

Abstract
The effects of two single-point cavity-forming mutations, F110S and I7S, on the internal dynamics of azurin from Pseudomonas aeruginosa were probed by the phosphorescence emission of Trp-48, deeply buried in the compact hydrophobic core of the macromolecule. Changes in flexibility of the protein matrix around the chromophore were monitored by the intrinsic phosphorescence lifetime (τ0) whereas more general effects on structural fluctuations were deduced from the phosphorescence acrylamide quenching rate constant (kq), which measures the diffusion of the solute through the protein fold. The results show a spectacular, 4–5 orders of magnitude, increase of kq emphasizing that large amplitude structural fluctuations permitting acrylamide migration to the protein core have been drastically enhanced in each azurin mutant. The large, 12–15 kcal/mol, decrease in the activation enthalpy associated to kq suggests that the rate enhancement is caused, rather than through a generalized increase of protein flexibility, by the elimination of an inner barrier to the diffusion process. According to τ0 the chromophore environment is more fluid with I7S but strikingly more rigid with F110S, demonstrating that when internal cavities are formed local effects on the mobility at the mutation site are unpredictable. Both τ0 and kq reveal a structure tightening role of bound Cd2+ that correlates with the increase in stability from apo- to holo-azurin. While these alterations in internal dynamics of azurin do not seem to play a role on electron transfer through the central region, the enhanced migration of acrylamide emphasizes that cavities may be critical for the rapid diffusion of substrates to buried, solvent inaccessible sites of enzymes.
INTRODUCTION
The static three-dimensional structure represents only a partial picture of proteins: it provides a description of the lowest energy state of the molecule, but macromolecular function, binding specificity, binding affinity, and thermodynamic stability are in many cases highly dependent on structural fluctuations and hence intimately coupled to flexibility. In the case of enzymes, for example, substrate molecules require access to catalytic sites that can be partly or even completely buried. Transient conformational rearrangements are necessary to create a path along which the substrate may enter into the binding pocket, involving the movement of both backbone and side-chain atoms. Evidence of protein structural fluctuations is available from many techniques, including thermal factors from x-ray crystallography (Artymiuk et al., 1979), fluorescence and phosphorescence quenching (Lakowicz and Weber, 1973; Vanderkooi, 1991; Schauerte et al., 1997; Strambini, 1989; Cioni and Strambini, 2002b), and anisotropy (Munro et al., 1979), nuclear magnetic resonance relaxation (Wagner and Wutrich, 1986; Palmer, 2001; Palmer et al., 2001), hydrogen exchange (Englander et al., 1996), neutron scattering (Zaccai, 2000), and molecular dynamics simulation (Brooks et al., 1988). However, few are the techniques able to monitor the slow dynamical processes (μs-ms) that are likely to be directly involved in binding of substrates and effectors to the site of enzymes, in allosteric transitions, and catalysis; consequently, relatively little is known about motion in this time domain. Recent NMR studies have provided evidence for dynamics on the microsecond-to-millisecond regime in structural regions of biomolecules that are known to have in vivo functions such as macromolecular recognition, ligand binding, and catalysis (Mulder et al., 2002; Akke, 2002). However, considerable uncertainty remains about the detailed connection between structural fluctuations and biological function.
Future efforts to design proteins with specific functions and stability may require the inclusion of structural features that allow dynamic excursions from the average structure. To include such features, a better understanding of the relationships between structure and chain flexibility is of paramount importance. In particular, cavities and connecting channels in the interior space of a protein are expected to play an important role in conferring the flexibility and alternative packing arrangements of the polypeptide that allow rapid transitions between structurally distinct states. Indeed, recent NMR studies on T4 lysozyme demonstrated that a cavity-forming mutant is able to accommodate molecules such as benzene and xenon in its interior, thanks to rapid transitions to a structurally open form of the protein (Quillin et al., 2000; Skrynnikov et al., 2001; Mulder et al., 2001, 2002).
This work inquires specifically on how relatively large internal cavities, created by the replacement of bulky side chains with smaller ones, may affect the dynamics of compact protein cores. Azurin from Pseudomonas aeruginosa was chosen as a model system for the wealth of structural (crystallographic, spectroscopic, and theoretical), thermodynamic, and kinetic data available on both native and mutated forms. Azurin is a 14-kDa copper-binding protein with an eight-stranded β-sandwich structure arranged in a double-wound Greek-key topology (Adman, 1991). The β-sandwich is closely packed and forms a highly hydrophobic core about the unique Trp residue (Trp-48) of the polypeptide (Nar et al., 1991), which serves as a natural probe of the local structure. The introduction of Ser in place of a bulky Phe (F110) or Ile (I7) creates (Fig. 1) a cavity of ∼40 Å3 in I7S and of 100 Å3 in F110S in the neighborhood of Trp-48, whereas for the rest the structure remains essentially native (Hammann et al., 1996). The two single-point mutations destabilize the globular fold by 3–4 kcal/mol relative to the WT (Mei et al., 1999). Although an inverse correlation has often observed between thermal stability (ΔGD) and conformational dynamics (Tang and Dill, 1998; Tsai et al., 2001) the effects of these mutations on the internal flexibility of the macromolecule are not fully understood. According to the crystallographic B-factors, greater conformational freedom is confined to the replacing side chain, Ser-7 or Ser-110, whereas neighboring groups such as the indole ring of Trp-48 remain firmly immobilized (Hammann et al., 1996). A similar conclusion is reached for the protein in solution by the modest decrease in the T1 NMR relaxation time of indole (Mei et al., 1996). Changes on the fluorescence emission of Trp-48 (spectral red-shifts, lifetimes, and anisotropy) have been interpreted in terms of a more heterogeneous and polar environment and in some cases they have been ascribed to a gain in structural mobility (Mei et al., 1996, 1999; Gilardi et al., 1994; Kroes et al., 1998). Comparative fluorescence studies with less stable, metal-free proteins (apo-azurins) have also pointed out that in these mutants the metal-ion has a direct influence on the structure of the protein core, an influence lacking in the WT protein (Gilardi et al., 1994), which also adopts a native β-sandwich structure with an empty metal-site or when copper is replaced by zinc (Nar et al., 1992). Available spectroscopic data report essentially on the effects of the mutation on the local mobility at the Trp site but little is known on the consequences of these cavities for the conformational dynamics of the macromolecule as whole.
FIGURE 1.

View of the α-carbon backbone of wild-type azurin from P. aeruginosa. The side chains of Trp-48 (W48), Ile-7 (I7), and Phe-110 (F110) are indicated in bold.
In recent years the phosphorescence emission of Trp has proven to be a remarkably sensitive probe of the fluidity of the protein environment, the long lifetime of the triplet state permitting us to detect slow conformational transitions occurring in the long submillisecond-to-second timescale (Saviotti and Galley, 1974; Vanderkooi, 1991; Schauerte et al., 1997; Strambini, 1989; Cioni and Strambini, 2002b). The phosphorescence of Trp-48 in apo-azurin is strong and long-lived even in buffer at ambient temperature. The delayed emission is effectively quenched in the native protein by the direct interaction, electron transfer/energy transfer, with the copper ion (Strambini and Gabellieri, 1991) but is equally intense in the apoprotein as when the copper is replaced by Zn, Cd, Hg, or Ag. The lifetime of apo- and holoproteins is similar indicating that the presence of a metal cation in the binding site does not affect the rigidity of the Trp domain (Strambini and Gabellieri, 1991; Hansen et al., 1996).
