

Abstract
Strong, integrin-mediated adhesion of neutrophils to endothelium during inflammation is a dynamic process, requiring a conformational change in the integrin molecule to increase its affinity for its endothelial counterreceptors. To avoid general activation of the cell, Mg2+ was used to induce the high-affinity integrin conformation, and micromechanical methods were used to determine adhesion probability to beads coated with the endothelial ligand ICAM-1. Neutrophils in Mg2+ bind to the beads with much greater frequency and strength than in the presence of Ca2+. An increase in adhesion strength and frequency was observed with both increasing temperature and contact duration (from 2 s to 1 min, 21 or 37°C). The dependence of adhesion probability on contact time or receptor density yielded estimates of the effective reverse rate constant, kr, and the equilibrium association constant, Ka, for binding of neutrophils to ICAM-1 coated surfaces in Mg2+: kr ≈ 0.7 s−1 and the product Kaρc ≈ 2.4 × 10−4, where ρc is the density of integrin on the cell surface.
INTRODUCTION
The firm adhesion of neutrophils to endothelium is a crucial, regulated step in the body's response to infection. This interaction is primarily mediated by binding of β2-integrin family members (LFA-1, Mac-1, p150/95) to their ligands, mainly ICAM-1, expressed on activated endothelium (Springer, 1994). An important step in this process is a change in the conformation of the integrin, which increases its affinity for its counterreceptor (ICAM-1) (Lupher et al., 2001; McDowall et al., 1998). In the natural state this change in conformation is brought about by intracellular signaling events in response to inflammatory stimuli and is accompanied by multiple intracellular events. In addition to changing the affinity of the integrin for its ligand, these intracellular signaling events also lead to cytoskeletal reorganization, changes in the association between integrins and actin binding proteins, and possible alterations in the distribution of integrins on the cell surface (Stewart et al., 1998; Leitinger et al., 2000). The activity state of integrins can be influenced by numerous factors, including integrin phosphorylation (Valmu et al., 1999), structural elements of the α- and β-subunit cytoplasmic tails (Sampath et al., 1998; Calderwood et al., 2000; Lub et al., 1997), or divalent cation sites on the α-subunits (Bazzoni et al., 1998; Masumoto and Hemler, 1993). Many details of the events associated with integrin activation continue to emerge. Correlating specific steps in the activation process with the formation and strength of adhesive contacts between neutrophils and endothelial ligands is critical for a complete understanding of the mechanisms leading to cell arrest and extravasation.
It is well known that the conformation and affinity of many integrins can be affected by the presence of different divalent cations in the extracellular medium. In particular it was observed that Mn2+ or Mg2+ plus the calcium chelator EGTA induces a conformation of LFA-1 that has a high affinity for ICAM-1, and that this effect is inhibited by the presence of millimolar Ca2+ (Dransfield et al., 1992; Leitinger et al., 2000; Labadia, et al., 1998). This feature provides an opportunity to explore the particular importance of integrin activation for adhesion in the absence of general activation of the cell. It is important to note that the effects of divalent ions on the affinity state of Mac-1 (the other major integrin found on neutrophils) is different from their effects on LFA-1. Early reports demonstrated that calcium and/or magnesium were essential for integrin affinity changes induced by inflammatory mediators, but that manganese can induce the active form of Mac-1 independent of cell activation (Altieri, 1991). These effects were confirmed in subsequent studies that correlated the high-affinity form of Mac-1 with the appearance of an epitope recognized by the monoclonal antibody CBRM1/5 (Diamond and Springer, 1993). Thus, whereas the effect of manganese on integrin affinity is similar for both LFA-1 and Mac-1, the effects of calcium and magnesium on the two molecules are different.
In this report we examine how divalent cations Mg2+ and Ca2+ affect the formation of adhesive contacts between neutrophils and the endothelial ligand ICAM-1 immobilized on the surfaces of beads. We show that ICAM-1 coated beads can firmly adhere to neutrophils in buffers containing Mg2+ and EGTA (where LFA-1 is in a high-affinity state) and do not adhere to neutrophils in buffers containing Ca2+ (where LFA-1 is in a low-affinity conformation) (Leitinger et al., 2000). From the dependence of the adhesion frequency on contact time and the density of ligand on the beads, we determine the effective kinetic rate constants for the formation of adhesive bonds between neutrophils and surfaces presenting the endothelial ligand ICAM-1.
MATERIALS AND METHODS
General procedures
Neutrophils were obtained from a drop of peripheral blood of healthy donors and diluted in 10 mM HEPES buffered saline (150 mM NaCl, 5 mM KCl, 2 mg/ml glucose in low endotoxin distilled water (Gibco BRL, Grand Island, NY)), to which was added 4% fetal calf serum (FCS, HyClone, Logan, UT) and either 5.0 mM Mg2+ plus 0.5 mM EGTA or 1.5 mM Ca2+, pH 7.4, 290 mOsm. Cells were placed on the microscope stage in a chamber consisting of two coverglasses separated by a plastic spacer ∼2 mm in thickness. Openings at opposite sides of the chamber provided access for two micropipettes, one of which was used to hold a ligand-coated bead, and the other of which was used to manipulate a neutrophil into contact with the bead.
Antibodies
MAb38 (anti-human CD11a, IgG2a), which recognizes the I domain of the integrin αL-subunit (CD11a) and blocks binding of ICAM-1 to LFA-1, and mAb IB4 (anti-human CD18, IgG2a), which recognizes the β2-integrin subunit (CD18) and blocks binding to ICAM-1 were purchased from Ancell (Bayport, MN). MAb CBRM1/5 (anti-human CD11b) was purchased from eBioscience (San Diego, CA). CBRM1/5 recognizes activation-specific epitope of human Mac-1, which is localized in the I-domain on the α-chain, and blocks Mac-1 dependent adhesion to ICAM-1 and fibrinogen.
Coating beads
Two methods were used to immobilize ICAM-1 on the beads. In one, ICAM-1 was coupled covalently to beads via tosyl linkage, producing random orientation of the molecules on the surface. In the other, Fc chimera was attached via protein G to create a more ordered molecular coating. In most experiments, tosylactivated paramagnetic M-450 Dynabeads (Dynal, Lake Success, NY) were used. The beads were coated with soluble recombinant forms of two members of the IgG superfamily of adhesion molecules: intercellular adhesion molecule-1 (ICAM-1, R&D Systems, Minneapolis, MN), and neural cell adhesion molecule (NCAM, Chemicon International, Temecula, CA). Briefly, 107 beads were washed twice in 1.0 ml of 0.1 M phosphate buffer (pH = 7.4). Then beads were incubated in 1.0 ml of phosphate buffer with 1.2–5.0 μg of the ligand at room temperature overnight. Unreacted tosyl groups were blocked by incubation with 0.25 M ethanolamine (EA) for 30 min at room temperature. Then beads were washed twice with 1% bovine serum albumin (BSA, Calbiochem-Novabiochem Corporation, La Jolla, CA) in phosphate buffered saline (PBS, BioWhittaker, Walkersville, MD) and stored in 0.1% BSA in PBS at 4°C.
