

Abstract
The diffusive motion of DNA-containing chromatin in live cells and isolated nuclei is investigated using a two-photon standing wave fluorescence photobleaching experiment with 100 nm spatial resolution. The chromatin is labeled using the minor groove binding dye Hoechst 33342. In live cells, the mean diffusion rate is 5 × 10−4 μm2/s, with considerable cell-to-cell variation. This diffusion is highly constrained and cannot be observed in a standard, single beam fluorescence recovery after photobleaching experiment. To determine the chemical origin of the diffusion, we study motion in isolated nuclei and vary the strength of the histone-DNA interactions by changing the ionic strength and using chemical and photocross-linking experiments. At higher NaCl concentrations, we see increased chromatin diffusion as the histone-DNA interaction is weakened due to ionic screening, whereas photocross-linking the core histones to the DNA results in a complete absence of diffusive motion. These trends are consistent with the 100 nm scale motion being correlated with the interactions of histone proteins with the DNA. If chromatin diffusion is connected to the nucleosomal dynamics on much smaller length scales, this may provide a way to assay biochemical activity in vivo based on larger scale macromolecular dynamics observed via fluorescence microscopy.
INTRODUCTION
DNA is perhaps the most widely studied macromolecule in biology, yet its properties in living cells are still not well understood. Results obtained in the last decade have revised the original picture of the cell nucleus as a static library for genomic DNA. Measurements of protein dynamics in live cell nuclei have revealed high mobilities and considerable variation in the motions, even for proteins thought to be strongly bound to the stationary DNA (Phair and Misteli, 2000; Misteli et al., 2000; Lever et al., 2000; Kimura and Cook, 2001). It is now recognized that the structure and dynamics of DNA itself in interphase cell nuclei also play a role in gene expression and other cellular processes. The best-known example of this is the distinction between transcriptionally active euchromatin and the inactive, densely packed heterochromatin (Lamond and Earnshaw, 1998). In situ hybridization experiments have revealed specific chromosomal domains in interphase cell nuclei, where previously the DNA had been thought to have adopted completely random conformations (Cremer and Cremer, 2001). In addition to these micron scale chromosomal domains, it now seems clear that there are structures on even smaller length scales within these larger domains, and that these structures may undergo time-dependent structural changes when nearby genes are expressed (Trumbar et al., 1999; Tsukamoto et al., 2000; Gunawardena and Rykowski, 2000). Faster, diffusive motions may be responsible for enabling the transient association of proteins and DNA, the initial chemical step in gene expression (Wolffe and Hansen, 2001; Marshall, 2002). Taken as a whole, these results demonstrate that chromatin is an active player in the biochemistry of gene expression. Because of this, the study of intranuclear molecular dynamics of both DNA and proteins has attracted a great deal of interest.
The study of chromatin dynamics in biological systems is complicated by several factors. The most obvious complication is that this motion is quite limited. In fact, under normal observation conditions in a fluorescence microscope, the nuclear DNA of live cells appears completely stationary. Fluorescence recovery after photobleaching (FRAP) experiments by Axelrod and co-workers looking at the motion of labeled DNA in 3T3 fibroblast cells saw no motion on a length scale of ∼0.5 μm over the course of minutes to hours (Abney et al., 1997). More recent experiments have taken advantage of the higher spatial resolution afforded by single particle tracking (SPT) techniques and fluorescent analogs of the lac repressor protein. The groups of Sedat and Gasser observed the motion of single genes labeled with a fluorescent lac repressor protein in both yeast and Drosophila cells, with the conclusion that random motion occurs with a diffusion constant on the order of 10−4 μm2/s within a 1 μm area in the nucleus, due to invisible constraints (Marshall et al., 1997; Heun et al., 2001). Further experiments have shown that these constraints are more apparent in regions near the nuclear envelope, leading to the hypothesis that physical attachment of the chromatin to the nuclear envelope controls the motional freedom of the chromatin and also the rate of gene expression (Vazquez et al., 2001; Chubb et al., 2002). Cremer and Cremer used a different method to label the chromatin, namely incorporation of fluorescently labeled nucleotides during DNA replication, which resulted in randomly spaced fluorescent spots throughout the nucleus (Bornfleth et al., 1999). In addition to measuring diffusion rates, this method also permits the investigation of spatially dependent diffusion within the same cell. They found considerable variation of the diffusion constant and attributed this variation to different local environments within a single nucleus.
Two main conclusions can be drawn from this work. First, chromatin motion in live cells is constrained to submicron length scales. The degree of constraint, at least in some cell types, depends on the stage of the cell cycle and location within the nucleus. The implications for biological function are as yet unclear, but limiting diffusion may affect the ability of the DNA to undergo transcription (Marshall, 2002). The second conclusion is that the motion of chromatin in live cells is quite variable. In addition to the changes in large-scale constrained motion mentioned above, the fast, short-range diffusion also varies by two orders of magnitude depending on cell type (Marshall et al., 1997), and even by one order of magnitude depending on location within a single nucleus (Bornfleth et al., 1999). The origin of the constraints on chromatin motion, the reason for its variability, and its molecular-level mechanism are all open questions in the field.
In this work, we are interested in the connection between the small-scale chromatin motion observed in live cells and molecular-level chemical events relevant to cellular biochemistry. In other words, what is the chemical origin of the short-range diffusion observed in live cells? To answer this question, we use a newly developed two-photon counterpropagating fluorescence recovery after patterned photobleaching (2P-c-FRAPP) experiment (Davis and Bardeen, 2002) to look at the short-range diffusion of fluorescently labeled intranuclear DNA in live Xenopus laevis XTC-2 cells and isolated nuclei. This experiment has spatial resolution similar to the SPT experiments, but with the advantage that it can be easily applied to a broad variety of systems, both in vivo and in vitro, whereas the SPT methods require conditions where a fluorescent lac repressor protein can be expressed and bind strongly to the chromosomal DNA. These conditions may not be fulfilled under our experimental conditions, for example, isolated nuclei in high salt solution (Schlax et al., 1995). For our experiments the flexibility afforded by Hoechst 33342 (H33342) labeling in conjunction with the 2P-c-FRAPP experiment is vital, since it allows us to compare dynamics in very different chemical environments. In live XTC-2 cells, we observe constrained diffusion rates similar to those observed previously in other cell types. This motion is largely absent in isolated cell nuclei at physiological ionic strength, which strongly suggests that the observed diffusion is not determined by some combination of structural constraints (like molecular crowding) and thermal fluctuations (like classical Brownian motion). By examining how the diffusion depends on ionic strength in isolated nuclei, and by using cross-linking to modify the protein-DNA interactions in both nuclei and live cells, we find that these motions follow similar trends as the interaction strength of core histone protein with the DNA. Although this is not conclusive evidence for the role of nucleosome sliding in chromatin diffusion, it does suggest a link between chromatin motion on the 100 nm length scale, which can be observed using fluorescence microscopy, and molecular-level histone-DNA dynamics, which are generally studied using biochemical methods for small model systems in vitro (Widom, 1998; Hansen, 2002). These dynamics control phenomena like nucleosome sliding and small-scale remodeling of the chromatin structure, which in turn allow for transcription factor binding and eventually gene expression (Becker, 2002; Luger, 2003; Vermaak and Wolffe, 1998).
