

Abstract
In the last few years, an increased attention has been focused on NAD+-dependent DNA ligases. This is mostly due to their potential use as antibiotic targets, because effective inhibition of these essential enzymes would result in the death of the bacterium. However, development of an efficient drug requires that the conformational modifications involved in the catalysis of NAD+-dependent DNA ligases are understood. From this perspective, we have investigated the conformational changes occurring in the thermophilic Thermus scotoductus NAD+-DNA ligase upon adenylation, as well as the effect of cofactor binding on protein resistance to thermal and chemical (guanidine hydrochloride) denaturation. Our results indicate that cofactor binding induces conformational rearrangement within the active site and promotes a compaction of the enzyme. These data support an induced “open-closure” process upon adenylation, leading to the formation of the catalytically active enzyme that is able to bind DNA. These conformational changes are likely to be associated with the protein function, preventing the formation of nonproductive complexes between deadenylated ligases and DNA. In addition, enzyme adenylation significantly increases resistance of the protein to thermal denaturation and GdmCl-induced unfolding, establishing a thermodynamic link between ligand binding and increased conformational stability. Finally, chemical unfolding of deadenylated and adenylated enzyme is accompanied by accumulation of at least two equilibrium intermediates, the molten globule and premolten globule states. Maximal populations of these intermediates are shifted toward higher GdmCl concentrations in the case of the adenylated ligase. These data provide further insights into the properties of partially folded intermediates.
INTRODUCTION
DNA ligases are ubiquitous enzymes required for important cellular processes such as DNA replication, DNA recombination, and DNA repair, in all three kingdoms of life (Lehman, 1974). They are divided into two broad classes, those requiring NAD+ as cofactor and those requiring ATP (Lehman, 1974). Regardless of their energy source, they catalyze the sealing of 5′-phosphate and 3′-hydroxyl termini at nicks in duplex DNA by means of three distinct catalytic events (Lehman, 1974). The first involves activation of the ligase through the formation of a covalent adenylated intermediate by transfer of the adenyl group of NAD+ or ATP to the ɛ-NH2 of a conserved lysine residue in the DNA ligase. In the second step, the AMP moiety is transferred from the ligase to the 5′-phosphate group at the single-strand break site, creating a new pyrophosphate bond. Finally, DNA ligase catalyzes the DNA ligation step with loss of free AMP.
NAD+-dependent ligases are of fairly uniform size (∼70 kDa) and display extensive amino acid sequence conservation throughout the entire protein. They are composed of four discrete domains (Doherty and Suh, 2000; Lee et al., 2000). Domain 1 comprises the subdomain 1a, required for ligase adenylation (Sriskanda and Shuman, 2002) and the subdomain 1b, called “Adenylation domain,” containing the lysine residue implicated in enzyme adenylation. The domain 2 is called “Oligomer-Binding (OB) fold domain.” The domain 3 contains a zinc finger (subdomain 3a) and a HhH (Helix-hairpin-Helix) (subdomain 3b) subdomains, whereas the domain 4 consists of a “BRCT domain” (BRCA-carboxy-terminal-related domain). ATP-dependent ligases are more diverse in size but share a common ligase domain (formed by domains 1 and 2) called “catalytic core.” This domain comprises six conserved sequence motifs, I, III, IIIa, IV, V-VI, that define a family of related nucleotidyltransferases including mRNA capping enzymes as well as RNA- and tRNA-ligases (Hakansson et al., 1997). Some ATP ligases are possibly flanked by additional domains that are likely to be implicated in the enzyme specialization, targeting the various ATP DNA ligases to different pathways in DNA repair and replication (for review, see Martin and MacNeill (2002); Timson et al. (2000)). Although there is a scant amino acid sequence similarity between NAD+- and ATP- DNA ligases, the tertiary structures of the catalytic cores are quite well conserved (Doherty and Suh, 2000; Singleton et al., 1999; Timson et al., 2000) and the adenylate binding pocket of NAD+ ligases is composed of the same six nucleotidyl transferase motifs described originally in the ATP-dependent enzymes (Aravind and Koonin, 1999). The conservation and similarity of these structural features strongly suggest that the two types of ligases have evolved from a common ancestor and are likely to have a similar catalytic mechanism. The divergence may have largely arisen from the need to accommodate different nucleotide cofactors (Aravind and Koonin, 1999).
Nick recognition requires ligases to be adenylated at the lysine residue located within the active site (Shuman, 1995). In fact, molecular modeling studies of T7 DNA ligase suggest that dsDNA binds predominantly in a positively charged interdomain cleft, between domain 1 and 2 (Doherty and Dafforn, 2000). Upon adenylation, the DNA binding domain 2, rotates around and exposes the DNA-binding face toward the active site (closed conformation), allowing DNA to bind. In the deadenylated enzyme, the proposed DNA-binding face is rotated away from the active site cleft (open conformation). Therefore, adenylation would act as a conformational switch, facilitating rotation of the DNA-binding surface of the OB fold toward the active site cleft. In this view, only adenylated ligase can bind to nicked DNA, preventing the formation of nonproductive ligase-DNA complexes. Interestingly, two crystal structures of PBCV-1 mRNA capping enzyme have provided conclusive evidence for such open-closed conformational switch during the guanylylation reaction (Hakansson et al., 1997), which is equivalent to the adenylation reaction catalyzed by DNA ligases (Shuman and Schwer, 1995). This involves a 13-Å movement of the C-terminal OB domain 2 toward domain 1. Such conformational modification has also been proposed for the Thermus filiformis NAD+-DNA ligase (Lee et al., 2000). In this case, domain 4 (BRCT domain) is very mobile in the open conformation but its mobility is restricted in the closed conformation, allowing a contact with domain 1a, which leads to an active toroidal conformation (Lee et al., 2000). Thus, despite their different amino acid sequences and specificities, all these enzymes are likely to undergo major domain rearrangements upon cofactor binding.
To date, NAD+-dependent DNA ligases have been predominantly isolated from eubacteria (Wilkinson et al., 2001). For that reason, they constitute an attractive target for broad-spectrum antibiotic therapy predicated on blocking the reaction of DNA ligase with NAD+, leading to an inactive enzyme and thus growth arrest of the eubacterium. The drug-binding site would ideally be unique to, and conserved among, NAD+-DNA ligases but absent from ATP-dependent ligases and other essential NAD+-requiring enzymes. This strategy implies that: i), the conformational modifications involved in the catalysis of NAD+-dependent DNA ligases are understood and ii), the structural components of NAD+-ligases interacting specifically with NAD+ are known. A recent study, in which the role of domain Ia of Escherichia coli DNA ligase was investigated, has provided major advances toward the elucidation of this later issue (Sriskanda and Shuman, 2002). This domain, essential for the reaction with NAD+ and unique to NAD+-dependent ligases, contains several conserved residues likely interacting with the nicotinamide mononucleotide (NMN) moiety of the NAD+ substrate. Because this domain is well conserved among all NAD+-ligases, it is expected that the role of domain Ia is common to all of them. To characterize the structural changes occurring upon adenylation, we have investigated the unfolding pathways of the deadenylated and adenylated Thermus scotoductus DNA ligase (Tslig). This enzyme possesses all the conserved motifs common to NAD+-dependent ligases and shows similar overall catalytic properties to other NAD+-dependent DNA ligases (Thorbjarnardóttir et al., 1995). In this paper, we show that cofactor binding induces conformational changes within the active site of Tslig, suggesting an open-closure mechanism upon adenylation. In addition, we provide strong evidences that cofactor binding also induces increased resistance to thermal and chemical denaturation. Finally, the results of chemical denaturation analysis indicate that the unfolding is a complex sequential process and goes through at least two intermediates, the molten globule and pre-molten globule states.
MATERIALS AND METHODS
Chemicals and enzymes
3-(1-pyridinio)-1-propane sulfonate and acrylamide were from Fluka (Buchs, Switzerland) β-NAD+, β-NMN, and 8-anilino-1-naphtalene sulfonic acid (ANS) were from Sigma-Aldrich NV/SA (St. Louis, MS). Guanidine Hydrochloride (GdmCl, ultra pure) was from ICN Biomedicals Inc. (Irvine, CA). Hi-Trap Heparin, MonoQ HR 5/5, Superdex 200 HR 10/30, and fast protein liquid chromatography (FPLC) systems were from Pharmacia LKB Biotechnology (Stockholm, Sweden). Water used for the experiments was purified over a Milli-Q water purification system from Millipore (Billerica, MA), and all solutions were filtered through 0.22 μm filters before use. Plasmid encoding T. scotoductus NAD+-DNA ligase (Tslig) was a kind gift from Z. O. Jónsson and G. Eggertsson (Reykjavik, Iceland) (Thorbjarnardóttir et al., 1995).
Three-dimensional modeling
The target sequence for modeling is the NAD+-dependent DNA ligase from T. scotoductus. The modeling, minimizations, and molecular dynamics were done using the molecular operating environment (MOE) program (Chemical Computing Group, Montreal, Canada). Optimal multiple sequence alignment was performed using the Match-Box program (Depiereux et al., 1997). The first step was a Protein Data Bank search using the target sequence to find the templates with the highest homology. Originally, a fast scan was performed using the Fasta methodology (Pearson, 1996). An expectation value (E-Value) was determined for each sequence. Then, the list was reduced using thresholds of the E-value (E-Value Cutoff and E-Value Accept). Only E-Values lower than E-Value Cutoff and with Z-score higher than 7 were accepted.
The best template found for T. scotoductus was 1DGS.A, the homologous NAD+-DNA-ligase of Thermus filiformis (expected value: 7.2 10−259), which does not contain a C-terminal BRCT domain (Lee et al., 2000). The five best templates were then aligned using the Match-Box software and evaluated using secondary structure prediction algorithm. The alignment was then introduced into the modeling process. Thus, 1DGS.A was used as a template for the modeling. The modeling program was configured to include outgaps and to build 10 intermediate models using a Boltzmann-weighted randomized modeling procedure adapted from Levitt (1992), combined with specialized logic for the proper handling of insertions and deletions (Fechteler et al., 1995), minimize them, and select the best intermediate. The final model is the one that scored best according to the packing quality function. The obtained model was then thoroughly minimized, using steepest gradient algorithm for 100 iterations, followed by the conjugate gradient algorithms for another 100 iterations, to end with the truncated Newton algorithm until the root-mean-square gradient went under 0.01.
The obtained model was then reviewed following the experimental data for the homologous proteins, and using MOE's stereochemical quality evaluation tools to confirm that the model's stereochemistry is reasonably consistent with the typical values found in crystal structures.