For some experiments, protein G beads (Dynal, Lake Success, NY) were used. The beads were coated with soluble recombinant forms of human ICAM-1/Fc chimera (R&D Systems, Minneapolis, MN) or human NCAM-L1/Fc chimera (R&D Systems, Minneapolis, MN). Briefly, 20 μl of the protein G bead suspension was sonicated for 5 min in 1 ml of BlockAid solution (Molecular Probes, Eugene, OR) to reduce nonspecific binding. Then 2.5 μg of ligand was added to the bead suspension and incubated for 40 min at room temperature on a rotating platform. The beads were washed twice in 0.2 M triethanolamine (Sigma, St. Louis, MO), pH = 8.2, and resuspended in 0.5 ml of 0.2 M triethanolamine, containing 20 mM dimethyl pimelimidate (Sigma, St. Louis, MO) to cross-link the chimera to the protein G. After a 30-min incubation at room temperature, the reaction was stopped by adding 0.5 ml of 50 mM Tris (Sigma, St. Louis, MO), pH = 7.5, and the beads were incubated for 15 min with rotational mixing. The cross-linked beads were washed twice in 0.1% BSA, 0.05% Tween 20 (Fisher Scientific, Fair Lawn, NJ) and 0.1% sodium azide in PBS and stored in the same washing buffer at 4°C. Using this protocol the beads were coated with either ICAM-1/Fc chimera, NCAM/Fc chimera, or a mixture of the two.
Flow cytometry
The density of ICAM-1 on ligand-coated beads was measured by flow cytometry. The beads were preincubated at 4°C overnight with FITC-conjugated antibody against human ICAM-1 (BBIG-I1, Ancell, Bayport, MN) or FITC-conjugated isotype control antibody (IgG1). To correlate fluorescence intensity with the number of bound antibodies on the beads, the fluorescence signal was calibrated using Quantum Simply Cellular Beads (Flow Cytometry Standards Corp., Fishers, IN). A suspension of simply cellular beads containing five different populations with known numbers of antibody binding sites was labeled to saturation with the same antibodies used to label the ligand-coated beads. The fluorescence intensity was converted to number of binding sites using software provided by the manufacturer. To correct for nonspecific binding, the number of nonspecific “sites” detected using isotype control antibody was subtracted from the total number of sites detected using the specific antibody.
Micropipette preparation
Micropipettes were made from glass capillary tubing (0.9 mm outside diameter × 0.2 mm wall thickness × 7 cm length; Friedrich & Dimmock Inc., Millville, NJ) using a vertical pipette puller (Model 730; David Kopf Instruments, Tujunga, CA) and a microforge consisting of a micromanipulator and a heated glass bead mounted on an inverted microscope. Pipettes were coated with 1% Surfasil solution (Pierce Chemical Corp., Rockford, IL) in reagent grade chloroform according to the manufacturer's protocol. Before beginning an experiment, pipettes were filled with HBSS without Ca2+ or Mg2+.
Micropipette technique
The experiments were performed on the stage of an inverted microscope. All experiments were performed either at room temperature or at 37°C as indicated. For experiments conducted at 37°C, an environmental box was used to enclose the stage and to keep humidity and temperature constant. Two micropipettes were positioned in a dual entry chamber mounted on the microscope stage. One stationary pipette was used to hold a bead coated with ligand, and another pipette was used to hold the neutrophil and to manipulate the cell (Fig. 1). For micropipette experiments, neutrophils were individually selected based on their polymorphonuclear structure, which was clearly identifiable under light microscopy. The bead and the neutrophil were held in contact for a user-specified length of time, then separated.
FIGURE 1.

Interaction between the bead and the cell during an experiment: (A) cell-bead contact; (B) no adhesion; (C) adhesion event. The drawn line in A indicates the measured projected length of the contact zone Lz. The adhesion event here resulted in the formation of a membrane strand between the cell and the bead. This occurred after some but not all the adhesive contacts.
The contact and separation of each neutrophil-bead pair was recorded on videotape and analyzed subsequently to determine the fraction of contacts resulting in adhesion and to measure the contact area. Adhesion was noted as a deformation of the cell surface as the bead and cell were separated. The adhesion probability was calculated as the total number of adhesive events divided by total number of touches.
Contact area determination
The projected length of the contact zone Lz was measured using a video caliper (Fig. 1 A) and the contact area was calculated from the relationship:
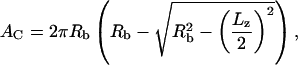 |
(1) |
where Rb is the bead radius and Lz is the contact zone length.
Correction of adhesion probability measurements
The adhesion probability (Padh) was measured and then converted into the expected number of bonds 〈n〉 through relationship (Chesla et al., 1998):
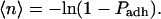 |
(2) |
In theory the number of bonds formed is expected to depend on the density of ligand on the bead ρb, density of the receptors on the cell ρc, the contact area Ac, the time of contact t, and the kinetic constants for ligand-receptor interaction (Chesla et al., 1998):
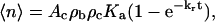 |
(3) |
where Ka is the equilibrium association constant and kr is the reverse rate constant for bond formation. To avoid biasing the data because of differences in contact area between different sets of measurements, the measured adhesion probability was converted to bond number (Eq. 2), and the bond number was corrected to a common area of contact (12 μm2, the mean value for all measurements reported). Then the corrected bond number was converted back to adhesion probability, and this value is reported in the figures that follow. In cases where comparisons were made between adhesion probabilities obtained with different bead preparations, a similar correction was made for the density of ICAM-1 on the beads (ρb). In determining the effective kinetic and equilibrium constants for adhesion, the data were further corrected to account for nonspecific adhesions as follows:
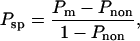 |
(4) |
where Pm is the measured adhesion probability to the ICAM-1 coated bead, Pnon is mean nonspecific adhesion (measured in the presence of calcium), and Psp is the receptor-specific adhesion probability. The specific adhesion probability was then converted to the expected number of bonds and plotted as 〈n〉/Ac (Ac = 12 μm2) as a function of contact duration t.
