

Abstract
Many dicationic amidine compounds bind in the DNA minor groove and have excellent biological activity against a range of infectious diseases. Para-substituted aromatic diamidines such as furamidine, which is currently being tested against trypanosomiasis in humans, and berenil, which is used in animals, are typical examples of this class. Recently, a meta-substituted diamidine, CGP 40215A, has been found to have excellent antitrypanosomal activity. The compound has a linear, conjugated linking group that can be protonated under physiological conditions when the compound interacts with DNA. Structural and molecular dynamics analysis of the DNA complex indicated an unusual AT-specific complex that involved water-mediated H-bonds between one amidine of the compound and DNA bases at the floor of the minor groove. To investigate this unique system in more detail DNase I footprinting, surface plasmon resonance biosensor techniques, linear dichroism, circular dichroism, ultraviolet-visible spectroscopy, and additional molecular dynamics simulations have been conducted. Spectrophotometric titrations of CGP 40215A binding to poly(dAT)2 have characteristics of DNA-binding-induced spectral changes as well as effects due to binding-induced protonation of the compound linker. Both footprinting and surface plasmon resonance results show that this compound has a high affinity for AT-rich sequences of DNA but very weak binding to GC sequences. The dissociation kinetics of the CGP 40215A–DNA complex are much slower than with similar diamidines such as berenil. The linear dichroism results support a minor-groove complex for the compound in AT DNA sequences. Molecular dynamics studies complement the structural analysis and provide a clear picture of the importance of water in mediating the dynamic interactions between the ligand and the DNA bases in the minor groove.
INTRODUCTION
Aromatic diamidines, such as berenil, pentamidine, and related compounds (Fig. 1), were discovered some years ago to have excellent activity against an array of infectious diseases from Pneumocystis carinii pneumonia to trypanosomiasis (Bell et al., 1990; Boykin et al., 1995, 1998; Das and Boykin, 1977; Francesconi et al., 1999; Rahmathullah et al., 1999; Reddy et al., 1999; Stephens et al., 2001; Tidwell and Boykin, 2003; Tidwell et al. 1990a,b). The serious human health problems caused by these diseases in developing countries as well as in developed countries after the spread of AIDS have stimulated research to find new drugs against the diseases. Furamidine (DB 75 in Fig. 1), which has a broad spectrum of activities against a range of infectious disease organisms (Boykin et al., 1995, 1998; Das and Boykin, 1977; Francesconi et al., 1999; Hopkins et al., 1998; Stephens et al., 2001; Tidwell and Boykin, 2003), is an example of a diamidine successfully developed as part of this expanded research effort (Tidwell and Boykin, 2003). An orally active prodrug of furamidine has passed Phase I clinical trials and is nearing the end of successful Phase II trials against trypanosomiasis (Tidwell and Boykin, 2003). It is clear that aromatic diamidines continue to present a promising route for development of drugs against organisms that cause infectious diseases.
FIGURE 1.

DNA hairpins and the structures of berenil, DB 75, DB 359, pentamidine, and CGP 40215A.
All of the biologically active aromatic diamidines studied to date have been found to bind strongly to AT-rich sequences in the minor groove of DNA, and it is clear that there is a correlation between DNA binding and the antiparasitic biological activities (Bell et al., 1991, 1993, 1990; Tidwell and Boykin, 2003). Design of dications that target the minor groove of DNA has evolved as a productive concept for discovering new antiparasitic drugs, and studies of the DNA complexes of these compounds have been conducted as part of the design effort. A number of diamidines have been crystallized with the DNA duplex d(CGCGAATTCGCG)2 and these structures provide valuable models for drug development in the diamidine series. Structures of DNA complexes of furamidine, berenil, and pentamidine, for example, reveal that they all bind in the DNA minor groove at the central AATT sequence. These compounds penetrate deeply into the groove and fit snugly between the walls of the groove. Their amidines form H-bonds with thymine-O2 and/or adenine-N3 acceptor groups on the edges of the bases at the floor of the groove (Edwards et al., 1992, Laughton et al., 1996, Neidle, 2002). The amino group of G protrudes into the minor groove and prevents the compounds from assuming their preferred orientation deep in the minor groove (Blackburn and Gait, 1997; Bloomfield et al., 2000; Saenger, 1984; Wemmer, 2000). In the same manner substituents placed on the inner face of the compound will clash with bases at the bottom of the groove and will not allow the compound to contact DNA in an optimal manner.
Compound curvature is also critical for complex formation and strong binding in the minor groove. The diamidines as well as other minor groove-binding agents either match the curvature of the DNA minor groove in their solution state or are able to adopt a low energy conformation to complement the DNA groove shape in the complex (Cory et al., 1992). Compounds that have too great or too small a curvature cannot optimize contacts in the minor groove and have reduced binding affinities. The importance of compound shape was recently demonstrated with the meta-furamidine analog, DB 359 (Fig. 1), in which the amidine groups have been moved from the para-position in furamidine to the meta-position on the phenyl ring (Nguyen et al., 2002b). The meta-analog cannot match the curvature of the minor groove and it binds significantly more weakly to AT DNA sequences than furamidine. The meta-compound also loses binding specificity and it binds to AT- and GC-rich sequences with similar, relatively low affinities. The compound also completely changes its DNA binding mode from a minor groove to an intercalation complex. Although furamidine and some related aromatic diamidines can also bind to DNA by intercalation in GC-rich sequences, this is a secondary and relatively weak interaction relative to the primary AT specific binding mode in the minor groove (Nguyen et al., 2002b). DNase I footprinting, for example, shows significant DNA protection with footprints in AT-rich sequences of at least four continuous AT basepairs for these compounds but no significant footprints in GC rich sequences (Boykin et al., 1995).
The research on diamidines has been successful in demonstrating a correlation between the structure and biological activity of these compounds with their ability to bind to AT sites in the minor groove (Tidwell and Boykin, 2003). Given this extensive and self-consistent data set, it was initially surprising that the meta-substituted diamidine, CGP 40251A, (Fig. 1) has excellent activity against Trypanosoma brucei rhodesiense (Brun et al., 1996). Preliminary results have shown that CGP 40215A binds strongly to the minor groove of the AATT sequence despite the fact that it lacks curvature compared to other related minor-groove binding diamidines and does not match the shape of DNA (Nguyen et al., 2002a). Unlike the meta-furamidine analog, however, CGP 40215A has an extended linker that assists in binding of the compound to the DNA minor groove. Because CGP 40215A represents a new structural type of biologically active diamidine for targeting of the DNA minor groove, we report here a detailed study of the DNA interactions of this compound. Biosensor surface plasmon resonance (SPR), DNase I footprinting, ultraviolet (UV) spectroscopy, linear, and circular dichroism methods have been utilized to study the binding specificity, the affinity, and kinetics as well as the binding-linked protonation, and the orientiation of the bound compound. To better understand these types of interactions at the molecular level, molecular dynamic simulations were performed and the results are compared with those from experiments. All of these findings not only provide useful information to better understand the DNA-binding properties of the atypical CGP 40215A molecule but also yield general ideas to help in the design of novel DNA binding meta-substituted diamidines utilizing the unique features of this complex.
MATERIALS AND METHODS
DNA and compounds
The double-stranded polymers poly(dAT)2 and poly(dGC)2 were purchased from Pharmacia (Pfizer, New York, NY), and they were used in UV-visible (UV-VIS) titration experiments. In SPR and CD experiments, 5′-biotin labeled DNA hairpins (Fig. 1) were used. The hairpins were purchased with HPLC purification (Midland Certified Reagent Co., Midland, TX) and used without further purification. The MES buffer used in these experiments contained 0.010 M [2-(N-morpholino) ethanesulfonic acid] (MES), 0.001 M EDTA, 0.1 M NaCl, pH 6.25. CGP 40215A was made available by the Pharmaceuticals Division, Ciba-Geigy Ltd (currently Novartis, Basel, Switzerland). Berenil (diminazene aceturate) was purchased from Calbiochem-Behring (La Jolla, CA).
Spectrophotometric titrations
Circular dichroism (CD) spectra were obtained with a computer-controlled Jasco J-710 spectrometer in a 1-cm quartz cell in MES buffer. The desired ratios of mol of compound to mol of DNA oligomer hairpin were obtained by adding the compound to a cell containing a constant amount of DNA. UV-VIS spectrophotometric spectra of the complexes at different DNA to compound ratios were collected with a Cary 4E spectrophotometer (Varian, Palo Alto, CA) at room temperature. Aliquots of DNA polymers (poly(dAT)2 or poly(dGC)2) were added to a cell containing a constant amount of compound to obtain different DNA to compound ratios.