At this step a molecular dynamics was done using Molarity, Volume, Temperature thermodynamic ensemble (number of particles, volume, and temperature are fixed). The equilibrium temperature chosen was 354°K, known as the optimal temperature of T. scotoductus. The heating, equilibrium, and cooling cycles were held for 1000 iterations each. The most stable conformation was chosen as a final model.
Protein purification
The recombinant wild-type T. scotoductus NAD+-DNA ligase (Tslig) was overexpressed as previously described (Thorbjarnardóttir et al., 1995), except that the growth was performed in TB medium (12 g/l tryptone, 24 g/l yeast extract, 4 ml/l glycerol, 12.54 g/l K2HPO4, 2.32 g/l KH2PO4 pH 7) containing 100 μg/ml ampicillin. Tslig DNA ligase was purified by two successive heatings (65°C and 80°C), followed by Hi-Trap Heparin and MonoQ HR chromatographic steps. Protein concentration was determined with the Coomassie protein assay reagent (Pierce, Rockford, IL), using BSA as standard. The final yield was ∼55 mg of protein per liter of culture. N-terminal sequencing confirmed the integrity of the recombinant protein. As previously reported in literature for NAD+-dependent DNA ligases (Barany and Gelfand, 1991; Brannigan et al., 1999; Georlette et al., 2000; Ishino et al., 1986; Kaczorowski and Szybalski, 1996; Modrich et al., 1973; Panasenko et al., 1978; Singleton et al., 1999; Takahashi and Uchida, 1986; Zimmerman and Pheiffer, 1983), the native recombinant enzyme is produced in an adenylated form: DSC, fluorescence, and circular dichroism profiles of the native and adenylated Tslig (after incubation with NAD+) are superimposable.
Deadenylation/adenylation of Tslig
Tslig stock solution was either deadenylated or adenylated according to Timson (Timson and Wigley, 1999) by adding excess of β-NMN or β-NAD+, respectively. The ratio [NMN/NAD+]/[Tslig] was ∼80. Deadenylated/adenylated mixtures were then heated at 65°C for 30 min and then cooled rapidly on ice. When required, protein solutions were dialyzed against appropriate buffer before experiments.
Differential scanning calorimetry
Measurements were performed using a MicroCal MCS-DSC instrument at a scan rate of 60 K/h and under 2 atm nitrogen pressure. Samples (∼4 mg/ml) were dialyzed overnight against 30 mM MOPS, 50 mM KCl, pH 7.6. To decrease aggregation, a nondetergent sulphobetaine (3-(1-pyridinio)-1-propane sulfonate) was added before differential scanning calorimetry (DSC) experiment (Goldberg et al., 1995), to a final concentration of 0.75 M. Thermograms were analyzed according to a non-two-state model in which Tm, ΔHcal, and ΔHeff of individual transitions are fitted independently using the MicroCal Origin software (version 2.9). The magnitude and source of the errors in the Tm and enthalpies values have been discussed elsewhere (Matouschek et al., 1994). All scans were found to be irreversible under the experimental conditions used for these studies.
GdmCl-induced unfolding transitions
Tslig deadenylated/adenylated samples were incubated overnight at 25°C in the presence of various concentrations of guanidine hydrochloride (GdmCl). Unfolding curves were determined by monitoring the intrinsic fluorescence emission or circular dichroism (CD) at 25°C. The pH was checked to ensure a constant value throughout the whole transition, and the denaturant concentration was determined from refractive index measurements (Pace, 1986), using a R5000 hand refractometer from Atago (Tokyo, Japan).
Fluorescence measurements
Both intrinsic and ANS fluorescence emission spectra were recorded on an Aminco SLM 8100 spectrofluorimeter. Excitation and emission slit widths were 2 and 4 nm, respectively, and the scan speed was 350 nm/min. Cuvettes with 1-cm pathlength were used. Intrinsic fluorescence measurements were performed using a protein concentration of 25 μg/ml (0.33 μM), with excitation at 280 nm, and emission spectra recorded from 300 to 440 nm. The buffer used was 20 mM phosphate sodium, 50 mM NaCl, pH 7.6, in the presence of desired GdmCl concentrations. With all samples, fluorescence spectra were corrected for the background fluorescence of the solution (buffer + denaturant). Two fluorescence parameters have been considered in this work: the fluorescence intensity at single excitation and emission wavelengths and the average emission wavelength (AEW). The AEW values were computed according to the following equation (Royer et al., 1993),
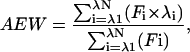 |
(1) |
where F is the fluorescence intensity and λ the wavelength. The AEW values were calculated between 300 and 440 nm.
ANS fluorescence measurements were performed with the samples used for intrinsic fluorescence measurements, with excitation at 390 nm, and emission spectra recorded from 420 to 600 nm. The fluorescence spectra were corrected for the background fluorescence of ANS. The ratio [ANS]/[Tslig] was ∼250 (ɛ350nm = 4950 M−1 cm−1 for ANS).
Circular dichroism measurements
Circular dichroism (CD) spectra were recorded at 25°C using a CD6 Jobin Yvon spectropolarimeter under constant nitrogen flow. In the far ultraviolet (UV) region, spectra were recorded in a 0.1-cm cell at protein concentrations of ∼0.25 mg/ml whereas in the near UV region, a 1.0-cm cell was used for protein concentrations of ∼1 mg/ml. The buffer used was 20 mM sodium phosphate, 50 mM NaCl, pH 7.6, in the presence of desired GdmCl concentrations. Spectra were acquired at a scan speed of 20 nm/min, with a 2-nm bandwidth and a 1-s integration time. Spectra were averaged over five scans and corrected for the buffer signal. Raw data were expressed in terms of the mean residue ellipticity [θ] using the known mature adenylated Tslig sequence (MW = 76855 Da) for calculation of the mean residue weight.
GdmCl unfolding curves of adenylated Tslig were recorded at 222 and 280 nm, using a 2-nm bandwidth. At all denaturant concentrations, at least 30 data points were acquired over 1 min (2-s integration time), and averaged. The resulting values were corrected for the contribution of the solvent. An estimate of the helical content (fH) of Tslig has been obtained from the following equation (Chen et al., 1972),
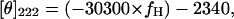 |
(2) |
where [θ]222 is the mean residue ellipticity (deg cm2 dmol−1) at 222 nm.
Data analysis
The transition curves obtained by fluorescence spectroscopy and CD were analyzed using Eqs. 3 and 4, assuming a two- (N↔U, where N is the native state and U is the fully unfolded state) or three-state model (N↔I↔U, where I is the thermodynamically stable intermediate) for the unfolding reaction. For N↔U transition (Pace, 1990),
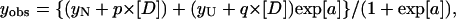 |
(3) |
where
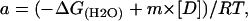 |
and where yobs is the measured variable parameter at a given denaturant concentration, whereas yN and yU represent the values of this parameter for the native and denatured states, respectively. ΔG(H2O) is the difference in free energy between folded and unfolded conformations under physiological concentrations and m is a measure of the dependence of the free energy on the denaturant concentration [D]; p and q are the slopes of the pre- and post-unfolding baselines, respectively, R is the gas constant and T is the absolute temperature. The midpoint of the denaturation curve ([U]/[N] = 1) is given by Cm = ΔG(H2O)/m. For N↔I↔U process (Vanhove et al., 1997),
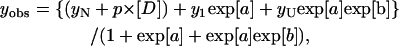 |
(4) |
where
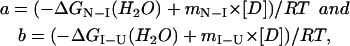 |
and where ΔGN−I(H2O) and ΔGI−U(H2O) are the differences in free energy between I and N and between U and I, respectively, in the absence of denaturant, and mN−I and mI−U are the slopes of the transitions.
Analysis of circular dichroism data using the“phase diagram” method
The “phase diagram” method analysis of spectroscopic data is extremely sensitive for the detection of intermediate states (Burstein, 1976; Bushmarina et al., 2001; Kuznetsova et al., 2002; Munishkina et al., 2003; Permyakov et al., 1980; Uversky et al., 2003). Although this method was developed for the analysis of fluorescence data (Burstein, 1976), it can be used with any spectroscopic technique. The essence of this method is to build up the diagram of Iλ1 vs. Iλ2, where Iλ1 and Iλ2 are the spectral intensity values measured at wavelengths λ1 and λ2 under different experimental conditions for a protein undergoing structural transformations. The relationship between Iλ1 and Iλ2 is described in the following equation,
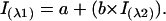 |
(5) |
The calculations allowing the determination of such equation, as well as the composition of a and b can be found elsewhere (Kuznetsova et al., 2002; Uversky et al., 2003). As a rule, λ1 and λ2 are arbitrary wavelengths of the spectrum, but in practice, such diagrams will be more informative if λ1 and λ2 will be on different slopes of the spectrum. If the wavelengths are from one slope or near the maximum, some transitions may remain undetected.
Size exclusion chromatography
Hydrodynamic dimensions (Stokes radius, RS) of deadenylated and adenylated Tslig in different conformational states were measured by gel filtration. Size exclusion chromatography (SEC) was performed on the Superdex 200 H/R 10/30 prepacked FPLC column calibrated according to (Uversky, 1993). Protein (∼0.25 mg/ml) containing the required concentrations of GdmCl was loaded onto the column equilibrated with the same buffer. The elution was carried out isocratically at a flow rate of 1.0–0.5 ml/min and monitored by the absorbance at 280 nm. All measurements were made at 25°C. Molecular Stokes radii (RS) were estimated from elution volume Vel measured according to the following equation,
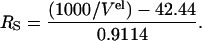 |
(6) |
The accuracy of determination of RS by this equation is ∼95%. Relative areas of chromatographic peaks were estimated from the elution profiles by their deconvolution using LabCalc. The accuracy of such deconvolution was ∼90%. Stages of unfolding of Tslig were determined as follows (Uversky and Ptitsyn, 1994, 1996),
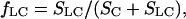 |
(7) |
where fLC represents the fraction of molecules that undergoes the transition from a compact (C) to a less compact state and is determined from relative areas (S) of corresponding FPLC peaks
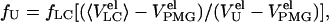 |
(8) |
where fU represents the fraction of unfolded molecules, and where 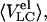
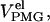 and
and 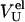 are the average elution volume of less compact molecules, the elution volume of molecules in the premolten globule state, and the elution volume of molecules in the unfolded state, respectively.
are the average elution volume of less compact molecules, the elution volume of molecules in the premolten globule state, and the elution volume of molecules in the unfolded state, respectively.