MATERIALS AND METHODS
Apparatus
The experimental set-up shown in Fig. 1 has been previously described (Davis and Bardeen, 2002) and is related to several methods developed by previous workers (Cicerone et al., 1995; Smith and McConnell, 1978; Davoust et al., 1982; Lanni and Ware, 1982). Briefly, a Ti:sapphire laser system is used to generate ∼45 fs, 800 nm pulses. The intensity of the pulses is varied by a Pockels cell (Conoptics 350-50, Danbury, CT), which is electronically switched by the output of a computer data acquisition (DAQ) board (National Instruments PCI-6024E, Austin, TX). An RG610 filter is used to eliminate background due to second harmonic generation in the Pockels cell. An interferometer is built around an inverted microscope (Olympus IX-70, Melville, NY) as shown in Fig. 1. Each beam of the interferometer is focused through one of two 40X 0.66 NA microscope objectives which are aligned 180° to each other (Bailey et al., 1993; Gustafsson et al., 1999; Hell et al., 1997). The beams are spatially and temporally overlapped in the sample, creating a standing wave. The position of the standing wave in the z-direction is controlled by a piezodriven translation stage which is controlled to better than 10-nm precision. The fluorescence is collected by one of the microscope objectives and is directed through a dichroic mirror (DC) and then into a photomultiplier. A hot mirror and BG28 filter are used to eliminate any infrared background from the fluorescence signal. To increase detection sensitivity, the excitation is modulated by an external chopper, and the signal is detected by a lock-in amplifier.
FIGURE 1.

Experimental apparatus to create a two-photon standing-wave at the focus of two microscope objective lenses, which allows a high-spatial resolution fluorescence recovery after patterned photobleaching experiment to be performed.
In a typical experiment, the piezo is scanned back and forth at the probe intensity (8 × 109 W/cm2). Then, movement of the piezo is stopped and the standing wave pattern is photobleached into the sample by increasing the intensity of the beam for 10 ms (2 × 1011 W/cm2). After photobleaching, the piezo is scanned back and forth again. This scanning of the probe fringe pattern over the photobleached fringe pattern results in an oscillatory fluorescence signal. As the sample undergoes diffusion, the total fluorescence signal generated by scanning the phase φ of the probe standing wave can be written as (Davis and Bardeen, 2002):
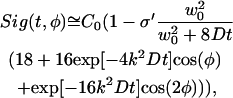 |
(1) |
where k = 2πn/λ, n is the index of refraction, λ is the wavelength, D is the diffusion coefficient, w0 is the beam radius, C0 is the initial concentration of chromophores, and σ′ is proportional to the bleach depth. Equation 1 contains three types of time-dependent terms: the cos(2φ) term, which makes a negligible contribution to the overall signal; the cos(φ) term which decays with a characteristic time τosc,
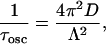 |
(2a) |
where Λ = λ/2n is the peak to peak fringe spacing, and a DC component which recovers with a half life τ1/2,
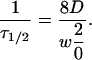 |
(2b) |
In our experiment, n = 1.33, and λ = 800 nm, resulting in a k value of 1.04 × 101 μm−1. This value results in a 1/e decay of the oscillations after a root-mean-square displacement of 110 nm in three dimensions, allowing us to observe motions well below the diffraction limit, as long as there is sufficient signal-to-noise to resolve a 1/e change in the oscillatory signal level. Equation 1 also predicts a second recovery due to normal FRAP recovery in the bleached spot with a timescale of τ1/2 which depends on the spot diameter. The characteristic timescales are proportional to the squares of the length scales,
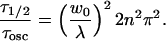 |
(3) |
Typically in our experiments the beam diameter is ∼1.2 μm, four times larger than the fringe spacing, so the characteristic timescales for the two decays differ by a factor of 50–100.
Data analysis
A full data scan is composed of several back and forth motions of the piezo, leading to abrupt phase changes in the signal. Therefore, the data are collected in subsets, each subset corresponding to uninterrupted motion of the piezo. The amplitude of the oscillations in each subset is obtained by performing a Fourier transform. A plot of the oscillation amplitude for each subset over time is fitted to an exponential decay for each full data scan. Determination of a statistical relationship between data sets collected on different days was made using Student's t-test at the 95% confidence level.
Live cell experiments were done on three different days under the same conditions. The data set for each day contains one scan for each of at least 20 cells, for a total of 63 scans. In 89% of the scans, the oscillations decayed exponentially, as would be expected for normal diffusive motion. All statistical analyses were performed using data from these scans. The other seven scans were excluded from statistical analysis because no reliable decay information could be extracted. In one case, the decay of the oscillations was complete before the second subscan of the piezo, giving only one data point for the exponential fit. One scan had a gradual increase in the prebleach signal level; a fast, partial decay of bleached oscillations; and then a nonhomogeneous decay of oscillations. The other five scans were excluded from statistical analysis because the oscillations did not decay fully, indicating that a certain fraction of the photobleached H33342 in the ∼4 fL excitation volume was attached to chromatin that was immobile during the scan.
