the geminate process. On the other hand, rapid-mixing kinetic experiments are giving kbim. Because for [L] = 10−3 M k12 × [L] ≪ k21 and k23 ≫ k32, according to Scheme A we can write:
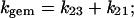 |
(3a) |
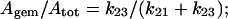 |
(3b) |
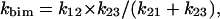 |
(3c) |
where Atot is the total absorbance change for CO rebinding, including both the geminate and the bimolecular process. The expression for kbim refers to the intrinsic second-order rate constant, which must be then multiplied by [L] for obtaining the actual observed rate. Equation 3, a–c, allows then to determine the individual parameters of Scheme A, according to the following relationships
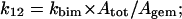 |
(3d) |
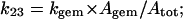 |
(3e) |
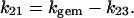 |
(3f) |
The same relationship also holds with respect to the apparent CO dissociation rate constant which, according to Scheme A, turns out to be
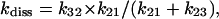 |
(3g) |
from which
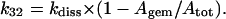 |
(3h) |
SCHEME A.

It is relevant, however, that kdiss does not play any role under the experimental conditions of laser photolysis experiments.
Fig. 4 shows the absorbance change at 440 nm relative to the CO recombination after a very rapid laser pulse (FWHM ≈ 5 ns). The progress curve (reported on a logarithmic timescale) displays two main events. The first one consists in a geminate process characterized by a CO-independent rate constant, kgem, and a signal amplitude, Agem, (see Eq. 3, a–c), that refers to a ligand molecule within the protein matrix (from state 2 to state 3; see Scheme A); the second one is the same bimolecular process observed by rapid mixing techniques and displaying the pH-dependent behavior reported in Fig. 2. This last process, which is characterized by a CO-dependent rate constant, kbim, and a signal amplitude (Atot − Agem) (see Eq. 3, a–c) refers to a ligand molecule originating from the bulk solvent (from state 1 to state 3; see Scheme A).
FIGURE 4.

CO rebinding to bovine lactoperoxidase at pH 7.0 after laser photolysis. T = 25°C. λ = 440 nm. Continuous line has been obtained by nonlinear least-squares fitting of data according to a two-exponentials reaction mechanism, as follows: OD = Agem × exp(−kgem×t) + Abim × exp(−kbim[CO]×t) (Eq. 4), where symbols have the same meaning as in Eq. 3. For further details, see text.
At this stage of knowledge, the application of the minimum scheme A appears appropriate; Table 1 reports the dynamic parameters obtained from CO recombination after laser photolysis of lactoperoxidase (according to Scheme A and Eq. 3, a–g). If we compare these parameters at pH 7.0 (mostly referring to the single-protonated form PH(II), see Scheme 1) with those determined for sperm whale myoglobin (also reported on Table 1; see Henry et al. (1983)), it turns out that all steps are at least two orders of magnitude faster in the case of Mb. This indicates that lactoperoxidase possesses a compact and tight structure that keeps the heme almost completely hidden from the bulk solvent. Further, the gate seems regulated by ionic interactions involving Arg-372 and His-226, whose protonation brings about a progressively increasing energy barrier.
TABLE 1.
Dynamic parameters for CO rebinding to lactoperoxidase and to sperm whale Mb at pH 7.0 after laser photolysis
| Lactoperoxidase | Sperm whale Mb | |
|---|---|---|
| kgem (s−1) | 2.32(±0.25) × 104 | 6.9(±0.1) × 105 |
| Agem/Atot | 0.25 ± 0.04 | 0.043 ± 0.006 |
| kbim (M−1s−1) | 2.75(±0.31) × 104 | 5.5(±0.8) × 106 |
| Abim/Atot | 0.75 ± 0.07 | 0.957 ± 0.011 |
| k12 (M−1s−1) | 1.08(±0.13) × 105 | 1.6(±0.3) × 107 |
| k21 (s−1) | 1.73(±0.24) × 104 | 5.3(±0.8) × 106 |
| k23 (s−1) | 5.91(±0.72) × 103 | 2.4(±0.6) × 105 |
CONCLUDING REMARKS
In summary, our results provide independent evidence that the LPO heme pocket is unusually narrow and that (at least) two residues play a key role in substrate binding and, possibly, in the catalytic process. Arg-372 and His-226 (distal His) result to be the most likely candidates.