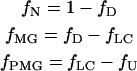 |
(9) |
where fN, fMG, fPMG, and fU are the fraction of molecules in native, molten-globule, premolten globule, and unfolded states, respectively.
Stern-Volmer quenching
The conformational state of enzymes was further characterized by acrylamide-induced fluorescence quenching. Samples were prepared in 20 mM sodium phosphate buffer, 50 mM NaCl, pH 7.6, and the protein concentrations were adjusted to provide an optical density at the excitation wavelength <0.1. Aliquots of a 1.2-M acrylamide stock solution were consecutively added to 1 ml protein solution to increase acrylamide concentration by ∼5 mM steps. Experiments were performed using excitation at 295 nm with fluorescence emission set at 333 nm (excitation and emission slit widths were 1 and 4, respectively) and the fluorescence intensities were recorded for 30 s. Experiments were performed in triplicate. The data were corrected for the dilution effects and for the absorptive screening caused by acrylamide (ɛ295nm = 0.25 M−1 cm−1 for acrylamide). Quenching data were plotted as the ratio of fluorescence in the absence of quencher (F0) to the intensity in the presence of quencher (F) against quencher concentration. The resulting data were fit to dynamic parameters according to the Stern-Volmer equation (Lakowicz, 1983),
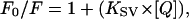 |
(10) |
where KSV is the Stern-Volmer quenching constant and [Q] the quencher concentration.
RESULTS AND DISCUSSION
Molecular model of adenylated Tslig
The sequences of T. scotoductus (Tslig) and T. filiformis (Tflig) DNA ligases display 80% identity, allowing to build a model of the former (Fig. 1 B). All residues implicated in nick sensing and ligation activities in Tflig (Lee et al., 2000) are conserved in Tslig. Like the Tflig template, Tslig is a monomeric enzyme folded into four discrete domains (Fig. 1, A and B). Domain I (residues 1-320) consists of two subdomains, subdomain Ia, essentially α-helical (residues 1-73) and the “adenylation” domain (Ib), comprising two mainly antiparallel β-sheets flanked by α-helices and containing an adenylation site (residues 74-320). Domain 2 or “OB fold domain” contains a five-stranded antiparallel β-barrel (residues 321-406). Domain 3 consists of two subdomains, where subdomain 3a is a Cys4-type zinc finger (residues 407-432) and subdomain 3b comprises four Helix-hairpin-Helix (HhH) motifs (residues 433-584). Domain 4 is a member of the BRCT (BRCA1 C-terminus) domain superfamily (residues 585-674). As shown for Tf DNA ligase (Lee et al., 2000), the circular arrangement of the four domains leads to a hole sufficiently large to hold a double-stranded DNA. In addition to 20 Phe (F) residues, Tslig contains 21 Tyr (Y) residues and 6 Trp (W) residues, indicated in Fig. 1 B. Four tryptophanes, W135, W246, W274, and W298, are located within the active site, whereas W375 and W405 are located within the domain 2. W246 and W298 could make contact with AMP within the active site, whereas W135 is located too far and W274 is “shielded” by two β-sheets (Fig. 1 B). In addition, the adenine base of AMP is stacked against the side chain of Y226, which is conserved among all known NAD+-dependent DNA ligases (Doherty and Suh, 2000; Lee et al., 2000). Finally, W135, W246, W274, and W405 are rather buried in the model, whereas W298 and W375 are relatively accessible to the solvent.
FIGURE 1.

Domain structure of T. scotodustus DNA ligase. (A) T. scotoductus DNA ligase (Tslig) is illustrated as a linear assortment of conserved structural domains (Ia, Adenylation, OB (Oligomer Binding) fold, Zn-binding, HhH (Helix-hairpin-Helix), and BRCT (BRCA1 C-terminus)). D1 to D4, domain 1 to domain 4. (B) Schematic view of the three-dimensional structure of Tslig. The tryptophan (W) and tyrosine (Y) residues are rendered as white and gray space filled structures, respectively. Tslig contains 6 W and 21 Y. The covalently bound AMP moiety is pointed out by an arrow.
Prediction of partially folded intermediates
Recently, it has been shown (Uversky, 2002a) that the competence of a protein to form equilibrium intermediate(s) I, may be determined by the bulk content of hydrophobic and charged amino acid residues. In fact, proteins unfolding through a N↔I(x)↔U scheme are specifically localized within a unique region of a charge-hydrophobicity space, with 〈H〉 = 0.446 ± 0.023 and 〈R〉 = 0.027 ± 0.022, where 〈H〉 and 〈R〉 are the mean hydrophobicity and the mean net charge of the protein, respectively. The mean hydrophobicity 〈H〉 is defined as the sum of the normalized hydrophobicities of all residues divided by the number of residues in the polypeptide. The mean net charge 〈R〉 is defined as the net charge at pH 7.0, divided by the total number of residues. Analysis of the Tslig amino acid sequence shows that this protein is characterized by 〈H〉 = 0.450 and 〈R〉 = 0.007, thus fulfilling the prediction for unfolding through intermediate states.
Effect of temperature on the structure of deadenylated and adenylated Tslig
The thermal stabilities of deadenylated and adenylated Tslig were compared by DSC. We have established that under conditions studied both forms unfold irreversibly, as demonstrated by the lack of ΔHcal recovery during a second denaturation scan and by the dependence of calorimetric traces on scan rates (not shown). For each form, two distinct heat absorption peaks were observed (Fig. 2). The deconvolution of the excess heat capacity (Cp) functions revealed two and three subsequent transitions for deadenylated and adenylated Tslig, respectively (Table 1). These results demonstrate that deadenylated and adenylated Tslig denature according to a non-two-state mechanism. The deviation from a two-state model was further confirmed by the fact that ΔHcal/ΔHeff ratio exceeds unity (not shown). Analysis of Table 1 also reveals that the adenylated enzyme displays an increase of ∼5°C and 80 kJ mol−1 in the Tmax and ΔHcal, respectively. Such results clearly indicate that cofactor anchoring at the active site increases markedly the conformational stability of the enzyme.
FIGURE 2.

Thermal unfolding of Tslig recorded by DSC. Adenylated Tslig is characterized by higher Tmax (top of the transition) and ΔHcal (area under the transition). All thermograms are baseline-subtracted and normalized for protein concentrations. Deadenylated Tslig (dashed line); adenylated Tslig (solid line).
TABLE 1.
Thermodynamic parameters of heat-induced unfolding of deadenylated and adenylated Tslig Parameters are determined from data shown in Fig. 2
| Tmax* (°C) | Transitions (n)† | Tm‡ (°C) | ΔHcal (kJ mol−1) | Σ ΔHcal (kJ mol−1) | |
|---|---|---|---|---|---|
| Deadenylated Tslig | 89.0 | 2 | 88.9 | 904 | 1649 |
| 96.6 | 96.5 | 745 | |||
| Adenylated Tslig | 95.2 | 3 | 91.8 | 301 | 1729 |
| 101.3 | 95.6 | 1009 | |||
| 101.4 | 417 |
Tmax corresponds to the temperature at the top of the peak.
n refers to the number of calorimetric domains identified by deconvolution of DSC thermograms.
Tm is the melting point of the unfolding transition.
Effect of GdmCl on the structure of deadenylated and adenylated Tslig
Several spectroscopic techniques have been applied to study the GdmCl-induced unfolding of deadenylated and adenylated Tslig. Unfolding of both forms was reversible because they regained native conformation following renaturation after complete denaturation in 7 M GdmCl (not shown).
Intrinsic fluorescence
The fluorescence emission spectra of the native forms of deadenylated and adenylated Tslig (λex 280 nm) show a single broad emission band with maxima at ∼332 nm (Fig. 3, A and B, and Table 4). Excitation at 295 nm did not dramatically modify the shape of emission spectra (λmax ∼333 nm), except that the shoulder near 306 nm (contribution of the Tyr residues) disappeared (see Fig. 3, A and Binsets). The λmax values of native enzymes indicate that the Trp residues in both forms are relatively buried and that adenylation does not change significantly the net polarity of the Trp environment within the “catalytic core” of the protein (domain 1 and 2, see Fig. 1 B). However, adenylation quenched the fluorescence of Tslig by ∼35% (Fig. 3, A and B), demonstrating that cofactor binding induces at least conformational rearrangement within the active site. Quenching of fluorescence after cofactor binding has been reported for numerous enzymes (Brandes et al., 1998; Candy et al., 1996; Diefenbach and Duggleby, 1991; Favilla et al., 2002; Goenka et al., 2001; Gupta and Kang, 1997; Marchal and Branlant, 1999; Morimatsu et al., 1995; Murataliev and Feyereisen, 2000; Sinha et al., 1999; Tang et al., 2001). The observed quenching of intrinsic Tslig fluorescence is likely due to an interaction of AMP with Y226 (the nucleotide is stacked against the side chain of Y226) and/or W246 and W298 (see also “Molecular model of adenylated Tslig”). In 6.5 M GdmCl, the wavelength of maximal emission is shifted to ∼347 nm (λex 280 nm) for both forms, indicating that they converge to an unfolded form exhibiting the same emissive properties with increased accessibility of the Trp residues to the bulk solvent (Fig. 3, A and B).
FIGURE 3.

Fluorescence emission spectra of deadenylated and adenylated Tslig. (A) Spectra of deadenylated Tslig recorded on excitation at 280 nm at 0 M (solid line), 2.5 M (long-dashed line), 3.5 M (dotted line), and 6.5 M (short-dashed line) GdmCl. Inset represents spectra recorded on excitation at 295 nm. (B) Spectra of adenylated Tslig recorded on excitation at 280 nm at 0 M (solid line), 3 M (long-dashed line), 4.4 M (dotted line), and 6.5 M (short-dashed line) GdmCl. Inset represents spectra recorded on excitation at 295 nm. Corresponding λmax are given in Table 4.
TABLE 4.
Stern-Volmer quenching constants (KSV) and λmax (nm) obtained for deadenylated and adenylated Tslig under different GdmCl concentrations
| Deadenylated Tslig
|
Adenylated Tslig
|
|||||
|---|---|---|---|---|---|---|
| GdmCl (M) | KSV (M−1)* |
†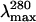
|
‡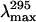
|
KSV (M−1)* |
†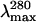
|
‡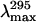
|
| 0 | 7.11 ± 0.11 | 332.2 | 333.0 | 5.66 ± 0.05 | 332.0 | 333.6 |
| 2.5 | 7.28 ± 0.10 | 333.6 | 333.6 | |||
| 3.0 | 5.29 ± 0.02 | 332.0 | 332.9 | |||
| 3.5 | 8.66 ± 0.12 | 338.2 | 339.9 | |||
| 4.4 | 7.87 ± 0.07 | 344.0 | 348.3 | |||
| 6.5 | 8.53 ± 0.08 | 347.0 | 350.0 | 7.81 ± 0.08 | 346.0 | 350.0 |
The Stern-Volmer quenching constants were obtained from the slopes of the lines for the plots of F0/F = 1 + (KSV × [Q]).