Live cells
Cell medium solutions used in all experiments were made from phenol red-free 70% DME/F12 medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY). Xenopus XTC-2 cells were grown in cell medium at room temperature. Cells suspended in cell medium were seeded onto collagen substrates (described below) and were allowed to attach overnight before staining. For staining, the gel-coated coverslips with attached cells were gently rinsed with a solution of 70% phosphate buffered saline (PBS, BioWhittaker, Walkersville, MD) in water. Cells were stained for 10 min at room temperature in a 9 μM solution of H33342 (Sigma). After staining, the samples were gently rinsed five times with 70% PBS and then covered with cell medium. Petri dishes containing the samples were parafilmed and stored at room temperature until use. All cells were clearly in interphase before the experiment, but no effort was made to synchronize them further.
Preparation of collagen substrates for live cell experiments
Collagen gel was made by combining 778 μL of 70% cell medium with 222 μL of 4.1 mg/mL rat tail collagen (type I) in 0.1 N acetic acid (Upstate Biotechnology, Charlottesville, VA). The mixture was chilled on ice, and then chilled 0.5 N NaOH was added to neutralize the solution. The final solution was kept on ice until use. A total of 300 μL of the collagen solution was pipetted onto a 50 × 45 mm microscope coverslip in a petri dish. The solution was spread evenly over the surface of the coverslip and allowed to gel for 15 min in a 37°C incubator. Collagen gel-coated coverslips were kept immersed in medium at room temperature until use.
Preparation of collagen substrates for isolated nuclei experiments
A 1.3 mg/mL solution of collagen was made by dilution with water at room temperature. A total of 300 μL was pipetted onto a 50 × 45 mm microscope coverslip in a petri dish, and the solution was spread evenly over the surface of the coverslip. A cotton swab saturated with 4% ammonium hydroxide was placed in the petri dish and the dish covered. The collagen was allowed to gel in the ammonia vapor for 10 min at room temperature, and then the swab was removed and the gel rinsed five times with and stored in 70% PBS. Immediately before the nuclei were added, the collagen film was rinsed three times with room temperature nuclear suspension solution.
Isolated nuclei
Cells were resuspended in cell media, and then rinsed with ice-cold 70% PBS. Subsequent steps were done on ice. After centrifugation, the cell pellet was loosened, and ∼250 μL mammalian cell lysis reagent (Pierce, Rockford, IL) was added. The sample was agitated for 30 s, and then the cell membranes were disrupted by resuspending the solution twice with a glass pipette. A total of 5 mL of cell media was added, and then the nuclei were centrifuged once and resuspended in the nuclear suspension solution with 9 μM Hoechst 33342. For chemical fixation, 4% formaldehyde was added to the unstained nuclear suspension, which was then incubated on ice for 15 min. The nuclei were rinsed three times with cell medium and then resuspended in the nuclear suspension solution with 9 μM Hoechst 33342. The nuclei were allowed to incubate and settle on the surface of the collagen-coated coverslips for 1 h before use.
Nuclear suspension solutions
Solutions were made with 70% cell medium, which had 80 mM Na+. Solid NaCl was added to cell medium to make a solution that had 2 M total Na+. This solution was diluted with cell medium to make the 400-mM total Na+ solution. The 20-mM Na+ solution was made by diluting cell medium with water.
RESULTS
Cell viability and photodamage
We first discuss the effects of our DNA probe, Hoechst 33342, on the XTC-2 cells used in this study. H33342 is known to be cytotoxic at sufficiently high concentrations and is also known to interfere with DNA-native protein interactions, in particular topoisomerase (Durand and Olive, 1982; Smith et al., 1990). Indeed, we find that the dye concentrations used in this study retard the growth of the cells, slowing the cell cycle down by roughly a factor of four, although the mortality of the XTC-2 cells is not increased at these concentrations or even at higher concentrations. When a stained cell is exposed to ultraviolet or two-photon excitation, however, H33342 can initiate photochemical cross-linking between proteins and DNA, which may also lead to cytotoxicity (Durand and Olive, 1982; Davis and Bardeen, 2003). Although the average number of excitations experienced by H33342 molecules in our photobleaching experiments is a factor of 1000 less than what is used for cross-linking (Davis and Bardeen, 2003), we still find that exposure to the laser fluences in our experiments leads to heightened mortality in our cells. In general, it is multiple exposures to the high power bleaching pulse that cause the fatal damage. After a single experiment, there is no measurable increase in cell mortality over the course of several days, as judged by the ability of the cells to remain adherent to the collagen surface. Thus we report in vivo results only for cells that have been exposed to a single photobleaching pulse. To see whether photodamage during a single experiment affected our results, in both cells and isolated nuclei, we did the following check. After performing one experiment, we repeated the experiment in the same spot to make sure that we obtained similar data. The data from a single spot was reproducible up to 3–4 cycles, after which we typically saw a slowdown in the recovery and eventually no recovery at all. Although H33342 has disadvantages as a marker for chromatin motion (it has significant phototoxicity, it nonspecifically labels all the double-stranded DNA in the nucleus, and it modifies the rates of protein-DNA reactions), it also has the advantage of very high specificity for double-stranded DNA. Green Fluorescent Protein fusion proteins, specifically core histones, have also been used to observe in vivo chromatin structure (Kanda et al., 1998; Sadoni et al., 2001), but there is experimental evidence that the core histones can detach and move independently of the DNA (Kimura and Cook, 2001; Siino et al., 2002). To measure the DNA dynamics in live cells without worrying about convolution with protein diffusion rates, H33342 is a reasonable choice so long as caution is used in the interpretation of the results.
Possible artifacts due to dye dissociation
In addition to the possible cytotoxic effects of our probe molecule H33342, we must also be concerned about experimental artifacts due to its binding and unbinding to the DNA during our measurements. Studies on closely related dyes like Hoechst 33258 obtain a dye-DNA binding constant on the order of 109 M−1, and a dissociation time on the order of 1 s (Loontiens et al., 1990). If the dye undergoes an unbinding-diffuse-rebinding sequence of events, this would contribute to the apparent diffusion of the fluorescent species, but would not reflect actual chromatin motion. It is not straightforward to estimate the magnitude of this effect, since the unbinding rate must be multiplied by the probability of escape from a given site. Otherwise, the dye will rebind to the same site within a nanosecond, with no net translational motion. We have performed several control experiments to rule out the role of free dye diffusion in our data. First, if free dye diffusion contributes to the observed 2P-c-FRAPP decays, then it should also lead to a normal FRAP recovery on the predicted timescale of roughly 10 min. This is because the dye should not be limited by the same constraints that force the much larger chromatin fibers to remain localized in the nucleus. Fig. 4 shows that there is no such recovery of the bleach spot, even after 60 min. Second, following Abney et al. (1997), we use formaldehyde to fix both cells and nuclei and then stain with H33342. Unlike what was previously observed for ethidium bromide, there is no fast recovery component with H33342 in these cross-linked systems, even at the highest salt concentrations used. Very long scans reveal that in these systems the oscillations do decay with a 1/e time of ∼800 s, which we estimate to be the rate due to dye diffusion alone. This timescale is too slow to affect the data analysis in this paper. Last, experiments on isolated nuclei were repeated with a chemically distinct DNA stain, YOYO-1, yielding results identical to those obtained with H33342.
