

Abstract
To understand ion permeation, one must assign correct ionization states to titratable amino acid residues in protein channels. We report on the effects of physical and methodological assumptions in calculating the protonation states at neutral bulk pH of titratable residues lining the lumen of the native Escherichia coli OmpF channel, and five mutants. We systematically considered a wide range of assumed protein dielectric constants and all plausible combinations of protonation states for electrostatically interacting side chains, and three different levels of accounting for solute shielding: 1), full nonlinear Poisson-Boltzmann; 2), linearized Poisson-Boltzmann; and 3), neglect of solute shielding. For this system we found it acceptable to neglect solute shielding, a result we postulate to be generalizable to narrow lumens of other protein channels. For the large majority of residues, the protonation state at neutral bulk pH was found to be independent of the assumed dielectric constant of the protein, and unambiguously determined by the calculation; for native OmpF only Asp-127 has a protonation state that is sensitive to the assumed protein dielectric constant. Our results are significant for understanding two published experimental observations: the structure of the narrow part of the channel, and the ionic selectivity of OmpF mutants.
INTRODUCTION
Porins, located in the outer membrane of Gram-negative bacteria (e.g., Escherichia coli), are channel proteins that facilitate uptake of nutrients (Jap and Walian, 1990; Nikaido, 1993). These proteins allow diffusion of ions and even large molecules of the size of several hundred daltons. OmpF, which belongs to this family of proteins, is slightly cation selective and has also been found to be voltage and pH sensitive. Under normal conditions, OmpF folds into homotrimeric structures (for reviews, refer to Schulz (1996), Delcour (1997), and Schirmer (1998)). Three-dimensional structures of several porins, including OmpF (Cowan et al., 1992; Weiss and Schulz, 1992), have been determined to high resolutions by x-ray crystallography. The crystal structure of OmpF (from E. coli) indicates that each monomer forms a rigid 16-stranded β-barrel with eight turns at the periplasmic side and eight loops (L1–L8) that narrow the structure at the cell surface and channel interior. Loop L3 in particular (Fig. 1), folds inside the barrel and creates a narrow region (diameter ∼6 Å) halfway through the membrane. This “constriction zone” comprises a noticeably large cluster of interacting titratable residues and has been thought to be responsible for the channel's charge specificity (Cowan et al., 1992). Loop L2, which connects one monomer to its neighbor, also comprises ionizable residues that form strong ionic networks with residues of the constriction zone of its neighbor. Other than the residues in the constriction zone, these barrels are also lined with several other titratable residues facing the permeation pathway. The titration states of these residues should thus be expected to play a significant role in modulating ion permeation through the channel.
FIGURE 1.

Partial view of the x-ray structure of OmpF-trimer highlighting titratable residues in the constriction zone. Several loops have been truncated for clarity. Our calculations described in text predict residues colored in red to be positively charged at neutral pH and residues colored in green to be negatively charged at neutral pH. E296 colored in orange is expected to be protonated. Charge assignment of D127 was found to be sensitive to the value of dielectric constant assumed for the protein. Other assignments are not sensitive to assumption of dielectric constant. This figure was created with RasMol Version 2.6.
Earlier in 1994, Karshikoff et . al. (1994) reported a calculation of the ionization states of all the 100 titratable residues of OmpF. Their calculations indicated several titratable residues in the constriction zone to have uncommon ionization states. In 1998, Tieleman and Berendsen (1998) used the set of ionization states published by Karshikoff et al. (1994) to generate a 1-ns molecular dynamics (MD) simulation of the trimer embedded in an explicit phospholipid bilayer. They reported significant deviations of the average dynamical structure relative to the crystallographic structure. Especially, loop 3 was found to move into the permeation pathway of the channel decreasing the pore radius at the constriction zone by a few angstroms. More recently, Im and Roux (2002b) used a different set of ionization states (setting only residues Glu-296 and Asp-312 to be uncharged and using default ionization states for all other titratable residues) and generated a 5-ns MD simulation of the trimer embedded in an explicit dimyristoylphosphatidylcholine (DMPC) membrane with explicit solvent. They too reported a significant narrowing of the constriction zone. A similar set of deviations, upon setting both Glu-296 and Asp-312 to be charged, were also observed in MD simulations conducted in our laboratory (Van Der Straaten et al., 2002). This failure of molecular dynamics simulations using a variety of charge assignments to replicate the crystal structure has motivated us to perform the present study in which we reexamine the physical assumptions and computational protocols underlying the charge assignments.
One of the major issues in computing ionization states is assuming a dielectric constant for the protein. Incremental additions from a value of 1 (the value for vacuum) occur due to electronic polarization, atomic polarization, and dipolar rotation. A reasonable increment for electronic polarization is 1.5–2.5 (CRC Handbook of Chemistry and Physics). A reasonable increment for atomic polarization is 0.05–0.3 (Bottcher, 1973). Calculations on folded proteins (Gilson and Honig, 1986; Simonson et al., 1991; Smith et al., 1993), mostly α-helical, show that the contribution due to dipole rotation has an average value between 2 and 3. Based on these results alone, the bulk dielectric constant of protein would be considered to lie in the range 4.55–6.8 (also supported by experiments on dry proteins (Takashima and Schwan, 1965). However, there is a large literature on what assumption of dielectric constant in the protein gives rise to the best results for pK and other electrostatic calculations on proteins. Depending on the context, it is possible to make cogent arguments based on agreement with experimental results for assuming dielectric constants anywhere from 4 to 20 (Warshel et al., 1984; Sternberg et al., 1987; King et al., 1991; Simonson et al., 1991, 1998; Pethig, 1992; Oberoi and Allewell, 1993; Smith et al., 1993; Yang et al., 1993; Antosiewicz et al., 1996b; Demchuk and Wade, 1996; Gibas and Subramaniam, 1996; Sham et al., 1997, 1998; Schutz and Warshel, 2001).