The first statement accords well with the LPO structural model (De Gioia et al., 1996) and with a number of reports (Ferrari et al., 1997; Hu et al., 1993; Ohlsson and Paul, 1983) asserting that the heme site is accessible to inorganic substrates (that, however, often undergo steric or angular strains once bound to the iron). This implies that bulky organic substrates cannot penetrate the LPO catalytic pocket (Ferrari et al., 1999; Ghibaudi et al., 2003; Modi et al, 1989b; Hosoya et al., 1989). A peculiar role seems to be played by Arg-372, which allows the insertion of small inorganic substrates but not that of large organic substrates. As a consequence, His-226 and Arg-372 seem to play a role in substrate binding, as confirmed in this study.
Acknowledgments
Authors want to express their thanks to Professor Luigi Casella for several stimulating discussions on the functional properties of bovine lactoperoxidase.
The financial support by the Italian Ministry of University and Research (MIUR COFIN MM03185591 to M.C. and R.P.F.) is gratefully acknowledged.
Abbreviations used: CCD, charged-coupled devices; FWHM, full width half maximum; LPO, bovine lactoperoxidase; NHE, normal hydrogen electrode; TBMPC, tributylmethyl phosphonium chloride polymer bound (polystyrene cross-linked with 1% divinyl benzene) to anionic exchange resin.
References
- Abu-Soud, H. M., and S. L. Hazen. 2001. Interrogation of heme pocket environment of mammalian peroxidases with diatomic ligands. Biochemistry. 40:10747–10755. [DOI] [PubMed] [Google Scholar]
- Arcovito, A., S. Gianni, M. Brunori, C. Travaglini Allocatelli, and A. Bellelli. 2001. Fast coordination changes in cytochrome c do not necessarily imply folding. J. Biol. Chem. 276:41073–41078. [DOI] [PubMed] [Google Scholar]
- Cavalieri, E. L., D. E. Stack, P. D. Devanesan, R. Todorovic, I. Dwivedy, S. Higginbotham, S. L. Johansson, K. D. Patil, M. L. Gross, J. K. Gooden, R. Ramanathan, R. L. Cerny, and E. G. Rogan. 1997. Molecular origin of cancer: catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. USA. 94:10937–10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta, M., P. Ascenzi, T. G. Traylor, and M. Brunori. 1985. Kinetics of carbon monoxide binding to monomeric hemoproteins. Role of the proximal histidine. J. Biol. Chem. 260:4151–4155. [PubMed] [Google Scholar]
- Coletta, M., F. Ascoli, M. Brunori, and T. G. Traylor. 1986. pH dependence of carbon monoxide binding to ferrous horseradish peroxidase. J. Biol. Chem. 261:9811–9814. [PubMed] [Google Scholar]
- Courtin, F., D. Deme, A. Virion, J. L. Michot, J. Pommier, and J. Nunez. 1982. The role of lactoperoxidase-H2O2 compounds in the catalysis of thyroglobulin iodination and thyroid hormone synthesis. Eur. J. Biochem. 124:603–609. [DOI] [PubMed] [Google Scholar]
- Crull, G. B., and H. M. Goff. 1993. NMR relaxation studies of the interaction of thiocyanate with lactoperoxidase. J. Inorg. Biochem. 15:181–192. [DOI] [PubMed] [Google Scholar]
- De Gioia, L., E. M. Ghibaudi, E. Laurenti, M. Salmona, and R. P. Ferrari. 1996. A theoretical three-dimensional model for lactoperoxidase and eosinophil peroxidase, built on the scaffold of the myeloperoxidase X-ray structure. JBIC. 1:476–485. [Google Scholar]
- DePillis, G. D., S. Ozaki, J. M. Kuo, D. A. Maltby, and P. R. Ortiz de Montellano. 1997. Autocatalytic processing of heme by lactoperoxidase produces the native protein-bound prosthetic group. J. Biol. Chem. 272:8857–8860. [DOI] [PubMed] [Google Scholar]
- Doerge, D. R., and C. J. Decker. 1994. Inhibition of peroxidase-catalyzed reactions by arylamines: mechanism for the anti-thyroid action of sulfamethazine. Chem. Res. Toxicol. 7:164–169. [DOI] [PubMed] [Google Scholar]
- Dolman, D., H. B. Dunford, D. M. Chowdhury, and M. Morrison. 1968. The kinetics of cyanide binding by lactoperoxidase. Biochemistry. 7:3991–3996. [DOI] [PubMed] [Google Scholar]
- Ferrari, R. P., E. Laurenti, L. Casella, and S. Poli. 1993. Oxidation of catechols and catecholamines by HRP and LPO: ESR spin stabilization approach combined with optical methods. Spectrochim. Acta. 49A:1261–1267. [Google Scholar]
- Ferrari, R. P., E. Laurenti, P. I. Cecchini, O. Gambino, and I. Sondergaard. 1995. Spectroscopic investigations on the highly purified lactoperoxidase Fe(III)-heme catalytic site. J. Inorg. Biochem. 58:109–127. [DOI] [PubMed] [Google Scholar]
- Ferrari, R. P., E. M. Ghibaudi, S. Traversa, E. Laurenti, L. De Gioia, and M. Salmona. 1997. Spectroscopic and binding studies on the interaction of inorganic anions with lactoperoxidase. J. Inorg. Biochem. 68:17–26. [DOI] [PubMed] [Google Scholar]
- Ferrari, R. P., S. Traversa, L. De Gioia, P. Fantucci, G. Suriano, and E. Ghibaudi. 1999. Catechol(amine)s as probes of lactoperoxidase catalytic site structure: spectroscopic and modeling studies. JBIC. 4:12–20. [DOI] [PubMed] [Google Scholar]
- Ferrari, R. P., and S. Traversa. 2000. In The Peroxidase Multigene Family of Enzymes. P. E. Petrides, W. M. Nauseef, editors. Chapter 15. Springer-Verlag, New York, NY. 114–121.