FIGURE 4.

Fluorescence images of a live cell nucleus, stained with Hoechst 33342, after exposure to a two-photon photobleaching pulse, which is indicated by the arrow in the 0-min panel. Over the course of 1 h, the bleached spot does not disappear. The large dark spot in the center of the nucleus is the nucleolus, and the scale bar is 10 μm.
Live cell data
Cells for all laser experiments were fully attached and were clearly in interphase as judged by the presence of a nucleolus. The scan shown in Fig. 2 a is done in a 1 μm diameter area in the middle of the cell shown in the inset. During the prebleach scan (t < 0), as the standing wave is translated back and forth across the sample (∼350 nm in each direction) by the piezo-driven mirror, there are no oscillations in the fluorescence signal. This indicates that the concentration of H33342 bound to chromatin is homogeneous on the length scale of the fringe spacing of the standing wave (∼300 nm). At t = 0, the motion of the piezo is stopped and the laser intensity increased by a factor of 20 for 10 ms, photobleaching the dye molecules at the peaks of the standing wave pattern. As the attenuated standing wave probe is translated over the sample, the peaks and valleys of the probe passing over the peaks and valleys of the photobleached pattern cause an oscillatory fluorescence signal. As the chromatin diffuses, the fringe pattern induced by the bleach decreases, and the signal oscillations die out within seconds. The abrupt phase changes in the signal are due to the back and forth motions of the piezo. Each uninterrupted scan of the piezo constitutes a subset of data. The amplitude of the oscillations in each subset is plotted versus time (Fig. 2 b), and the decay of this amplitude is fitted to a single exponential (solid line). The average τosc of 6 s and an average D of 5 × 10−4 μm2/s (Eq. 2a) is in good agreement with other reports for other cell types measured using single particle tracking (Marshall et al., 1997; Heun et al., 2001; Vazquez et al., 2001; Chubb et al., 2002; Bornfleth et al., 1999). There is considerable variation of the measured diffusion constant from cell to cell, and this is summarized in Fig. 3, which shows a histogram of 56 measurements on cells from samples measured on three different days. The distribution is approximately Gaussian, but with a tail extending to longer decay times. There are not sufficient statistics to determine whether the peak at τosc = 12 s indicates a slight bimodal distribution or is simply a statistical fluctuation. The important point is that this distribution is inherent in the cells, as opposed to being measurement noise, since it does not narrow after averaging multiple experiments. It is also present in multiple measurements on a single cell, and for different cell populations measured on different days.
FIGURE 2.

(a) Experimental data obtained from a 2P-c-FRAPP experiment on a live XTC-2 cell. A transmitted light image of the cell is shown in the inset, and the excitation occurs at the center of the cross-hairs. (b) Decay of the oscillation amplitude for the data in a, where each point represents the oscillation amplitude during a half cycle of the piezo phase shifter, along with an exponential fit to the data points.
FIGURE 3.

Histogram of the decay times for 56 live cell experiments, like the one shown in Fig. 2.
One concern with this data is that the measured dynamics may be strongly perturbed by the bleaching pulse, with different cells exhibiting different damage susceptibilities. Our multiple bleach experiments are evidence against such an effect, since one would expect that such damage would become progressively worse with each pulse in the same spot. Also, successive measurements at different locations within a single cell yield random diffusion rates, and not a progressive slowing down, as would be expected if accumulated photodamage were playing an important role.
In addition to the interfringe spacing, the FRAPP experiment has another inherent length scale, which is the two-photon FRAP length scale, ∼1 μm in our experiment. If the diffusion of chromatin were simple Brownian motion, the FRAP recovery would be halfway complete on a timescale ∼100 times longer than the τosc time of the fringe decay (Davis and Bardeen, 2002). In the case of chromatin diffusion in XTC-2 cells, this would predict a τ1/2 for the FRAP recovery of 8 min. We do not see this recovery in our experiments: for example, the bleach trace in Fig. 2 does not recover even after 10 min of scanning. This persistent bleach can be most clearly seen in Fig. 4, which shows a series of fluorescence images of a live XTC-2 cell nucleus after a spot was photobleached in the upper left corner. Over the course of an hour, the spot fades slightly but does not disappear. This lack of recovery is consistent with previous FRAP experiments (Abney et al., 1997), and shows how the extra spatial resolution afforded by the standing wave geometry is necessary to resolve the small displacements that are actually occurring in vivo.