The excitation wavelength was 280 nm.
The excitation wavelength was 295nm.
GdmCl-induced unfolding of deadenylated Tslig monitored by the characteristic changes in fluorescence intensity (λex 280 nm), FI333 and in AEW are consistent with a simple two-state model N↔U (Fig. 4, A and B). Satisfactory fits of the data could be obtained from Eq. 3, providing similar ΔG(H2O), m, and Cm (concentration of GdmCl required for half-denaturation of the enzyme) values (Table 2).
FIGURE 4.

GdmCl-induced structural rearrangements of Tslig, monitored by changes in fluorescence spectroscopy. Unfolding of (○) deadenylated and (•) adenylated Tslig recorded by the changes of the fluorescence intensity (λex 280 nm) at 333 nm (A), of the average emission wavelength (B), and of the fluorescence intensity (λex 280 nm) at 306 nm (C). Data were analyzed according to a two-state reaction, except in A for adenylated Tslig where a three-state pathway was used; the lines represent the best fits to Eqs. 3 (two states) and 4 (three states), calculated using the thermodynamic parameters in Table 2. A. U., arbitrary units. Measurements were carried out at pH 7.6, 25°C. Protein concentration was kept at 0.025 mg/ml.
TABLE 2.
Thermodynamic parameters* of GdmCl-induced unfolding of deadenylated and adenylated Tslig, as obtained from the analysis of the equilibrium transitions
Δ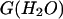 * (kJ mol−1) * (kJ mol−1) |
m* (kJ mol−1) | Cm (M) | Transition monitored† | |
|---|---|---|---|---|
| Deadenylated Tslig | ||||
Fluorescence 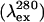
|
||||
| FI (333 nm) | 23.6 ± 2.5 | 6.8 ± 0.7 | 3.5 | MG↔U |
| AEW | 28.9 ± 2.0 | 8.0 ± 0.6 | 3.5 | MG↔U |
| Adenylated Tslig | ||||
Fluorescence 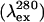
|
||||
| FIN−I (333 nm) | 18.2 ± 3.4 | 11.2 ± 1.8 | 1.6 | N↔MG |
| FII−U (333 nm) | 42.2 ± 4.0 | 10.2 ± 1.0 | 4.2 | MG↔PMG |
| AEW | 38.8 ± 1.8 | 9.3 ± 0.5 | 4.2 | MG↔PMG |
| Circular dichroism | ||||
| near (280 nm) | 12.3 ± 0.1 | 7.7 ± 0.1 | 1.6 | N↔MG |
| near (260 nm) | 31.2 ± 4.2 | 7.5 ± 1.1 | 4.2 | MG↔PMG |
| farN−I (222 nm) | 67.7 ± 12.7 | 19.0 ± 3.6 | 3.6 | MG↔PMG |
| farI−U (222 nm) | 27.4 ± 2.3 | 5.2 ± 0.4 | 5.2 | PMG↔U |
All thermodynamic parameters were determined according to Eq. 3 (see Materials and Methods) except for fluorescence intensity and far-UV CD of adenylated Tslig, in which thermodynamic parameters were calculated with the help of Eq. 4.
N, native state; MG, molten globule state; PMG, premolten globule state, and U, unfolded state.
Unlike the deadenylated form, GdmCl-induced unfolding of adenylated Tslig monitored by FI333 (λex 280 nm) supports a three-state unfolding model (N↔I↔U) in which a partially unfolded state (I) is significantly populated at intermediate GdmCl concentrations (∼2–4 M) (Fig. 4 A). Fitting of the experimental data to Eq. 4 provides ΔGN−I(H2O) of 18.2 ± 3.4 kJ mol−1, ΔGI−U(H2O) of 42.2 ± 4 kJ mol−1, CmN−I of 1.6 M and CmI−U of 4.2 M (Table 2). Interestingly, the presence of this intermediate is not recorded by the AEW data (Fig. 4 B). In this case, unfolding occurs as a single transition, with a Δ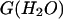 value of 38.8 ± 1.8 kJ mol−1 (which is significantly lower than the Σ[ΔGN−I(H2O) + ΔGI−U(H2O)] = 60.4 kJ mol−1) and a Cm of 4.2 M (Table 2). Therefore, the N↔I transition can be attributed to the enzyme adenylation and describes the elimination of the AMP-induced quenching of the intrinsic fluorescence at 333 nm, which is not accompanied by a significant change of the environment of the tryptophan residues (λmax is not red-shifted, see Fig. 3 B). In addition, comparison of the emission spectra (λex 280 nm) of native enzyme with that of the enzyme in 3 M GdmCl (Fig. 3 B) reveals a change in the 300-nm region, attributed to the contribution of Tyr residues. The analysis of the FI at 306 nm as a function of GdmCl concentrations (Fig. 4 C) clearly demonstrates an increase of the fluorescence intensity at low and moderate GdmCl concentrations, with a maximum reached at ∼2.5 M. One of the putative mechanisms responsible for this increase is a structural rearrangement of the protein, with a concomitant separation between one or some Tyr and Trp residue(s), leading to the suppression of the energy transfer from the former to the latter. Careful examination of Fig. 1 B reveals that some Tyr residues are located in the vicinity of the Trp residues, making such a hypothesis plausible. As a second possibility, AMP in the intermediate state, which was initially stacked against the side chain of Y226, could move away from this residue leading to a dequenching phenomenon and, thus to the observed increase in the tyrosine fluorescence emission. An interesting feature is that the AMP is not present in the deadenylated enzyme and that in this form no significant fluorescence increase at 306 nm is noticed at low and moderate GdmCl concentrations (Fig. 4 C).
value of 38.8 ± 1.8 kJ mol−1 (which is significantly lower than the Σ[ΔGN−I(H2O) + ΔGI−U(H2O)] = 60.4 kJ mol−1) and a Cm of 4.2 M (Table 2). Therefore, the N↔I transition can be attributed to the enzyme adenylation and describes the elimination of the AMP-induced quenching of the intrinsic fluorescence at 333 nm, which is not accompanied by a significant change of the environment of the tryptophan residues (λmax is not red-shifted, see Fig. 3 B). In addition, comparison of the emission spectra (λex 280 nm) of native enzyme with that of the enzyme in 3 M GdmCl (Fig. 3 B) reveals a change in the 300-nm region, attributed to the contribution of Tyr residues. The analysis of the FI at 306 nm as a function of GdmCl concentrations (Fig. 4 C) clearly demonstrates an increase of the fluorescence intensity at low and moderate GdmCl concentrations, with a maximum reached at ∼2.5 M. One of the putative mechanisms responsible for this increase is a structural rearrangement of the protein, with a concomitant separation between one or some Tyr and Trp residue(s), leading to the suppression of the energy transfer from the former to the latter. Careful examination of Fig. 1 B reveals that some Tyr residues are located in the vicinity of the Trp residues, making such a hypothesis plausible. As a second possibility, AMP in the intermediate state, which was initially stacked against the side chain of Y226, could move away from this residue leading to a dequenching phenomenon and, thus to the observed increase in the tyrosine fluorescence emission. An interesting feature is that the AMP is not present in the deadenylated enzyme and that in this form no significant fluorescence increase at 306 nm is noticed at low and moderate GdmCl concentrations (Fig. 4 C).
Finally, analysis of m values, as well as Δ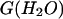 and Cm values estimated for apo- and holoforms emphasizes two important points. First, based on an expected theoretical m value of 60 kJ mol−1 (Myers et al., 1995), the low m values reported for deadenylated and adenylated Tslig (Table 2) are likely to reflect deviation from a two-state mechanism (Myers et al., 1995; Pace, 1986). Second, comparison of Δ
and Cm values estimated for apo- and holoforms emphasizes two important points. First, based on an expected theoretical m value of 60 kJ mol−1 (Myers et al., 1995), the low m values reported for deadenylated and adenylated Tslig (Table 2) are likely to reflect deviation from a two-state mechanism (Myers et al., 1995; Pace, 1986). Second, comparison of Δ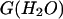 and Cm values clearly indicates cofactor stabilization, as the adenylated enzyme shows a 9.9 kJ mol−1 increase in the stabilization energy and a 0.7 M increase in Cm values (Table 2).
and Cm values clearly indicates cofactor stabilization, as the adenylated enzyme shows a 9.9 kJ mol−1 increase in the stabilization energy and a 0.7 M increase in Cm values (Table 2).
ANS fluorescence
Changes in ANS fluorescence are frequently used to detect accumulation of nonnative partially folded intermediates in globular proteins (Ptitsyn, 1995; Semisotnov et al., 1991). Such intermediates are characterized by the presence of solvent-exposed hydrophobic clusters, resulting in a pronounced blue shift of the fluorescence emission maximum (∼525–480 nm) and a considerable enhancement of ANS fluorescence intensity at ∼480 nm (Semisotnov et al., 1991). With native deadenylated and adenylated Tslig, no binding of ANS could be observed. Fig. 5 shows that in the case of deadenylated Tslig, an increase in GdmCl concentrations leads to considerable changes in the ANS fluorescence intensity at 480 nm, with the maximal fluorescence observed at 3.2 M GdmCl. This demonstrates that albeit the denaturation followed by changes in the intrinsic fluorescence can be described as a cooperative two-state process N↔U (Fig. 4, A and B), ANS binding experiments show that the chemical unfolding of the deadenylated enzyme is accompanied by accumulation of at least one intermediate with solvent-accessible nonpolar clusters. Unlike the deadenylated ligase, GdmCl-induced unfolding of adenylated Tslig does not show any blue shift (not shown) and the ANS fluorescence at 480 nm is low and constant throughout the entire unfolding transition (Fig. 5), suggesting that no binding of the dye occurred. However, in this case, presence of AMP could cause alterations in the microenvironment of the potential ANS binding site, preventing any binding between the intermediate state and the dye. In fact, displacement of ANS into a more polar environment upon cofactor binding has been already reported for various enzymes (D'Auria et al., 1999; Favilla et al., 2002; Kube et al., 1987; Shepherd and Hammes, 1976). Therefore, in adenylated Tslig, it is likely that: i), the ANS binding site is at, or close to, the cofactor-binding site, i.e., the active site, and ii), the absence of ANS binding upon the whole unfolding transition is due to a steric constraint rather than to the absence of unfolding intermediates.