To obtain the pKa values of CGP 40215A, the pH titrations of the free (in water or with 0.1 M NaCl) and bound (in MES buffer, 0.1 M NaCl with poly(dAT)2) ligand were performed by adding concentrated NaOH solutions. An initial low-pH condition was obtained by addition of HCl solution. The absorbance spectra were collected with a Cary 3E or 4E spectrophotometer, and the pH values were recorded with an Accumet pH Meter 910 (Fisher Scientific, Hampton, NH) after each addition of NaOH. The DNA concentration (in basepairs) was 25 times higher than that of the ligand to assure complete complex formation.
Electric linear dichroism
The theory of electric linear dichroism (ELD) and its applications have been discussed in the literature (Bailly et al., 1992; Bloomfield et al., 2000; Cantor and Schimmel, 1980; Colson et al., 1996; Rodger, 1993; Rodger and Nordén, 1997). In brief, this technique probes the orientation of the ligand relative to the DNA helical axis. Groove-binding ligands generally give positive reduced LD values whereas stacking ligands yield negative reduced LD values. The concentrations of calf thymus DNA and the double-stranded polymers poly(dAT)2 and poly(dGC)2 (Pharmacia) were determined with molar extinction coefficients of 6600, 6600, and 8400 M−1cm−1 (Pachter et al., 1982; Schmechel and Crothers, 1971; Wells et al., 1970), respectively. For the ELD experiments, calf thymus DNA was deproteinized with sodium dodecyl sulfate (protein content <0.2%) and all nucleic acids were dialyzed against 1 mM sodium cacodylate buffered solution pH 7.0. ELD measurements were performed using a computerized optical measurement system and the procedures as previously outlined (Houssier, 1981). The optical setup incorporating a high sensitivity T-jump instrument equipped with a Glan polarizer was used under the following conditions: bandwidth 3 nm, sensitivity limit 0.001 in ΔA/A, response time 3 μs. All experiments were conducted at 20°C with a 10-mm pathlength Kerr cell having 1.5-mm electrode separation (Colson et al., 1996).
DNase I footprinting
In addition to the two DNA fragments of 117 bp and 265 bp (prepared by 3′-[32P]-end labeling of the EcoRI-PvuII double digest of the plasmid pBS using α-[32P]-dATP (Amersham, Amersham, UK) and AMV reverse transcriptase (Roche, Basel, Switzerland)), the two 198-bp fragments were obtained from plasmids pMS1 and pMS2 (kindly provided by Dr. K. R. Fox, University of Southampton, Southampton,UK) after digestion with the restriction enzymes HindIII and XbaI (Lavesa and Fox, 2001). In each case, the labeled digestion products were separated on a 6% polyacrylamide gel under nondenaturing conditions in TBE buffer (89 mM Tris-borate pH 8.3, 1 mM EDTA). After autoradiography, the requisite band of DNA was excised, crushed, and soaked in water overnight at 37°C. This suspension was filtered and the DNA was precipitated with ethanol. DNase I footprinting experiments were performed essentially as previously described (Waring and Bailly, 1997). Briefly, samples (3 μl) of the labeled DNA fragments were incubated with 5 μl of the buffered solution containing the ligand at appropriate concentration. After 30 min incubation at 37°C to ensure equilibration of the binding reaction, the digestion was initiated by the addition of 2 μl of a DNase I solution whose concentration was adjusted to yield a final enzyme concentration of ∼0.01 unit/ml in the reaction mixture. After 3 min, the reaction was stopped by freeze drying. Samples were lyophilized and resuspended in 5 μl of an 80% formamide solution containing tracking dyes. The DNA samples were then heated at 90°C for 4 min and chilled in ice for 4 min before electrophoresis on an 8% polyacrylamide gel under denaturating conditions (8-M urea). A Molecular Dynamics 425E PhosphorImager was used to collect data from the storage screens exposed to dried gels overnight at room temperature (Sunnyvale, CA). Baseline-corrected scans were analyzed by integrating all the densities between two selected boundaries using ImageQuant version 3.3 software (Molecular Dynamics). Each resolved band was assigned to a particular bond within the DNA fragments by comparison of its position relative to sequencing standards generated by treatment of the DNA with dimethylsulphate followed by piperidine-induced cleavage at the modified guanine bases in DNA (G-track).
Surface plasmon resonance
The experiments were performed on a BIACORE 2000 instrument and Biacore SA sensor chips were used for DNA immobilization (Uppsala, Sweden). Manual injections were performed for samples of 1–2 nM biotinated DNA hairpins onto the SA chip at a flow rate of 5 μL/min. The amount of DNA captured was in the range from 250 to 300 resonance units (RUs). One flow cell was left blank and used as reference. The sensorgrams for the interaction (RUs versus time) were collected at 25°C with a flow rate of 20 μL/min. The compound was dissolved in degassed MES buffer with 5 × 10−3% v/v Surfactant P20 (Biacore). This buffer was also used in surface regeneration. The RU values were averaged over a 1-min time span in the steady-state response region and were converted to r (mol of compound bound per mol of DNA hairpin) for binding analysis. This was done by dividing the response units by the predicted RUmax as previously described (Davis and Wilson, 2000). The binding constants of the compounds were obtained from fitting a plot of r versus compound concentration. The plot was fitted with a one- or two-site model (for one-site model, K2 = 0):
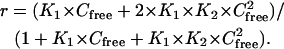 |
The kinetic information was obtained by directly fitting the response units versus time using predefined models accompanied with the BIAevaluation 3.0 program (Biacore, 1997). The association and dissociation rate constants (ka and kd) were obtained by global fitting using simultaneous ka/kd, 1:1 (Langmuir) binding with mass transport effects (Myszka et al., 1998).
Molecular dynamics simulations
The dynamics of the complex formed between CGP 40215A and d(CGCGAATTCGCG) duplex was explored using three molecular dynamics (MD) simulations. All simulations were carried out using AMBER 5.1 suites of program (Case et al., 1997) and the Cornell et al. force field (Cornell et al., 1995). The force-field parameters of CGP40215A were developed in analogy to existing parameters in the force field of Cornell et al. (1995) and based on the ab initio calculations of small molecules using Gaussian98 (Frisch et al., 1998). The atomic charges of CGP 40215A were determined using the restrained electrostatic potential fitting procedure (RESP) (Bayly et al., 1993) at the HF/6-31G* level of theory. In the first simulation, the d(CGCGAATTCGCG) duplex obtained from the crystal structure of pentamidine (Edwards et al., 1992) was used. CGP 40215A was placed in the minor groove at around the central AT step at ∼3 Å from the floor of the groove with the amino moiety pointing out of the groove. No specific hydrogen bonds were initiated. The CGP 40215A–DNA complex was solvated with ∼4100 TIP3P water molecules (Jorgensen et al., 1983) to fill a periodic box size of ∼45 Å × 60 Å × 45 Å. Twenty-nine Na+ and 10 Cl− ions were placed around the complex to obtain electrostatic neutrality and a NaCl concentration of ∼0.1 M. The systems were equilibrated with a standard multistage equilibration protocol (Hamelberg et al., 2000, 2001). After the equilibration, this simulation was continued for an additional 10 ns in an isothermal-isobaric ensemble at a temperature of 300 K and a pressure of 1 bar. The SHAKE (Ryckaert et al., 1977) algorithm was applied to all bonds and an integration time step of 2.0 fs was used for the integration of Newton's equation. Lennard-Jones interactions were subjected to a 9-Å cutoff, the nonbonded pair lists were updated every 10 steps, and the trajectories were sampled at every 1-ps interval. A second simulation was carried out, similar to the one described above, starting with the canonical B-form DNA.
To evaluate the specificity of CGP 40215A and the proposed binding site, a third simulation was carried out using the canonical B-form d(CGCGAATTCGCG) duplex but at 400 K to increase the rate of dynamic movements of the ligand. The compound was initially placed in the minor groove centered over the GA step rather than over the central AT step. No specific hydrogen bonds were formed and CGP 40215A was initially placed ∼3 Å away from the floor of the minor groove. After equilibration, this simulation was carried out for an additional 3 ns under similar simulation conditions as described above.
RESULTS
UV-VIS and pH titration
UV-VIS titration spectra of CGP 40215A with poly(dAT)2 exhibit strong hypochromicity at 350 nm with increases in compound absorbance below 340 nm (online supplement, Fig. S1). A very different pattern is observed for the interaction of this compound with poly(dGC)2. No significant spectral shifts are seen in the GC DNA titration but hypochromicity is observed over a broad region (300–400 nm) (online supplement, Fig. S1).