Isolated nuclei
To investigate the origins of the DNA diffusion observed in live cells, we made identical measurements on isolated nuclei. Fig. 5 shows the results of the 2P-c-FRAPP and normal one-beam FRAP experiments done on nuclei at different ionic strengths. The top panels show fluorescence images of the nuclei as the concentration of NaCl is varied from 20 mM to 2 M. At physiological ionic strength (80 mM, Fig. 5 b), the nucleus retains its overall structure as compared to the live cell nucleus in Fig. 2. The motion observed in the live cell is completely absent in the isolated nucleus, however. At lower salt concentrations, when the chromatin adopts a “beads-on-a-string” structure due to less screening of the nucleosomes (Gerchman and Ramakrishnan, 1987; Clark and Kimura, 1990) (20 mM, Fig. 5 a), the heterogeneity visible in Fig. 5 b disappears, and the nuclear DNA appears homogeneous. Despite this loss of structure, there is still no motion on the experimental timescale. As the ionic strength is increased, the chromatin again becomes more diffuse, as shown in Fig. 5, c and d, at 400 and 2 M NaCl concentrations. At these higher salt concentrations, when the histone-DNA coulomb interaction begins to be screened, we begin to see considerable motion. The fluorescence recovery rate increases rapidly, and it becomes impossible to detect a photobleached pattern on our experimental timescale. The 2P-c-FRAPP experiments become simple FRAP experiments. The data shown for both 400 mM and 2 M NaCl solutions are averages of single FRAP scans on 20 different nuclei. At 400 mM, both the core histones and the linker histones H1 are still associated with the DNA (Thorne et al., 1998; Spadafora et al., 1979). If we expose the nuclei to 400 mM NaCl and then return them to physiological ionic strength, the chromatin is again stationary, and the nuclear appearance visible in Fig. 5 b is recovered. This shows that there is no irreversible loss of a specific protein or structural component that occurs at high salt concentrations. The measured diffusion constant at 400 mM is ∼1 μm2/s, almost four orders of magnitude faster than that of a live cell. At salt concentrations between 80 mM and 400 mM NaCl, we do not see an incremental increase in the diffusion rate with increasing salt concentration. Instead, it appears that the motion is “all or nothing.” Within the same scan, there can be either fast moving or stationary components, or both, but nothing that is as slow as the motion we see in live cells. At 2 M NaCl, where the histones are completely extracted from the chromatin (Thorne et al., 1998), the FRAP recovery occurs on a timescale similar to that seen at 400 mM. The lack of complete recovery at this NaCl concentration is not understood—it may be due to clumping of the DNA at these very high ionic strengths. It is important to note that although both low and high salt concentrations result in a loss of nuclear structure, only higher salt concentrations lead to faster diffusion.
FIGURE 5.

(Top) Fluorescence image of isolated nucleus. (Bottom) 2P-c-FRAPP experiment in nucleus at (a) 20 mM NaCl, (b) 80 mM NaCl (physiological); normal FRAP experiment at (c) 400 mM NaCl, and (d) 2 M NaCl.
Cross-linking experiments
To further investigate the chemical origin of the observed chromatin motion, we have used both chemical and photochemical cross-linking to see whether the association of proteins, with each other or with DNA, affects the dynamics. Fig. 6 shows results for nuclei in 400 mM NaCl before (Fig. 6 a) and after cross-linking (Fig. 6, a and b). Both formaldehyde and UV exposure with H33342 staining lead to the same results: where previously the DNA had undergone rapid diffusion, it is now completely stationary. Formaldehyde is rather nonspecific, forming DNA-DNA, DNA-protein, and protein-protein cross-links (Jackson, 1999), whereas H33342 appears to generate predominantly histone-protein-DNA cross-links (Davis and Bardeen, 2003). This suggests that DNA-protein cross-linking is the major contributor to the freezing of the motion in these high ionic strength samples. To investigate this further, the experiment was repeated in nuclei in which all the core histone proteins had been extracted using a 2 M NaCl solution. In this sample, shown in Fig. 7, UV exposure does not have a significant effect on the motion. However, in a 600-mM NaCl solution, when the linker histone H1 is dissociated from the DNA (Spadafora et al., 1979), UV exposure was still able to completely freeze the motion, similar to what is seen at 400 mM.
FIGURE 6.

Fluorescence recovery signal for isolated nucleus stained with H33342 (a) at 400 mM NaCl, (b) for same nucleus as in a but after exposure to ∼20 J/cm2 of 365 nm cross-linking light, (c) at 400 mM after exposure to 400 mM formaldehyde cross-linking conditions.
FIGURE 7.

(a) Fluorescence recovery signal for isolated nuclei stained with H33342 at 2 M NaCl, where the proteins are completely dissociated from the DNA. (b) Same as in a but after exposure to ∼20 J/cm2 of 365 nm cross-linking light. Both a and b are averages of five scans.
The fact that cross-linking freezes chromatin motion in isolated nuclei led us to try similar experiments in live cells. The results of the cross-linking experiments are shown in Fig. 8 for a single cell. The 5-s decay of the fringes seen in the live cell (Fig. 8 a) becomes too slow to detect after UV exposure cross-links the histones to the DNA (Fig. 8 b). As in isolated nuclei, protein-DNA cross-linking freezes the 100 nm scale motion of chromatin in live cells.
FIGURE 8.

2P-c-FRAPP experiment on (a) a live cell before exposure to ∼20 J/cm2 of 365 nm cross-linking light, and (b) the same cell after exposure.
DISCUSSION
Origin of short-range chromatin diffusion
There are several candidates for the chemical origin of chromatin's diffusive motion on the 100 nm length scale in live cells. The simplest scenario is that the observed dynamics result from thermal fluctuations that drive the motion of a semiflexible random polymer. Such motions could be understood solely in the context of regular polymer physics in dense solutions, e.g., in terms of reptation or the Rouse-Zimm model. If this were the case, studying the diffusive motion of chromatin would yield information on its local environment and persistence length, but not on the chemical interactions that are of the most interest in terms of nucleosomal rearrangements that promote transcription. The evidence for thermal diffusion of chromatin in live cells is mixed, and comes mainly from looking at whether the motion depends on the metabolic state of the cell. Although one group saw no effect on diffusion in yeast cells upon exposure to sodium azide, which blocks metabolism in cells (Marshall et al., 1997), later experiments by a different group on the same cell type did observe a complete cessation of motion after exposure (Heun et al., 2001). If the dynamics of chromatin in live cells were due to simple Brownian motion, we would expect to observe similar dynamics in isolated nuclei at physiological ionic strength. These nuclei retain their overall morphology and internal structure, according to our fluorescence images, but many of the nuclear proteins are expected to leak out through the large nuclear pores, which should diminish crowding and increase the diffusion rate. But instead of more rapid diffusion, at 80 mM NaCl in cell media we see no motion at all. A similar lack of diffusion is observed in dead cells which have detached from the collagen substrate, even when they are next to live cells which show measurable diffusive decays. Under physiological conditions, in both isolated nuclei and nonliving, intact cells, there is no observable DNA motion. In agreement with Gasser and co-workers (Heun et al., 2001), our data indicate that some degree of cellular activity is required for short-range chromatin motion.