The present report addresses the issue of cavity effects on the internal dynamics of azurin by means of two sensitive parameters related to the phosphorescence emission of Trp-48. In brief, the intrinsic room-temperature phosphorescence lifetime (τ0) reports on the local flexibility of the protein matrix around the chromophore (Gonnelli and Strambini, 1995; Strambini and Gonnelli, 1995), whereas the bimolecular rate constant (kq) for the quenching of phosphorescence by acrylamide in solution relates to the diffusion of the solute through the protein fold to the chromophore's site and is correlated to the structural flexibility of the macromolecule (Cioni and Strambini, 1998). Recently, these parameters have proven instrumental in detecting variations of protein dynamics induced by a variety of perturbations, among which are chemical denaturants (Cioni and Strambini, 1998), hydrostatic pressure (Cioni and Strambini, 1996), and heavy water (Cioni and Strambini, 2002a). The results obtained with the two single-point mutants, I7S and F110S, point out that the cavity formed in the core of the azurin causes an unprecedented increase in the rate of acrylamide migration to Trp-48, implying a drastic enhancement of large-amplitude structural fluctuations. Despite a more flexible protein fold in the mutant proteins, local effects on the mobility of the Trp environment can be of opposite sign, and probably are specific for the site of mutation. Lastly, both τ0 and kq confirm a greater plasticity of the mutant structures and a tightening role of bound Cd2+.
MATERIALS AND METHODS
All chemicals were of the highest purity grade available from commercial sources. Acrylamide (>99.9% electrophoretic purity) was from Bio-Rad Laboratories (Richmond, CA). Water, doubly distilled over quartz, was purified by Milli-Q Plus system (Millipore, Bedford, MA). All glassware used for sample preparation was conditioned in advance by standing for 24 h in 10% HCl suprapur (Merck, Darmstadt, Germany).
The procedure of isolation and purification of wild-type P. aeruginosa azurin has been described by van de Kamp et al. (1990). Details about site-directed mutagenesis procedure, the protein expression, the isolation, and the purification of the I7S and F110S mutants have been described elsewhere (Gilardi et al., 1994). The apoproteins were prepared from holo-azurins by adding potassium cyanide and EDTA to final concentrations of 0.1 M potassium cyanide and 1 mM EDTA in 0.15 M Tris-HCl, pH 8, followed by column chromatography (Van de Kamp et al., 1990).
Cd-azurins were formed from apo-azurins by the addition of CdCl2 in the molar ratio of 2:1. Complex formation was verified by looking at the Trp fluorescence intensity in competition experiments with Cu2+ as Cu-azurin is strongly quenched (Strambini and Gabellieri, 1991). Within the first 2 h from the addition of Cu2+ no change in fluorescence was observed in the Cd-azurin samples.
For phosphorescence measurements in fluid solutions, it is paramount to rid the solution of all O2 traces. The samples were placed in 5 × 5 square quartz cuvettes especially designed to allow thorough removal of O2 by the alternative application of moderate vacuum and inlet of ultrapure N2 (Strambini and Gonnelli, 1995). All experiments were conducted in 2 mM Tris-HCl, pH 7.5, at a protein concentration of 2 to 5 μM. For measurements in low temperature glasses the solvent was 50/50 (V/V) propylene glycol/ buffer (20 mM potassium phosphate, pH 7.5) and the protein concentration was 10 μM. Acrylamide quenching experiments were carried out as described before (Cioni and Strambini, 1998).
Fluorescence and phosphorescence measurements
All luminescence measurements were conducted on homemade instrumentation. Briefly, for emission spectra, continuous excitation is provided by a Cermax xenon lamp (LX150UV, ILC Technology, Sunnyvale, CA) whose output is selected (6-nm bandpass) by a 0.23-m double-grating monochromator (SPEX 1680, Spex Industries, Edison, NJ) optimized for maximum stray-light rejection. The emission collected at 90° from the excitation is dispersed by a 0.25-m grating monochromator (Jobin-Yvon, Model H-25, Milan, Italy) set to a bandpass of 1 nm. A two-position light chopper intersects either the excitation beam only (fluorescence mode) or both excitation and emission beams in alternate fashion in such a way that only delayed emission gets through to the detector (phosphorescence mode). The photomultiplier (9635QB, Thorn EMI, Rockaway, NJ) current was fed to a low-noise current preamplifier (Standford, Model SR570, Sunnyvale, CA) followed by a lock-in amplifier (ITHACO Model 393, Ithaca, NJ) operated at the chopper frequency. The output is digitized and stored by multifunction board (PCI-20428W, Intelligent Instrumentation, Tucson, AZ) utilizing visual Designer software (PCI-20901S, Ver. 3.0, Intelligent Instrumentation). Spectra are acquired at a scan rate of 0.2 nm s−1 and with a time constant (lock-in amplifier) of 125 ms.
For phosphorescence decays, pulsed excitation is provided by a frequency-doubled, Nd/Yag-pumped dye laser (Quanta Systems, Milan, Italy; λex = 293 nm) with a pulse duration of 5 ns and a typical energy per pulse of 0.5 to 1 mJ. The phosphorescence emitted at 90° from the excitation is selected by an interference filter (DTblau, Balzer, Milan, Italy) with a transmission window between 410 and 450 nm. A gating circuit that inverts the polarity of dynodes 1 and 3, for up to 1.5 ms after the laser pulse, protects the photomultiplier (Hamamatsu Model R928, Hamamatsu City, Japan) from the intense fluorescence light pulse. As for spectral measurements, the photocurrent signal is amplified, digitized, and multiple sweeps averaged by the same computer-scope system. All phosphorescence decays were analyzed in terms of a sum of exponential components by a nonlinear least-squares fitting algorithm (GLOBALS Unlimited, LFD, University of Illinois, Urbana-Champaign, IL).
RESULTS
Inasmuch as native copper, in both reduced and oxidized states, fully quenches the phosphorescence of azurin, this report examines apo- (metal-free) and holo-azurins in which native Cu has been substituted with nonquenching Cd2+, a metal known to form an equally stable complex with the protein (Engeseth and McMillin, 1986).