FIGURE 5.

ANS fluorescence intensity. GdmCl dependence of ANS binding by Tslig, measured by the intensity of ANS fluorescence at 480 nm (λex 390 nm). (○) deadenylated and (•) adenylated Tslig. A. U., arbitrary units. Measurements were carried out at pH 7.6, 25°C. Protein concentration was kept at 0.025 mg/ml.
Circular dichroism
Further insights on the unfolding pathways of adenylated Tslig were obtained by monitoring near- and far-UV CD spectra. It is worth mentioning that CD measurements were only performed for the adenylated form of Tslig, because the remaining traces β-NMN in the deadenylated enzyme disturbed CD signal, preventing any accurate recording.
The near-UV CD spectrum of a protein reflects the asymmetric environment of its aromatic residues and thus probes the protein tertiary structure. Accordingly, denaturation is accompanied by the loss in intensity and fine structure of the near-UV CD signals. Fig. 6 A represents the near-UV spectra of adenylated Tslig measured at different GdmCl concentrations. The native protein is characterized by a pronounced near-UV CD spectrum with a negative band in the 280-nm region due to the contribution of Tyr and Trp residues and an additional positive band around 260 nm, likely due to AMP contribution. The intensity of these bands decreased dramatically with the increase in GdmCl concentration, reflecting the denaturant-induced distortion of rigid tertiary structure. The change in [θ]260 (Fig. 6 B, top panel) and [θ]280 (Fig. 6 B, bottom panel) show that the GdmCl-induced denaturation of protein is described by a simple sigmoidal curve, consistent with a two-state transition. Interestingly, Cm calculated for the [θ]260 vs. [GdmCl] dependence is 4.2 M (Table 2), which corresponds to the Cm of the second transition detected by changes in intrinsic fluorescence (CmI−U). On the other hand, the calculated Cm for the transition monitored by changes at 280 nm is 1.6 M (Table 2), which is in a good agreement with Cm of the first transition detected by changes in the fluorescence intensity (CmN−I). These discrepancies between the CD denaturation profiles at 260 and 280 nm clearly demonstrate that the loss of the tertiary structure does not occur according to a simple N↔U model, but follows a more complex pathway involving at least one intermediate state.
FIGURE 6.

GdmCl-induced unfolding transitions of adenylated Tslig followed by near/far UV CD changes. (A) CD spectra (smoothed data) in the near UV region of Tslig. (B) Equilibrium unfolding transitions of Tslig followed by near-UV CD measurements at 260 nm (top panel) and at 280 nm (bottom panel). (C) CD spectra (smoothed data) in the far UV region of Tslig; inset represents equilibrium unfolding transitions followed by CD measurements at 222 nm. Data were analyzed on the basis of a two-state (Eq. 3) and three-state model (Eq. 4) for near- and far-UV CD, respectively; the lines represent the best fits, calculated using the thermodynamic parameters in Table 2. Measurements were carried out at pH 7.6, 25°C. In the near UV, protein concentration was 1 mg/ml in a 1-cm cell. In the far UV, protein concentration was 0.25 mg/ml in a 0.1-cm cell.
The far-UV spectra of adenylated Tslig (Fig. 6 C) were also used to analyze the conformation of its polypeptide backbone. The native form exhibits double minima at 208 nm and 222 nm, typical of proteins containing α-helical secondary structures. Based on Eq. 2, its α-helical content was estimated to be ∼19%. The GdmCl-resistance of the enzyme was also investigated by far-UV CD measurements (Fig. 6 C). At low GdmCl concentrations, a slight decrease of the ellipticity is observed, which maintains until ∼3 M GdmCl. This decrease could be attributed to: i), a stabilization effect of GdmCl because it is known that NAD+-dependent DNA ligases are stabilized by salt or ii), to the formation of an intermediate state such as a molten globule (MG). The second hypothesis is supported by the fact that MG with far-UV spectra more pronounced than those of native proteins have been observed in a number of proteins (for review, see Vassilenko and Uversky (2002)). No significant changes in far-UV CD spectra is noticed up to 3.5 M GdmCl, pointing out that the formation of the intermediate state (Cm 1.6 M), detected by fluorescence and near-UV (280 nm) studies, is not accompanied by noticeable changes in protein secondary structure. In addition, the analysis of the GdmCl dependence of ellipticity at 222 nm (Fig. 6 C, inset) reveals that adenylated Tslig unfolding can be described by two successive sigmoidal curves, separated by a short plateau around 4 M GdmCl. The Cm of the first transition is 3.6 M (Table 2) whereas that of the second transition is 5.15 M, which is higher than that observed with the fluorescence intensity (CmI−U of 4.2 M). Finally, the calculated α-helical content at 1.6 M, 3.6 M, 4.2 M, and 5.1 M GdmCl was shown to be ∼19%, 17.3%, 11.5%, and 2.8%, respectively. All these results point out that adenylated Tslig unfolds through at least two intermediates. The first intermediate is characterized by a loosely packed protein core and native-like content of secondary structure, i.e., it has properties similar to those of the molten globule state (Ptitsyn, 1995). As for the second intermediate, it is less compact and less structured than the first one but still much more compact and more structured than the fully unfolded protein, displaying properties of the premolten globule state (Ptitsyn, 1995). We must emphasize here that this state is no longer considered as a “folding exception” because it has been reported for many enzymes (for review, see Uversky and Fink (2002)).
“Phase diagram” analysis of circular dichroism data
Representation of CD data in a form of “phase diagrams” (see Materials and Methods) provided an additional support to the idea that GdmCl-induced unfolding of adenylated Tslig is an exceptionally complex process. Application of this method to protein unfolding predicts that the dependence of I(λ1)= f(I(λ2)) will be linear if changes in the protein environment lead to all-or-none transition between two different conformations. On the other hand, nonlinearity of this function reflects the sequential character of structural transformations. Moreover, each linear portion of the I(λ1)= f(I(λ2)) dependence will describe an individual all-or-none transition.
Fig. 7 illustrates the phase diagrams for the GdmCl unfolding of adenylated Tslig based on the analysis of near- and far-UV CD. In the near-UV CD (Fig. 7 A), the phase diagram consists of three linear parts, reflecting the existence of at least three independent transitions separating four different conformational states, i.e., N, an additional intermediate N* in the range of 0–1.0 M GdmCl, MGsw (swollen MG), and U. In the case of the analysis based on far-UV CD (Fig. 7 B), the phase diagram consists of four linear parts, reflecting the existence of five different conformational states, i.e., N, N*, MG, premolten globule (PMG) (see “Size exclusion chromatography” section), and U. Finally, all conformational states occurring upon unfolding were highlighted when the analysis was based on the comparison of near- and far-UV CD. The appearance of the N* intermediate (Fig. 7) could be due to a salt effect of GdmCl on structure of adenylated Tslig, as previously mentioned (see “Circular dichroism” section). Compaction of the native conformation and/or enhanced stability of the protein due to Gdm+ binding to negatively charged moieties of protein has been reported for multimeric (Akhtar et al., 2002) as well monomeric (Mayr and Schmid, 1993) proteins.
FIGURE 7.

Phase diagram based on near- and far-UV CD spectra of adenylated Tslig and representing the enzyme unfolding induced by an increase in GdmCl concentration. Denaturant concentration values are indicated in the vicinity of the corresponding symbol. Each straight line represents an all-or-one transition between two conformers. (A) Phase diagram based on near-UV CD ([θ]280 vs. [θ]260); (B) phase diagram based on far-UV CD ([θ]222 vs. [θ]232); (C) phase diagram based on near/far-UV CD ([θ]222 vs. [θ]260).
Analysis of unfolding intermediates by size exclusion chromatography
To obtain information about the effect of GdmCl on the hydrodynamic dimensions of deadenylated and adenylated Tslig and their unfolding intermediates, the gel-filtration behavior of the proteins under different experimental conditions was studied. SEC separates proteins by differences in their hydrodynamic dimensions rather than by their molecular masses (Ackers, 1970). This approach has been successfully applied to determine the Stokes radius (RS) values for proteins in different conformational states (Uversky, 1993; Uversky and Ptitsyn, 1994, 1996). Fig. 8 presents FPLC profiles of deadenylated (A) and adenylated (B) Tslig (at 25°C) in solutions with different concentrations of GdmCl.
FIGURE 8.

Elution profiles of Tslig on size-exclusion chromotography. Deadenylated (A) and adenylated (B) Tslig (0.25 mg/ml) were loaded on a Superdex 200 H/R column at 25°C in 20 mM sodium phosphate buffer, 250 mM NaCl, pH 7.6, at different GdmCl concentrations.
Native deadenylated Tslig elutes as a single peak, whose elution volume corresponds to a protein with a RS of 32.5 Å (Table 3). This value is in excellent agreement with the RS calculated for a native globular protein with a molecular mass of 76.5 kDa (Table 3) (Uversky, 2002b). When the GdmCl concentration increases, the peak is substantially shifted to smaller elution volumes, corresponding to the transition to a slightly more expanded compact denatured state (molten globules) and the swelling of the MG state (MGsw) (Figs. 8 A and 9 A). In the vicinity of 3–3.5 M GdmCl, the appearance of a separate peak indicates the transformation of the MG into a less compact state (LC) (Figs. 8 A and 9 A). In addition, as shown on Fig. 9 A, the elution volume of the LC state strongly depends on the GdmCl concentration, and this dependence is much stronger than that for the unfolded molecules (LC approaches its baseline only at ∼4 M GdmCl). Concerning the native adenylated Tslig, it also elutes as a single peak (Fig. 8 B), whose elution volume corresponds to a protein with RS of 33.4 Å (Table 3). In the presence of denaturant, the elution profiles of the adenylated enzyme (Figs. 8 B and 9 B) display the same trends as that of the deadenylated protein, except that changes in elution profile occur at higher GdmCl concentrations, reflecting cofactor-induced stabilization of the enzyme.
TABLE 3.