RESULTS
Basic characterization of magnesium-induced neutrophil adhesion to ICAM-1
Bead characterization
The distribution of intensities for anti-ICAM-1 labeled beads is shown in comparison with those labeled with an isotype control antibody in Fig. 2 A, and the conversion from fluorescence intensity to the number of bound antibodies is illustrated in Fig. 2 B. We calculated the number of sites on the beads to be 26 K/bead (∼360 sites/μm2), 21 K/bead (∼290 sites/μm2), 18 K/bead (∼250 sites/μm2) or 17 K/bead (∼240 sites/μm2) for the preparations used in this study.
FIGURE 2.

Immunofluorescent flow cytometry analysis of ICAM-1 coated beads. (A) The left peak corresponds to the antibody-binding capacity of nonspecific IgG; the right peak corresponds to the binding capacity of antibody against ICAM-1. (B) Calibration curve to convert intensity to the number of bound antibodies. After subtracting the number of nonspecific antibodies bound, the number of antibodies bound specifically to ICAM-1 was calculated to be 26,000 sites/bead for this case.
Magnesium induces neutrophil adhesion to ICAM-1
The coated beads were then tested for their adhesiveness to neutrophils. In the first series of experiments, fifteen bead-cell pairs from each of seven different donors were tested using ICAM-1-coated beads in the presence of Mg2+ plus EGTA or Ca2+, and 24 bead-cell pairs total from three different donors were tested using NCAM-coated beads in the presence of Mg2+. Cells were perturbed as little as possible. A drop of whole blood was dispersed in buffer and cells were selected individually for testing based on their multilobular nuclear structure. Each cell was placed in contact with a bead for one minute at 37° and then separated. For ICAM-1 in the presence of Mg2+, a high number of adhesive contacts was observed, and in the presence of Ca2+, few adhesive contacts were observed (Fig. 3). Beads coated with NCAM were also tested as a control. The probability of adhesion to those beads (in the presence of Mg2+ and EGTA) was significantly lower than adhesion to the ICAM-1 coated beads.
FIGURE 3.

Adhesion probability between neutrophils and beads coated with ICAM-1. Each ICAM-1 column represents data from 105 cell-bead pairs total from seven donors, and the NCAM column represents 24 cell-bead pairs from three donors. Experiments were performed at 37°C, and the duration of the contact between the cell and the bead was 1 min. The number of binding sites for ICAM-1 was 26,000/bead. Error bars indicate standard deviation for all measurements treated as a single population. The shaded area in the graph indicates a subjective assessment of the occurrence of strong adhesion between cell and bead, as indicated by wide attachment sites between the bead and the cell and substantial cell deformation during separation.
Adhesion increases with temperature and contact time
Integrin/ICAM-1 interactions are modulated by both temperature and the duration of contact. Cells from four donors were tested for short (2 s) and long (1 min) contact times at room temperature (RT ≈ 21°C) and 37°C. For 2-s contacts, 10 cell-bead pairs were contacted 25 times each for each donor. For 1-min contacts, each of 10 cell-bead pairs from each donor were contacted once. Cells contacted briefly (2 s) with the ICAM-1-coated beads at RT showed an adhesion probability of 66%, but this probability increased to nearly 100% at increased temperature (37°C) or contact duration (Fig. 4). For long contact times the probability of adhesion was >90% regardless of temperature.
FIGURE 4.

Time and temperature-dependent adhesion probability between ICAM-1 coated beads and neutrophils. Each bar represents data from 10 cell-bead pairs from each of three donors (total of 30 cell-bead pairs). For the 2-s contacts each cell-bead pair was contacted 25 times. The number of binding sites for ICAM-1 was 26,000/bead. Error bars represent standard deviation. The *** denotes a statistically significant difference in adhesion probability (p < 0.001).
Antibody-blocking experiments
Reports in the literature indicate that LFA-1 but not Mac-1 should be activated in the presence of Mg2+ plus EGTA. This is supported by results shown in Fig. 5. Cells were contacted to ICAM-1 coated beads for 2 s, and 25 touches were observed for evidence of adhesion. Approximately four cells from each of four different donors were tested for each case. Data were normalized for a bead surface density of 290 sites/μm2. For these values, “baseline” adhesion probability in the presence of millimolar calcium was ∼14%, but in the presence of Mg2+ plus EGTA, the probability increased to ∼59%. In Mg2+ plus EGTA, the presence of mAb38, which has been shown to bind to LFA-1 and block LFA-1 mediated adhesion (Landis et al., 1994; Dransfield and Hogg, 1989), reduced adhesion to near baseline. Thus the increased adhesion in Mg2+ plus EGTA is largely attributable to LFA-1. Interestingly, addition of antibody CBRM1/5 that specifically blocks activated Mac-1 (Oxvig et al., 1999; Weber et al., 1996; Diamond and Springer, 1993) further reduced the level of adhesion from ∼23% to ∼9.5%, in close agreement with results obtained for the blocking antibody IB4 (Padh ∼ 12%), which binds to the common β2-subunit of LFA-1 and Mac-1 (Landis et al., 1994; Berman et al., 1996; Mesri et al., 1998). Thus, a portion of the baseline adhesion observed in millimolar calcium may be attributable to Mac-1. At longer contact duration (20 s) the effects of exchanging Ca2+ for Mg2+ or of including LFA-1 blocking antibody were similar to what was observed for 2-s contacts. Baseline adhesion in calcium buffer was ∼26%, compared to ∼71% for Mg2+ plus EGTA. Nearly all of the adhesion induced by Mg2+ plus EGTA was eliminated in the presence of LFA-1 blocking antibody (Padh ∼ 32%). Surprisingly, the presence of blocking antibodies that recognized the common β2-subunit or that specifically targeted Mac-1 frequently caused an elevation of adhesion for long contact times (20 s). The corrected adhesion probability for 20-s contacts in Mg2+ plus EGTA in the presence of IB4 or mAb38 plus CBRM1/5 was ∼47%. This increase appeared to be the result of cell activation caused by antibodies binding to Mac-1. The increase was variable from individual to individual, and was often accompanied by morphological signs of cell activation, such as the extension of pseudopodia. These data suggest that antibodies against Mac-1 may tend to sensitize the cell to activation.
FIGURE 5.

Effect of blocking antibodies on neutrophil adhesion to ICAM-1. Experiments were performed in 1.5 mM Ca2+ or 5.0 mM Mg2+plus EGTA. The number of binding sites for ICAM-1 was corrected to 290/μm2 and the contact area was corrected to 12 μm2. Experiments were performed at room temperature, contact duration for all experiments was 2 s. Each cell-bead pair was contacted 25 times. Error bars represent standard deviation. Adhesion probability in Mg2+ was significantly different from the rest of the experiments performed (p < 0.001). Statistical differences (p < 0.05) among the groups are indicated by the brackets.