Presumably the underlying physical reason this issue has not been resolved is that the dielectric properties of the protein are not well described by any single dielectric constant, but rather require a spatially variant dielectric tensor for a correct description. Demchuk and Wade (1996) have analyzed the effect of dielectric constant on pKa calculations and have demonstrated that continuum electrostatics calculations can reproduce pKa for almost all ionizable residues provided a site-specific dielectric tensor is known a priori. In the absence of having such a tensor, in this paper we will repeat our calculations for a wide range of dielectric constants, to identify which features of the protein protonation are likely to be sensitive to approximations in the description of the protein dielectric.
In addition to the choice of dielectric constant, another methodological issue is how to calculate shielding due to ions in the electrolyte. In considering electrodiffusion-limited associations at the surfaces of proteins, it has been established that it is important to take full account of the shielding by solving the nonlinear Poisson-Boltzmann equation (Kozack et al., 1995). It is less clear what the shielding terms mean when applied to the narrow lumen of an ion channel (as we do in this paper), where the dimensions of the cavity are of the same order as the Debye-Huckel shielding distance. To ascertain the importance of this issue, computations were done in three ways: 1), with the full nonlinear Poisson-Boltzmann equation at physiological concentration; 2), with the linearized Poisson-Boltzmann equation at physiological concentration; and 3), with a very small concentration (1 mM), so that the shielding terms were essentially ignored. Another significant methodological choice is whether to use a single-charge model, in which the protonic charge is added at one site, or to use a model in which the charge is spread over the entire group. In a comparative study on this issue (Antosiewicz et al., 1996a), pK values as calculated by the single-charge and full-charge model were compared for four different proteins. Of the 52 different titratable residues for which experimental pK values were available, the single-charge model gave better results for 17, the full-charge model for 19, and the other 16 showed no significant difference (less than or equal to 0.1 pH units). The charge distribution in the full-charge model is calculated based on the assumption that there are no other charged residues in the near vicinity, and is thus expected to be valid in this circumstance. In the lumen of the porin channel, on the other hand, there are many titratable residues in close proximity, with mutual electrostatic interactions enhanced even further than normal by being surrounded by low dielectric protein. Because the prior results of Antosiewicz et al. (1996a) provide no compelling reason to prefer the full-charge model, and the specific situation in the lumen does not fulfill the assumptions underlying the full-charge model, and because the single-charge model is much more computationally efficient to implement, we have chosen to use the single-charge model.
There is a particular issue with respect to Asp and Glu, each of which potentially has two protonation sites. To efficiently deal with all the possibilities, in this paper we will first identify all those residues whose ionization states at physiological pH depends on the choice of protonation sites on acidic residues Asp and Glu, by virtue of spatial proximity. From the remaining set of residues, we will then identify all those residues whose ionization states at physiological pH are a function of the assumed protein dielectric constant. This will give us only a small number of residues with dependence on protein dielectric constant, or on choice of protonation site, that we can then address individually by exhaustive consideration of the possible variations. Such a strategy, we believe, not only eliminates most uncertainty about ionization states but also clearly defines the residual uncertainty.
In addition to the native OmpF, we will use the same strategy to compute also the ionization states of residues in mutant OmpF's that have been shown to be different from native OmpF in conductance, selectivity, and gating properties (Phale et al., 2001). In particular, we consider the mutants D113G, E117Q, NQ, NQAAA, and R42C.
Because of the large number of combinations we consider, (multiple dielectric constants, multiple treatments of the electrolyte screening effect, multiple van der Waals radii, multiple combinatorics of protonation sites, and multiple porin mutants), the calculation grows to several thousand processor hours of computation time. To deal with this, we implemented a spatial decomposition strategy for the electrostatics program UHBD (Madura et al., 1995) that permitted us to distribute the computation over many processors on the SGI Origin 2000 at the National Center for Supercomputing Applications, so that the computations could be done in a reasonable time.
THEORY
pKa values of titratable amino acids depend on the electrostatic environment in which they are embedded. Current theories (Tanford and Roxby, 1972; Bashford and Karplus, 1990; Beroza et al., 1991; Oberoi and Allewell, 1993; Yang et al., 1993; Antosiewicz et al., 1994; Karshikoff, 1995; Sham et al., 1997) divide the calculation of pKa values in two parts. The first part, “Single-site titration,” deals with the calculation of the effect of electrostatic interactions between each titrating site in the protein and all other partial charges, dielectric boundaries, and solvent effects of the protein system. Such a calculation yields an “intrinsic pKa” of a titratable site, the pKa of a titrating group in an uncharged protein assuming nontitratability of all other sites. The second part, “Multiple-site titration,” deals with the calculation of the effect of the presence of other titratable sites in the protein. The pKa of a titratable site incorporating all the above effects is called its “apparent pKa.” In our work we do this two-part calculation utilizing an adaptation of the UHBD software package release 5.1 (Madura et al., 1995), which is based on a single-point charge model.
Single-site titration (intrinsic pKa)
The first part is theoretically tackled by considering a thermodynamic cycle (Fig. 2) for ionizing a titratable residue in the presence and absence of the protein system. The difference between free energies of ionization of the residue in water and protein system 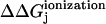 is then
is then
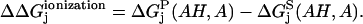 |
(1) |
FIGURE 2.

Thermodynamic cycle for ionizing a titratable residue in the presence and absence of protein system. Subscript (s) denotes water and subscript (p) denotes protein system (protein in solvent).
Assuming a single-site protonation model, it has been shown (Antosiewicz et al., 1994) that the above ionization free energies can be written as
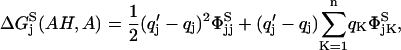 |
(2) |
and
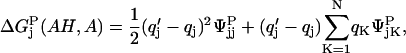 |
(3) |
where, φjk and ψjk are the potentials at the location of charge k created by a unit positive charge at the site of titratable group j. q′K is the charge of the site after ionization and qK is that before ionization. n and N are, respectively, the atoms in the amino acid and the protein.