Molecular dimensions of deadenylated and adenylated Tslig in different conformational states
| Deadenylated Tslig
|
Adenylated Tslig
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| State | GdmCl (M) | Vel (ml) | RS, exp* (Å) | RS, theo† (Å) | RS/(RS)N | GdmCl (M) | Vel (ml) | RS, exp* (Å) | RS, theo‡ (Å) | RS/(RS)N |
| Native (N) | 0 | 13.88 | 32.5 | 34.6 | 1.00 | 0 | 13.72 | 33.4 | 34.7 | 1.00 |
| Molten globule (MG)‡ | 2.50 | 12.44 | 41.6 | 37.9 | 1.28 | 3.06 | 12.01 | 44.8 | 37.9 | 1.32 |
| Pre-molten globule (LCmax) (PMG) | 3.04 | 11.20 | 51.4 | 50.6 | 1.58 | 3.06 | 10.99 | 53.3 | 50.7 | 1.60 |
| Unfolded (U) | 4.32 | 10.40 | 58.9 | 84.9 | 1.81 | 6.30 | 9.50 | 68.9 | 85.1 | 2.06 |
RS values calculated using the experimental equation RS = (1000/Vel − 42.44)/0.9114.
RS values calculated using the equations described in Uversky (2002b): for N state, log(RS) = −0.204 + 0.357 log (M); for MG state, log(RS) = −0.053 + 0.334 log (M); for PMG state, log(RS) = −0.21 + 0.392 log (M), and for U state, log(RS) = −0.723 + 0.543 log (M).
Maximum population of MG (see also Fig. 9, E and F).
FIGURE 9.

GdmCl-induced unfolding transitions of Tslig monitored by size-exclusion chromatography. Dependence of elution volumes Vel of deadenylated (A) and adenylated (B) Tslig on GdmCl concentrations. 〈VC〉, elution volumes of “compact” molecules and 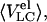 elution volumes of “compact” molecules and 〈VLC〉, elution volumes of “less compact” molecules
elution volumes of “compact” molecules and 〈VLC〉, elution volumes of “less compact” molecules 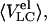 elution volumes of “less compact” molecules. LCmax, maximal value of Vel for less compact molecules. N, native state; MGSW, swollen molten globule state; PMG, premolten globule state; U, unfolded state. Stages of unfolding (four-state transition) of deadenylated (C) and adenylated (D) Tslig. (C) Fraction of denatured molecules fD was obtained from the relative change of elution volumes from 0 to 3.53 M GdmCl (N↔MG transition) (▪); fLC, fraction of less compact molecules (▴); fU, fraction of unfolded molecules (•); fluorescence average emission wavelength, (dash-dotted line). (D) Fraction of denatured molecules fD obtained from the relative change of near UV ellipticity [θ]280 (□) and the relative change of elution volumes from 0 to 4.34 M GdmCl (N↔MG transition) (▪); fLC, (▴); fU, (•); relative change of far UV ellipticity [θ]222 (dashed line). Multiple-state GdmCl-induced N↔U transition in deadenylated (E) and adenylated (F) Tslig presented in terms of fractions of different states: fN, fraction of molecules in the native state; fMG, fraction in the molten-globule state; fPMG, fraction in the premolten globule state, and fU, fraction in the unfolded state.
elution volumes of “less compact” molecules. LCmax, maximal value of Vel for less compact molecules. N, native state; MGSW, swollen molten globule state; PMG, premolten globule state; U, unfolded state. Stages of unfolding (four-state transition) of deadenylated (C) and adenylated (D) Tslig. (C) Fraction of denatured molecules fD was obtained from the relative change of elution volumes from 0 to 3.53 M GdmCl (N↔MG transition) (▪); fLC, fraction of less compact molecules (▴); fU, fraction of unfolded molecules (•); fluorescence average emission wavelength, (dash-dotted line). (D) Fraction of denatured molecules fD obtained from the relative change of near UV ellipticity [θ]280 (□) and the relative change of elution volumes from 0 to 4.34 M GdmCl (N↔MG transition) (▪); fLC, (▴); fU, (•); relative change of far UV ellipticity [θ]222 (dashed line). Multiple-state GdmCl-induced N↔U transition in deadenylated (E) and adenylated (F) Tslig presented in terms of fractions of different states: fN, fraction of molecules in the native state; fMG, fraction in the molten-globule state; fPMG, fraction in the premolten globule state, and fU, fraction in the unfolded state.
Fig. 9, C and D, also demonstrates the existence of three different stages in GdmCl-induced unfolding of deadenylated and adenylated Tslig. The first stage (fD) reflects protein denaturation, whereas two others stages (fLC and fU) correspond to two conformational transitions of an already-denatured protein. Three stages of unfolding correspond to the formation of at least two intermediates between the native and unfolded states, i.e., it is a four-state process. The first intermediate displays all the properties of the molten globule. It is almost as compact as the native protein, with a ∼30% RS increase (Table 3). Such increase is quite high compared to the usual 15–20% increase of RS associated with MG formation (Ptitsyn, 1995; Uversky, 1993): for both forms the expected RS is 37.9 Å (Table 3). Importantly, Fig. 9, C and D, show that the hydrodynamic dimensions of MG Tslig (in particular that of the adenylated form) increase with the increase in denaturant concentration; i.e., the molten globule swells considerably. For the adenylated enzyme, extrapolation of the plateau after the N→MG transition to 0 M GdmCl gives an elution volume corresponding to a RS of nonswollen MG, 36.2 Å. Thus, the actual increase in RS is ∼9%, which coincides with the usually observed increase of RS associated with the MG formation. The first intermediate is also characterized by the lack of rigid tertiary structure but possesses a pronounced secondary structure, as demonstrated by near- and far-UV CD spectra for adenylated Tslig in Fig. 6. Finally, in the presence of moderate concentrations of GdmCl the deadenylated enzyme is able to bind ANS (see above), reflecting an increased solvent accessibility of hydrophobic patches on the surface of the protein.
The second intermediate possesses all the properties of the PMG state. It is less compact than the molten globule but much more compact than the unfolded state (Table 3). In addition, the experimental RS (51.4 Å and 53.3 Å for dea- and adenylated Tslig, respectively) are in agreement with the theoretical RS for PMG (50.7 Å for both forms). It also still contains substantial secondary structure, as demonstrated by far-UV CD spectra for adenylated Tslig (Fig. 9 D). Analysis of fLC and fU of deadenylated and adenylated Tslig (Fig. 9, C and D) points out that in the case of the deadenylated enzyme the LC→U transformation occurs in a narrower range of denaturant concentrations, assessing that cofactor stabilization affects all stages of unfolding.
Finally, at high GdmCl concentrations, both forms are considerably unfolded and characterized by RS of 58.9 and 68.9 Å for deadenylated and adenylated enzyme, respectively (Table 3). Those values are however lower than the expected RS values, i.e., 84.9 Å and 85.1 Å for deadenylated and adenylated Tslig, respectively.
The four-state N↔U transition can be visualized by plotting the fractions of molecules in all four states (N, MG, PMG, and U) as a function of GdmCl concentrations (Fig. 9, E and F). The plots illustrate that the molten globule state appears at the first step of unfolding at the expense of the native state and that it transforms first into the premolten globule and then into the unfolded state. The maximum of population of the MG state corresponds to 2.5 M GdmCl for deadenylated Tslig and 3.1 M GdmCl for adenylated Tslig, whereas the maximum of population of the PMG state corresponds to 3.5 M GdmCl for deadenylated Tslig and 4.4 M GdmCl for adenylated Tslig. For the deadenylated enzyme, the U is reached at ∼4 M denaturant although it is only reached at ∼6 M for the adenylated ligase. All together, these results nicely illustrate that AMP binding leads to conformational changes protecting the enzyme against denaturation.
Fluorescence quenching
To gain additional information on the structural changes associated with cofactor binding and on the different folding intermediates accumulated during unfolding, a dynamic quenching investigation was performed on both preparations, using acrylamide as a quencher. Due to its polar nature, this molecule quenches the surface-exposed and partially buried Trp residues. A rigorous approach to probe Trp accessibility in the different conformers usually requires determination of the bimolecular rate constant kq (kq = KSV τ0, where KSV is the Stern-Volmer constant and τ0 the fluorescence lifetime). However Tslig contains 6 Trp therefore impairing the determination of individual τ0. As generally accepted in such cases, conformational changes were investigated by comparing KSV constants rather than kq constants. Fig. 10 represents the Stern-Volmer plots for deadenylated (Fig. 10 A) and adenylated (Fig. 10 B) Tslig, under different experimental conditions and Table 4 shows the KSV of the fitted curves. Analysis of KSV of native deadenylated (7.11 ± 0.11 M−1) and adenylated (5.66 ± 0.05 M−1) enzymes clearly demonstrates a greater solvent accessibility for deadenylated Tslig. Based on the structure of the Tf DNA ligase, the adoption of an open form in the deadenylated state and a closed form in the adenylated state has been suggested (Cherepanov and de Vries, 2002; Lee et al., 2000). Our results provide a direct evidence for the occurrence of an open-closure mechanism of the NAD+-DNA ligase upon adenylation, as tryptophanes in the closed form are less accessible to the quencher, leading to a decreased KSV value.
FIGURE 10.

Trp fluorescence quenching by acrylamide. Quenching experiments of deadenylated (A) and adenylated (B) Tslig were conducted as described under “Experimental Procedures.” (A) Fluorescence intensity of deadenylated Tslig (λex 295 nm, λem 333 nm) at 0 M (1), 2.5 M (2), 3.5 M (3), and 6.5 M GdmCl (4). (B) Fluorescence intensity of adenylated Tslig recorded at 0 M (1), 3.06 M (2), 4.4 M (3), and 6.5 M (4) GdmCl. The quenching constant KSV values corresponding to the plot slope (Eq. 10) are given in Table 4.