Calculation of effective rate constants for adhesion
Time dependence of adhesion at room temperature
The time dependence of the probability of adhesion can be used to calculate effective forward and reverse rate constants for bond formation in the interface. To assess the kinetics of adhesive contact formation we measured the adhesion probability for 1-, 2-, 5-, 10-, and 20-s contacts between cells and ICAM-1 coated beads. Adhesion in the presence of 1.5 mM calcium served as a control (Fig. 6 A). Adhesion levels in the presence of calcium were similar to adhesion levels in Mg2+ plus EGTA to NCAM, a molecule structurally related to ICAM-1 that is not known to bind to neutrophils. Experiments were performed at room temperature because the adhesion probability at 37°C approached 100% within 2 s, and the time resolution of our apparatus was not sufficient to clearly resolve a sufficient number of shorter contact durations. Each cell-bead pair was tested at all contact times.
FIGURE 6.

(A) Dependence of adhesion probability on contact time. The experiments were performed as repeated tests of 20 touches for each time point. Each point represents data from four cell-bead pairs from each of four donors: (•) experiments performed in 5.0 mM Mg2+plus EGTA with ICAM-1 coated beads (ρb = 290/μm2); (□) experiments performed in 1.5 mM Ca2+ with ICAM-1 coated beads (ρb = 360/μm2, data corrected to ρb = 290/μm2); (▾) experiments performed in 5.0 mM Mg2+plus EGTA with NCAM coated beads. Error bars represent standard deviation. The horizontal line corresponds to baseline adhesion probability, the average of all data in the presence of 1.5 mM Ca2+. (B) Kinetic model fit to experimental data. The total adhesion probability, as shown in A, was converted to the probability of specific adhesion (nonspecific adhesion was measured in 1.5 mM Ca2+), then converted into the number of bonds 〈n〉 and plotted as 〈n〉/Ac as a function of time. Model used for the fit: y = A(1 − exp(−B x)), where A = 0.076 ± 0.004 μm−2, B = 0.70 ± 0.15 s−1. Error bars represent standard error. Experiments were performed at room temperature.
Before calculating the expected bond number, the adhesion probability shown in the Fig. 6 A was corrected (Eq. 4) for the mean baseline adhesion in the presence of calcium (21%). Then the expected bond number 〈n〉 for specific interactions was calculated via Eq. 2, and is shown as a function of time in Fig. 6 B. The average bond number is related to the molecular properties as given in Eq. 3. This relationship was fit by least squares regression to the data. For ICAM-1-neutrophil interaction kr = 0.70 ± 0.15 s−1 and the product Kaρc was 2.6 ± 0.14 × 10−4 (ρb = 290 sites/μm2).
Ligand surface concentration dependence of adhesion
Another approach that yields an estimate of the kinetic rate constants is to measure the dependence of bond formation rate on ligand density ρb. Increasing the number of binding sites on the bead surface should lead to a linear increase in the expected number of bonds 〈n〉 and a corresponding increase in the adhesion probability for a given contact duration. Experiments in which adhesion probability was measured using different preparations of beads with different site densities confirm this expectation (Fig. 7). Two different types of beads were used for the experiments: tosyl beads coated with either recombinant human ICAM-1 or NCAM, or protein G beads coated with different proportions of recombinant human ICAM-1/Fc chimera and NCAM/Fc chimera. The density of ICAM-1 on the beads was measured by flow cytometry. Surface densities of ICAM-1 ranged from 90 sites/μm2 to 1660 sites/μm2. The expected exponential dependence of adhesion probability on site density was observed, as indicated in Fig. 7 A. The dependence of adhesion probability on receptor density was similar for the two different chemistries used to attach ICAM-1 to the beads, indicating that flow cytometry provided an accurate measure of the number of accessible ICAM-1 adhesion sites on the beads. Anti-β2 blocking antibody reduced adhesion probability to background levels. (Contact duration was 2 s.)
FIGURE 7.

(A) Adhesion probability as a function of number of binding sites per square micron. Experiments were performed at room temperature, contact duration for all experiments was 2 s. Error bars represent standard deviation. Adhesion was blocked in the presence of β2-integrin blocking Ab IB4 (▾). (B) The expected number of bonds as a function of number of binding sites per square micron. The data for densities less than 400/μm2 from A were converted into 〈n〉 using Eq. 2. The line shows a linear regression to the data. (○) data for tosyl-activated beads; (▪) data for protein G beads (see Methods). The intercept A = 0.03 ± 0.097 and the slope B = 0.002 ± 0.0004 μm2.
To estimate Kaρc, the adhesion probability was converted to the expected number of bonds and plotted as a function of receptor density on the bead surface (Fig. 7 B). We restricted the fit to site densities below 400/μm2, because at higher densities the high adhesion probability and the logarithmic relationship between adhesion probability and bond number made the determination of 〈n〉 unreliable. If the reverse rate constant is known, the dependence of 〈n〉 on ρb can be used to obtain an independent estimate of Kaρc via Eq. 3. For these measurements, the mean contact area was 12 μm2, the contact duration was 2 s, and taking kr = 0.70 s−1, we obtain a value of Kaρc = 2.2 × 10−4 for neutrophil-ICAM-1 interactions, in reasonable agreement with the value obtained using measurements of adhesion probability at different contact durations.
DISCUSSION
Interaction of leukocytes with endothelial surfaces is a dynamic process, and therefore the time dependence of adhesive contact formation, and mechanisms that affect these dynamics, are important physiologically. It is well established that a key step for firm attachment of neutrophils to endothelium is a change in the affinity of β2-integrins and bond formation with their principal counterreceptor on the endothelial surface, ICAM-1. A number of studies have characterized the kinetics of LFA-1–ICAM-1 interactions using systems in which one of the ligands is immobilized to a surface, and the other is in solution. Tominaga and colleagues (1998) and Lupher and colleagues (2001) used surface plasmon resonance to measure kinetic rate constants for genetically engineered LFA-1 constructs binding to immobilized ICAM-1. Both studies gave values for a forward rate constant kf = 2 × 105 M−1s−1 and a reverse rate constant kr = 0.1 s−1. Others have examined the binding of soluble forms of ICAM-1 with immobilized LFA-1 (Woska et al., 1996; Labadia et al., 1998) and both studies gave a value for Kd of 1.3 × 10−7 M (kf = 2.2 × 105 M−1s−1 and kr = 0.03 s−1). Although there is some discrepancy in the off-rate depending on which molecule is immobilized on the surface, these studies show a consistent behavior for the LFA-1–ICAM-1 interaction kinetics when one of the molecules is free in the solution.