The intrinsic pKa of group j, which is the pKa of a titrating group in a discharged protein, is then related to the change in free energy of ionization (Tanford and Kirkwood, 1957) as
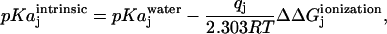 |
(4) |
where, 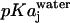 is the pKa of group j in water.
is the pKa of group j in water.
The calculation of intrinsic pKa of titratable group j thus requires the calculation of potentials φjk and ψjk, which can be obtained as part of the solution of the Poisson-Boltzmann equation (PBE) describing the protein system. It is important to note that the values of these potentials are only due to the charge at the titration site of group j, reducing the number of fixed charges in the system to only the one at the titration site. The values of these potentials can thus be obtained by performing two numerical solutions of the PBE. φjk can be determined by setting up a PBE with a single-unit charge at the ionization site of the group in water (represented as a continuum dielectric) and ψjk can be determined by setting up another PBE with a single-unit charge at the ionization site of the same group in the protein system.
Clearly, such a calculation methodology would bring about shifts from model pKa due to dielectric boundary effects and solvent effects and from protein charges in close proximity to the group whose pKa is being computed. Specifically, protein charges in close proximity, negative in case of acids and positive in case of bases, would bring about a positive (negative) shift in pKa values for acids (bases). On the other hand, positive (negative) protein charges in proximity would bring about negative (positive) shifts in pKa values of acids (bases). The magnitude of these computed shifts would also depend upon an assumption of protein dielectric constant and solvent concentrations used in these calculations.
Multiple-site titration (apparent pKa)
The second part of determining the effect of the presence of multiple titratable groups in the protein can be treated as a problem of ligand binding with multiple interacting ligands. Titratable residues in proximity contend for the acquisition/release of hydrogen ions giving rise to interdependent titration behaviors. The activity that each residue exhibits in this behavior depends on three factors. Firstly, it depends upon the availability of hydrogen ions, which is nothing but the pH of the solvent. Secondly, it depends upon the intrinsic pKa values of all the participating residues, which is taken from the first part of our calculation. And thirdly, it depends upon the proximity of these residues, which may be related to the electrostatic potential on a participating residue created by a unit charge on the titration site of another residue (coupling constant). A statistical distribution that incorporates these effects can be based on a probability that is proportional to the Boltzmann-weighted sum over all protonation states at each pH (Bashford and Karplus, 1990, 1991). The fraction of molecules having site i protonated, or a point in the titration curve of site i for a given pH is then:
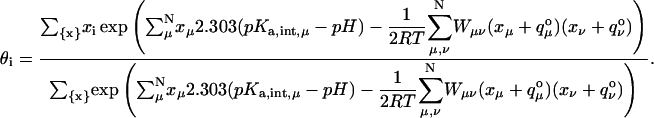 |
(5) |
Where, x is an N-element protonation state vector whose elements, xμ, are zero or one according to whether site μ is protonated or unprotonated, 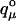 is the charge of the unprotonated state of site μ {x} indicates summation over all M = 2N possible protonation states and Wνμ are the interaction energies between titrating sites ν and μ.
is the charge of the unprotonated state of site μ {x} indicates summation over all M = 2N possible protonation states and Wνμ are the interaction energies between titrating sites ν and μ.
For a protein with N titratable sites, this equation has 2N terms in the summation. Solving this equation for a range of pH values, one can generate titration curves for any residue. To get a clearer picture of the effect of coupling constant on the titration behavior of residues, consider a protein with two cation sites with intrinsic pKa values of pKa1 and pKa2 coupled by an interaction of strength W. Expansion of all terms in Eq. 5 yields:
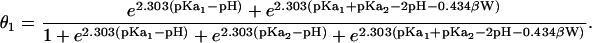 |
(6) |
A rearrangement yields a modified form of Henderson-Hesselbach equation:
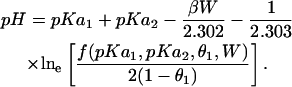 |
(7) |
Such a representation clearly shows that the effect of interaction energy would not only be visible in the shape of titration curves, but would also be visible as a shift in the entire titration curve, the extent of shift being directly proportional to the strength of interaction. Positive interaction energies would thus tend to lower the pKa of a residue and negative interaction energies would tend to increase the pKa of a residue. For this system, if titratable sites of the two residues are separated by a distance of 4 Å, a simple calculation at a dielectric constant of 4 yields a pKa shift of the order of 15 units. Because the magnitude of interaction energy W also depends upon the protein dielectric (PD) constant, we can see that an assumption of PD constant also plays a significant role in driving multiple-site titration. It should be noted that the strict validity of Eqs. 5–7 depend on the linear addition of potentials due to charges, which is not true for the higher order terms in the full nonlinear solution to the Poisson-Boltzmann equation. The issue of how much that compromises our results will be dealt with below.
IMPLEMENTATION
Atomic coordinates
The coordinates of OmpF trimer was taken from the x-ray crystallographic structure determined by Cowan et al. (1992), PDB entry 2OMF. The structure had several missing titratable hydrogens, which were added based on an OPLS force-field definition of amino acids (Jorgensen et al., 1996). To emulate the low dielectric environment (dielectric constant, 4) around the protein, a layer of neutral nonpolar spheres (extending at least 17 A from all boundaries of the protein) was placed around the protein. The thickness of this layer was set to 24 Å and was limited between the two rings of aromatic residues found around the protein (Schabert et al., 1995; Van Der Straaten et al., 2002) (Fig. 3). The coordinates of OmpF mutants D113G, R42C, and NQAAA were taken directly from their respective x-ray structures, PDB entries 1GFM, 1GFP, and 1HXT. Structures of mutants E117Q and NQ were modeled by modifying the structure of wild-type OmpF. As indicated by Phale et al. (2001), a 3-Å resolution x-ray of structures of these mutants showed virtually no change as compared to the structure of wild-type OmpF. Thus, the E117Q mutant was modeled by a simple replacement of one carboxylic oxygen of Glu-117 with an amine group. The NQ mutant was then modeled from the structure of E117Q by replacing a carboxylic oxygen of residue Asp-113 with another amine group.