In the course of these experiments, it was observed that the absorbance spectrum of CGP 40215A at pH 6.25 is significantly different than that at pH 7.0. To better understand the spectral shifts of this compound on the addition of DNA, spectrophotometric pH titrations were conducted at room temperature (Fig. 2). Starting under acidic conditions where the CGP 40215A nitrogen linker and the amidine groups are protonated, the absorbance at 350 nm increases and it decreases at 307 nm as the pH is raised. The plots of absorbance versus pH (Fig. 2) yield a typical titration curve with a best-fit pKa value of 5.4 in water without salt and 6.3 in 0.1 M NaCl. The amidines are known to have pKa values >10, and thus, remained protonated. To evaluate the pKa shift upon complex formation with the AT DNA polymer, the CGP 40215A–poly(dAT)2 complex was titrated in the same manner. As expected from the above experiment, the absorbance intensity >350 nm increased as the pH was raised from the acidic region. The pKa of the nitrogen linker underwent a large shift to a value of 8.9 (Fig. 2), an increase of 2.6 pKa units between the free and bound ligand at the same salt concentration. In summary, the spectral changes on complex formation with the AT sequences indicate protonation of the CGP 40215A linker group in the DNA complexes at neutral pH. The spectral results for titration of protonated CGP 40215A with the AT DNA are similar to those observed for para-dicationic diamidine compounds that form minor groove complexes with neutral linkers.
FIGURE 2.

Titration spectra of CGP 40215A free and bound to poly(dAT)2. Spectral shifts of CGP 40215A in water are shown under different pH conditions (top). Under acidic conditions, the absorbance at 350 nm is low and it increases as the pH increases. Similar effects were observed for bound ligand (middle). Fits of the normalized absorbance data (at 347 nm for free and 373 nm for bound ligand) yield pKa values 5.4 and 8.9, respectively (bottom).
CD titration
CD titration spectra of berenil with an AT DNA sequence (Fig. 3) show strong induced positive CD signals >300 nm in agreement with published results (Laughton et al., 1996). Isoelliptic behavior is observed near 300 nm with minimal changes at 248 nm but larger effects at 270 nm. The strong positive induced CD signal of berenil when titrated with the AT sequences is the usual effect that is observed with diamidine derivatives with the amidines in para-positions (Fig. 1) (Laughton et al., 1996). The induced CD signals at 327 nm and 390 nm are plotted against ligand/DNAbp ratios in Fig. 3. The break at the ratio of ∼0.2 is equivalent to two compound binding sites per hairpin. This agrees with the SPR and DNase I footprinting (shown below) as well as structural studies that indicate berenil covers from three to four AT basepairs in the DNA minor groove (Braithwaite and Baguley, 1980; Portugal and Waring, 1987).
FIGURE 3.

CD titration spectra of the AT hairpin with berenil or CGP 40215A. Strong positive-induced CD is observed above 300 nm with berenil (top), but weak negative CD (at low ligand/DNA) is observed for CGP 40215A (middle). The induced CD signals at 327 nm and 390 nm for berenil and at 320 nm for CGP 40215A are plotted against the ligand/DNA(basepairs) ratios (bottom). The break at ∼0.2 corresponds to two berenil molecules per one AT hairpin whereas the most induced negative CD signals are observed at a ratio (r = 0.1) equivalent to one CGP molecule per one AT hairpin.
The CD titration spectra of CGP 40215A on addition of AT DNA sequences are quite different than the results with berenil. In contrast to the large and positive-induced signals with berenil, weak negative-induced CD signals are seen on initial titration of the AT hairpin DNA with CGP 40215A. As shown in Fig. 3, over the same range of compound to DNA ratios, the induced signal at 320 nm is most negative when the ratio of CGP 40215A to DNAbp is 0.1 (or one ligand per hairpin). Above this ratio, the induced CD begins to shift in the positive direction, presumably due to weak secondary bindings. Large CD changes in the DNA region near 248 nm are also observed in the CGP 40215A titration. In contrast to the results with AT sequences, no significant induced CD signals are observed when CGP 40215A is titrated with the GC hairpin (online supplement, Fig. S2). This agrees with the UV spectral results that suggest a weak nonspecific interaction between CGP 40215A and GC DNA. In summary, neither berenil nor CGP 40215A has significant induced CD signals with GC DNA. Both compounds have induced CD spectra on the binding to AT DNA but the induced shifts are positive for berenil and negative for CGP 40215A at low ligand/DNA ratios.
Electric linear dichroism
When CGP 40215A binds to calf thymus DNA, positive-reduced dichroism values (ΔA/A) in the 300–350 nm absorption band were obtained (online supplement, Fig. S3 A). The ΔA/A values vary linearly with the electric field strength at 350 nm to reach a maximum of +0.2 at 14 kV/cm (online supplement, Fig. S3 B) for a ligand to DNA(basepairs) of 0.05 (equivalent to DNA/ligand ratio of 40) (online supplement, Fig. S3 C). Such positive ELD signals show that the CGP 40215A molecules are bound in one of the DNA grooves at low ratios where the binding is predominantly at AT sites. However, the ΔA/A values measured with CGP 40215A are weaker than those obtained with berenil. Complementary measurements with the alternating polymers poly(dAT)2 and poly(dGC)2 provide additional information on the CGP 40215A–DNA complex. Positive ΔA/A values were recorded with the AT polymer whereas negative values were measured with the GC polymer (Fig. 4). These results indicate that the binding mode of CGP 40215A is minor-groove binding at AT as observed with other diamidines (Nguyen et al., 2002b). The GC mode could be weak intercalation or an extended stacked external complex due to the low ionic strength condition required for the ELD measurements.
FIGURE 4.

Variations of the electric linear dichroism (ΔA/A) with electric field strength for CGP40215A and berenil bound to poly(dGC)2 or poly(dAT)2. Measurements were conducted at a drug/DNA(basepairs) ratio of 0.1 (100 μM DNA(basepairs), 10 μM drug), 1 mM sodium cacodylate buffer, pH 7.0, at wavelengths of 330 nm and 360 nm for CGP 40215A and berenil, respectively.
Footprinting
DNase I footprinting was used to compare the sequence selectivity of CGP 40215A with that of berenil and pentamidine, diamidines that bind preferentially to AT-rich sequences (Jenkins et al., 1993). Four DNA restriction fragments were prepared by 3′-end radiolabeling with 32P. Two fragments of 117 and 265 bp (referred to as pBS fragments) without designed DNA sequences have been previously used to study sequence recognition by a variety of minor groove binders, including diphenylfurans, pentamidine, and berenil (Bailly et al., 1994; Nguyen et al., 2002a). Two designed fragments of 198 bp, referred to as universal footprinting substrates (Lavesa and Fox, 2001), contain all 136 distinguishable tetranucleotide sequences [(44)/2 + (44/2)/2 = 136]. These two fragments, MS1 and MS2, contain the same sequence of 136 bp but cloned in opposite orientation, 5′→3′ and 3′→5′. With each fragment, the products of digestion by DNase I in the absence and presence of the test drugs were resolved by polyacrylamide gel electrophoresis (online supplement, Fig. S4).
For comparison purposes, the band intensities in the gels of all fragments were quantified by phosphor imaging to obtain the differential cleavage plots (Fig. 5). The sites protected from DNase I cleavage in the presence of the drugs (negative values) all coincide with AT tracts. Very little difference can be seen between the profile obtained with CGP 40215A and that of berenil, indicating that the two drugs essentially recognize the same sequences. Similar results were obtained with the two 198-bp fragments. Here again, the footprinting patterns of CGP 40215A and berenil are quite similar. The two drugs, as well as pentamidine to a lower extent, bind preferentially to a number of AT tracts such as 5′-AATTA, 5′-TTTT, 5′-AAAT, and 5′-TATT. In summary, the footprinting results indicate that CGP 40215A, which combines the structural features of DB359 (meta-amidine groups) with a nitrogen rich linker chain, can be unambiguously classified to as an AT-specific groove binder. As with berenil, no significant footprints are observed with CGP 40215A and GC sequences.
FIGURE 5.

Differential cleavage plots comparing the susceptibility of the different DNA restriction fragments to DNase I cutting in the presence of CGP 40215A (•), berenil (□), or pentamidine (○) at 5 μM. Negative values correspond to a ligand-protected site and positive values represent enhanced cleavage. Vertical scales are in units of ln(fa) − ln(fc), where fa is the fractional cleavage at any bond in the presence of the drug and fc is the fractional cleavage of the same bond in the control, given closely similar extents of overall digestion. Only the region of the restriction fragment analyzed by densitometry is shown.