In the case of cell adhesion, the adhesion molecules are confined to an irregularly shaped membrane and, in addition, are likely to have nonuniform distribution and restricted lateral mobility (Abitoraby et al., 1997; Erlandsen et al., 1993). Thus, kinetic constants measured when one of the molecular partners is in solution provide only part of what is needed to understand the dynamics and regulation of the adhesiveness of the cell for substrate. The different factors that contribute to the substantial differences between rates of reaction between molecules in solution and rates of reaction between molecules confined to surfaces are summarized in excellent reviews by Dustin et al. (2001) and Pierres et al. (1995). Even when the same molecules are confined to opposing surfaces, reaction rates can be markedly different depending on the topography, alignment, and molecular diffusivities of the two surfaces (Williams et al., 2001). For example, equilibrium dissociation constants measured using fluorescent techniques to detect the formation of bonds between a cell and molecules confined to a planar bilayer are orders of magnitude different from dissociation constants measured by mechanical methods like the one used in the present study, where the molecules are confined to the surface of another cell or bead (Dustin et al., 2001). Recognition of this complexity provided motivation for the present study in which the effective kinetics of bond formation were measured under conditions in which one molecular partner is positioned in situ, and the other is confined to a surface.
An important prerequisite for β2-integrins to bind to ICAM-1 is a change in conformation leading to an increase in affinity for the endothelial ligand (Diamond and Springer, 1993; Stewart et al., 1998; Leitinger et al., 2000). This change in affinity is induced in vivo by “inside-out” signaling mechanisms resulting from chemical or mechanical stimulation of the cells in the vasculature. It can be effected in vitro by changing the divalent ion composition in the extracellular environment. Dransfield and colleagues (1992) demonstrated that either manganese or magnesium (in the presence of EGTA) induces the high affinity form of LFA-1, one of the two principle integrins found on the neutrophil surface. Our original goal in using magnesium in the present study, rather than other more physiological activators of neutrophils, was to study the dynamics of integrin/ICAM-1 interactions without complications that typically result from chemical stimulation of the cell, such as cytoskeletal reorganization, changes in cell shape, and changes in adhesion molecule expression on the cell surface. Except as noted for the case of Mac-1 blocking antibodies, neutrophils suspended in the magnesium solutions used in the present study showed no evidence of membrane ruffling or other changes in cell morphology that are characteristic of cytoskeletal reorganization that accompanies cell activation. Prior studies in our laboratory (Spillmann et al., 2002) have shown that the presence of magnesium also has no effect on L-selectin or β2-integrin expression on the cell surface. (Important early signs of neutrophil activation are an upregulation of expression of the β2-integrin Mac-1, and downregulation of the surface expression of L-selectin (Borregaard et al., 1994; Anderson et al., 2000; Taylor et al., 1996)). Thus, the effects on neutrophils of suspension in Mg2+-containing buffers, other than an increased integrin affinity, are minimal. Yet the increased integrin affinity caused by Mg2+ is sufficient to have measurable effects on cell adhesion. In addition to the evidence presented in this report, we demonstrated in a prior study, that magnesium in the absence of calcium increases homotypic adhesion between neutrophils. Furthermore, this increase was inhibitable by calcium or by blocking β2-integrin-mediated adhesion using Fab fragments of anti-β2 antibodies (Spillmann et al., 2002).
An additional consequence of using magnesium to induce integrin high affinity is that we can attribute the increased adhesion we observe to the activity of LFA-1, not Mac-1. The original studies demonstrating an effect of Mg2+ or Mn2+ on the β2-integrins were performed on lymphocytes, where LFA-1 is the predominant form (Dransfield et al., 1992). Interestingly, other investigators have shown that manganese, but not magnesium, will induce the high affinity form of Mac-1 (Altieri, 1991; Diamond and Springer, 1993). Using the commercially available antibody CBRM1/5, which recognizes the activated conformation of Mac-1, we also found little or no change in CBRM1/5 binding in the presence or absence of Mg2+ (E. B. Lomakina, unpublished results). Furthermore, we demonstrate in the present study that blocking antibody against LFA-1 alone eliminates essentially all of the increase in neutrophil adhesion induced by magnesium plus EGTA. Reports in the literature reveal that the relative importance of Mac-1–ICAM-1 binding in neutrophil-endothelial attachment is unclear. An early report by Lo and colleagues (1989) demonstrated that neutrophil interactions with endothelium were mediated by both LFA-1 and Mac-1, but that only LFA-1 acted as the counterreceptor for ICAM-1. Subsequent studies from the Springer laboratory (Diamond and Springer, 1991) provided convincing evidence that ICAM-1 is in fact a ligand for Mac-1, but also that only a small proportion of Mac-1 molecules bind to ICAM-1, even when cells were activated and CBMR1/5 binding was clearly evident (Diamond and Springer, 1993).
This has implications for estimating the effective forward rate constant for β2-integrin-mediated adhesion because the surface concentration of adhesion molecules on the cell surface is needed to calculate the effective equilibrium association constant Ka. The total number of Mac-1 (CD11b) on the resting neutrophil is ∼45,000/cell, and the number of LFA-1 molecules (CD11a) is ∼24,000 (Bikoue et al., 1996). The mean spherical area of a neutrophil is ∼240 μm2, but the surface area of the membrane bilayer is approximately two times that (Ting-Beall et al., 1993). Thus the density of LFA-1 on the surface is ∼50/μm2. Using this value and the value of 2.4 × 10−4 for Kaρc, we obtain an effective equilibrium association constant Ka of 4.8 × 10−6 μm2 and a forward rate constant kf of 3.4 × 10−6 μm2/s.
Measurement error
There were two potentially important sources of measurement error in these experiments. One relates to the reliance on visual observation of the contacting surfaces during separation to detect adhesion. Adhesive events that are too weak or too short-lived to produce an observable deflection of the membrane would not be counted. This is an inherent limitation of our method. Although it is difficult to estimate what cannot be observed, indications are that we are not missing significant numbers of adhesive events. In reviewing the video recordings of the separations, we noted that there were 1–2% of contacts where there was a “questionable” adhesion event. These accounted for a very small fraction of the total adhesion events observed.
A second potentially important source of error involved measurements of the macroscopic contact area. One potential error is due to the limitations of optical microscopy in locating the true edges of the cell and bead in the diffraction band of the contact zone. The accuracy of edge location was improved by observing the deformation of the cell as it contacted the bead and determining the point within the diffraction band where contact occurred. Thus, we believe that these errors were minimal and random. A more serious concern results from the possibility that the cell and the bead were not perfectly aligned on axis. In this case, the contact area may be overestimated because the images of the cell and bead overlap before actual contact is made. In a separate study, we have estimated that such errors could result in a systematic overestimation of the average area of contact of ∼3.0 μm2. Importantly, all of the cases tested should be affected similarly in magnitude and in the same direction, minimizing the effect of such errors on calculated parameters. To estimate the potential effect these errors could have on the calculation of the effective kinetic coefficients, we applied a uniform decrease of 3.0 μm2 to all contact areas and recalculated the coefficients. The result was less than a 20% increase in kf and a 5.5% increase in kr.