FIGURE 3.

Dielectric model of OmpF used in pKa calculations is shown embedded in a low dielectric slab (width, 24 Å) composed of neutral nonpolar spheres (radii, 2 A). The position of the slab was postulated to lie between Tyr-180 and Trp-214.
Parameters in simulation
As we have seen earlier, the calculation of intrinsic pKa depends upon the solution of the PBE. To numerically solve a continuous PBE it is imperative to discretize it in a manner such that the assumptions arising out of the process do not alter the purpose of the solution. For our calculations that include all the nonlinear terms in the solution to the PBE, we have used the nonlinear PBE solver of UHBD (Holst et al., 1994; Madura et al., 1995), which discretizes the PBE via a finite difference (FD) schema. We have used a series of four focusing FD grids (97×97×97; 30×30×30; 36×36×36; and 50×50×50 with spacings of 2 Å, 1.0 Å, 0.5 Å, and 0.25 Å, respectively, centered on the titrating site of interest) to represent and then refine the solution of PBE at the site of interest. Such strategy has been shown to work well in these cases where PBE calculations involve only a single-source charge (Gilson et al., 1988; Mohan et al., 1992; Yang et al., 1993; Antosiewicz et al., 1994). The next step toward discretization deals with the representation of atoms on grids. Mohan et al. (1992) had reported that the use of one-half the OPLS (Jorgensen and Tirado-Rives, 1988) Lennard-Jones σ values for atomic radii worked better than then-available charge-radii parameters for finite difference Poisson Boltzmann (FDPB)-based calculations of small molecule solvation energies. More recently, a comparison of force fields in titration curve calculations by Gibas (1996) also supports the same. We have thus used one-half the OPLS Lennard-Jones σ values for atomic radii to set up the dielectric boundary between protein and its surrounding system. It is important to note that although the OPLS force fields use 0.0 as the σ value for polar hydrogens, we have used a value of 2.4 A in our calculations. Following the works of Gibas (1996) and others (Antosiewicz et al., 1994, 1996b; Forsyth et al., 1998), we have used the united-atom charge parameter set from CHARMm version 22.0 (Molecular Simulations Inc., Waltham, MA). The next step deals with the construction of protein-solvent dielectric boundary. This was constructed on the basis of a solvent-accessible surface definition by probing the molecular surface with a probe of 1.4-Å radius, which is the size of a water molecule (Richards, 1977). Based on this dielectric boundary, every grid point is then assigned an ionic strength. The PBE solver uses a masking function λ(r) that is set to a value of 0 wherever ions cannot penetrate and a value of 1 where they can. In all our calculations, we have used a low-ionic strength of 100 mM. The PBE solver of UHBD also allows an incorporation of a Stern layer, a region at the solvent-molecule interface, which mobile solute molecules cannot physically occupy. Introduction of an ion-exclusion layer prevents the mobile ions from overlapping with the atoms of the molecule. This ion-exclusion layer was set to extend one ion radius of 2 Å beyond the van der Waals radii of the atoms. The system was then solved to obtain the potential grid.
To confirm that our findings were not highly sensitive to charge-radii sets used in these simulations, we also redid the calculations for native OmpF with an assumed dielectric constant of 4 using PARSE (Sitkoff et al., 1994) charge-radii parameter sets. We found only minor differences in the pK shift and no difference at all in the protonation state of the porin at neutral pH.
In case of multiple-site-titration, the solution of Eq. 5 for each residue was determined using a cluster algorithm (Gilson, 1993). (This code can be obtained from http://gilsonlab.umbi.umd.edu/index.html). Because interaction energies Wij can be obtained by performing a PBE calculation with a single-source charge at titratable site i, these energies were also obtained from results of previous PBE calculations.
In the cluster algorithm, small clusters of titratable sites are initially created on the basis of the strength of charge-charge correlations (calculated from intrinsic pKa and Wij) and independently solved treating intercluster interactions by a mean field approximation. New charges from this cluster are then used in the computation of fractional charges of the next cluster. All clusters are similarly updated and the process is iterated to self-consistency. The convergence criterion of the algorithm is defined by setting a maximum tolerance for change in fractional charge (0.05 in our case). In cases of strongly coupled titrating sites, as pointed out by Bashford and Karplus (1991), a mean field approximation fails to correctly predict titration behaviors. Thus, to make sure that strongly coupled sites do not fall in different clusters, one must have larger cluster sizes. But given the algorithm's potential to get into infinite oscillatory loops for very large clusters, we tested this algorithm for several cluster sizes. For wild-type OmpF, we found that the algorithm did not converge for some pH values when we set the maximum cluster size to 20. We also found results for cluster sizes in the range 15–19 to be identical. We thus set 15 as the maximum cluster size for all our calculations.
We have explored the degree of nonlinearity of the explicit charge-reaction field interaction by doing a subset of the computations with a linearized Poisson-Boltzmann solver. We did the linearized calculations at an assumed protein dielectric constant of 4 and compared results with the full nonlinear solution. We found the difference to be quite small, far below what would affect any of the conclusions of the paper. We did not repeat the linearized computations for larger assumed protein dielectric constants, on the grounds that any discrepancy would be maximized at the lowest value of the dielectric constant, because this condition maximizes the Born energy from inserting the residue into the protein and the energy of residue-residue interactions. The results we present are all using the full nonlinear Poisson-Boltzmann equation for maximum accuracy, but the relatively small difference between the linear and nonlinear solutions suggests that Eq. 5 is a conceptually reasonable way to understand how the different components of the interaction energies are summed.