Biosensor SPR: affinity and kinetics
Sensorgrams for the interaction of berenil and CGP 40215A with the AT hairpin (online supplement, Fig. S5) indicate significant differences in the association and dissociation kinetic patterns between these two ligands. CGP 40215A exhibits slow on/off kinetics in contrast to the fast on/off rate for berenil at all concentrations. The averaged response units at the steady state were converted to r versus the free compound concentrations (as described in the Materials and Methods section) and were fitted with a one- or two-site binding model (online supplement, Fig. S6). For berenil, a much weaker (two orders of magnitude) secondary binding constant is required for satisfactory fitting of SPR results (Table 1), but this is not necessary for CGP 40215A. Very weak association of the ligand with the GC hairpin was observed, but the binding constants are too low to be accurately determined at the concentrations used in the SPR experiments. The sensorgrams for the interaction of CGP 40215A and berenil with an AATT DNA sequence have been previously reported (Nguyen et al., 2002a) and their binding constants are also summarized in Table 1.
TABLE 1.
Binding constants of berenil and CGP 40215A to AT or AATT hairpins
| Steady-state* | Kinetics† | |||||
|---|---|---|---|---|---|---|
| K (M−1) | ka (M−1 s−1) | kd (s−1) | ka/kd (M−1) | |||
| AT hairpin–berenil | 3.3 × 107, 4.3 × 105 | – | – | – | ||
| AATT hairpin–berenil | 1.1 × 107 | – | – | – | ||
| AT hairpin–CGP 40215A | 1.7 × 108 | 7.7 × 106 | 3.4 × 10−2 | 2.3 × 108 | ||
| AATT hairpin–CGP 40215A | 7.0 × 107 | 1.5 × 106 | 2.1 × 10−2 | 7.3 × 107 | ||
SPR experiments were conducted at 25°C in MES buffer at pH 6.25.
Obtained from nonlinear fitting of steady-state responses versus concentrations. The errors from fittings are <5%.
Obtained from global kinetics fits of three lowest concentration responses with a mass transport term (Myszka et al., 1998). The errors from fittings are <14%. Berenil kinetics are too fast to be measured under these conditions (see text).
The data for CGP 40125A was well fit with a single-site model for the interaction with either the AT or AATT hairpins (online supplement, Fig. S6) (Nguyen et al., 2002a). Although the AT hairpin has eight consecutive A/T basepairs and the AATT has four consecutive A/T basepairs, no difference in stoichiometry was observed. Any secondary binding of CGP 40215A to the AT hairpin must be too weak for detection at the concentrations used. In addition, as seen with berenil, CGP 40215A binds more strongly to the AT hairpin than to the AATT hairpin (Table 1). As observed from the results described above, very weak association with the GC hairpin was observed for CGP 40215A. The preferential binding to alternating AT DNA of CGP 40125A is similar to that of berenil, but the binding is about five times stronger than that of berenil. The strong binding and AT specificity are typical characteristics of many DNA minor-groove binders. The binding constants (107–108 M−1) are comparable to other AT minor-groove binders of the para-amidine class such as furamidine. The SPR results confirm the observations from UV and CD experiments that suggest the interaction with AT is stronger than with GC sequences.
To evaluate the CGP 40215A binding kinetics, the rate constants were obtained by global fitting of the sensorgrams with a 1:1 binding model that includes mass transport effects (Fig. 6). Visual inspection of the residual plots reveals that there is not a significant systematic deviation in the data and that any baseline drifts are within the noise levels. In addition, the RUmax value obtained from the fitting is in agreement with the experimental value. Both of these factors indicate that the model for the kinetic fittings of the interaction of CGP 40215A with the AT or AATT hairpins is correct. The binding constants obtained from the ratios of association to dissociation rate constants are in very good agreement with those obtained from steady-state fitting (Table 1). Because of timescale limitations in SPR, the fast kinetics for berenil association and dissociation cannot be obtained with this method.
FIGURE 6.

Kinetic fitting plots of CGP 40215A with the AATT hairpin (top) and the AT hairpin (bottom). Sensorgrams for the three lowest concentrations along with the global fitting curves (solid lines) are shown. The rate constants are summarized in Table 1.
Possible conformations of CGP 40215A
Three possible low-energy conformations of CGP 40215A in solution are possible due to rotation about the phenyl-linker carbon bond (Fig. 1). Each of these possible conformations was minimized at the HF/6-31G* level of theory. Conformation (i) has the lowest energy, followed by ii, and iii has the highest energy. The energy of conformation (iii) is 5.3 kcal/mol greater than that of i, whereas the energy of ii is 2.3 kcal/mol greater than that of conformation i. Assuming that CGP 40215A only exists significantly in these three states, the Boltzman probabilities of finding these conformations in solution are calculated to be 0.96, 0.04, and <0.01, respectively.
Furthermore, the transition of CGP 40215A from conformation i to ii was explored by rotating one of the phenyl groups at 30° intervals and calculating the single-point energy of each conformation (online supplement, Fig. S7). In the transition from conformation i to ii, the major energy barriers are at the 90° and 270° points. Conformation i is, thus, a global minimum in the energy profile.
Dynamics of the DNA-CGP complex
The initial docked CGP 40215A–DNA complex was built as described in the Materials and Methods section. After the initial equilibration in the MD simulation, the complex relaxed quickly to an energy minimum and remained stable for the entire simulation. Within the first few hundred picoseconds of the MD simulation specific hydrogen bonds were formed between CGP 40215A and the floor of the minor groove (Fig. 7). To study the type of interactions between CGP 40215A and the AATT duplex, some of these specific hydrogen bond distances were monitored during the entire simulation (Fig. 7 and Fig. 8, A–D). As shown from 500 ps, H44 from one of the amidine groups of CGP 40215A forms an interaction with the O2 of T20, and H21 on the nitrogen of the linker closest to that amidine group interacts with the O2 of T7 as can be seen in Fig. 7, and schematically in Fig. 8 A. This hydrogen-bonding pattern is an energy minimum and remains intact for the next 4 ns. At ∼4 ns the amidine group quickly rotates 180°, forming a new but similar hydrogen bond between H47 of CGP 40215A and O2 of T20. At ∼5.4 ns, CGP 40215A starts to slide toward the other end of the AATT duplex and forms hydrogen bonds between the other amidine group of the compound and the floor of the minor groove. The new complex is quite similar to the previous complex as can be seen in Fig. 7 and Fig. 8 B. At ∼9.5 ns, the ligand starts to move again back into the position shown in Fig. 8 A. The interaction can be viewed as a back and forth “seesaw” movement with a basepair shift during the transition. Similar interactions were observed with simulations started from DNA crystal structures or from a canonical B-form DNA. One direct effect associated with the movement of the ligand on the DNA duplex is seen on the minor groove width. When the ligand is on one end of the DNA duplex (Fig. 8 A), the minor groove width around the TT step narrows as shown in Fig. 8 C. Similarly, the minor groove width around the AA step narrows (Fig. 8 D) when the ligand moves to the other end of the DNA duplex (Fig. 8 B).
FIGURE 7.

H-bond distances between some CGP 40215A groups and DNA bases as predicted from MD simulations at 300 K. The top panel illustrates the H-bond interaction between the O2 of T20 and hydrogens from one of the amidine groups. The rapid interchange at ∼4 ns indicates a rapid rotation of the amidine group. The second panel illustrates the H-bond interaction between the O2 of T7 and H21 on the linker. The third and fourth panels illustrate the simultaneous moments occurring at the other end of the binding site with the other amidine interacting with O2 of T8 as well as H15 on the linker with O2 of T19. At ∼5.3 ns, the ligand moves from one end of the binding site to the other as evident from the increase in H-bond distances of the departing amidine (top panel) and the decrease in H-bond distances of the approaching amidine (third panel).
FIGURE 8.

Schematics of the movement of CGP 40215A within the binding site. (A) When one of the amidine groups is in direct contact with T20, the center of the linker induces a narrower groove width at the T7 and T8 site as indicated in C. The opposite effect is observed when the ligand moves to the other end of the binding site (B and D). The minor groove is narrow in direct correlation with the movement and the position of the ligand.
The simulation at 400 K (online supplement, Fig. S8) was started with the compound initially placed in the minor groove centered over the GA step rather than over the central AT step. At ∼300 ps into the simulation, the ligand moves to the central AT step of the DNA duplex and forms an interaction with the minor groove of the DNA in a similar manner to the interactions seen in the 300 K simulations. The hydrogen bond patterns observed are also identical to those of the 300 K simulations. Moreover, the transition from one type of complex to the other is similar to those at lower temperature but is observed multiple times during this simulation, as expected from the higher temperature dynamics.