Comparison with other measurements
The values we obtain for off and on rates for neutrophil–ICAM-1 interactions are similar to off and on rates measured for other molecular pairs by mechanical approaches. For example, a recent report by Long et al. (2001) gives rate constants for associations between E selectin immobilized onto red blood cell surfaces and unknown carbohydrate ligands presented by HL-60 cells or a colon adenocarcinoma cell line. Off rates were measured in the range of 0.5–1.0 s−1. Forward rate constants determined in that study are not directly comparable to ours, because they are expressed as the product of the rate constant and the unknown surface density of counterreceptor. Chesla and colleagues (1998) report off-rates on the order of 0.4 s−1 and effective forward rates of 0.8–1.7 × 10−6 μm2/s for IgG interacting with Fc receptor. In calculating this last value, as well as the values we report here for LFA-1–ICAM-1 interactions, the macroscopic contact area was taken as the area where molecules can interact to form bonds, and the concentration of receptor was taken as the mean surface concentration for molecules over the entire cell surface. We recognize that the true reaction space, that is, the area where the two surfaces are in molecularly close contact, may be substantially smaller than the macroscopic contact area. For example, Williams et al. (2001) estimated that for ruffled cell surfaces, the portion of the membrane in close contact with substrate may be as little as 3% of the macroscopic contact area. We also recognize that nonuniform distribution of molecules over the surface could result in surface concentrations within these regions of close contact that are substantially different from the mean concentration for the whole cell. With this in mind, we have been careful to refer to these coefficients as “effective” forward and reverse rate constants because they not only reflect contributions from intrinsic rates of reaction but also the effects of surface topography and lateral distribution of molecules. Given these complications, and the fact that we are dealing with different, structurally unrelated molecules, the relative agreement between our values and those reported by Chesla et al. (1998) is both remarkable and interesting. It suggests the possibility that for these two cases, the kinetics of adhesion between cells and substrate may be dominated by extrinsic factors, such as surface topography and receptor mobility, and relatively insensitive to molecular kinetics per se.
Blocking antibodies and the choice of control conditions
The main focus of this work is the effect on cell adhesion of upregulation of integrin affinity. Our choice to use magnesium plus EGTA to effect this upregulation enables us to attribute the observed increases as contributions specific to the integrin LFA-1. This attribution is based on reports in the literature that LFA-1 (Dransfield et al, 1992; Labadia et al., 1998) but not Mac-1 (Altieri, 1991; Diamond and Springer, 1993) undergoes a marked increase in affinity for ICAM-1 under these conditions. It is further supported by our own observations that the levels of adhesion measured in the presence of millimolar calcium are similar to levels of adhesion observed in magnesium plus EGTA when LFA-1 binding is blocked by a monoclonal antibody. Therefore, we have chosen the calcium data as our baseline against which magnesium/EGTA-induced adhesion is measured. Interestingly, for contact times of 2 s, this baseline can be further reduced when binding via Mac-1 is also inhibited, either by a blocking antibody against the common β2-subunit or by antibodies specific for the Mac-1 α-chain. This suggests that at least a portion of the background adhesion we observe is attributable to Mac-1, a finding consistent with early reports that Mac-1 makes minor contributions to cell adhesion to ICAM-1 (Lo et al, 1989; Diamond and Springer, 1993).
An unexpected and somewhat mysterious finding of the present study was the elevation of adhesion probability for long contact times in the presence of antibodies that recognize Mac-1. It appears that such antibodies cause a gradual activation of the cells that is manifested as a tendency for the cells to produce pseudopodia and an increase in adhesion to ICAM-1-coated beads. The effect is mysterious, first, because it is highly variable from donor to donor, and second, because it remains unclear what might be mediating the adhesion, given that it is occurring in the presence of β2-blocking antibodies. This phenomenon awaits further study.
There is a slight increase in the baseline adhesion when the contact time is increased from 2 to 20 s. In the calculation of the effective kinetic coefficients, the baseline was assumed to be constant. If instead, a gradual increase in the baseline is assumed, slightly different values for the effective kinetic coefficients are obtained: for ICAM-1–neutrophil interaction kr = 1.1 ± 0.2 s−1 and the product Kaρc was 2.6 ± 0.1 × 10−4 (ρb = 290 sites/μm2). It seems likely, given the low levels of adhesion to NCAM-coated beads at long times, that this gradual increase in baseline might be due to a gradual increase in Mac-1 binding. Unfortunately, the anomalous effects of anti-Mac-1 antibodies at long times, make this possibility difficult to test.
Density and accessibility of ICAM-1 on the bead surface
The range of molecular densities on the beads used in this study (90–1660 per μm2) is within the range of ligand densities on endothelium that have been reported. Dustin and Springer (1988) reported 5 × 106 sites/cell on recombinant TNF-α-stimulated endothelium from human umbilical cord (HUVEC). A “typical” endothelial cell has a luminal surface area of ∼800 μm2 (Barbee et al., 1993), giving a mean concentration on activated endothelium of ∼6,000 molecules/μm2. Hentzen and colleagues (2000) reported 2.3 × 105 sites per cell on resting human umbilical vein endothelial cells in culture, corresponding to ∼290 sites/μm2, and 8.5 × 105 sites per cell after 4-h stimulation with IL-1, or ∼1000 sites/μm2. A number of groups have reported heterogeneity in adhesion molecule expression both in vivo and in vitro (Iigo et al., 1997; Panes et al., 1995) suggesting that for individual cells in the endothelium, the density may be significantly higher or lower than these mean values. Thus, over the physiological range of ICAM-1 densities, the probability of adhesion mediated by activated β2-integrin can vary significantly.