High resolution phosphorescence spectra in glass matrices at 140 K
The crystallographic structure of the azurin mutants I7S and F110S reports a fold essentially identical to that of the wild-type protein. In dilute aqueous solutions proteins often exhibit multiple stable conformations and therefore mutations may alter both the dominant form and the conformational distribution. Information on alterations of both the local protein structure and on conformational heterogeneity can be provided by the high resolution phosphorescence spectrum of Trp-48, information obtained in low temperature glasses. Fig. 2 shows that the phosphorescence spectrum of wild-type apo-azurin in (60/40, V/V) glycerol/buffer (20 mM potassium phosphate, pH 7.5) at 140 K exhibits a well-resolved vibrational structure. The peak wavelength of the 0,0 vibronic band (λ0,0) and its bandwidth (BW = the width at half-height) are normally taken as indicators of spectral energy and spectral broadness. The former parameter is related to the polarity of the medium about the indole ring and the latter to the homogeneity of its microenvironment. For the apo-WT, λ0,0 is 409.9 ± 0.1 nm, a value consistent with a rather nonpolar site (411 nm; Galley, 1976). The BW is relatively large (6.5 nm) when compared to structurally homogeneous protein sites (BW ≤ 3.25 nm; Gabellieri et al., 1996) and attests to conformational heterogeneity in the Trp microenvironment. The replacement of either I7 or F110 with Ser causes in each case a moderate blue-shift of the spectrum (twice larger with I7S) and a further increase in BW (Table 1). The change in spectral energy is indicative of a more polar environment in the mutant proteins relative to the WT whereas the enhanced BW indicates greater conformational freedom. As the bulky, nonpolar side chains of I7 and F110 are both in direct contact with the indole ring, the spectral blue-shift and increased BW induced by their substitution with the smaller and polar Ser are fully accounted for by the enhanced polarity and looser packing of the Trp-48 environment.
FIGURE 2.

High resolution phosphorescence spectrum of apo-WT azurin in a glycerol/buffer (60/40, w/w) glass at 140 K (heavy line). The spectrum of glyceraldehyde-3 phosphate dehydrogenase, GAPDH (Strambini and Gabellieri, 1998), is included as reference (light line). The buffer is potassium phosphate 20 mM, pH 7.5. The protein concentration is 14 μM; λex = 295 nm.
TABLE 1.
Peak wavelength (λ0–0) and bandwidth (BW) of the 0–0 vibronic band of the phosphorescence spectrum of WT and mutant azurins in the apo- and Cd-form
|
λ0–0 (nm)
|
BW (nm)
|
|||
|---|---|---|---|---|
| Protein | Apo | Cd2+ | Apo | Cd2+ |
| WT | 409.9 ± 0.1 | 410.0 ± 0.1 | 6.5 ± 0.05 | 6.4 ± 0.05 |
| F110S | 409.4 ± 0.1 | 410.2 ± 0.1 | 7.5 ± 0.05 | 7.3 ± 0.05 |
| I7S | 408.8 ± 0.1 | 409.2 ± 0.1 | 7.3 ± 0.05 | 6.9 ± 0.05 |
Binding Cd2+ to the apoproteins red-shifts the spectrum and enhances its resolution (Table 1), changes that are consistent with a decreased polarity and conformational heterogeneity, respectively. These effects of metal complexation are small, practically negligible in the WT, indicating that in the native protein the environment of Trp-48 is quite similar in both apo and holo structures.
Intrinsic phosphorescence lifetime of azurins in buffer at room temperature
In fluid solutions the mobility of the protein/solvent matrix about the triplet probe promotes radiationless processes that can drastically reduce the phosphorescence lifetime, τ0, relative to the upper limit of ∼6–7 s of rigid glasses (Strambini and Gonnelli, 1995). As a consequence, long-lived phosphorescence (τ ∼ 0.1–1 s) from proteins in buffer at ambient temperature is detected exclusively from Trp residues buried in rigid sites, and their lifetime is a direct monitor of the local flexibility of the polypeptide (Gonnelli and Strambini, 1995), provided no intramolecular quenching occurs with proximal Cys, His, or Tyr side chains. Proximal residues of this type are indeed absent in the case of Trp48 in azurin.
Sample phosphorescence decays of WT and the mutant azurins, apo- and Cd-forms, in buffer (2 mM Tris-HCl buffer, pH 7.5) at 20°C, are shown for comparison in Fig. 3. We find that each mutation affects both the phosphorescence lifetime and the decay homogeneity. As opposed to the WT emission, which is uniform and well represented by a single lifetime, for F110S and I7S it requires at least two lifetime components to fit the data. The derived amplitudes (αi) and lifetimes (τi) are collected in Table 2 and show that in both mutants a fraction of the intensity, ranging from 17 to 48%, exhibits a lifetime that is 2–3-fold shorter than the main component. The lifetime multiplicity of the mutant azurins provides unequivocal evidence that in solution there exist multiple protein conformations sufficiently stable to remain distinct even in the long timescale of phosphorescence (∼1 s). Interestingly, the two cavity-creating mutations have opposite effects on the average phosphorescence lifetime, τav = ∑αiτi, τav decreasing 3.6/2.2-fold for apo-/holo-I7S but increasing by 1.7/6-fold for apo-/holo-F110S.
FIGURE 3.

Comparison of phosphorescence decays of WT, I7S, and F110S mutants in Tris-HCl 2 mM pH 7.5 at 20°C in both apo- (top panel) and Cd- (bottom panel) forms. The protein concentration was typically 2–5 μM.
TABLE 2.
Lifetimes and amplitudes relative to the phosphorescence decay in buffer at 20°C of apo- and Cd-azurins
| Protein | τ1 (s) | α1 | τ2 (s) | τav (s) | ητav(cp) | ΔH#(1/ητav) (kcal mol−1) |
|---|---|---|---|---|---|---|
| Apo-WT | 0.63 ± 0.02 | 1.0 | – | 0.63 ± 0.02 | 5.0 × 104 | 24 ± 1.5 |
| Apo-F110S | 1.49 ± 0.05 | 0.48 ± 0.02 | 0.81 ± 0.03 | 1.14 ± 0.05 | 1.6 × 105 | 38 ± 2.0 |
| Apo-I7S | 0.18 ± 0.01 | 0.81 ± 0.01 | 0.06 ± 0.003 | 0.16 ± 0.01 | 3.7 × 103 | 22 ± 1.0 |
| Cd-WT | 0.55 ± 0.01 | 1.0 | – | 0.55 ± 0.01 | 3.8 × 104 | 24 ± 1.5 |
| Cd-F110S | 3.36 ± 0.10 | 1.0 | – | 3.36 ± 0.10 | 2.3 × 107 | 33 ± 1.5 |
| Cd-I7S | 0.41 ± 0.02 | 0.17 ± 0.01 | 0.26 ± 0.01 | 0.25 ± 0.02 | 7.7 × 103 | 21 ± 1.0 |
η(τav) has been calculated according to Gonnelli and Strambini (1995). ΔH#(ητav) are the activation enthalpies obtained from the temperature dependence of 1/η(τav).
Estimations of the local fluidity of the Trp environment based merely on the relationship between τ0 and solvent viscosity derived with model compounds (Strambini and Gonnelli, 1995), without taking into account any potential effect of H-bonding to the indole ring, are included in Table 2. According to this empirical indicator the structure about Trp-48, relative to WT, is far more fluid in I7S (∼8- and 4-fold reduction in the local effective viscosity (ητ) for apo and holo forms, respectively) but considerably more rigid in F110S (∼23- and 605-fold enhancement for apo and holo forms, respectively). Because the creation of a cavity, either empty or filled with water, is expected to enhance local structural fluctuations the result obtained with the F110S mutant is totally unexpected and probably hints to the formation of a specific bonding arrangement resulting in the complete immobilization of the indole ring. We note that the lifetime of 3.35 s of holo-F110S is the largest reported to date for any protein in solution at 20°C—a value approaching the τ0 ≈ 6 s typical of glass matrices. Lastly, a considerable increase of τ0 between apo- and holoproteins is observed only with the mutant proteins, indicating a structure tightening role of the metal ion in these less stable azurin variants.