Structural dynamics and hydration patterns of the CGP 40215A–DNA complex
To evaluate in detail the role that water molecules play in the interaction of the ligand with the AATT binding site, hydration density maps, at approximately three times the density of bulk water, were plotted at several regions of the MD simulations (Fig. 9). The first hydration density plot (Fig. 9 A) illustrates the complex with the ligand at one end of the DNA duplex. The second plot (Fig. 9 B) is for the complex at the transition region as the ligand moves from one to the other end of the DNA duplex binding site. The third density plot (Fig. 9 C) shows the hydration density with the ligand at the other end of the binding site as shown in Fig. 8 B. These hydration patterns illustrate the two limits of the seesaw interactions of the ligand with the AATT site as well as the transition region where both amidines are away from the floor of the groove. When one of the amidine groups of the ligand is directly interacting with the floor of the minor groove through hydrogen bonds (Fig. 8, A and B), the amidine group on the other end makes contact with the minor groove bases through water molecules (Fig. 9, A and C). Both amidine groups also form water bridges that extend from the ends of the molecule and help to stabilize the complex (Fig. 9 A). A similar complex, but with the amidine-base interactions switched, is seen when the ligand moves to the other end of the binding site (Fig. 9 C). By moving from one end of the DNA duplex binding site to the other, the ligand goes through a transition state wherein neither amidine group can interact directly with the floor of the minor groove due to its linear structure. The interaction between both amidine groups of the ligand and the minor groove is entirely via water molecules in this intermediate state (Fig. 9 B).
FIGURE 9.

Stereo views of the hydration profile (water density in red) associated with the movement of CGP 40215A within the AATT minor groove. (A) The ligand forms direct H-bond contacts with the O2 of T; a small amount of water is also involved at this end. There is no direct H-bond contact between the DNA bases and the other amidine (the higher end in the figure); more water molecules are seen mediating the interaction between the ligand and the DNA bases at this end. (B) At the transition, both amidine groups make no direct H-bond contacts with the DNA bases; water molecules are observed mediating the interaction at both ends of the ligand. (C) When the ligand moves to the other end, opposite effects to A are observed.
DISCUSSION
As described in the Introduction, a number of para-substituted diphenylamidine compounds, such as berenil and furamidine, bind strongly in the minor groove of DNA and have clinically useful antiparasitic activities. Meta-substituted analogs, however, generally bind relatively weakly to DNA and by binding modes, such as intercalation, that are different from the minor groove complexes of the para-derivative (Nguyen et al., 2002b). The meta-analogs generally do not show useful antiparasitic activities. One of the key concepts that has been developed with regard to minor groove complexes is that the bound small molecule either has or can adopt a molecular curvature that is complementary to that of the DNA minor groove (Bailly et al., 1994; Cory et al., 1992; Fairley et al., 1993). This complementary structure enhances surface contacts and aligns groups for favorable interactions with DNA. The only meta-substituted diphenylamidine to exhibit significant antiparasitic activity to date is CGP 40215A (Fig. 1) (Brun et al., 1996). The activity of this compound raises several intriguing questions about its molecular target and its possible DNA interactions, as outlined in the Introduction. The experiments reported here were designed to address those questions and build on the binding and structural results previously published (Nguyen et al., 2002a). We specifically wish to know if the hypothesis that the DNA minor groove is a primary target for the biological action of diamidines must be revised.
A crystal structure of CGP 40215A bound to an AATT duplex sequence that has been used with several other minor groove binding diamidines has been determined (Nguyen et al., 2002a). In this structure the ligand is offset from the central region of the duplex and interacts differently with the two ends of the AATT binding site. One amidine and an adjacent −NH group of the linker form direct H-bonds to DNA basepairs whereas the other amidine at the opposite end of the molecule interacts indirectly through a tightly bound water molecule. MD simulations of the AATT complex reveal that the ligand can form similar complexes at either end of the AATT site, but that the central alignment of the ligand is not favorable. The H-bond patterns that were seen in the x-ray structures are similar to those of the two limiting MD structures. The two MD structures, if rotated 180° end-to-end, have excellent overlap with each other and with the x-ray structure.
The linear conjugated nitrogen-rich linker of CGP 40215A, with its ability to accept a proton and gain a positive charge, is a feature that makes this compound unique in the diamidine series. UV-VIS spectroscopic titrations of the compound as a function of pH show that under neutral pH conditions the nitrogen linker is not significantly protonated (pKa = 6.3 in 0.1 M NaCl). An increase of 2.6 pKa units is observed, however, when the ligand binds to poly(dAT)2. This remarkable increase in pKa value and significant spectral changes on complex formation suggest that the protonated nitrogen linker is heavily involved in the strong binding of the ligand to AT DNA sequences. When protonated, the molecule becomes symmetric and can form additional electrostatics and H-bond interactions with the minor groove. Because the amidine groups have pKa values >10, the compound has a charge of ∼2, as with other diamidines (Fig. 1), under physiological conditions and passes through cell membranes in a similar manner, presumably via amidine transporters that have been characterized in parasites (De Koning, 2001; De Koning and Jarvis, 2001).
Because of the combined effects of protonation and binding, the observed spectral changes on titration of CGP 40215A with DNA are complex. The hypochromicity observed near 350 nm when CGP 40215A is titrated with AT or GC DNA above pH 6 is similar to the reduction in absorbance at 350 nm on protonation of the CGP 40215A. Protonation of CGP 40215A causes an increase in absorbance below 340 nm and an increase is also seen in the titration with alternating AT DNA. The increase in absorbance is, however, smaller than in the pH titration. All of these results suggest that the protonated species binds to the AT DNA with some hypochromicity, and this hypochromicity reduces the absorbance increase that is observed on protonation. These results also indicate that the anionic and acidic DNA minor groove environment causes a pKa shift and induces protonation with an additional positive charge (+3) on the compound as a result of complex formation (Jones and Wilson, 1981; Lamm and Pack, 1990). Examples of similar protonation of other ligands on interaction with DNA are relatively rare (Gimenez-Arnau et al., 1998a,b; Renault et al., 1997). The protonation enhances DNA interactions, does not interfere with the free ligand entering into cells, and it illustrates a potentially useful drug design strategy.
The LD results with CGP 40215A–DNA complexes allow a clear prediction of the sequence-dependent binding modes of the compound in solution. Positive reduced dichroism values were obtained for the interaction of CGP 40215A with AT DNA sequences in agreement with the binding observed with the AATT sequence in x-ray studies (Nguyen et al., 2002a). The reduced dichroism values of CGP 40215A are lower than those of berenil and this may be due to the linear structure of CGP 40215A, its dynamics in the complex (see below), as well as its meta-substitution. The orientation of berenil and CGP 40215A relative to the DNA helical axis can be estimated with a few reasonable assumptions based on related structures (Jansen et al., 1993; Kim et al., 1993; Rodger and Nordén, 1997): i), the transition moment(s) (near 330 nm for CGP and 360 nm for berenil) are aligned along the molecular long axis of the ligands, ii), the DNA orientation factor remains the same for bound DNA and free DNA, iii), the DNA basepairs make an angle of ∼90° with the helix axis. With these assumptions berenil makes an angle of ∼33° whereas CGP 40215A makes an angle of 39° with the helix axis in the AT polymer. These angles strongly support a minor groove binding mode for berenil and CGP 40215A in solution (Brown et al., 1990; Jenkins et al., 1993; Laughton et al., 1996; Nguyen et al., 2002a; Trent et al., 1996).
Large positive-induced CD changes are typically observed for minor groove complexes in AT sequences. The weak negative induced CD observed for CGP 40215A up to a 1:1 molar ratio when interacting with AT DNA is a very unusual result for strong-binding aromatic diamidines. Given the linear structure and resulting weaker interaction of the aromatic system of the ligand with the DNA base transition moments, the weaker CD signals relative to other minor groove binders such as berenil are expected. Negative-induced CD changes for ligand-DNA interactions have also been observed for some polyamide minor groove binding agents (Lee et al., 1993a,b). Theoretical treatments of ligand-DNA-induced CD spectra indicate that the induced CD depends strongly on the conformation and relative position of the bound chromophore and that negative-induced CD for groove binders is expected in some bound configurations (Lyng et al., 1992). In summary, the CD changes indicate strong binding of CGP 40215A to AT DNA sequences but do not provide clear information on the binding mode in these sequences. This illustrates the need for several complementary biophysical methods to establish a compound-DNA binding mode.