A concern that is often raised when dealing with immobilized ligand on an artificial surface is whether or not the molecules are properly oriented to allow reaction to occur. To address this concern we used two different methods for immobilizing ICAM-1 on beads. The tosylactivated beads form covalent bonds with any available primary amino or sulfhydryl groups. Therefore, the population of molecules on the surface is expected to be randomly oriented, some with the binding site bonded near the surface, and some with the binding site freely mobile. In the case of protein G coated beads, only the Fc portion of the Fc-ICAM-1 chimera binds to the bead surface, such that all of the binding sites on these molecules should be free from the bead surface and capable of sampling orientations suitable for ligand binding. Our finding that the probability of adhesion for these two types of beads is similar when the density of ligand is the same indicates that molecular orientation is not a significant issue in the present study. This result is somewhat surprising, as one would expect the accessibility of the ligand binding site should be important in determining rates of reaction. There are several possible explanations for the result. One is that even though molecules may not be oriented perfectly, rates of molecular motion are fast compared to other constraints on the rate of reaction, such that reorientation of the binding site is not rate limiting. A second consideration is that the tosyl-bound ligand that is recognized immunologically in flow cytometry may correspond only to that portion of the molecules on the bead surface that are properly folded and oriented to allow reaction with receptor. Anecdotally, we did find that allowing the beads to “cure” for 24–48 h after coating typically resulted in an increase in the immunologically detectable number of sites on the bead surface, suggesting that there may be a gradual refolding of molecules that may have denatured upon initial interaction with the bead surface.
CONCLUSIONS
The effective kinetic coefficients that we have determined here reflect contributions from multiple mechanisms operating at molecular and microscopic scales. Thus, although one important contributor to the effective rate is the molecular rate constants for bond formation, the coefficients we determine here are not readily comparable to coefficients obtained for molecular interactions. The formation of an adhesive contact at the cell-bead interface may depend on a number of additional factors, including surface topography in the contact zone, and the lateral distribution and mobility of adhesive molecules in the cell membrane. By determining forward rates for adhesion in the native membrane, we have encompassed all of these effects in our reported values. Thus, the coefficients we report may be more directly applied to predict cell-substrate interactions. The disadvantage of our approach is that the reported values are situation specific: namely, the rate constant under conditions where integrin activation has been induced by magnesium plus EGTA. Nevertheless, by defining the conditions under which the interactions are measured we lay the groundwork for understanding how different factors and different activation mechanisms can lead to different phenomenological behavior.
Acknowledgments
The authors thank Richard Bauserman and Donna Brooks for technical assistance; Joanne Schultz for assistance with flow cytometry; and Christopher Spillmann for scientific discussions. This work was supported by the U.S. Public Health Service, National Institutes of Health, grant No. HL18208.
References
- Abitoraby, M. A., R. K. Pachynski, R. E. Ferrando, M. Tidswell, and D. J. Erle. 1997. Presentation of integrins on leukocyte microvilli: a role for the extracellular domain in determining membrane localization. J. Cell Biol. 139:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri, D. C. 1991. Occupancy of CD11b/CD18 (Mac-1) divalent ion binding site(s) induces leukocyte adhesion. J. Immunol. 147:1891–1898. [PubMed] [Google Scholar]
- Anderson, S. I., N. A. Hotchin, and G. B. Nash. 2000. Role of the cytoskeleton in rapid activation of CD11b/CD18 function and its subsequent down regulation in neutrophils. J. Cell Sci. 113:2737–2745. [DOI] [PubMed] [Google Scholar]
- Barbee, K. A., P. F. Davies, and R. Lal. 1993. Shear stress-induced reorganization of the surface topography of living endothelial cells imaged by atomic force microscopy. Circ. Res. 74:163–171. [DOI] [PubMed] [Google Scholar]
- Bazzoni, G., L. Ma, M.-L. Blue, and M. E. Hemler. 1998. Divalent cations and ligands induce conformational changes that are highly divergent among beta1 integrins. J. Biol. Chem. 273:6670–6678. [DOI] [PubMed] [Google Scholar]
- Berman, M. E., Y. Xie, and W. A. Muller. 1996. Roles of platelet/endothelial cell adhesion molecule-1 (PECAM-1, CD31) in natural killer cell transendothelial migration and beta 2 integrin activation. J. Immunol. 156:1515–1524. [PubMed] [Google Scholar]
- Bikoue, A., F. George, P. Poncelet, M. Mutin, G. Janossy, and J. Sampol. 1996. Quantitative analysis of leukocyte membrane antigen expression: normal adult values. Cytometry. 26:137–147. [DOI] [PubMed] [Google Scholar]
- Borregaard, N., L. Kjeldsen, H. Sengelov, M. S. Diamond, T. A. Springer, H. C. Anderson, T. K. Kishimoto, and D. F. Bainton. 1994. Changes in subcellular localization and surface expression of L-selectin, alkaline phosphatase, and Mac-1 in human neutrophils during stimulation with inflammatory mediators. J. Leuk. Biol. 56:80–87. [DOI] [PubMed] [Google Scholar]
- Calderwood, D. A., S. J. Shattil, and M. H. Ginsberg. 2000. Integrins and actin filaments: reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 275:22607–22610. [DOI] [PubMed] [Google Scholar]
- Chesla, S. E., P. Selvaraj, and C. Zhu. 1998. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 75:1553–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond, M. S., and T. A. Springer. 1991. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 65:961–971. [DOI] [PubMed] [Google Scholar]
- Diamond, M. S., and T. A. Springer. 1993. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 120:545–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dransfield, I., and N. Hogg. 1989. Regulated expression of Mg2+ binding epitope on leukocyte integrin alpha subunits. EMBO. 8:3759–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dransfield, I., C. Cabanas, A. Craig, and N. Hogg. 1992. Divalent cation regulation of the function of the leukocyte integrin LFA-1. J. Cell Biol. 116:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin, M. L., and T. A. Springer. 1988. Lymphocyte function-associated antigen-1 (LFA-1) interaction with intercellular adhesion molecule-1 (ICAM-1) is one of at least three mechanisms for lymphocyte adhesion to cultured endothelial cells. J. Cell Biol. 107:321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin, M. L., S. K. Bromley, M. M. Davis, and C. Zhu. 2001. Identification of self through two-dimensional chemistry and synapses. Annu. Rev. Cell Dev. Biol. 17:133–157. [DOI] [PubMed] [Google Scholar]