We have also redone the calculations at low dielectric constant with a very low ionic strength, 1 mM, which is effectively zero ionic strength. The results for pK values were slightly different from those with 100 mM ionic strength, but in no case were they so different as to modify the prediction of the ionization states at neutral pH. We conclude that for the special case of computing the fields in the narrow lumen of a protein channel in the course of determining the ionization state of titratable sites, the terms in the solution of the Poisson-Boltzmann equation due to electrolyte shielding are effectively negligible.
Parallel computing in pKa calculation
Wild-type OmpF and its mutants are large transmembrane trimers (∼100 kDa) with over 100 titratable residues per monomer. Along with the low-dielectric membrane, each protein system contained over 17,000 atoms. As we have seen, the calculation of intrinsic pKa for each residue requires two FD-based PBE solutions, implying that a calculation of intrinsic pKa values along with their interaction energies with other titratable residues for each trimer would require 2×3×100×4 PBE solutions (from: 2 PBE per site, 100 sites per monomer, 3 monomers, and 4 series of focusing grids). To speed up these calculations we parallelized the existing protocol. The nondependence of the calculation of intrinsic pKa values and interaction energies upon each other provided the opportunity to spread the entire calculation over multiple processors. A combination of PERL, C, and MPI scripts were written as a wrapper around the PBE solver of UHBD to decompose the calculation of the entire matrix of ΔΔGionization (diagonal terms) and interaction energies Wij (nondiagonal terms) over a desired number of processors. For a series of test cases, we found timescales for the entire calculation to decrease linearly with increase in number of processors (data not shown).
All simulations for this paper have been done on the SGI Origin 2000 supercomputer provided by the National Center for Supercomputing Application (NCSA), Urbana-Champaign (Urbana, IL).
RESULTS AND DISCUSSION
This section is presented in three parts. In the first part we present a calculation of the ionization states of all residues lining the pore of wild-type OmpF. In the second part, we present the effect of mutations D113G, E117Q, NQ, NQAAA, and R42C on the ionization states of these residues. In the third section, based on all these results, we propose a heuristic for analyzing and correlating pKa results from calculations assuming different dielectric constants for the protein.
Ionization states at physiological pH in wild-type OmpF
Using the methodology described above, intrinsic and apparent pKa values of all residues in the homotrimer were calculated at different protein dielectric (PD) constants of 4, 6, 10, 20, 40, and 80. PKa values of residues lining the pore of the channel (shown in Table 1) were then obtained by averaging their respective values over all three monomers. Because the structure has a three-fold rotational symmetry, standard deviations obtained during averaging will detect the presence of artifacts related to how the grid is superimposed on the protein structure in PBE calculations. Magnitudes of these standard deviations were negligible for protein dielectric constants of 10 and 20 and were far too small at lower dielectric constants to affect ionization states of any residue at neutral pH.
TABLE 1.
Comparison of pKa values of residues calculated at different protein dielectric constants
| pKa
|
||||||||
|---|---|---|---|---|---|---|---|---|
|
PD = 4
|
PD = 10
|
PD = 20
|
||||||
| Charge at pH = 7 | Residues | Model | Intrinsic | Apparent | Intrinsic | Apparent | Intrinsic | Apparent |
| −1 | Asp-12 | 4 | 7.2 | 2.4 | 5.4 | <1 | 4.7 | <1 |
| −1 | *Asp-37 | 4 | 7.2 | 6.9 | 5.3 | <1 | 4.6 | <1 |
| −1 | Asp-74 | 4 | 3.7 | <1 | 3.7 | <1 | 3.7 | <1 |
| −1 | Asp-107 | 4 | 6.4 | <1 | 5.1 | <1 | 4.6 | 1.5 |
| −1 | Asp-113 | 4 | 4.8 | 3.5 | 4.8 | 3.1 | 4.7 | 3.0 |
| −1 | Asp-121 | 4 | 2.0 | <1 | 3.3 | 1.4 | 3.6 | 2.0 |
| −1 | Asp-126 | 4 | 7.6 | <1 | 5.3 | <1 | 4.5 | <1 |
| ? | *Asp-127 | 4 | 13.2 | >14 | 7.6 | 7.2 | 5.7 | 4.9 |
| −1 | Asp-221 | 4 | 6.5 | 4.2 | 5.3 | 3.5 | 4.8 | 3.3 |
| −1 | Glu-2 | 4.4 | 6.7 | 6.1 | 5.3 | 4.8 | 4.8 | 4.2 |
| −1 | Glu-48 | 4.4 | 6.9 | <1 | 5.6 | 2.0 | 5.1 | 2.3 |
| −1 | Glu-62 | 4.4 | 8.0 | <1 | 6.0 | <1 | 5.3 | <1 |
| −1 | Glu-71 | 4.4 | 12.1 | <1 | 7.7 | <1 | 6.1 | <1 |
| −1 | Glu-181 | 4.4 | 6.8 | 5.6 | 5.4 | 4.6 | 4.9 | 4.2 |
| 0 | Tyr-32 | 9.6 | 13.2 | >14 | 11.1 | >14 | 10.3 | >14 |
| 0 | Tyr-40 | 9.6 | >14 | >14 | 13.2 | >14 | 11.6 | >14 |
| 0 | Tyr-63 | 9.6 | >14 | >14 | 13.2 | >14 | 11.2 | >14 |
| 0 | Tyr-102 | 9.6 | >14 | >14 | 12.0 | 11.5 | 10.9 | 11.0 |
| 0 | Tyr-106 | 9.6 | 13.1 | >14 | 11.5 | >14 | 10.8 | >14 |
| 0 | Tyr-124 | 9.6 | >14 | >14 | 12.8 | >14 | 11.1 | >14 |
| 0 | Tyr-302 | 9.6 | 14.0 | >14 | 11.8 | >14 | 10.9 | 13.1 |
| +1 | Lys-10 | 10.4 | 9.1 | 13.1 | 9.8 | 13.0 | 10.1 | 13.1 |
| +1 | Lys-16 | 10.4 | 10.0 | 11.6 | 10.4 | 13.6 | 10.5 | >14 |
| +1 | Lys-46 | 10.4 | 9.4 | >14 | 10.1 | >14 | 10.3 | >14 |
| +1 | Lys-80 | 10.4 | 10.2 | 13.6 | 10.6 | 12.6 | 10.6 | 12.9 |
| +1 | Lys-89 | 10.4 | 7.3 | 11.8 | 9.1 | 12.7 | 9.8 | 13.1 |
| +1 | Lys-219 | 10.4 | 8.4 | >14 | 9.6 | >14 | 10.0 | >14 |
| +1 | Lys-243 | 10.4 | 9.0 | 10.9 | 9.9 | 12.0 | 10.2 | 12.4 |
| +1 | Arg-42 | 12 | 13.7 | >14 | 13.0 | >14 | 12.7 | >14 |
| +1 | *Arg-82 | 12 | 14.0 | >14 | 13.1 | >14 | 12.8 | >14 |
| +1 | Arg-100 | 12 | 6.9 | >14 | 10.2 | >14 | 11.3 | >14 |
| +1 | Arg-132 | 12 | 12.1 | >14 | 12.3 | >14 | 12.3 | >14 |
| +1 | Arg-167 | 12 | 12.1 | >14 | 12.0 | >14 | 12.0 | >14 |
| +1 | Arg-168 | 12 | 9.6 | 10.7 | 11.0 | >14 | 11.5 | >14 |
| +1 | Arg-270 | 12 | 12.1 | >14 | 12.3 | >14 | 12.3 | >14 |
Indicates residues whose ionization state calculations have been dealt with independently.