DNase I footprinting results with CGP 40215A are very similar to those of berenil, furamidine, and other para-substituted minor groove binding compounds but are very different from other meta-substituted diamidines. The results from footprinting experiments indicate that CGP 40215A binds strongly to AT-rich sequences and that the binding site is 4–6 basepairs. As seen in Fig. 5, CGP 40215A produces similar but generally stronger footprints in AT sequences than pentamidine, a compound of similar size. All of these results strongly support a minor groove complex of CGP 40215A with AT DNA sequences in solution as observed in the crystallographic structure with an AATT sequence (Nguyen et al., 2002a).
SPR-biosensor experiments provide quantitative information about CGP 40215A–DNA complexes that is in complete agreement with results from other methods. The binding of both CGP 40215A and berenil to GC DNA sequences, at the salt and compound concentrations used in the SPR experiments, is too weak to allow quantitative determination of binding constants. Both compounds bind very strongly to AT sequences as expected for minor groove complex formation. Surprisingly, the meta-substituted CGP 40215A binds to AT sequences more strongly than berenil and related para-substituted diamidines and it binds much more strongly to AT DNA than other meta-substituted compounds (Nguyen et al., 2002b). A binding stoichiometry of one compound per AATT hairpin (Fig. 1) was found for berenil and CGP 40215A as expected from their size. The longer eight-basepair AT hairpin can bind two berenil molecules, one strongly and one significantly more weakly (Table 1), but fitting results indicate that only one CGP 40215A molecule binds significantly to the same hairpin. The difference in stoichiometry between berenil and CGP 40215A with the longer AT sequence is probably due in part to their difference in size (CGP 40215A is ∼20% longer than berenil) and charge. The additional charge on CGP 40215A increases intermolecular electrostatic repulsion of bound molecules and inhibits two CGP 40215A molecules from binding next to each other. In SPR kinetics studies berenil dissociates too fast for quantitative analysis but the dissociation rate of CGP 40215A from AT complexes is significantly slower. It is estimated from previous studies (Laughton et al., 1996) that the dissociation rate constant for berenil is >1 s−1 whereas the typical range in SPR (BIACORE 2000) is from 10−6 to 10−1 s−1 (Biacore, 1994; Myszka, 1997). Binding constants of CGP 40215A with the AT or AATT hairpin obtained from global fitting of the association and dissociation SPR kinetics curves are in good agreement with those obtained from the steady-state SPR method (Table 1). In both cases (with AT and AATT hairpins), the scatter in the residual plots from global kinetics fits is within the instrumental noise indicating good quality fits. The SPR results thus clearly show that the CGP 40215A complex with AT DNA sequences is very favorable for a diamidine and much more favorable than for any other meta-substituted derivative. The slower kinetics of CGP 40215A are reasonable given the structural and dynamic results discussed below.
Both x-ray and MD studies show that the ligand can form direct and indirect H-bonds with the DNA bases where water-mediated H-bonds compensate for the lack of curvature of this ligand. The water molecules extend the ligand structure as part of the complex and play an essential role as mediators between the ligand and the DNA surfaces. The water molecules form a part of a continuous array of van der Waals and H-bond interactions with the minor groove as the ligand dynamics transfer the structure between two equivalent structural binding modes. The MD structures suggest that although there are no direct H-bond contacts between the amidines and the DNA bases in the seesaw transition state, the H-bond interactions between the linker of CGP 40215A with the cross-strand Ts (T7 and T19) as well as solvent interactions help to keep the ligand from dissociating at this stage. A recent study on the dynamic role of water in the Hoechst 33258–DNA complex has shown that ordered water molecules reside in the DNA groove at the ends of the complex (Pal et al., 2003). Such ordered water molecules also serve as mediators between CGP 40215A and DNA bases as observed in both x-ray and MD structures. The MD simulations also show that the minor-groove width at the binding site is more narrow in the region where the ligand binds. The minor-groove width is most narrow at the position where the center of the linker is located. The additional positive charge of the linker may contribute to narrowing the groove width by reducing repulsion between the negatively charged phosphate groups across the minor groove. As the ligand moves back and forth from the ends of the AATT site, the movement of this central charge leads to correlated shifts in minor-groove width at the two ends of the binding site (Fig. 8).
The results from spectroscopic, footprinting, biosensor, x-ray experiments, and MD simulations have provided a clear picture of the CGP 40215A-AT complex. The strong binding of CGP 40215A to AT DNA is associated with a large increase of the linker pKa. A tricationic CGP 40215A and a water molecule thus form a complex in the DNA minor groove that is very similar to, but stronger than, complexes observed with similar, para-substituted aromatic diamidines. The CGP 40215A–DNA complex is, however, very different from those formed by meta-substituted aromatic diamidines such as DB359 (Fig. 1). These structural and dynamics results also provide a better understanding of the slower kinetics of CGP 40215A relative to similar diamidines such as berenil. The CGP 40215A binding mode involves a simultaneous insertion of the compound into the minor groove of the AT sequence and i), protonation of the compound, ii), direct binding of a water molecule at one amidine–DNA contact, iii), the usual ligand/DNA/solvent rearrangement required for a strong minor-groove complex formation. Once all contacts are established and the ligand is protonated, the interaction is quite strong and the dissociation reaction is much slower than with similar amidine compounds.
This nitrogen-rich compound (9 nitrogen atoms for 17 carbons) possesses an unusual linker for DNA recognition that at first looks unfavorable, due to its linear structure, but is actually a very good DNA-binding module. This linker becomes protonated upon penetrating the minor groove of the double helix and, with a strongly bound water molecule, provides an example of a recognition process where the receptor and associated water amplify the binding properties of the ligand in a truly cooperative complex. Intrinsically, because of its meta-substitution pattern, CGP 40215A would appear to be not particularly well shaped for DNA recognition but the DNA-mediated protonation of the linker enhances the H-bond array in the minor groove. This type of protonation-assisted binding has been previously observed with DNA binding proteins (Kunne et al., 1998; Lagunavicius et al., 1997; Lundback et al., 2000). It has also been reported with some intercalating agents such as ethidium, ellipticine, and acridine derivatives (Gimenez-Arnau et al. 1998a,b, Renault et al., 1997) but rarely with a minor-groove binder. This is, therefore, the first detailed example of a DNA-controlled change in the protonation state of a small molecule that facilitates AT-selective insertion into the minor groove of DNA.
The work reported here raises opportunities for the design of new types of AT-specific drugs. We have discovered the first meta-substituted diamidine capable of forming tight and stable complexes within the minor groove of AT-rich DNA sequences. The critical new component of CGP 40215A-DNA complexes is obviously its linker chain that forms strong interactions with DNA bases and allows the dynamic movement of the compound between two limiting and equivalent structures at the ends of the AATT binding site. The DNA-induced protonation of the linker provides tight anchoring of the cationic molecule in DNA AT sites. This unusual DNA-dependent pH effect can be exploited further to enhance the DNA binding capacities of small molecules.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Alexandra Joubert (INSERM U524) for helping with the DNase I footprinting experiments.
This work was supported by grants from the National Institutes of Health, Georgia Research Alliance (D.W.), Gates Foundation (D.W. and R.B.), Ligue Nationale Contre le Cancer (C.B.), and Cancer Research UK (S.N.).
References
- Bailly, C., I. O. Donkor, D. Gentle, M. Thornalley, and M. J. Waring. 1994. Sequence-selective binding to DNA of cis- and trans- butamidine analogues of the anti-Pneumocystis carinii pneumonia drug pentamidine. Mol. Pharmacol. 46:313–322. [PubMed] [Google Scholar]
- Bailly, C., J. P. Henichart, P. Colson, and C. Houssier. 1992. Drug-DNA sequence-dependent interactions analysed by electric linear dichroism. J. Mol. Recognit. 5:155–171. [DOI] [PubMed] [Google Scholar]
- Bayly, C. I., P. Cieplak, W. Cornell, and P. A. Kollman. 1993. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem. 97:10269–10280. [Google Scholar]
- Bell, C. A., M. Cory, T. A. Fairley, J. E. Hall, and R. R. Tidwell. 1991. Structure-activity relationships of pentamidine analogs against Giardia lamblia and correlation of antigiardial activity with DNA-binding affinity. Antimicrob. Agents Chemother. 35:1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, C. A., C. C. Dykstra, N. A. Naiman, M. Cory, T. A. Fairley, and R. R. Tidwell. 1993. Structure-activity studies of dicationically substituted bis- benzimidazoles against Giardia lamblia: correlation of antigiardial activity with DNA binding affinity and giardial topoisomerase II inhibition. Antimicrob. Agents Chemother. 37:2668–2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, C. A., J. E. Hall, D. E. Kyle, M. Grogl, K. A. Ohemeng, M. A. Allen, and R. R. Tidwell. 1990. Structure-activity relationships of analogs of pentamidine against Plasmodium falciparum and Leishmania mexicana amazonensis. Antimicrob. Agents Chemother. 34:1381–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacore. 1994. BIACORE 2000: Instrument Handbook. Biacore AB, Uppsala, Sweden.