Quenching of folding intermediates was also investigated and revealed that the adenylation state influences the sensitivity of the protein to the quencher. For the deadenylated enzyme, the MG state (2.5 M GdmCl) displays almost the same KSV as the native enzyme. Furthermore, examination of the fluorescence emission maximum, 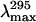 (Fig. 3 A) does not reveal any significant change in the exposure of Trp residues to the solvent. Upon PMG formation (3.5 M GdmCl), the accessibility of Trps to the quenching agent and solvent increases rendered by a higher KSV and a red shift of the
(Fig. 3 A) does not reveal any significant change in the exposure of Trp residues to the solvent. Upon PMG formation (3.5 M GdmCl), the accessibility of Trps to the quenching agent and solvent increases rendered by a higher KSV and a red shift of the 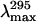 (∼6 nm, see Fig. 3 A and Table 4). Finally, the KSV of the U state (6.5 M GdmCl) was close to that of the PMG, whereas the
(∼6 nm, see Fig. 3 A and Table 4). Finally, the KSV of the U state (6.5 M GdmCl) was close to that of the PMG, whereas the 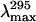 still increased (350 nm), reflecting the full exposure of Trp residues at high denaturant concentrations. Interestingly, the MG state of the adenylated enzyme (3.06 M GdmCl) shows a slightly decreased KSV compared to the native protein (Fig. 10 B). The main cause for this result cannot be explained by a significant burial of the Trp residues in a hydrophobic environment because no major blue shift of the fluorescence emission maximum,
still increased (350 nm), reflecting the full exposure of Trp residues at high denaturant concentrations. Interestingly, the MG state of the adenylated enzyme (3.06 M GdmCl) shows a slightly decreased KSV compared to the native protein (Fig. 10 B). The main cause for this result cannot be explained by a significant burial of the Trp residues in a hydrophobic environment because no major blue shift of the fluorescence emission maximum, 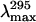 was observed (Fig. 3 B and Table 4). In contrast, the PMG state of the adenylated Tslig (4.4 M GdmCl) is characterized by an increased KSV compared to the deadenylated enzyme and comparable to that of the unfolded state (Table 4). These results suggest that Trp residues in those forms are substantially solvated and accessible to the quencher molecules. This conclusion was further confirmed by the analysis of the position of maximal fluorescence,
was observed (Fig. 3 B and Table 4). In contrast, the PMG state of the adenylated Tslig (4.4 M GdmCl) is characterized by an increased KSV compared to the deadenylated enzyme and comparable to that of the unfolded state (Table 4). These results suggest that Trp residues in those forms are substantially solvated and accessible to the quencher molecules. This conclusion was further confirmed by the analysis of the position of maximal fluorescence, 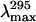 (Fig. 3 B and Table 4), which demonstrates concomitant changes in the exposure of Trps to solvent. Thus, the results of quenching experiments indicate that the Trp residues in the MG states of apo- and holoenzymes are in the same global environment as those of the native enzyme. On the other hand, in the PMG state Trp residues are significantly exposed to the solvent, suggesting more pronounced conformational changes than in the case of the MG.
(Fig. 3 B and Table 4), which demonstrates concomitant changes in the exposure of Trps to solvent. Thus, the results of quenching experiments indicate that the Trp residues in the MG states of apo- and holoenzymes are in the same global environment as those of the native enzyme. On the other hand, in the PMG state Trp residues are significantly exposed to the solvent, suggesting more pronounced conformational changes than in the case of the MG.
CONCLUSION
In this report, we provide new insights into the conformational changes occurring upon adenylation of NAD+-dependent DNA ligases. Although previous works had suggested such changes (Cherepanov and de Vries, 2002; Doherty and Suh, 2000; Lee et al., 2000), our biophysical analyses on the deadenylated and adenylated Tslig firmly demonstrate that the transformation of the cofactor-free enzyme into its catalytically active form causes a conformational rearrangement within its active site accompanied by the compaction of the enzyme. These results support an “open-closure” induced-mechanism avoiding the formation of nonproductive ligase-DNA complexes. Mutational studies performed on aromatic residues located within the active site will be instrumental in the identification of the amino acid(s) involved in the conformational changes associated with adenylation.
The structural modifications occurring upon adenylation have also profound effects on the conformational stability of the enzyme. Upon adenylation, the conformational stability of the DNA ligase significantly increases, as demonstrated by differential scanning microcalorimetry and guanidine hydrochloride denaturation. Cofactor molecules have long been known to stabilize enzymes specifically by stabilizing their active site. Our results obviously indicate that this observation is also valid for Tslig, establishing a thermodynamic link between cofactor binding and conformational stability. Furthermore, the effect of DNA binding on the conformational stability of the enzyme is expected to give supplementary information on the substrate-induced modulation of enzyme stability.
To gain information about the deadenylated and adenylated conformers, the unfolding pathways of deadenylated and adenylated Tslig have been followed by a combination of different biophysical methods. The GdmCl-induced equilibrium unfolding of both forms can be described by a four-state transition model, N↔MG↔PMG↔U, shifted toward higher GdmCl concentrations in the case of adenylated enzyme.
Acknowledgments
We thank N. Gérardin and R. Marchand for their skillful technical assistance. We also thank F. Biemar, C. Houssier, A. Matagne, A. R. Merrill, and M. Vanhove for helpful assistance and discussions.
This work was supported by the European Union (Grant CT970131), the Région Wallonne (Grants Bioval 981/3860, Bioval 981/3848, Initiative 114705), the Fonds National de la Recherche Scientifique Belgium (Grant 2.4515.00), and the Institut Polaire Français. The purchase of the Jasco 810 equipment was supported in part by a grant from the Fonds de la Recherche Fondamentale et Collective (contract 2.4545.01).
Abbreviations used: AEW, average emission wavelength; ANS, 8-anilino-1-naphtalene sulfonic acid; GdmCl, guanidine hydrochloride; FI, fluorescence intensity; FPLC, fast protein liquid chromatography; NAD+, nicotinamide adenine dinucleotide; NMN, nicotinamide mononucleotide; MG, molten globule; PMG, premolten globule; SEC, size exclusion chromatography; Tslig, Thermus scotoductus DNA ligase.
References
- Ackers, G. K. 1970. Analytical gel chromatography of proteins. Adv. Protein Chem. 24:343–446. [DOI] [PubMed] [Google Scholar]
- Akhtar, M. S., A. Ahmad, and V. Bhakuni. 2002. Guanidinium chloride- and urea-induced unfolding of the dimeric enzyme glucose oxidase. Biochemistry. 41:3819–3827. [DOI] [PubMed] [Google Scholar]
- Aravind, L., and E. V. Koonin. 1999. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 287:1023–1040. [DOI] [PubMed] [Google Scholar]
- Barany, F., and D. H. Gelfand. 1991. Cloning, overexpression and nucleotide sequence of a thermostable DNA ligase-encoding gene. Gene. 109:1–11. [DOI] [PubMed] [Google Scholar]
- Chen, Y. H., J. T. Yang, and H. M. Martinez. 1972. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry. 11:4120–4131. [DOI] [PubMed] [Google Scholar]
- Brandes, H. K., F. W. Larimer, T. Y. Lu, J. Dey, and F. C. Hartman. 1998. Roles and microenvironments of tryptophanyl residues of spinach phosphoribulokinase. Arch. Biochem. Biophys. 352:130–136. [DOI] [PubMed] [Google Scholar]
- Brannigan, J. A., S. R. Ashford, A. J. Doherty, D. J. Timson, and D. B. Wigley. 1999. Nucleotide sequence, heterologous expression and novel purification of DNA ligase from Bacillus stearothermophilus1. Biochim. Biophys. Acta. 1432:413–418. [DOI] [PubMed] [Google Scholar]
- Burstein, E. A. 1976. Intrinsic Protein Fluorescence: Origin and Applications. Series Biophysics, Vol. 7, Viniiti, Moscow.
- Bushmarina, N. A., I. M. Kuznetsova, A. G. Biktashev, K. K. Turoverov, and V. N. Uversky. 2001. Partially folded conformations in the folding pathway of bovine carbonic anhydrase II: a fluorescence spectroscopic analysis. Chembiochem. 2:813–821. [DOI] [PubMed] [Google Scholar]
- Candy, J. M., J. Koga, P. F. Nixon, and R. G. Duggleby. 1996. The role of residues glutamate-50 and phenylalanine-496 in Zymomonas mobilis pyruvate decarboxylase. Biochem. J. 315:745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov, A. V., and S. de Vries. 2002. Dynamic mechanism of nick recognition by DNA ligase. Eur. J. Biochem. 269:5993–5999. [DOI] [PubMed] [Google Scholar]
- D'Auria, S., P. Herman, M. Rossi, and J. R. Lakowicz. 1999. The fluorescence emission of the apo-glucose oxidase from Aspergillus niger as probe to estimate glucose concentrations. Biochem. Biophys. Res. Commun. 263:550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depiereux, E., G. Baudoux, P. Briffeuil, I. Reginster, X. De Bolle, C. Vinals, and E. Feytmans. 1997. Match-Box_server: a multiple sequence alignment tool placing emphasis on reliability. Comput. Appl. Biosci. 13:249–256. [DOI] [PubMed] [Google Scholar]
- Diefenbach, R. J., and R. G. Duggleby. 1991. Pyruvate decarboxylase from Zymomonas mobilis. Structure and re-activation of apoenzyme by the cofactors thiamin diphosphate and magnesium ion. Biochem. J. 276:439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty, A. J., and T. R. Dafforn. 2000. Nick recognition by DNA ligases. J. Mol. Biol. 296:43–56. [DOI] [PubMed] [Google Scholar]
- Doherty, A. J., and S. W. Suh. 2000. Structural and mechanistic conservation in DNA ligases. Nucleic Acids Res. 28:4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favilla, R., M. Goldoni, A. Mazzini, P. Di Muro, B. Salvato, and M. Beltramini. 2002. Guanidinium chloride induced unfolding of a hemocyanin subunit from Carcinus aestuarii. I. Apo form. Biochim. Biophys. Acta. 1597:42–50. [DOI] [PubMed] [Google Scholar]
- Fechteler, T., U. Dengler, and D. Schomburg. 1995. Prediction of protein three-dimensional structures in insertion and deletion regions: a procedure for searching data bases of representative protein fragments using geometric scoring criteria. J. Mol. Biol. 253:114–131. [DOI] [PubMed] [Google Scholar]
- Georlette, D., Z. O. Jonsson, F. Van Petegem, J. Chessa, J. Van Beeumen, U. Hubscher, and C. Gerday. 2000. A DNA ligase from the psychrophile Pseudoalteromonas haloplanktis gives insights into the adaptation of proteins to low temperatures. Eur. J. Biochem. 267:3502–3512. [DOI] [PubMed] [Google Scholar]
- Goenka, S., B. Raman, T. Ramakrishna, and C. M. Rao. 2001. Unfolding and refolding of a quinone oxidoreductase: α-crystallin, a molecular chaperone, assists its reactivation. Biochem. J. 359:547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, M. E., N. Expert-Bezançon, L. Vuillard, and T. Rabilloud. 1995. Non-detergent sulphobetaines: a new class of molecules that facilitate in vitro protein renaturation. Fold. Des. 1:21–27. [PubMed] [Google Scholar]
- Gupta, G. S., and B. P. Kang. 1997. LDH-C4-substrate binary complexes studied by intrinsic fluorescence method. Indian J. Biochem. Biophys. 34:307–312. [PubMed] [Google Scholar]
- Hakansson, K., A. J. Doherty, S. Shuman, and D. B. Wigley. 1997. X-ray crystallography reveals a large conformational change during guanyl transfer by mRNA capping enzymes. Cell. 89:545–553. [DOI] [PubMed] [Google Scholar]
- Ishino, Y., H. Shinagawa, K. Makino, S. Tsunasawa, F. Sakiyama, and A. Nakata. 1986. Nucleotide sequence of the lig gene and primary structure of DNA ligase of Escherichia coli. Mol. Gen. Genet. 204:1–7. [DOI] [PubMed] [Google Scholar]
- Kaczorowski, T., and W. Szybalski. 1996. Co-operativity of hexamer ligation. Gene. 179:189–193. [DOI] [PubMed] [Google Scholar]
- Kube, D., T. V. Esakova, M. V. Ivanov, A. I. Gromov, and N. K. Nagradova. 1987. Detection of ligand-induced conformation changes in lactate dehydrogenase by using fluorescent probes. Biokhimiia. 52:179–187. [PubMed] [Google Scholar]
- Kuznetsova, I. M., O. V. Stepanenko, K. K. Turoverov, L. Zhu, J. M. Zhou, A. L. Fink, and V. N. Uversky. 2002. Unraveling multistate unfolding of rabbit muscle creatine kinase. Biochim. Biophys. Acta. 1596:138–155. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. 1983. Fluorescence quenching. In Principles of Fluorescence Spectroscopy. J. R. Lakowicz, editor. Plenum Press, New York. 257–301.