- Erlandsen, S. L., S. R. Hasslen, and R. D. Nelson. 1993. Detection and spatial distribution of the β2 integrin (Mac-1) and L-selectin (LECAM-1) adherence receptors on human neutrophils by high-resolution field emission SEM. J. Histochem. Cytochem. 41:327–333. [DOI] [PubMed] [Google Scholar]
- Hentzen, E. R., S. Neelamegham, G. S. Kansas, J. A. Benanti, L. V. McIntire, L. C. W. Smith, and S. I. Simon. 2000. Sequential binding of CD11a/CD18 and CD11b/CD18 defines neutrophil capture and stable adhesion to intercellular adhesion molecule-1. Blood. 95:911–920. [PubMed] [Google Scholar]
- Iigo, Y., M. Suematsu, T. Higashida, J. Oheda, K. Matsumoto, Y. Wakabayashi, Y. Ishimura, M. Miyasaka, and T. Takashi. 1997. Constitutive expression of ICAM-1 in rat microvascular systems analyzed by laser confocal microscopy. Am. J. Physiol. 273: H138–147. [DOI] [PubMed] [Google Scholar]
- Labadia, M. E., D. D. Jeanfavre, G. O. Caviness, and M. M. Morelock. 1998. Molecular regulation of the interaction between leukocyte function-associated antigen-1 and soluble ICAM-1 by divalent metal cations. J. Immunol. 161:836–842. [PubMed] [Google Scholar]
- Landis, R. C., A. McDowall, C. L. Holness, A. J. Littler, D. L. Simmons, and N. Hogg. 1994. Involvement of the “I” domain of LFA-1 in selective binding to ligands ICAM-1 and ICAM-3. J. Cell Biol. 126:529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitinger, B., A. McDowall, P. Stanley, and N. Hogg. 2000. The regulation of integrin function by Ca2+. Biochim. Biophys. Acta. 1498:91–98. [DOI] [PubMed] [Google Scholar]
- Lo, S. K., G. A. Van Seventer, S. M. Levin, and S. D. Wright. 1989. Two leukocyte receptors (CD11a/CD18 and CD11b/CD18) mediate transient adhesion to endothelium by binding to different ligands. J. Immunol. 143:3325–3329. [PubMed] [Google Scholar]
- Long, M., H. Zhao, K. S. Huang, and C. Zhu. 2001. Kinetic measurements of cell surface E-selectin/carbohydrate ligand interactions. Ann. Biomed. Eng. 29:935–946. [DOI] [PubMed] [Google Scholar]
- Lub, M., S. J. van Vliet, S. P. Oomen, R. A. Pieters, M. Robinson, C. G. Figdor, and Y. van Kooyk. 1997. Cytoplasmic tails of beta 1, beta 2, and beta 7 integrins differentially regulate LFA-1 function in K562 cells. Mol. Biol. Cell. 8:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupher, M. L., E. A. S. Harris, C. R. Beals, L. Sui, R. C. Liddington, and D. E. Staunton. 2001. Cellular activation of leukocyte function-associated antigen-1 and its affinity are regulated at the I domain allosteric site. J. Immunol. 167:1431–1439. [DOI] [PubMed] [Google Scholar]
- Masumoto, A., and M. E. Hemler. 1993. Mutation of putative divalent cation sites in the alpha 4 subunit of the integrin VLA-4: distinct effects on adhesion to CS1/fibronectin, VCAM-1, and invasin. J. Cell Biol. 123:245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowall, A., B. Leitinger, P. Stanley, P. A. Bates, A. M. Randi, and N. Hogg. 1998. The I domain of integrin leukocyte function-associated antigen-1 is involved in a conformational change leading to high affinity binding to ligand intercellular adhesion molecule 1 (ICAM-1). J. Biol. Chem. 273:27396–27403. [DOI] [PubMed] [Google Scholar]
- Mesri, M., J. Plescia, and D. C. Altieri. 1998. Dual regulation of ligand binding by CD11b I domain. Inhibition of intercellular adhesion and monocyte procoagulant activity by a factor X-derived peptide. J. Biol. Chem. 273:744–748. [DOI] [PubMed] [Google Scholar]
- Oxvig, C., C. Lu, and T. A. Springer. 1999. Conformational changes in tertiary structure near the ligand binding site of an integrin I domain. Proc. Natl. Acad. Sci. USA. 96:2215–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panes, J., M. A. Perry, D. C. Anderson, A. Manning, B. Leone, G. Cepinskas, C. L. Rosenbloom, M. Miyasaka, P. R. Kvietys, and D. N. Granger. 1995. Regional differences in constitutive and induced ICAM-1 expression in vivo. Am. J. Physiol. 269:H1955–1964. [DOI] [PubMed] [Google Scholar]
- Pierres, A., A.-M. Benoliel, and P. Bongrand. 1995. Measuring the lifetime of bonds made between surface-linked molecules. J. Biol. Chem. 270:26586–26592. [DOI] [PubMed] [Google Scholar]
- Sampath, R., P. J. Gallaher, and F. M. Pavalko. 1998. Cytoskeletal interactions with the leukocyte integrin beta2 cytoplasmic tail. Activation-dependent regulation of associations with talin and alpha-actinin. J. Biol. Chem. 273:33588–33594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillmann, C., D. Osorio, and R. E. Waugh. 2002. Integrin activation by divalent ions affects neutrophil homotypic adhesion. Ann. Biomed. Eng. 30:1002–1011. [DOI] [PubMed] [Google Scholar]
- Springer, T. A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- Stewart, M. P., A. McDowall, and N. Hogg. 1998. LFA-1-mediated adhesion is regulated by cytoskeletal restraint and by a Ca2+-dependent protease, calpain. J. Cell Biol. 140:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, A. D., S. Neelamegham, J. D. Hellums, C. W. Smith, and S. I. Simon. 1996. Molecular dynamics of the transition from L-selectin- to beta 2-integrin-dependent neutrophil adhesion under defined hydrodynamic shear. Biophys. J. 71:3488–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting-Beall, H. P., D. Needham, and R. M. Hochmuth. 1993. Volume and osmotic properties of human neutrophils. Blood. 81:2774–2780. [PubMed] [Google Scholar]
- Tominaga, Y., Y. Kita, A. Satoh, S. Asai, K. Kato, K. Ishikawa, T. Horiuchi, and T. Takashi. 1998. Affinity and kinetic analysis of the molecular interaction of ICAM-1 and leukocyte function-associated antigen-1. J. Immunol. 161:4016–4022. [PubMed] [Google Scholar]
- Valmu, L., T. J. Hilden, G. van Willigen, and C. G. Gahmberg. 1999. Characterization of beta2 (CD18) integrin phosphorylation in phorbol ester-activated T lymphocytes. Biochem. J. 339:119–125. [PMC free article] [PubMed] [Google Scholar]
- Weber, C., J. Kitayama, and T. A. Springer. 1996. Differential regulation of beta 1 and beta 2 integrin avidity by chemoattractants in eosinophils. Proc. Natl. Acad. Sci. USA. 93:10939–10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, T. E., S. Nagarajan, P. Selvaraj, and C. Zhu. 2001. Quantifying the impact of membrane microtopology on effective two-dimensional affinity. J. Biol. Chem. 276:13283–13288. [DOI] [PubMed] [Google Scholar]
- Woska, J. R., Jr., M. M. Morelock, D. D. Jeanfavre, and B. J. Bormann. 1996. Characterization of molecular interactions between intercellular adhesion molecule-1 and leukocyte function-associated antigen-1. J. Immunol. 156:4680–4685. [PubMed] [Google Scholar]