To estimate the activation barrier of the structural fluctuations underlying the triplet state relaxation, the lifetime in buffer was monitored across the 0–40°C temperature range. Above 50°C the phosphorescence of apoproteins is rapidly quenched and the lifetime drops to the ms-range and below, consistent with extensive unfolding of the globular structure. Lifetimes are converted into effective local “viscosities,” using the empirical relationship between τ and solvent viscosity (η) (Strambini and Gonnelli, 1995), so that the parameter (1/ηprot) is an indirect measure of the rate of the structural fluctuations about Trp-48. Arrhenius plots of ln(1/ηprot) versus 1/T are substantially linear (Fig. 4), and the activation enthalpies, ΔH#(1/ηprot), derived from the slope are collected in Table 2. As a whole, activation enthalpies are large, >20 kcal/mol, suggesting that the barrier to local motions about Trp-48 involves in each case costly rearrangements of the surrounding β-sheet. It is interesting to note that, relative to the WT, differences in the activation barrier in the mutant proteins are correlated to the rigidity of their structure (τ0), increasing by ∼14/9 kcal/mol (apo/holo) for tighter F110S and decreasing, by two-thirds, kcal/mol (apo/holo) for looser I7S.
FIGURE 4.

Arrhenius plots of 1/ητav obtained over the 0–40°C temperature interval. The proteins samples are (▴) WT, (▪) F110S, and (•) I7S, in the apo-form (top panel) and Cd-form (bottom panel). Other experimental conditions are as in Fig. 3.
Quenching of phosphorescence by acrylamide
A totally independent assessment of the flexibility of the macromolecule can be obtained from the ease with which neutral quenching molecules like acrylamide diffuse through the globular fold to the site of the chromophore and quench its phosphorescence by a relatively short-range interaction (Cioni and Strambini, 1998). Quenching experiments involve the measurement of the phosphorescence lifetime at increasing acrylamide concentrations and the bimolecular rate constant (kq) of the reaction is obtained from the gradient of the lifetime Stern-Volmer plot (1/τ = 1/τ0 + kq [acryl]). The mutants phosphorescence emission is intrinsically heterogeneous and, especially for apo-F110S, was found to remain so even when the average phosphorescence lifetime is considerably reduced by acrylamide. The heterogeneity reflects the presence of more than one stable conformation of the macromolecule, each with its τ0 and quenching rate constant. For convenience, lifetime Stern-Volmer plots were constructed from the average lifetime, τav, obtained in general from a biexponential fitting of the phosphorescence decay. Thus, the value of the rate constant derived from these plots is an average quantity.
In these experiments the maximum [acryl] was 0.25 M for the WT and sufficient to cause a 50–100-fold reduction of τ in the mutant proteins. In all cases a linear dependence of 1/τ on [acryl] was found. Representative lifetime Stern-Volmer plots for quenching at 20°C are shown in Fig. 5 and the magnitude of kq calculated from the slope is given in Table 3. The accessibility of a large solute like acrylamide to the tightly packed core of azurin is expected to be greatly hindered and, in fact, kq is reduced by >7/9 (apo/holo) orders of magnitude in the native protein, compared to a solvent-exposed Trp residue (Kerwin et al., 2002). The picture changes significantly with the mutants as the substitution of either Phe-110 or Ile-7 with less bulky Ser causes a dramatic enhancement, 4–5 orders of magnitude, of acrylamide diffusion through the protein core in apo- and holo-azurins alike. It should be recalled that because acrylamide quenching can occur through space (Cioni and Strambini, 1998), increased quenching rates could potentially be accounted for by structural alterations in the mutant proteins that bring the chromophore close to the protein surface, where through-space interactions with acrylamide in the solvent dominate the quenching reaction. However, an estimate of the bimolecular rate constant based on the distance dependence of the indole-acrylamide interaction (Cioni and Strambini, 1998) show that a value of kq ≈ 104 M−1 s−1corresponds to a separation of W48 from the aqueous interface by <0.2 nm, compared to 0.8 nm in the native fold. Since the crystallographic structures of I7S and F110S show that neither the general fold of the protein nor the position of W48 relative to the solvent are affected by the mutation this alternative explanation is ruled out. Such a dramatic increase in acrylamide permeability by a single mutation emphasizes that close packing of the core region surrounding the aromatic ring is probably crucial for blocking those structural fluctuations permitting acrylamide migration to the interior. Of the two mutants I7S is 3–4 times more readily permeable to acrylamide, a finding which is consistent with the greater flexibility inferred for this mutant also by τ0. Again, acrylamide quenching constants confirm the important role of the metal ion in tightening the apo structure. Relative to apoproteins, acrylamide diffusion in holoproteins is slowed down 6–7-fold in the mutants and ∼40-fold in the WT.
FIGURE 5.

Lifetime Stern-Volmer plots for the quenching of azurin phosphorescence by acrylamide. The proteins samples are: (▪) WT, (•) F110S, and (▴) I7S, in the apo-form (top panel) and Cd-form (bottom panel). The azurin concentration is 2–5 μM in Tris-HCl (2 mM, pH 7.5) at 20°C. Each point is the average of at least three independent experiments and the error bars indicate the range of τav variations.
TABLE 3.
Acrylamide bimolecular phosphorescence quenching constants at 20°C of apo- and Cd-azurins
|
kq (M−1 s−1)
|
ΔH#(kq) (kcal mol−1)
|
ΔGD* (kcal mol−1)
|
||||
|---|---|---|---|---|---|---|
| Sample | Apo | Cd2+ | Apo | Cd2+ | Apo | Cd2+ |
| NATA | 1.5 × 109 | 5 | – | |||
| WT | 3.2 ± 0.5 × 101 | 0.8 ± 0.2 | 22.0 ± 1.5 | 22.1 ± 1 | 6.4 | 9.4 |
| F110S | 8.9 ± 0.5 × 104 | 1.6 ± 0.2 × 104 | 10.7 ± 0.6 | 11.6 ± 0.8 | 2.7 | 4.8 |
| I7S | 5.0 ± 0.4 × 105 | 7.0 ± 0.6 × 104 | 7.2 ± 0.5 | 6.8 ± 0.6 | 3.4 | 5.8 |
ΔH#(kq) is the activation enthalpy obtained from the temperature dependence of kq and ΔGD is the denaturation free energy.
As with τ0, kq was determined over the 0–40°C temperature range and the activation enthalpies obtained from essentially linear Arrhenius plots (not shown) are given in Table 3. The results indicate that the remarkable increase in acrylamide diffusion caused by the mutations is accompanied by a sizable, 2–3-fold reduction of the activation barrier (ΔH#kq) which, in keeping with a looser fold, is smallest for I7S. The barrier is similar between apo- and holoproteins, indicating that Cd coordination to the metal binding site reduces kq mostly by an unfavorable entropic contribution.