Refer to Table 2 for residues Tyr-22, Tyr-310, Glu-117, Glu-296, and Asp-312.
Glutamic and aspartic acids offer two sites for protonation. It is important to note that the choice of protonation site for these acids affect only the intrinsic pKa of its neighbors and does not affect either their own intrinsic pKa or residue pairwise interaction energies unless the protonation process alters the protein-solvent dielectric boundary. In most cases, the choice of protonation site in a particular residue did not affect the protein-solvent dielectric boundary and thus was only expected to affect intrinsic pKa of its neighbors. In cases where the choice of protonation site did affect dielectric boundary definitions, residues were found to be isolated and thus were not expected to affect intrinsic pKa of their respective neighboring residues. Calculations at a PD constant of 4 indicated that the choice of protonation site generated large differences in pKa (∼1 pKa unit) for some titratable residues. Among the affected residues, other than Tyr-22, Arg-82, Glu-117, Glu-296, Tyr-310, and Asp-312, the differences in intrinsic pKa calculated with different protonation sites were not found to be significant enough to affect their ionization state probabilities at neutral pH. To identify all those residues that could play a major role in affecting pKa of these residues, we calculated pairwise interaction energies of these residues with all other titratable residues in the protein. Using a cutoff of 1 kcal/mol (calculated with PD constant of 4), we found that Arg-82 interacts strongly with residues Arg-42, Arg-100, Arg-132, Glu-62, and Glu-71 of the neighboring monomer implying that different protonation sites in Glu-62 and Glu-71 may be responsible for yielding different pKa for Arg-82. The fact that there are also several other arginines strongly interacting with Arg-82, a choice of alternate protonation sites on these glutamates can also affect its apparent pKa by altering the relative differences between the intrinsic pKa of arginines. We thus need to explore and justify the choice of the protonation sites of these glutamates before predicting the most probable ionization state of Arg-82. We deal with Arg-82 a little later. In the cases of Tyr-22, Glu-117, Glu-296, Tyr-310, and Asp-312, we found that these residues belong to a single cluster of strongly interacting residues (for convenience, we now refer to it as the loop 3 cluster). These residues are spatially proximal to each other and in their protonated states can be expected to interact via H-bond networks that can adopt several possible configurations. It has been shown (Alexov and Gunner, 1997; Nielsen and Vriend, 2001; Georgescu et al., 2002) that a choice of H-bond network can have a significant impact on guiding the results of pKa calculations. Thus, before predicting the most probable ionization states of any of the residues belonging to this cluster along with the aforementioned residue Arg-82, it becomes imperative to exhaustively explore not only the effects of the assumptions of PD constants but also for the choice of acidic protonation sites. We have thus dealt with these residues independently (read further).
Among the remaining residues we now identify all those residues whose ionization states at physiological pH depend on the assumption of PD constant. Data in Table 1 show only two residues, Asp-37 and Asp-127, whose ionization states depend upon a choice of the PD constant. Calculations using a PD constant of 4 predict Asp-37 to be partially charged whereas calculations using any higher PD constant predict it to have a higher probability of being fully charged at physiological pH. Calculations using PD constants of either 4 or 8 predict Asp-127 to have a higher probability of being protonated whereas calculations assuming any higher PD constants predict it to have a higher probability of being fully charged at physiological pH.
Thus regardless of the assumption of PD constant or choice of protonation sites on acids (Asp and Glu), other than residues Tyr-22, Tyr-310, Asp-37, Asp-127, Asp-312, Glu-117, Glu-296, and Arg-82, all other residues lining the pore of the channel can be expected to have an ionization state characteristic of their bulk value at neutral pH. We now address each of these residues individually and present possible scenarios of their ionization states.
Asp-37
The intrinsic pKa of Asp-37 calculated at a PD of 4 was found to be high indicating either its proximity to negative charges in the protein or a substantial change in solvent exposed area. Because Asp-37 is not a buried residue, this shift was more likely due to the presence of proximal negative charges. The OH atom of Tyr-98 belonging to a neighboring monomer was found to be ∼4.2 Å away from the titratable atom of Asp-37. This increased the free energy of the ionized state of Asp-37 and was reflected as a positive shift in intrinsic pKa. Because Asp-37 is not a buried residue, its side chain may have conformational flexibility. Such cases have been shown by Sham et al. (1998) to be best treated with higher protein dielectric constants that mimic relaxation in ionization processes. Representing protein with a dielectric constant of 6 resulted in lowering its intrinsic pKa to 6.3 and its apparent pKa to 5.1 indicating Asp-37 to have a very high probability of being fully charged at physiological pH. Needless to mention, the use of any larger PD constants further lowers the apparent pKa, making it more probable to be ionized at neutral pH. We thus expect Asp-37 to be fully charged at neutral pH.