- Biacore. 1997. BIAevaluation Version 3.0: Software Handbook. Biacore AB, Uppsala, Sweden.
- Blackburn, G. M., and M. J. Gait. 1997. Nucleic Acids in Chemistry and Biology. Oxford University Press, New York.
- Bloomfield, V. A., D. M. Crothers, and J. Ignacio Tinoco. 2000. Nucleic Acids: Structures, Properties, and Functions. University Science Books, Sausalito, CA.
- Boykin, D. W., A. Kumar, J. Spychala, M. Zhou, R. J. Lombardy, W. D. Wilson, C. C. Dykstra, S. K. Jones, J. E. Hall, R. R. Tidwell, C. Laughton, C. M. Nunn, and S. Neidle. 1995. Dicationic diarylfurans as anti-Pneumocystis carinii agents. J. Med. Chem. 38:912–916. [DOI] [PubMed] [Google Scholar]
- Boykin, D. W., A. Kumar, G. Xiao, W. D. Wilson, B. C. Bender, D. R. McCurdy, J. E. Hall, and R. R. Tidwell. 1998. 2,5-bis[4-(N-alkylamidino)phenyl]furans as anti-Pneumocystis carinii agents. J. Med. Chem. 41:124–129. [DOI] [PubMed] [Google Scholar]
- Braithwaite, A. W., and B. C. Baguley. 1980. Existence of an extended series of antitumor compounds which bind to deoxyribonucleic acid by nonintercalative means. Biochemistry. 19:1101–1106. [DOI] [PubMed] [Google Scholar]
- Brown, D. G., M. R. Sanderson, J. V. Skelly, T. C. Jenkins, T. Brown, E. Garman, D. I. Stuart, and S. Neidle. 1990. Crystal structure of a berenil-dodecanucleotide complex: the role of water in sequence-specific ligand binding. EMBO J. 9:1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun, R., Y. Buhler, U. Sandmeier, R. Kaminsky, C. J. Bacchi, D. Rattendi, S. Lane, S. L. Croft, D. Snowdon, V. Yardley, G. Caravatti, J. Frei, J. Stanek, and H. Mett. 1996. In vitro trypanocidal activities of new S-adenosylmethionine decarboxylase inhibitors. Antimicrob. Agents Chemother. 40:1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor, C. R., and P. R. Schimmel. 1980. Biophysical Chemistry. W. H. Freeman and Company, New York.
- Case, D. A., D. A. Pearlman, J. W. Caldwell, T. E. Cheatham, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, D. M. Ferguson, R. J. Radmer, G. L. Seibel, U. C. Singh, P. K. Weiner, and P. A. Kollman. 1997. The Amber Software Suite. University of California, San Francisco, CA.
- Colson, P., C. Bailly, and C. Houssier. 1996. Electric linear dichroism as a new tool to study sequence preference in drug binding to DNA. Biophys. Chem. 58:125–140. [DOI] [PubMed] [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Cory, M., R. R. Tidwell, and T. A. Fairley. 1992. Structure and DNA binding activity of analogues of 1,5-bis(4- amidinophenoxy)pentane (pentamidine). J. Med. Chem. 35:431–438. [DOI] [PubMed] [Google Scholar]
- Das, B. P., and D. W. Boykin. 1977. Synthesis and antiprotozoal activity of 2,5-Bis(4-guanylphenyl)furans. J. Med. Chem. 20:531–536. [DOI] [PubMed] [Google Scholar]
- Davis, T. M., and W. D. Wilson. 2000. Determination of the refractive index increments of small molecules for correction of surface plasmon resonance data. Anal. Biochem. 284:348–353. [DOI] [PubMed] [Google Scholar]
- De Koning, H. P. 2001. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by three distinct transporters: implications for cross-resistance with arsenicals. Mol. Pharmacol. 59:586–592. [DOI] [PubMed] [Google Scholar]
- De Koning, H. P., and S. M. Jarvis. 2001. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by the P2 adenosine transporter and at least one novel, unrelated transporter. Acta Trop. 80:245–250. [DOI] [PubMed] [Google Scholar]
- Edwards, K. J., T. C. Jenkins, and S. Neidle. 1992. Crystal structure of a pentamidine-oligonucleotide complex: implications for DNA-binding properties. Biochemistry. 31:7104–7109. [DOI] [PubMed] [Google Scholar]
- Fairley, T. A., R. R. Tidwell, I. Donkor, N. A. Naiman, K. A. Ohemeng, R. J. Lombardy, J. A. Bentley, and M. Cory. 1993. Structure, DNA minor groove binding, and base pair specificity of alkyl- and aryl-linked bis(amidinobenzimidazoles) and bis(amidinoindoles). J. Med. Chem. 36:1746–1753. [DOI] [PubMed] [Google Scholar]
- Francesconi, I., W. D. Wilson, F. A. Tanious, J. E. Hall, B. C. Bender, R. R. Tidwell, D. McCurdy, and D. W. Boykin. 1999. 2,4-Diphenyl furan diamidines as novel anti-Pneumocystis carinii pneumonia agents. J. Med. Chem. 42:2260–2265. [DOI] [PubMed] [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foreman, J. Ciolkowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, A. L. G. Liu, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle, and J. A. Pople. 1998. Gaussian 98. Gaussian, Inc., Pittsburgh, PA.
- Gimenez-Arnau, E., S. Missailidis, and M. F. Stevens. 1998a. Antitumour polycyclic acridines. Part 2. Physicochemical studies on the interactions between DNA and novel polycyclic acridine derivatives. Anticancer Drug Des. 13:125–143. [PubMed] [Google Scholar]
- Gimenez-Arnau, E., S. Missailidis, and M. F. Stevens. 1998b. Antitumour polycyclic acridines. Part 4. Physico-chemical studies on the interactions between DNA and novel tetracyclic acridine derivatives. Anticancer Drug Des. 13:431–451. [PubMed] [Google Scholar]
- Hamelberg, D., L. McFail-Isom, L. D. Williams, and W. D. Wilson. 2000. Flexible structure of DNA: ion dependence of minor-groove structure and dynamics. J. Am. Chem. Soc. 122:10513–10520. [Google Scholar]
- Hamelberg, D., L. D. Williams, and W. D. Wilson. 2001. Influence of the dynamic positions of cations on the structure of the DNA minor groove: sequence-dependent effects. J. Am. Chem. Soc. 123:7745–7755. [DOI] [PubMed] [Google Scholar]
- Hopkins, K. T., W. D. Wilson, B. C. Bender, D. R. McCurdy, J. E. Hall, R. R. Tidwell, A. Kumar, M. Bajic, and D. W. Boykin. 1998. Extended aromatic furan amidino derivatives as anti-Pneumocystis carinii agents. J. Med. Chem. 41:3872–3878. [DOI] [PubMed] [Google Scholar]
- Houssier, C. 1981. Investigating nucleic acids, nucleoproteins, polynucleotides, and their interactions with small ligands by electro-optical systems. In Molecular Electro-Optics. S. Krause, editor. Plenum Publishing Corporation, New York. 363–398.