Isolated nuclei are clearly different from live cells—the next question concerns the origin of this difference. Chromatin diffusion could result from interactions between the genomic DNA and the large number of nuclear proteins, whose task is to physically rearrange and remodel nucleosomes to facilitate processes like transcription and replication. Most of these proteins are absent in isolated nuclei, and it may be that their absence leads to stationary histone proteins and a lack of nucleosome motions. In this context, it is worth noting that ATP-dependent nucleosome sliding has been observed in isolated nuclei (Varga-Weisz et al., 1995). If this is the case, then it suggests that the 100-nm fluctuations reflect protein-DNA interactions on much smaller length scales and may provide information about molecular-level dynamics like nucleosome sliding. Rather than attempt to investigate the role of individual chromatin remodeling factors, we have concentrated on whether the most basic protein-DNA interaction, namely that of the core histones with the DNA, can affect the chromatin diffusion in a systematic way. We used several methods to change the strength of the histone binding to DNA. The most straightforward approach is to vary the ionic strength of the solution, which has been shown to affect histone-DNA binding kinetics and nucleosome sliding for in vitro systems. As the ionic strength is lowered, the chromatin partially unfolds to a “beads-on-a-string” structure due to nucleosome-nucleosome coulomb repulsion which is no longer effectively screened. In these samples, despite the diffuse nature of the unfolded chromatin, we see that most of the chromatin remains stationary. Less screening leads to the unfolding of the fiber to separate the nucleosomes, but does not decrease the strength of the histone-DNA interaction. At higher ionic strength (400 mM NaCl), where the histone-DNA interactions become screened, the chromatin structure also disappears, but now the diffusion is similar to the free DNA diffusion seen in 2 M solution. At 400 mM, even though the histone octamers are not fully dissociated from the DNA, their ability to move along the DNA strand is significantly enhanced. This enhancement is reversible, as would be expected for noncovalent interactions of this type. Our results are consistent with in vitro results that observe increased nucleosome sliding, using gel electrophoresis, at similar ranges of ionic strength (Meersseman et al., 1992; Weischet, 1979). Furthermore, at these ionic strengths, increased rotational diffusion of chromatin in isolated nuclei has been observed as well (Selvin et al., 1990).
It is unclear why the slow motion seen in live cells is not recovered by gradually increasing the NaCl concentration in isolated nuclei. This may be because the mechanisms of motion in high salt nuclei and live cells are probably completely different. The specific protein modifications that live cells use to control chromatin conformation are much more local and nonperturbing than the large swings in ionic strength employed in these experiments, which average over all the DNA-DNA and DNA-histone interactions. The strength of the DNA-histone interactions can also vary with DNA sequence, and some may require more screening to loosen the contacts. It is possible that once the histone-DNA interactions are screened to the point where sliding can occur, the sliding motion is relatively fast. In our experiment, one bleached spot samples thousands of nucleosomes. The histone-DNA interactions that are not loosened collectively appear as the stationary component, whereas the sliding nucleosomes show diffusive motion, until there is enough NaCl to screen even the strongest of the DNA-histone contacts, and all of the nucleosomes are sliding, making it impossible to detect a photobleached pattern.
A second piece of evidence that the observed diffusion is due to nucleosomal dynamics is the fact that motion is frozen in both live cells and isolated nuclei when the core histone proteins are cross-linked to the DNA. Although the formaldehyde cross-links are nonspecific, the UV-induced cross-linking of chromatin stained with H33342 is more selective for the core histones (Davis and Bardeen, 2003). At 400 mM NaCl, the large-scale, free DNA-like motions are completely stopped after UV irradiation. At 600 mM NaCl, where histone H1 and nonhistone proteins are dissociated from the DNA, UV irradiation can still completely stop the motion. At 2 M NaCl, however, where the core histones are completely dissociated from the DNA, the motion is barely affected by UV irradiation. The small effect that is observed may be due to incomplete dissociation of the core histones, or a small number of dissociated histones cross-linking with DNA bases that happen to be close enough. Clearly, however, covalently attaching proteins to the DNA freezes the chromatin dynamics we are measuring in isolated nuclei. Although we know that the UV cross-linking leads to attachment of the core histones, we cannot rule out the possibility that other proteins, which would still remain in the nucleus at 400 mM, are also cross-linked and play a role in stopping the motion. Obvious candidates would be the protein components of a nuclear matrix, if it exists. As in the isolated nuclei, the motion observed in live cells can be completely stopped by UV irradiation. This fact indicates that at least some of the proteins responsible for the motion in live cells are still present in nuclei at high salt concentrations.
Chromatin dynamics in live cells
In live cells, the absolute rate of diffusion (in our case, 5 × 10−4 μm2/s) probably depends on many factors, including the degree of attachment of the chromatin to a nuclear matrix, the amount of macromolecular crowding, and the local density of chromatin itself. Presumably, cell-to-cell variations in these factors contribute to the distribution of diffusion rates we observe in Fig. 3. There is at least one additional factor, however, that is not related to the static structure of the nucleus. The dependence of the chromatin diffusion on variables like ionic strength and cross-linking is consistent with this diffusion arising from the motility of its constituent histones. The mechanism for this motility in live cells is an open question. As mentioned earlier, there is no shortage of nuclear proteins designed to modify chromatin structure. The difference between the dynamics of chromatin in live cells and in isolated nuclei at physiological ionic strength is likely due to the presence of chromatin remodeling factors in live cells (Caserta et al., 2002; Becker, 2002). Examples of such factors present in interphase nuclei, which often work through selective histone deacetylation (Tong et al., 1998), include ATP-dependent protein complexes like NURF (Hamiche et al., 1999), and various members of the SWI/ISWI family like Mi-2 and ACF which have been observed in X. laevis cells (Guschin et al., 2000a,b). All of these complexes can cause small-scale rearrangements of the nucleosomes along the DNA as it undergoes transcription and replication throughout the nucleus. Whether the total activity of these factors is sufficient to produce the chromatin motion observed in live cells is unknown, since even the fraction of the chromatin that is undergoing remodeling is still an area of active research. Other unknown factors may also contribute to chromatin diffusion; for example, if the chromatin is attached to a nuclear matrix and those attachments are affected by the activity of other types of proteins, that would provide a completely different mechanism for the 100 nm scale fluctuations we observe. It is probably unreasonable to expect that the dynamics of the extremely large and tangled chromatin fibers in cell nuclei can be understood in terms of a single parameter like the histone sliding rate, in units of base pairs per second. But our results do indicate that it may be fruitful to try to quantitatively connect 100 nm scale diffusive motion of chromatin to nucleosomal dynamics and the detailed biochemistry occurring in a small region of the nucleus. This would provide cell biologists a diagnostic tool to characterize local transcriptional activity in live cells using a noninvasive fluorescence microscopy technique.