Asp-127
Calculations with a PD of 4 predict intrinsic pKa of Asp-127 to be 13.1. As also pointed out by Karshikoff (1994), the oxygen atom of Ala-237, which is ∼3.6 A away from the titratable atom of Asp-127, might be acting as a proton acceptor in an H-bond with this residue and thus may be responsible for this shift. But given the distance of this hydrogen bond, the increased stabilization of the protonated state of Asp-127 induced by it is not enough to shift the intrinsic pKa by >9 units. Thus there have to be other reasons for such a large shift. The x-ray structure of OmpF indicates that Asp-127 is sandwiched between the barrel and loop L3 that forms the constriction zone of the channel. In our protein dielectric model, the protein-solvent dielectric boundary was defined using a solvent-accessible surface definition by probing the molecular surface with a probe of 1.4 Å. Such a surface definition buries this residue in the low dielectric protein making it inaccessible to the protein-solvent boundary. This increases the free energy of the ionized state of the residue and is reflected as a large positive shift in its intrinsic pKa. On representing the protein with a van der Waal's surface, we found that it leaves Asp-127 accessible to the protein-water interface. Thermal fluctuations of residues nearby, corresponding to a B-value of ∼25 (taken from crystal structure), indicate possibilities of water penetration into the pocket formed between the barrel and loop L3. Water penetration has been suggested (Dwyer et al., 2000) to have an effect of increasing the effective dielectric constant of the region from 4 to ∼12. If we assume a case of water penetration and redo calculations at a PD of 12, we find that the intrinsic pKa of Asp-127 comes out to be 6.9 and the apparent pKa comes out to be 6.3. The relative negative shift in apparent pKa observed over intrinsic pKa is due to the presence of Arg-167, the titratable atom of Arg-167 being 5.4 A away from that of Asp-127. Assuming a case of water penetration thus indicates Asp-127 to have a higher probability of being ionized at neutral pH.
Loop 3 cluster: Tyr-22, Tyr-310, Glu-117, Glu-296, and Asp-312
As shown earlier, these residues strongly interact with each other. A choice of protonation sites on residues Glu-117, Glu-296, and Asp-312 were found to affect the calculation of their intrinsic as well as apparent pKa. It is important to note that a choice of protonation sites in each acidic group did not affect the protein-solvent dielectric boundary. The affects on intrinsic pKa were thus only a consequence of changes in relative distances from protons and because all the residues in this cluster are acids, changes in apparent pKa were a direct consequence of changes in relative differences between their intrinsic pKa. To explore possible protonation states, we adopted a strategy of systematically considering all possible H-bonding networks, and computing pKa values for each postulated network.
Along with Ser-272, a residue proximal to this cluster, we were able to create several H-bond networks with varying numbers of hydrogen bonds. For our calculations, we chose only two networks, Network-1 and Network-2 shown, respectively, in Fig. 4, a and b, that had the maximum number of hydrogen bonds (minimum local energy). Performing pKa calculations using these two networks at a PD constant of 4 yielded two different sets of pKa (refer to Table 2), the most distinct difference being that one predicted Asp-312 to be partially charged at neutral pH whereas the other predicted it to be fully charged at neutral pH. The possible final configuration of Network-1 can be described by Fig. 4 a with the exception of the absence of the proton on Glu-117 and the possible final conformation resulting from a calculation using Network-2 is described in Fig. 4 c where residues Glu-117 and Asp-312 have been shown in their deprotonated states. Calculations with an elevated PD constant of 10 shifted intrinsic pKa of all residues toward their respective model pKa and decreased the interaction energy between titratable sites. This had an effect of lowering the apparent pKa of Glu-117 and Asp-312 in both cases, making them highly probable of being fully charged at neutral pH, and making Fig. 4 c the only possible H-bonding network for the part of the channel for the higher assumed PD constant. The apparent pKa of Asp-296 still remained >14 for both cases. To resolve the issue of two possibilities (Fig. 4, a and c), we performed a similar set of calculations on OmpF mutant E117Q (described in a later section).
FIGURE 4.

Network-1 is shown in Fig. 4 a whereas Network-2 is shown in Fig. 4 b. Fig. 4 c shows the H-bond configuration resulting upon accounting for the most probable ionization states of all residues belonging to the loop 3 cluster predicted using Network-2. These figures were created with RasMol Version 2.6.
TABLE 2.