- Jansen, K., P. Lincoln, and B. Nordén. 1993. Binding of DAPI Analogue 2,5-Bis(4-amidinophenyl)furan to DNA. Biochemistry. 32:6605–6612. [DOI] [PubMed] [Google Scholar]
- Jenkins, T. C., A. N. Lane, S. Neidle, and D. G. Brown. 1993. NMR and molecular modeling studies of the interaction of berenil and pentamidine with d(CGCAAATTTGCG)2. Eur. J. Biochem. 213:1175–1184. [DOI] [PubMed] [Google Scholar]
- Jones, R. L., and W. D. Wilson. 1981. Effect of ionic strength on the pKa of ligands bound to DNA. Biopolymers. 20:141–154. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kim, S. K., S. Eriksson, M. Kubista, and B. Nordén. 1993. Interaction of 4′,6-Diamidino-2-phenylindole (DAPI) with Poly[d(G-C)2] and Poly[d(G-m5C)2]: evidence for major groove binding of a DNA Probe. J. Am. Chem. Soc. 115:3441–3447. [Google Scholar]
- Kunne, A. G. E., M. Sieber, D. Meierhans, and R. K. Allemann. 1998. Thermodynamics of the DNA binding reaction of transcription factor MASH- 1. Biochemistry. 37:4217–4223. [DOI] [PubMed] [Google Scholar]
- Lagunavicius, A., S. Grazulis, E. Balciunaite, D. Vainius, and V. Siksnys. 1997. DNA binding specificity of MunI restriction endonuclease is controlled by pH and calcium ions: involvement of active site carboxylate residues. Biochemistry. 36:11093–11099. [DOI] [PubMed] [Google Scholar]
- Lamm, G., and G. R. Pack. 1990. Acidic domains around nucleic acids. Proc. Natl. Acad. Sci. USA. 87:9033–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughton, C. A., F. Tanious, C. M. Nunn, D. W. Boykin, W. D. Wilson, and S. Neidle. 1996. A crystallographic and spectroscopic study of the complex between d(CGCGAATTCGCG)2 and 2,5-bis(4-guanylphenyl)furan, an analogue of berenil. Structural origins of enhanced DNA-binding affinity. Biochemistry. 35:5655–5661. [DOI] [PubMed] [Google Scholar]
- Lavesa, M., and K. R. Fox. 2001. Preferred binding sites for [N-MeCYs(3), N-MeCys(7)]TANDEM determined using a universal footprinting substrate. Anal. Biochem. 293:246–250. [DOI] [PubMed] [Google Scholar]
- Lee, M., A. L. Rhodes, M. D. Wyatt, M. D'Incalci, S. Forrow, and J. A. Hartley. 1993a. In vitro cytotoxicity of GC sequence directed alkylating agents related to distamycin. J. Med. Chem. 36:863–870. [DOI] [PubMed] [Google Scholar]
- Lee, M., A. L. Rhodes, M. D. Wyatt, S. Forrow, and J. A. Hartley. 1993b. Design, synthesis, and biological evaluation of DNA sequence and minor groove selective alkylating agents. Anticancer Drug Des. 8:173–192. [PubMed] [Google Scholar]
- Lundback, T., S. van Den Berg, and T. Hard. 2000. Sequence-specific DNA binding by the glucocorticoid receptor DNA-binding domain is linked to a salt-dependent histidine protonation. Biochemistry. 39:8909–8916. [DOI] [PubMed] [Google Scholar]
- Lyng, R., A. Rodger, and B. Norden. 1992. The CD of ligand-DNA systems. 2. Poly(dA-dT) B-DNA. Biopolymers. 32:1201–1214. [DOI] [PubMed] [Google Scholar]
- Myszka, D. G. 1997. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr. Opin. Biotechnol. 8:50–57. [DOI] [PubMed] [Google Scholar]
- Myszka, D. G., X. He, M. Dembo, T. A. Morton, and B. Goldstein. 1998. Extending the range of rate constants available from BIACORE: interpreting mass transport-influenced binding data. Biophys. J. 75:583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neidle, S. 2002. Nucleic Acid Structure and Recognition. Oxford University Press, New York.
- Nguyen, B., M. P. Lee, D. Hamelberg, A. Joubert, C. Bailly, R. Brun, S. Neidle, and W. D. Wilson. 2002a. Strong binding in the DNA minor groove by an aromatic diamidine with a shape that does not match the curvature of the groove. J. Am. Chem. Soc. 124:13680–13681. [DOI] [PubMed] [Google Scholar]
- Nguyen, B., C. Tardy, C. Bailly, P. Colson, C. Houssier, A. Kumar, D. W. Boykin, and W. D. Wilson. 2002b. Influence of compound structure on affinity, sequence selectivity and mode of binding to DNA for unfused aromatic dications related to furamidine. Biopolymers. 63:281–297. [DOI] [PubMed] [Google Scholar]
- Pachter, J. A., C. H. Huang, V. H. DuVernay, Jr., A. W. Prestayko, and S. T. Crooke. 1982. Viscometric and fluorometric studies of deoxyribonucleic acid interactions of several new anthracyclines. Biochemistry. 21:1541–1547. [DOI] [PubMed] [Google Scholar]
- Pal, S. K., L. Zhao, and A. H. Zewail. 2003. Water at DNA surfaces: ultrafast dynamics in minor groove recognition. Proc. Natl. Acad. Sci. USA. 100:8113–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portugal, J., and M. J. Waring. 1987. Comparison of binding sites in DNA for berenil, netropsin and distamycin. A footprinting study. Eur. J. Biochem. 167:281–289. [DOI] [PubMed] [Google Scholar]
- Rahmathullah, S. M., J. E. Hall, B. C. Bender, D. R. McCurdy, R. R. Tidwell, and D. W. Boykin. 1999. Prodrugs for amidines: synthesis and anti-Pneumocystis carinii activity of carbamates of 2,5-bis(4-amidinophenyl)furan. J. Med. Chem. 42:3994–4000. [DOI] [PubMed] [Google Scholar]
- Reddy, B. S., S. M. Sondhi, and J. W. Lown. 1999. Synthetic DNA minor groove-binding drugs. Pharmacol. Ther. 84:1–111. [DOI] [PubMed] [Google Scholar]
- Renault, E., M. P. Fontaine-Aupart, F. Tfibel, M. Gardes-Albert, and E. Bisagni. 1997. Spectroscopic study of the interaction of pazelliptine with nucleic acids. J. Photochem. Photobiol. B. 40:218–227. [DOI] [PubMed] [Google Scholar]
- Rodger, A. 1993. Linear dichroism. Methods Enzymol. 226:232–258. [DOI] [PubMed] [Google Scholar]
- Rodger, A., and B. Nordén. 1997. Circular Dichroism and Linear Dichroism. Oxford University Press, New York.
- Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- Saenger, W. 1984. Principles of Nucleic Acid Structure. Springer-Verlag, New York.
- Schmechel, D. E., and D. M. Crothers. 1971. Kinetic and hydrodynamic studies of the complex of proflavine with poly A-poly U. Biopolymers. 10:465–480. [DOI] [PubMed] [Google Scholar]
- Stephens, C. E., F. Tanious, S. Kim, W. D. Wilson, W. A. Schell, J. R. Perfect, S. G. Franzblau, and D. W. Boykin. 2001. Diguanidino and “reversed” diamidino 2,5-diarylfurans as antimicrobial agents. J. Med. Chem. 44:1741–1748. [DOI] [PubMed] [Google Scholar]
- Tidwell, R. R., and D. W. Boykin. 2003. Dicationic DNA minor-groove binders as antimicrobial agents. In DNA and RNA Binders: From Small Molecules to Drugs. M. Demeunynck, C. Bailly, and W. D. Wilson, editors. WILEY-VCH, Weinheim, Germany. 414–460.
- Tidwell, R. R., S. K. Jones, J. D. Geratz, K. A. Ohemeng, C. A. Bell, B. J. Berger, and J. E. Hall. 1990a. Development of pentamidine analogues as new agents for the treatment of Pneumocystis carinii pneumonia. Ann. N. Y. Acad. Sci. 616:421–441. [DOI] [PubMed] [Google Scholar]
- Tidwell, R. R., S. K. Jones, J. D. Geratz, K. A. Ohemeng, M. Cory, and J. E. Hall. 1990b. Analogues of 1,5-bis(4-amidinophenoxy)pentane (pentamidine) in the treatment of experimental Pneumocystis carinii pneumonia. J. Med. Chem. 33:1252–1257. [DOI] [PubMed] [Google Scholar]
- Trent, J. O., G. R. Clark, A. Kumar, W. D. Wilson, D. W. Boykin, J. E. Hall, R. R. Tidwell, B. L. Blagburn, and S. Neidle. 1996. Targeting the minor groove of DNA: crystal structures of two complexes between furan derivatives of berenil and the DNA dodecamer d(CGCGAATTCGCG)2. J. Med. Chem. 39:4554–4562. [DOI] [PubMed] [Google Scholar]
- Waring, M. J., and C. Bailly. 1997. The influence of the exocyclic amino group characteristic of GC base pairs on molecular recognition of specific nucleotide sequences in DNA by berenil and DAPI. J. Mol. Recognit. 10:121–127. [DOI] [PubMed] [Google Scholar]
- Wells, R. D., J. E. Larson, R. C. Grant, B. E. Shortle, and C. R. Cantor. 1970. Physicochemical studies on polydeoxyribonucleotides containing defined repeating nucleotide sequences. J. Mol. Biol. 54:465–497. [DOI] [PubMed] [Google Scholar]
- Wemmer, D. E. 2000. Designed sequence-specific minor groove ligands. Annu. Rev. Biophys. Biomol. Struct. 29:439–461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.