CONCLUSIONS
By using the vital DNA stain Hoechst 33342 and a two-photon standing wave photobleaching experiment with 100 nm spatial resolution, we have characterized the variation of the diffusive motion of chromatin in live cells. By varying the ionic strength and using chemical and photocross-linking experiments, we have shown that this motion follows the same qualitative trends as expected for the mobility of histone proteins along the DNA strands. Other interactions may also play a role, but our data are consistent with the simple picture that nucleosomal motion leads to more flexibility in the chromatin fibers and thus more diffusive motion. This observation provides evidence that in vivo chromatin diffusion on a 100 nm length scale, as studied by cell biologists, may be connected to the in vitro nucleosomal dynamics studied by biochemists. This connection may lead to new ways of assaying localized biochemical activity in live cells by observing larger scale macromolecular dynamics via fluorescence microscopy.
Acknowledgments
We acknowledge support from the Research Corporation (RIA 0436) and the University of Illinois Research Board. We also acknowledge the help of the School of Chemical Sciences Cell Media Facility.
References
- Abney, J. R., B. Cutler, M. L. Fillbach, D. Axelrod, and B. A. Scalettar. 1997. Chromatin dynamics in interphase nuclei and its implications for nuclear structure. J. Cell Biol. 137:1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, B., D. L. Farkas, D. L. Taylor, and F. Lanni. 1993. Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation. Nature. 366:44–48. [DOI] [PubMed] [Google Scholar]
- Becker, P. B. 2002. Nucleosome sliding: facts and fiction. EMBO J. 21:4749–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornfleth, H., P. Edelmann, D. Zink, T. Cremer, and C. Cremer. 1999. Quantitative motion analysis of subchromosomal foci in living cells using four-dimensional microscopy. Biophys. J. 77:2871–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caserta, M., L. Verdone, and E. Di Mauro. 2002. Aspects of nucleosomal positional flexibility and fluidity. Chembiochem. 3:1172–1182. [DOI] [PubMed] [Google Scholar]
- Chubb, J. R., S. Boyle, P. Perry, and W. A. Bickmore. 2002. Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. 12:439–445. [DOI] [PubMed] [Google Scholar]
- Cicerone, M. T., F. R. Blackburn, and M. D. Ediger. 1995. Anomalous diffusion of probe molecules in polystyrene: evidence for spatially heterogeneous segmental dynamics. Macromolecules. 28:8224–8232. [Google Scholar]
- Clark, D. J., and T. Kimura. 1990. Electrostatic mechanism of chromatin folding. J. Mol. Biol. 211:883–896. [DOI] [PubMed] [Google Scholar]
- Cremer, T., and C. Cremer. 2001. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2:292–301. [DOI] [PubMed] [Google Scholar]
- Davis, S. K., and C. J. Bardeen. 2002. Using two-photon standing waves and patterned photobleaching to measure diffusion from nanometers to microns in biological systems. Rev. Sci. Inst. 73:2128–2135. [Google Scholar]
- Davis, S. K., and C. J. Bardeen. 2003. Cross-linking of histone proteins to DNA by UV illumination of chromatin stained with Hoechst 33342. Photochem. Photobiol. 77:675–679. [DOI] [PubMed] [Google Scholar]
- Davoust, J., P. F. Devaux, and L. Leger. 1982. Fringe pattern photobleaching, a new method for the measurement of transport coefficients of biological macromolecules. EMBO J. 1:1233–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand, R. E., and P. L. Olive. 1982. Cytotoxicity, mutagenicity and DNA damage by Hoechst 33342. J. Histochem. Cytochem. 30:111–116. [DOI] [PubMed] [Google Scholar]
- Gerchman, S. E., and V. Ramakrishnan. 1987. Chromatin higher order structure studied by neutron scattering and scanning transmission electron microscopy. Proc. Natl. Acad. Sci. USA. 84:7802–7806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena, S., and M. C. Rykowski. 2000. Direct evidence for interphase chromosome movement during the mid-blastula transition in Drosophila. Curr. Biol. 10:285–288. [DOI] [PubMed] [Google Scholar]
- Guschin, D., T. M. Geiman, N. Kikyo, D. J. Tremethick, A. P. Wolffe, and P. A. Wade. 2000a. Multiple ISWI ATPase complexes from Xenopus laevis. J. Biol. Chem. 275:35248–35255. [DOI] [PubMed] [Google Scholar]
- Guschin, D., P. A. Wade, N. Kikyo, and A. P. Wolffe. 2000b. ATP-dependent histone octamer mobilization and histone deacetylation mediated by the Mi-2 chromatin remodeling complex. Biochemistry. 39:5238–5245. [DOI] [PubMed] [Google Scholar]
- Gustafsson, M. G. L., D. A. Agard, and J. W. Sedat. 1999. I5M: 3D widefield light microscopy with better than 100 nm axial resolution. J. Microsc. 195:10–16. [DOI] [PubMed] [Google Scholar]
- Hamiche, A., R. Sandaltzopoulos, D. A. Gdula, and C. Wu. 1999. ATP-dependent histone octamer sliding mediated by the chromatin remodeling complex NURF. Cell. 97:833–842. [DOI] [PubMed] [Google Scholar]
- Hansen, J. C. 2002. Conformational dynamics of the chromatin fiber in solution. Annu. Rev. Biophys. Biomol. Struct. 31:361–392. [DOI] [PubMed] [Google Scholar]
- Hell, S. W., M. Schrader, and H. T. van der Voort. 1997. Far-field fluorescence microscopy with three-dimensional resolution in the 100-nm range. J. Microsc. 187:1–7. [DOI] [PubMed] [Google Scholar]
- Heun, P., T. Laroche, K. Shimada, P. Furrer, and S. M. Gasser. 2001. Chromosome dynamics in the yeast interphase nucleus. Science. 294:2181–2186. [DOI] [PubMed] [Google Scholar]
- Jackson, V. 1999. Formaldehyde cross-linking for studying nucleosomal dynamics. Methods. 17:125–139. [DOI] [PubMed] [Google Scholar]