- Ferri, T., A. Poscia, and R. Santucci. 1998. Direct electrochemistry of membrane-entrapped horseradish peroxidase. Part I. A voltammetric and spectroscopic study. Bioelectrochem. Bioenerg. 44:177–181. [Google Scholar]
- Furtmüller, P. G., U. Burner, and C. Obinger. 1998. Reaction of myeloperoxidase compound I with chloride, bromide, iodide, and thiocyanate. Biochemistry. 37:17923–17930. [DOI] [PubMed] [Google Scholar]
- Ghibaudi, E. M., E. Laurenti, P. Beltramo, and R. P. Ferrari. 2000. Can estrogenic radicals, generated by lactoperoxidase, be involved in the molecular mechanism of breast carcinogenesis? Redox Rep. 5:229–235. [DOI] [PubMed] [Google Scholar]
- Ghibaudi, E., E. Laurenti, C. Pacchiardo, G. Suriano, N. Moguilevsky, and R. P. Ferrari. 2003. Organic and inorganic substrates as probes for comparing native bovine lactoperoxidase and recombinant human myeloperoxidase. J. Inorg. Biochem. 94:146–154. [DOI] [PubMed] [Google Scholar]
- Henry, E. R., J. H. Sommer, J. Hofrichter, and W. A. Eaton. 1983. Geminate recombination of carbon monoxide to myoglobin. J. Mol. Biol. 166:443–451. [DOI] [PubMed] [Google Scholar]
- Hosoya, T., J. Sakurada, C. Kurokawa, R. Toyoda, and S. Nakamura. 1989. Interaction of aromatic donor molecules with lactoperoxidase probed by optical difference spectra. Biochemistry. 28:2639–2644. [DOI] [PubMed] [Google Scholar]
- Hu, S., R. W. Treat, and J. R. Kincaid. 1993. Distinct heme active-site structure in lactoperoxidase revealed by resonance Raman spectroscopy. Biochemistry. 32:10125–10130. [DOI] [PubMed] [Google Scholar]
- Josephy, P. D. 1996. The role of peroxidase-catalyzed activation of aromatic amines in breast cancer. Mutagenesis. 11:3–7. [DOI] [PubMed] [Google Scholar]
- Kooter, I. M., A. J. Pierik, and R. Wever. 1997a. Difference Fourier transform infrared evidence for ester bonds linking the heme group in myeloperoxidase, lactoperoxidase, and eosinophil peroxidase. J. Am. Chem. Soc. 119:11542–11543. [Google Scholar]
- Kooter, I. M., N. Moguilevsky, A. Bollen, N. M. Sijtsema, C. Otto, and R. Wever. 1997b. Site-directed mutagenesis of Met243, a residue of myeloperoxidase involved in binding of the prosthetic group. JBIC. 2:191–197. [Google Scholar]
- Lukat, G. S., K. R. Rodgers, and H. M. Goff. 1987. Electron paramagnetic resonance spectroscopy of lactoperoxidase complexes: clarification of hyperfine splitting for the NO adduct of lactoperoxidase. Biochemistry. 26:6927–6932. [DOI] [PubMed] [Google Scholar]
- Lukat, G. S., B. M. Doran, L. M. Utschig, and H. M. Goff. 1993. Magnetic resonance spectroscopy, calcium content, and anion coordination studies of bovine and goat lactoperoxidase. J. Inorg. Biochem. 50:157–171. [DOI] [PubMed] [Google Scholar]
- Manthey, J. A., N. J. Boldt, D. F. Bocian, and S. I. Chan. 1986. Resonance Raman studies of lactoperoxidase. J. Biol. Chem. 261:6734–6741. [PubMed] [Google Scholar]
- Metodiewa, D., K. Reszka, and H. B. Dunford. 1989a. Oxidation of the substituted catechols dihydroxyphenylalanine methyl ester and trihydroxyphenylalanine by lactoperoxidase and its compounds. Arch. Biochem. Biophys. 274:601–608. [DOI] [PubMed] [Google Scholar]