- Lee, J. Y., C. Chang, H. K. Song, J. Moon, J. K. Yang, H. K. Kim, S. T. Kwon, and S. W. Suh. 2000. Crystal structure of NAD+-dependent DNA ligase: modular architecture and functional implications. EMBO J. 19:1119–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman, I. R. 1974. DNA ligase: structure, mechanism, and function. Science. 186:790–797. [DOI] [PubMed] [Google Scholar]
- Levitt, M. 1992. Accurate modeling of protein conformation by automatic segment matching. J. Mol. Biol. 226:507–533. [DOI] [PubMed] [Google Scholar]
- Marchal, S., and G. Branlant. 1999. Evidence for the chemical activation of essential cys-302 upon cofactor binding to nonphosphorylating glyceraldehyde 3-phosphate dehydrogenase from Streptococcus mutans. Biochemistry. 38:12950–12958. [DOI] [PubMed] [Google Scholar]
- Martin, I. V., and S. A. MacNeill. 2002. ATP-dependent DNA ligases. Genome Biol. 3:3005.1–3005.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matouschek, A., J. M. Matthews, C. M. Johnson, and A. R. Fersht. 1994. Extrapolation to water of kinetic and equilibrium data for the unfolding of barnase in urea solutions. Protein Eng. 7:1089–1095. [DOI] [PubMed] [Google Scholar]
- Mayr, L. M., and F. X. Schmid. 1993. Stabilization of a protein by guanidinium chloride. Biochemistry. 32:7994–7998. [DOI] [PubMed] [Google Scholar]
- Modrich, P., Y. Anraku, and I. R. Lehman. 1973. Deoxyribonucleic acid ligase. Isolation and physical characterization of the homogeneous enzyme from Escherichia coli. J. Biol. Chem. 248:7495–7501. [PubMed] [Google Scholar]
- Morimatsu, K., T. Horii, and M. Takahashi. 1995. Interaction of Tyr103 and Tyr264 of the RecA protein with DNA and nucleotide cofactors. Fluorescence study of engineered proteins. Eur. J. Biochem. 228:779–785. [DOI] [PubMed] [Google Scholar]
- Munishkina, L. A., C. Phelan, V. N. Uversky, and A. L. Fink. 2003. Conformational behavior and aggregation of a-synuclein in organic solvents: modeling the effects of membranes. Biochemistry. 42:2720–2730. [DOI] [PubMed] [Google Scholar]
- Murataliev, M. B., and R. Feyereisen. 2000. Functional interactions in cytochrome P450BM3. Evidence that NADP(H) binding controls redox potentials of the flavin cofactors. Biochemistry. 39:12699–12707. [DOI] [PubMed] [Google Scholar]
- Myers, J. K., C. N. Pace, and J. M. Scholtz. 1995. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4:2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C. N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131:266–280. [DOI] [PubMed] [Google Scholar]
- Pace, C. N. 1990. Measuring and increasing protein stability. Trends Biotechnol. 8:93–98. [DOI] [PubMed] [Google Scholar]
- Panasenko, S. M., R. J. Alazard, and I. R. Lehman. 1978. A simple, three-step procedure for the large scale purification of DNA ligase from a hybrid lambda lysogen constructed in vitro. J. Biol. Chem.253:4590–4592. [PubMed] [Google Scholar]
- Pearson, W. R. 1996. Effective protein sequence comparison. Methods Enzymol. 266:227–258. [DOI] [PubMed] [Google Scholar]
- Permyakov, E. A., V. V. Yarmolenko, V. I. Emelyanenko, E. A. Burstein, J. Closset, and C. Gerday. 1980. Fluorescence studies of the calcium binding to whiting ( Gadus merlangus) parvalbumin. Eur. J. Biochem. 109:307–315. [DOI] [PubMed] [Google Scholar]
- Ptitsyn, O. B. 1995. Molten globule and protein folding. Adv. Protein Chem. 47:83–229. [DOI] [PubMed] [Google Scholar]
- Royer, C. A., C. J. Mann, and C. R. Matthews. 1993. Resolution of the fluorescence equilibrium unfolding profile of trp aporepressor using single tryptophan mutants. Protein Sci. 2:1844–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semisotnov, G. V., N. A. Rodionova, O. I. Razgulyaev, V. N. Uversky, A. F. Gripas, and R. I. Gilmanshin. 1991. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 31:119–128. [DOI] [PubMed] [Google Scholar]
- Shepherd, G. B., and G. G. Hammes. 1976. Fluorescence energy transfer measurements between ligand binding sites of the pyruvate dehydrogenase multienzyme complex. Biochemistry. 15:311–317. [DOI] [PubMed] [Google Scholar]
- Shuman, S. 1995. Vaccinia virus DNA ligase: specificity, fidelity, and inhibition. Biochemistry. 34:16138–16147. [DOI] [PubMed] [Google Scholar]
- Shuman, S., and B. Schwer. 1995. RNA capping enzyme and DNA ligase: a superfamily of covalent nucleotidyl transferases. Mol. Microbiol. 17:405–410. [DOI] [PubMed] [Google Scholar]
- Singleton, M. R., K. Hakansson, D. J. Timson, and D. B. Wigley. 1999. Structure of the adenylation domain of an NAD+-dependent DNA ligase. Structure. 7:35–42. [DOI] [PubMed] [Google Scholar]
- Sinha, K. M., M. Ghosh, I. Das, and A. K. Datta. 1999. Molecular cloning and expression of adenosine kinase from Leishmania donovani: identification of unconventional P-loop motif. Biochem. J. 339:667–673. [PMC free article] [PubMed] [Google Scholar]
- Sriskanda, V., and S. Shuman. 2002. Conserved residues in domain Ia are required for the reaction of Escherichia coli DNA ligase with NAD+. J. Biol. Chem. 277:9695–9700. [DOI] [PubMed] [Google Scholar]
- Takahashi, M., and T. Uchida. 1986. Thermophilic HB8 DNA ligase: effects of polyethylene glycols and polyamines on blunt-end ligation of DNA. J. Biochem. 100:123–131. [DOI] [PubMed] [Google Scholar]
- Tang, C. K., C. E. Jeffers, J. C. Nichols, and S. C. Tu. 2001. Flavin specificity and subunit interaction of Vibrio fischeri general NAD(P)H-flavin oxidoreductase FRG/FRase I. Arch. Biochem. Biophys. 392:110–116. [DOI] [PubMed] [Google Scholar]
- Thorbjarnardóttir, S. H., Z. O. Jónsson, O. S. Andresson, J. K. Kristjánsson, G. Eggertsson, and A. Pálsdóttir. 1995. Cloning and sequence analysis of the DNA ligase-encoding gene of Rhodothermus marinus, and overproduction, purification and characterization of two thermophilic DNA ligases. Gene. 161:1–6. [DOI] [PubMed] [Google Scholar]
- Timson, D. J., M. R. Singleton, and D. B. Wigley. 2000. DNA ligases in the repair and replication of DNA. Mutat. Res. 460:301–318. [DOI] [PubMed] [Google Scholar]
- Timson, D. J., and D. B. Wigley. 1999. Functional domains of an NAD+-dependent DNA ligase. J. Mol. Biol. 285:73–83. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N. 1993. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry. 32:13288–13298. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N. 2002a. Cracking the folding code. Why do some proteins adopt partially folded conformations, whereas other don't? FEBS Lett. 514:181–183. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N. 2002b. What does it mean to be natively unfolded? Eur. J. Biochem. 269:2–12. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N., and A. L. Fink. 2002. The chicken-egg scenario of protein folding revisited. FEBS Lett. 515:79–83. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N., L. N. Garriques, I. S. Millett, S. Frokjaer, J. Brange, S. Doniach, and A. L. Fink. 2003. Prediction of the association state of insulin using spectral parameters. J. Pharm. Sci. 92:847–858. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N., and O. B. Ptitsyn. 1994. Partly folded state, a new equilibrium state of protein molecules: four-state guanidinium chloride-induced unfolding of β-lactamase at low temperature. Biochemistry. 33:2782–2791. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N., and O. B. Ptitsyn. 1996. Further evidence on the equilibrium “pre-molten globule state”: four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. J. Mol. Biol. 255:215–228. [DOI] [PubMed] [Google Scholar]
- Vanhove, M., G. Guillaume, P. Ledent, J. H. Richards, R. H. Pain, and J. M. Frere. 1997. Kinetic and thermodynamic consequences of the removal of the Cys-77-Cys-123 disulphide bond for the folding of TEM-1 β-lactamase. Biochem. J. 321:413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilenko, K. S., and V. N. Uversky. 2002. Native-like secondary structure of molten globules. Biochim. Biophys. Acta. 1594:168–177. [DOI] [PubMed] [Google Scholar]
- Wilkinson, A., J. Day, and R. Bowater. 2001. Bacterial DNA ligases. Mol. Microbiol. 40:1241–1248. [DOI] [PubMed] [Google Scholar]
- Zimmerman, S. B., and B. H. Pheiffer. 1983. Macromolecular crowding allows blunt-end ligation by DNA ligases from rat liver or Escherichia coli. Proc. Natl. Acad. Sci. USA. 80:5852–5856. [DOI] [PMC free article] [PubMed] [Google Scholar]