- Kanda, T., K. F. Sullivan, and G. M. Wahl. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385. [DOI] [PubMed] [Google Scholar]
- Kimura, H., and P. R. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamond, A. I., and W. C. Earnshaw. 1998. Structure and function in the nucleus. Science. 280:547–553. [DOI] [PubMed] [Google Scholar]
- Lanni, F., and B. R. Ware. 1982. Modulation detection of fluorescence photobleaching recovery. Rev. Sci. Inst. 53:905–908. [Google Scholar]
- Lever, M. A., J. P. H. Th'ng, X. Sun, and M. J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin of living human cells. Nature. 408:873–876. [DOI] [PubMed] [Google Scholar]
- Loontiens, F. G., P. Regenfuss, A. Zechel, L. Dumortier, and R. M. Clegg. 1990. Binding characteristics of Hoechst 33258 with calf thymus DNA, Poly[d(A-T)], and d(CCGGAATTCCGG): multiple stoichiometries and determination of tight binding with a wide spectrum of site affinities. Biochemistry. 29:9029–9039. [DOI] [PubMed] [Google Scholar]
- Luger, K. 2003. Structure and dynamic behavior of nucleosomes. Curr. Opin. Genet. Dev. 13:127–135. [DOI] [PubMed] [Google Scholar]
- Marshall, W. F. 2002. Order and disorder in the nucleus. Curr. Biol. 12:R185–R192. [DOI] [PubMed] [Google Scholar]
- Marshall, W. F., A. Straight, J. F. Marko, J. Swedlow, A. Dernburg, A. Belmont, A. W. Murray, D. A. Agard, and J. W. Sedat. 1997. Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. 7:930–939. [DOI] [PubMed] [Google Scholar]
- Meersseman, G., S. Pennings, and E. M. Bradbury. 1992. Mobile nucleosomes—a general behavior. EMBO J. 11:2951–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T., A. Gunjan, R. Hock, M. Bustin, and D. T. Brown. 2000. Dynamic binding of histone H1 to chromatin in living cells. Nature. 408:877–881. [DOI] [PubMed] [Google Scholar]
- Phair, R. D., and T. Misteli. 2000. High mobility of proteins in the mammalian cell nucleus. Nature. 404:604–609. [DOI] [PubMed] [Google Scholar]
- Sadoni, N., K. F. Sullivan, P. Weinzierl, E. H. K. Stelzer, and D. Zink. 2001. Large-scale chromatin fibers of living cells display a discontinuous functional organization. Chromosoma. 110:39–51. [DOI] [PubMed] [Google Scholar]
- Schlax, P. J., M. W. Capp, and M. T. Record. 1995. Inhibition of transcription initiation by lac repressor. J. Mol. Biol. 245:331–350. [DOI] [PubMed] [Google Scholar]
- Selvin, P. R., B. A. Scalettar, J. P. Langmore, D. Axelrod, M. P. Klein, and J. E. Hearst. 1990. A polarized photobleaching study of chromatin reorientation in intact nuclei. J. Mol. Biol. 214:911–922. [DOI] [PubMed] [Google Scholar]
- Siino, J. S., I. B. Nazarov, M. P. Svetlova, L. V. Solovjeva, R. H. Adamson, I. A. Zalenskaya, P. M. Yau, E. M. Bradbury, and N. V. Tomilin. 2002. Photobleaching of GFP-labeled H2AX in chromatin: H2AX has low diffusional mobility in the nucleus. Biochem. Biophys. Res. Commun. 297:1318–1323. [DOI] [PubMed] [Google Scholar]
- Smith, P. J., S. M. Bell, A. Dee, and H. Sykes. 1990. Involvement of DNA topoisomerase II in the selective resistance of a mammalian cell mutant to DNA minor groove ligands: ligand-induced DNA-protein crosslinking and responses to topoisomerase poisons. Carcinogenesis. 11:659–665. [DOI] [PubMed] [Google Scholar]
- Smith, B. A., and H. M. McConnell. 1978. Determination of molecular motion in membranes using periodic pattern photobleaching. Proc. Natl. Acad. Sci. USA. 75:2759–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spadafora, C., P. Oudet, and P. Chambon. 1979. Rearrangement of chromatin structure induced by increasing ionic strength and temperature. Eur. J. Biochem. 100:225–235. [DOI] [PubMed] [Google Scholar]
- Thorne, A. W., P. D. Cary, and C. Crane-Robinson. 1998. Extraction and separation of core histones and non-histone chromosomal proteins. In Chromatin—A Practical Approach. H. Gould, editor. Oxford University Press, New York, NY. 35–58.
- Tong, J. K., C. A. Hassig, G. R. Schnitzler, R. E. Kingston, and S. L. Schreiber. 1998. Chromatin deacetylation by an ATP-dependent nucleosome remodeling complex. Nature. 395:917–921. [DOI] [PubMed] [Google Scholar]
- Trumbar, T., G. Sudlow, and A. S. Belmont. 1999. Large-scale chromatin unfolding and remodeling induced by VP16 acidic activation domain. J. Cell. Biol. 145:1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto, T., N. Hashiguchi, S. M. Janicki, T. Tumbar, A. S. Belmont, and D. L. Spector. 2000. Visualization of gene activity in living cells. Nat. Cell Biol. 2:871–878. [DOI] [PubMed] [Google Scholar]
- Varga-Weisz, P. D., T. A. Blank, and P. B. Becker. 1995. Energy-dependent chromatin accessibility and nucleosome mobility in a cell-free system. EMBO J. 14:2209–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez, J., A. S. Belmont, and J. W. Sedat. 2001. Multiple regimes of constrained chromosome motion are regulated in the interphase Drosophila nucleus. Curr. Biol. 11:1227–1239. [DOI] [PubMed] [Google Scholar]
- Vermaak, D., and A. P. Wolffe. 1998. Chromatin and chromosomal controls in development. Dev. Genet. 22:1–6. [DOI] [PubMed] [Google Scholar]
- Weischet, W. O. 1979. On the de novo formation of compact oligonucleosomes at high ionic strength. Evidence for nucleosomal sliding in high salt. Nucleic Acids Res. 7:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widom, J. 1998. Structure, dynamics, and function of chromatin in vitro. Annu. Rev. Biophys. Biomol. Struct. 27:285–327. [DOI] [PubMed] [Google Scholar]
- Wolffe, A. P., and J. C. Hansen. 2001. Nuclear visions: functional flexibility from structural instability. Cell. 104:631–634. [DOI] [PubMed] [Google Scholar]