DISCUSSION
Trp-48 is located roughly at the center of the β-barrel, the indole ring surrounded by nonpolar side chains that form a compact hydrophobic core (Fig. 1). By all criteria, crystallographic B-factors (12), T1 NMR relaxation times (Mei et al., 1996), H/D exchange rates (van de Kamp et al., 1992), and molecular dynamic simulations (Arcangeli et al., 1999, 2001), the aromatic ring is highly immobilized in an unusually rigid domain. In accord with a rigid environment, Trp-48 exhibits a long phosphorescence lifetime in buffer at ambient temperature and the rate of acrylamide migration to the interior is one of the smallest ever found in proteins (Cioni and Strambini, 1998). The crystallographic structure of the mutants (Hammann et al., 1996) shows that by replacing I7 or F110 with the smaller and polar Ser a cavity (of 40 and 100 Å3 for I7S and F110S, respectively) has been engineered in proximity of Trp-48 that is expected to have important consequences both on the mobility of local side chains as well as on global conformational dynamics.
Local effects of cavities
The phosphorescence spectrum (λ00, BW) and the intrinsic lifetime report on the immediate environment of Trp-48, in particular on its structural uniformity and fluidity. The introduction of Ser in place of I7 or F110 has, in either case, broadened and blue-shifted the spectrum, a reflection of increased polarity and structural disorder about Trp-48. Unambiguous evidence for the existence of distinct stable conformations of azurin in mutant samples is provided by multiple lifetimes in buffer at ambient temperature, which is an indication that these states are long-lived species differing from one another in the local flexibility (τ0) about the chromophore. Enhanced polarity and structural heterogeneity of the Trp environment are consistent with available crystallographic and spectroscopic data. The mutant crystal structures show that thanks to the contribution of Ser and, in the case of F110S, also of 1–3 water molecules in the cavity, the environment about Trp-48 becomes more polar. Greater local disorder is inferred from the large B-factors of Ser-7 or of Ser-110 in addition to a variable number of water molecules in the cavity (Hammann et al., 1996). A more polar-disordered surrounding was also deduced from a red shifted fluorescence spectrum, a wider distribution of fluorescence lifetimes and ground state energies (Mei et al., 1996; Gilardi et al., 1994; Kroes et al., 1998) and the loss of the near-UV CD signal (Mei et al., 1996).
Although spectroscopic data and crystal structures alike are in concord that these cavity-forming mutations have introduced structural heterogeneity in the hydrophobic core of azurin, the consensus on whether the local disorder is static or dynamic is not unanimous among different approaches. The sensitivity of τ0 to the fluidity of the embedding protein matrix emphasizes that the response is remarkably different between the two mutants. In the case of I7S the shortening of τ0 attests to a much more fluid Trp environment, relative to the WT, amounting to roughly a 13- and 5-fold decrease in the local “viscosity” for apo- and holoproteins, respectively. The gain in flexibility apparently does not involve greater conformational freedom of the aromatic ring itself as the low crystallographic B-factor (9 Å2), its long NMR spin lattice relaxation time (T1) (Mei et al., 1996), and constant fluorescence anisotropy (Kroes et al., 1998), all suggest that the indole ring remains rigidly held in the molecular frame. It is therefore likely that fluidity is conferred to the side chains lining the cavity or is due to water molecules in the cavity that are too mobile to give a diffraction pattern (Buckle et al., 1996; Bakowies and van Gunsteren, 2002).
Contrary to I7S, and expectations based on a larger, water-containing cavity (100 compared to 40 Å3), and the unusually large B-factor of Ser-110 (27 Å2), all of which would presuppose an even greater fluidity in F110S than in I7S, the local structure in F110S turns out to be more rigid than in the native protein. The considerable lengthening of τ0, relative to the WT, translates into an increase in local viscosity by a factor of 3.2 and 605 for apo- and holoprotein, respectively. Indeed, the lifetime of Cd-F110S (3.36 s) approaches the limiting value of 6 s observed for the chromophore in rigid glass matrices and is the largest ever reported for a protein in buffer at ambient temperature. A glassy-like state for the Trp environment calls for an extensive and rigid H-bonding network involving local side chains and water molecules in the cavity. The crystal structure of Cu-F110S (Hammann et al., 1996) does detect immobilized water molecules in the cavity. These are H-bonded to Ser-110 which, in turn, forms an H-bond to the aromatic ring nitrogen of Trp-48, further restricting independent motions of the chromophore. An immobilized indole ring was also inferred from the B-factors, T1 relaxation time, and fluorescence anisotropy. In contrast, a large fluorescence red-shift and a wide distribution of fluorescence lifetimes have been ascribed to a looser structure (Gilardi et al., 1994). The plurality of lifetimes of F110S points out that alternative strong H-bonding arrangements can form in the cavity and that this static disorder can account for the large B-factors of Ser-110 as well as for the broad fluorescence emission.
The above discrimination between fluid and rigid cores is also supported by the activation energies governing the structural fluctuations underlying the triplet state relaxation. Relative to the WT, the activation barrier is lower by 2–3 kcal/mol for the more fluid I7S but increases by an unusually large amount, 9–12 kcal/mol, in the case of F110S to indicate that, in the latter case, segmental motions involve the breaking of stronger and cooperative bonds.
Effects of cavities on large amplitude structural fluctuations
An important consequence of either cavity-forming mutation is the dramatic increase in accessibility of acrylamide to an otherwise impermeable protein core. Before interpreting the 4–5 orders of magnitude enhancement of kq in terms of an increased internal flexibility of the mutated proteins, it is important to rule out alternative more trivial ways to enhance the quenching rate. For example, the cavity could open a direct channel to the solvent, either void or filled with unstructured water, through which the quencher might diffuse rapidly to the interior without significant disruption of the local fold. At the other extreme, the native fold could be essentially impermeable to acrylamide and quenching would occur only upon transient exposure of the chromophore to the solvent by global unfolding-like transitions. In that case the large quenching rate of cavity mutants would simply reflect the increase in the kinetic unfolding rate constant (ku) of the less stable azurin variants (ΔGD = RT ln(kf/ku), kf the refolding rate constant). As neither of these hypotheses is consistent with available experimental data we will argue that kq is a measure of relatively large amplitude structural fluctuations occurring in the globular fold. Indeed, the crystallographic structure of the two mutants show a cavity in proximity of Trp-48 but an unaltered compact packing of the polypeptide throughout the thick spacer separating the cavity from the solvent, with no direct channel linking the cavity to the aqueous interface. Further, quenching by a global unfolding mechanism is to be excluded, for it predicts extremely small quenching rates and early saturation effects in the lifetime Stern-Volmer plot (Fig. 5). Quenching by this mechanism is described by the reaction scheme
 |
where ku and kf have the usual meaning and 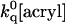 ≈ 109[acryl] s−1 is the rate of quenching of solvent-exposed Trp (Kerwin et al., 2002). This mechanism predicts quenching saturation; that is, a downward curving Stern-Volmer plot, to set in as soon as
≈ 109[acryl] s−1 is the rate of quenching of solvent-exposed Trp (Kerwin et al., 2002). This mechanism predicts quenching saturation; that is, a downward curving Stern-Volmer plot, to set in as soon as 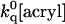 > kf and quenching rate = (ku × ′kq)/(kf + ′kq), ≤ku with ′kq =
> kf and quenching rate = (ku × ′kq)/(kf + ′kq), ≤ku with ′kq = 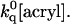 For apo-WT, recently-determined unfolding-refolding kinetics at 20°C have yielded ku = 1.3 × 10−2 s−1 and kf = 140 s−1 (Pozdnyakova et al., 2002). These values predict quenching saturation to become manifest very early, at [acryl] >10−7 M, and the maximum rate not to exceed 1.3 × 10−2 s−1. Experiments conducted in the 0–0.25 M concentration range show, on the contrary, no deviation from linearity in the S-V plot up to a quenching rate 8 s−1. The large discrepancy between prediction and experiment points out that acrylamide quenching occurs in the globular state of azurin and that structural fluctuations of much higher frequency (>10 s−1) than global unfolding are responsible for the internal migration of acrylamide to Trp-48. The same conclusions apply to the mutant proteins. Assuming the worst-case scenario, that the decrease in stability (ΔΔGD) of apo mutants, relative to the WT, is owed entirely to the enhancement of ku, one finds that ku(I7S) = 1.9 s−1 (ΔΔGD = 3.0 kcal/mol) and ku(F110S) = 6.1 s−1 (ΔΔGD = 3.7 kcal/mol). Again the global unfolding mechanism would predict saturation in the S-V plot at unrealistically low acrylamide concentrations ([acryl] > 10−7 M) and maximum quenching rates far smaller than those actually measured (102 s−1).