- Metodiewa, D., K. Reszka, and H. B. Dunford. 1989b. Evidence for a peroxidatic oxidation of norepinephrine, a catecholamine, by lactoperoxidase. Biochem. Biophys. Res. Commun. 160:1183–1188. [DOI] [PubMed] [Google Scholar]
- Modi, S., D. V. Becere, and S. Mitra. 1989a. Binding of thiocyanate to lactoperoxidase: 1H and 15N nuclear magnetic resonance studies. Biochemistry. 28:4689–4694. [DOI] [PubMed] [Google Scholar]
- Modi, S., D. V. Becere, and M. Samaresh. 1989b. Binding of aromatic donor molecules to lactoperoxidase: proton NMR and optical difference spectroscopic studies. Biochim. Biophys. Acta. 996:214–225. [DOI] [PubMed] [Google Scholar]
- Monzani, E., A. L. Gatti, A. Profumo, L. Casella, and M. Gullotti. 1997. Oxidation of phenolic compounds by lactoperoxidase. Evidence for the presence of a low-potential compound II during catalytic turnover. Biochemistry. 36:1918–1926. [DOI] [PubMed] [Google Scholar]
- Oakley, G. G., U. Devanaboyina, L. W. Robertson, and R. C. Gupta. 1996. Oxidative DNA damage induced by activation of polychlorinated biphenyls (PCBs): implications for PCB-induced oxidative stress in breast cancer. Chem. Res. Toxicol. 9:1285–1292. [DOI] [PubMed] [Google Scholar]
- Ohlsson, P. I., and K. G. Paul. 1983. The reduction potential of lactoperoxidase. Acta Chem. Scand. B37:917–921. [DOI] [PubMed] [Google Scholar]
- Rae, T. D., and H. M. Goff. 1996. Lactoperoxidase heme structure characterized by paramagnetic proton NMR spectroscopy. J. Am. Chem. Soc. 118:2103–2104. [Google Scholar]
- Ramakrishna, N. V. S., K. M. Li, E. J. Rogan, E. L. Cavalieri, M. George, R. L. Cerny, and M. L. Gross. 1993. Adducts of 6-methylbenzo[a]pyrene and 6-fluorobenzo[a]pyrene formed by electrochemical oxidation in the presence of deoxyribonucleosides. Chem. Res. Toxicol. 6:837–845. [DOI] [PubMed] [Google Scholar]
- Sakurada, J., S. Takahashi, and T. Hosoya. 1987. Proton nuclear magnetic resonance studies on the iodide binding by horseradish peroxidase. J. Biol. Chem. 262:4007–4010. [PubMed] [Google Scholar]
- Sievers, G., J. Peterson, P. A. Gadsby, and A. J. Thomson. 1984. The nitrosyl compound of ferrous lactoperoxidase. Biochim. Biophys. Acta. 785:7–13. [Google Scholar]
- Sipe, H. J., S. J. Jordan, P. M. Hanna, and R. P. Mason. 1994. The metabolism of 17 beta-estradiol by lactoperoxidase: a possible source of oxidative stress in breast cancer. Carcinogenesis. 15:2637–2643. [DOI] [PubMed] [Google Scholar]
- Suriano, G., S. Watanabe, E. M. Ghibaudi, A. Bollen, R. P. Ferrari, and N. Moguilevsky. 2001. Glu375Gln and Asp225Val mutants: about the nature of the covalent linkages between heme group and apo-Protein in bovine lactoperoxidase. Bioorg. Med. Chem. Lett. 11:2827–2831. [DOI] [PubMed] [Google Scholar]
- Wolf, S. M., R. P. Ferrari, S. Traversa, and K. Biemann. 2000. Determination of the carbohydrate composition and the disulfide bond linkages of bovine lactoperoxidase by mass spectrometry. J. Mass Spectrom. 35:210–217. [DOI] [PubMed] [Google Scholar]
- Wolfson, L. M., and S. S. Sumner. 1993. Antibacterial activity of the lactoperoxidase system: a review. J. Food. Protect. 56:887–892. [DOI] [PubMed] [Google Scholar]
- Zhang, H., and H. B. Dunford. 1993. Hammett rho-sigma correlation of lactoperoxidase compound II with phenols. Can J. Chem. 71:1990–1994. [Google Scholar]