For apo-WT, recently-determined unfolding-refolding kinetics at 20°C have yielded ku = 1.3 × 10−2 s−1 and kf = 140 s−1 (Pozdnyakova et al., 2002). These values predict quenching saturation to become manifest very early, at [acryl] >10−7 M, and the maximum rate not to exceed 1.3 × 10−2 s−1. Experiments conducted in the 0–0.25 M concentration range show, on the contrary, no deviation from linearity in the S-V plot up to a quenching rate 8 s−1. The large discrepancy between prediction and experiment points out that acrylamide quenching occurs in the globular state of azurin and that structural fluctuations of much higher frequency (>10 s−1) than global unfolding are responsible for the internal migration of acrylamide to Trp-48. The same conclusions apply to the mutant proteins. Assuming the worst-case scenario, that the decrease in stability (ΔΔGD) of apo mutants, relative to the WT, is owed entirely to the enhancement of ku, one finds that ku(I7S) = 1.9 s−1 (ΔΔGD = 3.0 kcal/mol) and ku(F110S) = 6.1 s−1 (ΔΔGD = 3.7 kcal/mol). Again the global unfolding mechanism would predict saturation in the S-V plot at unrealistically low acrylamide concentrations ([acryl] > 10−7 M) and maximum quenching rates far smaller than those actually measured (102 s−1).
The above considerations point out that the two cavity-forming mutations drastically accelerate acrylamide migration through the globular fold of azurin. It may be instructive to inquire whether the phenomenon can simply be explained in terms of an overall loosening of the mutants' structure, as a result of their 3–4-kcal/mol lower stability, or whether a cavity in the core region plays a specific role in lowering the diffusion barrier. The following indirect evidence seems to point to a direct role of the cavity. According to the crystallographic B-factors the structures of the mutant proteins show no sign of greater flexibility, enhanced thermal motions/disorder being limited to the mutation sites (Hammann et al., 1996). Further, stability arguments alone appear inadequate to explain the large effect of the mutations on the rate of acrylamide diffusion, even if we note an inverse correlation between kq and ΔGD (Table 3). An upper bound of the effects of ΔΔGD on kq may be obtained by assuming that the decreased stability is due entirely to an enhanced unfolding rate constant ku, as recently observed in an unfolding-refolding kinetics study on H117G and H46G cavity-forming mutants of azurin (Pozdnyakova et al., 2002), and that this maximum enhancement factor predicted for ku applies equally to structural transitions of smaller amplitude than global unfolding. As the rate constant kq is proportional to the frequency of these fluctuations the increase predicted by ΔΔGD is by a factor of 1.5 × 102 for I7S and of 4.7 × 102 for F110S, both of which are much smaller than the corresponding factors of 1.5 × 104 and 2.3 × 103 actually observed for kq. Moreover, stability alone would wrongly predict that kq (I7S) < kq(F110S). In addition, relatively large destabilization of other proteins by chemical denaturants (Cioni and Strambini, 1998) or hydrostatic pressure (Cioni and Strambini, 2002b) have far smaller consequences on kq than these cavity-forming mutations, again suggesting that the creation of a cavity in the compact core of azurin exerts a huge influence on large-amplitude structural fluctuations, beyond that normally expected from the weakening of the folded state. Recently, the creation of a cavity in T4 lysozyme was shown to give rise to large amplitude structural fluctuations in the ms-timescale that permit the migration of molecules such as benzene and xenon to the inner pocket (Quillin et al., 2000; Skrynnikov et al., 2001; Mulder et al., 2001, 2002).
The above considerations point out that the accessibility of a large solute like acrylamide to the compact core of azurin is sharply modulated by the presence of an internal cavity with a volume threshold, smaller or equal to 40 Å3, the cavity estimated in I7S. A plausible rationalization of this behavior is offered by the structure of azurin as it points out that Trp-48 is surrounded by densely packed side chains rigidly held in place by an enveloping β-sheet scaffold. An extremely small value of kq for the WT protein is likely to reflect the scarce probability for acrylamide to cross this compact inner shell as it requires concerted motions of large domains. A large activation barrier for kq (ΔH± = 22 kcal/mol) similar to that for the relaxation of the triplet state (ΔH± = 24 kcal/mol) does suggest that the rate-limiting step in the migration of acrylamide to Trp-48 involves fluctuations in its core structure. Replacements of a bulky side chain in this inner shell with a smaller one will generate a local cavity and drastically abate the diffusion barrier. For the mutant proteins the activation barrier to acrylamide diffusion drops to 11 kcal/mol with F110S and to 7 kcal/mol with I7S, correctly predicting more rapid diffusion in the latter protein. It should be noted, however, that based on the decrease of ΔH± one foresees an even larger increase of kq than actually found. It is possible therefore that the overall impact on kq of lowering the barrier height of the rate-limiting step is partly attenuated by negative entropic contributions to the diffusion process, as might be expected if in the cavity mutants the reaction is dominated by a limited number of low energy pathways.
Role of Cd2+on the internal dynamics of WT and mutant azurins
Bound copper stabilizes the native fold by 3, 2.1, and 2.4 kcal/mol for WT, F110S, and I7S, respectively (Mei et al., 1999). Analogous ΔGD data is not available for Cd proteins but the stabilization is expected to be similar to that of Cu for the exchange of Cu2+ with Cd2+ maintains an equally large ΔHD and high thermal unfolding temperature (Engeseth and McMillin, 1986). The phosphorescence probe reports that binding of Cd to the apoproteins reduces the conformational heterogeneity (improved spectral resolution and narrower lifetime distributions) and tightens the globular fold (longer lifetimes and smaller accessibility to acrylamide). Locally, at the level of the Trp environment metal complexation affects only mutated proteins (Table 2) showing that the cavity confers a plasticity to the hydrophobic core that is lacking in the WT.
With respect to the accessibility of acrylamide, Cd proteins are more impermeable, kq decreasing 40-fold in the WT, 5.6-fold in F110S, and 7.1-fold in I7S. Interestingly, the activation enthalpy associated to kq is independent of metal complexation (Table 3) suggesting that the barrier, and plausibly the rate limiting step, to diffusion is practically the same in apo- and holoproteins. Hence, the rate reduction is apparently due to negative entropic contributions, as would happen from limiting the number of alternative migration pathways. In the WT the shortest route between W48 and the solvent is through the deep metal-binding pocket. By virtually blocking this route of access to the core, metal coordination per se is likely to make the major contribution to the reduction kq from apo- to holoprotein. The cavity engineered in mutant proteins opens alternative low energy paths to the surface, which might explain why the reduction of kq on metal binding is smaller here than in the WT.
In summary, the present results emphasize that cavities engineered in compact globular folds drastically enhance large-amplitude structural fluctuations and, consequently, the permeability of the macromolecule to large solutes. The outcome need not represent a generalized increase in protein flexibility; instead, it could be related to the removal of region-specific barriers to the diffusion process. The results also point out that when internal cavities are formed, local effects on the mutation site are unpredictable. An example is F110S, for which the region about W48 becomes unexpectedly rigid—probably through generation of extended H-bonding networks comprising water molecules. At a functional level, the above alterations in internal dynamics of azurin do not seem to play a role in electron transfer processes through the central region, inasmuch as the rate is similar in WT and mutants (Farver et al., 1996). The findings with acrylamide stress that cavities may, instead, be critical for the rapid diffusion of substrates to buried solvent-inaccessible enzyme sites.
References
- Adman, E. T. 1991. Copper protein structures. Adv. Protein Chem. 42:144–197. [DOI] [PubMed] [Google Scholar]
- Akke, M. 2002. NMR methods for characterizing microsecond to millisecond dynamics in recognition and catalysis. Curr. Opin. Struct. Biol. 12:642–647. [DOI] [PubMed] [Google Scholar]
- Arcangeli, C., A. R. Bizzarri, and S. Cannistraro. 1999. Long-term molecular dynamics simulation of copper azurin: structure, dynamics and functionality. Biophys. Chem. 78:247–257. [DOI] [PubMed] [Google Scholar]
- Arcangeli, C. A., R. Bizzarri, and S. Cannistraro. 2001. Concerted motions in copper plastocyanin and azurin: an essential dynamics study. Biophys. Chem. 90:45–56. [DOI] [PubMed] [Google Scholar]
- Artymiuk, P. J., C. C. F. Blake, D. E. P. Grace, S. J. Oatley, D. C. Phillips, and M. J. Sternberg. 1979. Crystallographic studies of the dynamic properties of lysozyme. Nature (Lond.). 280:563–568. [DOI] [PubMed] [Google Scholar]
- Bakowies, D., and W. F. van Gunsteren. 2002. Water in protein cavities: a procedure to identify internal water and exchange pathways and application to fatty acid-binding protein. Prot. Struct. Funct. Genet. 47:534–545. [DOI] [PubMed] [Google Scholar]
- Brooks, C. L., M. Karplus, and B. M. Pettitt. 1988. Proteins: a Theoretical Perspective of Dynamics, Structure, and Thermodynamics. Wiley, New York.
- Buckle, A. M., P. Cramer, and A. R. Fersht. 1996. Structural and energetic responses to cavity-creating mutations in hydrophobic cores: observation of a buried water molecule and the hydrophilic nature of such hydrophobic cavities. Biochemistry. 35:4298–4305. [DOI] [PubMed] [Google Scholar]
- Cioni, P., and G. B. Strambini. 1998. Acrylamide quenching of protein phosphorescence as a monitor of structural fluctuations in the globular fold. J. Am. Chem. Soc. 120:11749–11757. [Google Scholar]
- Cioni, P., and G. B. Strambini. 2002a. Effect of heavy water on protein flexibility. Biophys. J. 82:3246–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioni, P., and G. B. Strambini. 2002b. Tryptophan phosphorescence and pressure effects on protein structure. B.B.A. Prot. Struct. Mol. Biol. 1595:116–130. [DOI] [PubMed] [Google Scholar]
- Cioni, P., and G. B. Strambini. 1996. Pressure effects on the structure of oligomeric proteins prior to subunit dissociation. J. Mol. Biol. 263:789–799. [DOI] [PubMed] [Google Scholar]
- Engeseth, R. H., and D. R. McMillin. 1986. Studies of thermally induced denaturation of azurin and azurin derivatives by differential scanning calorimetry: evidence for copper selectivity. Biochemistry. 25:2448–2455. [DOI] [PubMed] [Google Scholar]
- Englander, S. W., T. R. Sosnick, J. J. Englander, and L. Mayne. 1996. Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 6:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farver, O., L. K. Skov, G. Gilardi, G. van Pouderoyen, G. W. Canters, S. Wherland, and I. Pecht. 1996. Structure-function correlation of intramolecular electron transfer in wild-type and single-site mutated azurins. Chem. Phys. 204:271–277. [Google Scholar]
- Gabellieri, E., S. Rahuel-Clermont, G. Branlant, and G. B. Strambini. 1996. Effects of NAD+ binding on the luminescence of tryptophan 84 of Glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus. Biochemistry. 35:12549–12559. [DOI] [PubMed] [Google Scholar]
- Galley, W. C. 1976. Heterogeneity in protein emission spectra. In Concepts of Biochemical Fluorescence, Chapter 8. R. F. Chen and H. Edelhoch, editors. Marcel Dekker, New York.
- Gilardi, G., G. Mei, N. Rosato, G. W. Canters, and A. Finazzi-Agrò. 1994. Unique environment of Trp-48 in Pseudomonas aeruginosa azurin as probed by site-directed mutagenesis and dynamic fluorescence spectroscopy. Biochemistry. 33:1425–1432. [DOI] [PubMed] [Google Scholar]
- Gonnelli, M., and G. B. Strambini. 1995. Phosphorescence lifetime of tryptophan in proteins. Biochemistry. 34:13847–13857. [DOI] [PubMed] [Google Scholar]
- Hammann, C., A. Messerschmidt, R. Huber, H. Nar, G. Gilardi, and G. W. Canters. 1996. X-ray crystal structure of the two site-specific mutants Ile7Ser and Phe110Ser of azurin from Pseudomonas aeruginosa. J. Mol. Biol. 255:362–366. [DOI] [PubMed] [Google Scholar]
- Hansen, J. E., D. G. Steel, and A. Gafni. 1996. Detection of pH-dependent conformational change in azurin by time-resolved phosphorescence. Biophys. J. 71:2138–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerwin, B. A., B. S. Chang, C. V. Gegg, M. Gonnelli, T. Li, and G. B. Strambini. 2002. Interactions between PEG and type I soluble tumor necrosis factor receptor: modulation by pH and by PEGylation at the N-terminus. Protein Sci. 11:1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroes, S. J., G. W. Canters, G. Gilardi, A. van Hoek, and J. W. G. Visser. 1998. Time-resolved fluorescence study of azurin variants: conformational heterogeneity and tryptophan mobility. Biophys. J. 75:2441–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowicz, J. R., and G. Weber. 1973. Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry. 12:4171–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei, G., A. Di Venere, F. Malvezzi Campeggi, G. Gilardi, N. Rosato, F. De Matteis, and A. Finazzi-Agrò. 1999. The effect of pressure and guanidine hydrochloride on azurins mutated in the hydrophobic core. Eur. J. Biochem. 265:619–625. [DOI] [PubMed] [Google Scholar]
- Mei, G., G. Gilardi, M. Venanzi, N. Rosato, G. W. Canters, and A. Finazzi-Agrò. 1996. Probing the structure and mobility of Pseudomonas aeruginosa azurin by circular dichroism and dynamic fluorescence anisotropy. Protein Sci. 5:2248–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder, F. A. A., N. R. Skrynnikov, B. Hon, F. W. Dahlquist, and L. E. Kay. 2001. Measurements of slow (microseconds-milliseconds) time scale dynamics in protein side chains by15N relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 123:967–975. [DOI] [PubMed] [Google Scholar]
- Mulder, F. A. A., B. Hon, A. Mittermaier, F. W. Dahlquist, and L. E. Kay. 2002. Slow internal dynamics in proteins: application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 124:1443–1451. [DOI] [PubMed] [Google Scholar]
- Munro, I. M., I. Pecht, and L. Stryer. 1979. Subnanosecond motions of tryptophan residues in proteins. Proc. Natl. Acad. Sci. USA. 76:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nar, H., R. Huber, A. Messerschmidt, A. C. Filippou, M. Barth, M. van de Kamp, and G. W. Canters. 1992. Characterization and crystal structure of zinc azurin, a by-product of heterologous expression in E. coli of Pseudomonas aeruginosa copper azurin. Eur. J. Biochem. 205:1123–1129. [DOI] [PubMed] [Google Scholar]
- Nar, H., A. Messerschimdt, R. Huber, M. van de Kamp, and G. W. Canters. 1991. Crystal structure analysis of oxidized Pseudomonas aeruginosa azurin at pH 5.5 and pH 9.0. J. Mol. Biol. 221:765–772. [DOI] [PubMed] [Google Scholar]
- Nar, H., A. Messerschmidt, R. Huber, M. van de Kamp, and G. W. Canters. 1992. Crystal structure of Pseudomonas aeruginosa apo-azurin at 1.85 Å resolution. FEBS Lett. 306:119–124. [DOI] [PubMed] [Google Scholar]
- Palmer III, A. G. 2001. NMR probes of molecular dynamics: overview and comparison with other techniques. Annu. Rev. Biophys. Biomol. Struct. 30:129–155. [DOI] [PubMed] [Google Scholar]
- Palmer III, A. G., C. D. Kroenke, and P. J. Loria. 2001. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 339:204–238. [DOI] [PubMed] [Google Scholar]
- Pozdnyakova, I., J. Guidry, and P. Wittung-Stafshede. 2002. Studies of Pseudomonas aeruginosa azurin mutants: cavities in β-barrel do not affect refolding speed. Biophys. J. 82:2645–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillin, M. L., W. A. Breyer, I. J. Griswold, and B. W. Matthews. 2000. Size versus polarizability in protein-ligand interactions: binding of noble gases within engineered cavities in phage T4 lysozyme. J. Mol. Biol. 302:955–977. [DOI] [PubMed] [Google Scholar]
- Saviotti, M. L., and W. C. Galley. 1974. Room temperature phosphorescence and the dynamic aspects of protein structure. Proc. Natl. Acad. Sci. USA. 71:4154–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauerte, J. A., D. G. Steel, and A. Gafni. 1997. Time-resolved room temperature tryptophan phosphorescence in proteins. Methods Enzymol. 278:49–71. [DOI] [PubMed] [Google Scholar]
- Skrynnikov, N. R., F. A. A. Mulder, B. Hon, F. W. Dahlquist, and L. E. Kay. 2001. Probing slow time scale dynamics at methyl-containing side chains in proteins by relaxation dispersion NMR measurements: application to methionine residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 123:4556–4566. [DOI] [PubMed] [Google Scholar]
- Strambini, G. B., and E. Gabellieri. 1991. Phosphorescence from Trp-48 in azurin: influence of Cu(II), Cu(I), Ag(I) and Cd(II) at the coordination site. J. Phys. Chem. 95:4352–4356. [Google Scholar]
- Strambini, G. B. 1989. Tryptophan phosphorescence as monitor of protein flexibility. J. Mol. Liq. 42:155–165. [Google Scholar]
- Strambini, G. B., and M. Gonnelli. 1995. Tryptophan phosphorescence in fluid solution. J. Am. Chem. Soc. 117:7646–7651. [Google Scholar]
- Strambini, G. B., and E. Gabellieri. 1998. Tyrosine quenching of tryptophan in glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus. Biophys. J. 74:3165–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, K. E. S., and K. A. Dill. 1998. Native protein fluctuations: the conformational-motion temperature and the inverse correlation of protein flexibility with protein stability. J. Biomol. Struct. Dyn. 16:397–411. [DOI] [PubMed] [Google Scholar]
- Tsai, A. M., J. U. Terrence, and D. A. Neumann. 2001. The inverse relationship between protein dynamics and thermal stability. Biophys. J. 81:2339–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Kamp, M., G. W. Canters, S. S. Wijmenga, A. Lommen, C. W. Hilbers, H. Nar, A. Messerschmidt, and R. Huber. 1992. Complete sequential 1H nuclear magnetic resonance assignments and solution secondary structure of the blue copper protein azurin from Pseudomonas aeruginosa. Biochemistry. 31:10194–10207. [DOI] [PubMed] [Google Scholar]
- Van de Kamp, M., M. C. Silvestrini, M. Brunori, J. van Beeumen, F. C. Hali, and W. C. Canters. 1990. Involvement of the hydrophobic patch of azurin in the electron-transfer reactions with cytochrome-C551 and nitrite reductase. Eur. J. Biochem. 194:109–118. [DOI] [PubMed] [Google Scholar]
- Vanderkooi, J. M. 1991. Tryptophan phosphorescence from proteins at room temperature. In Topics in Fluorescence Spectroscopy, Vol. 3, Biochemical Applications. J. R. Lakowicz, editor. Plenum Publishing, New York. 113–136.
- Wagner, G., and K. Wutrich. 1986. Observation of internal mobility of proteins by nuclear magnetic resonance in solution. Methods Enzymol. 131:307–326. [DOI] [PubMed] [Google Scholar]
- Zaccai, G. 2000. How soft is a protein? A protein dynamics force constant measured by neutron scattering. Science. 288:1604–1607. [DOI] [PubMed] [Google Scholar]