Comparison of pKa of residues belonging to the loop 3 cluster
| Network-1
|
Network-2
|
|||||||
|---|---|---|---|---|---|---|---|---|
|
Protein dielectric = 4
|
Protein dielectric = 20
|
Protein dielectric = 4
|
Protein dielectric = 20
|
|||||
| Residues | pKaintrinsic | pKaapparent | pKaintrinsic | pKaapparent | pKaintrinsic | pKaapparent | pKaintrinsic | pKaapparent |
| Tyr-22 | 12.6 | >14 | 10.6 | >14 | 13.4 | >14 | 10.8 | >14 |
| Asp-117 | 4.3 | 1.4 | 4.8 | 4.1 | 4.3 | 6.2 | 4.8 | 4.4 |
| Glu-296 | 8.6 | >14 | 5.1 | 8.4 | 8.0 | >14 | 4.9 | 8.8 |
| Tyr-310 | >14 | >14 | 11.4 | >14 | >14 | >14 | 11.3 | >14 |
| Asp-312 | 2.9 | 7.5 | 3.7 | 2.4 | 1.4 | <1 | 3.4 | 1.4 |
Arg-82
Arg-82 strongly interacts with several proximal titratable residues Arg-42, Glu-62, Arg-100, Arg-132, and Glu-71 of neighboring monomer. We found earlier that the ionization state of Arg-82 was sensitive to the choice of protonation sites on residues Glu-62 and Glu-71. To select an appropriate set of protonation sites for Glu-71 and Glu-62, we used the following strategy. We first randomly selected protonation sites on Glu-62 and Glu-71. Keeping the CγOH angle (109.5 from an sp3 hybridization) and the length of the OH bond (1 A) fixed, a trajectory of the Glu-71 proton was created by rotating it about the CγO bond axis. A similar trajectory was created for the alternate protonation site on Glu-71. The PBE was solved for each position of the proton in the trajectory. The position corresponding to the minimum energy was then set as the position of the proton on Glu-71. Given a randomly selected protonation site on Glu-62, this yielded an optimum position for adding a proton on Glu-71. A similar set of trajectory points was then created for the protonation of Glu-62 and a series of PBE were solved to optimize its position. These set of calculations were iterated until the sum of energies for adding the two protons was a minimum. Fig. 5 is a partial view of the configuration that we finally obtained and used in our calculation. Arg-132 is truncated and Arg-42 is omitted from the figure. Calculations at a PD constant of 4 predicted the apparent pKa of all arginines Arg-42, Arg-82, Arg-100, and Arg-132 to be >14 and all glutamates, Glu-62 and Glu-71, to be <4; i.e., all of these residues will be fully charged at neutral pH. Calculations at elevated PD constants produced essentially the same result.
FIGURE 5.

The H-bond network used in calculations involving Arg-82. R132 is truncated and R42 is not shown to clearly depict the H-bond network. This figure was created with RasMol Version 2.6.
Effect of mutations on wild-type OmpF
OmpF mutant E117Q
The x-ray structure of E117Q mutant, as indicated by Phale et al. (2001), is unaltered from the wild-type crystal structure. As can be seen from Fig. 4, an absence of Glu-117 can affect pKa of all the residues shown in the network. We performed calculations (PD, 4) on both H-bond networks (as part of the mutant trimer) and found that in either case, a replacement of Glu-117 with glutamine predicted Asp-312 to have an apparent pKa lower than 2. If Asp-312 is charged in this mutant then according to the results of our calculations on wild-type OmpF, this mutation should either keep the net charge at the constriction zone equal to that in wild-type OmpF or increase it by 1 e.u. (absence of Glu-117). An analysis of ion selectivity variation experiments (Schirmer and Phale, 1999) indicates that the net charge at the constriction zone of wild-type OmpF is lower than that of its mutant E117Q. This increase in charge in the mutant is then only explainable if we consider Asp-312 to be charged in wild-type OmpF, which brings us to believe that Asp-312 should also be charged in wild-type OmpF; i.e., that the configuration of Fig. 4 c is the correct choice. We note that Fig. 4 c was also indicated by our calculations using a higher assumed PD (10 instead of 4). Recent MD simulations (Im and Roux, 2002b; Van Der Straaten et al., 2002) report water penetration between barrel and loop 3. This could have the effect of elevating the dielectric constant in this region, lending further support to Fig. 4 c as the correct choice for the hydrogen-bonding network in the native channel.
OmpF mutant D113G
Calculations on wild-type OmpF showed Asp-113 to have strong interactions only with Tyr-106 and predicted its apparent and intrinsic pKa to be close to its model pKa. Given that the x-ray structure of this mutant is very similar to that of wild-type OmpF (Lou et al., 1996), a mutation of this residue with a glycine, was thus only expected to affect the charge state of Tyr-106 in the whole protein. On redoing calculations for the mutant trimer at a PD of 4 we found Tyr-106 to have an apparent pKa >14, predicting it to be protonated at neutral pH.
OmpF mutant NQ (E117Q with D113N mutation)
As mentioned earlier, this double mutation also leaves the crystal structure unaltered from the wild-type crystal structure. A pKa calculation at a low PD constant of 4 indicated nothing more than a combined effect of mutants E117Q and D113G on ionization states of all other residues. The net charge of this mutant at neutral pH should thus be 2 e.u. more than the net charge of wild-type OmpF.
OmpF mutant NQAAA (NQ with R42A, R82A, and R132A mutations)
As can be projected from Fig. 6, Arg-100 and Arg-132 form strong salt bridges with the ionized form of Glu-71. Glu-71, which belongs to loop 2 of neighboring monomer, has been shown (Phale et al., 1998) to play a big role in stabilizing the trimeric structure of the protein. The absence of Arg-132 was thus expected to have an effect on pKa of Glu-71 and Arg-100. Fig. 6 also indicates the possibility of an H-bond formation between residues Glu-62 and Arg-82. The absence of Arg-82 was thus thought to affect the pKa of Glu-62. To resolve these issues, we calculated pKa of all residues of this mutant trimer. Calculations at a PD constant of 4 showed that the absence of the arginines did not affect the ionization states of either glutamates or Arg-100 at neutral pH. Both glutamates were found to have an apparent pKa <2 and Arg-100 was found to have an apparent pKa >14. Because the calculations were done with a PD of 4, we can safely conclude that the pKa of the two glutamates is less than their model pKa of 4.4 and that of Arg-100 is at least greater than its model pKa of 12.0.
FIGURE 6.

Truncated view of OmpF mutant R42C showing the side chains of residues C42, R82, and R132 located at the constriction zone of the channel.
OmpF mutant R42C
This mutation also belongs to the constriction-zone set of residues. We found Cys-42 to be uncharged for all assumed PD constants from 4 to 80. This was initially somewhat unexpected by us and previously by others (Schirmer and Phale, 1999), because Arg-82 is close enough to make a hydrogen bond to a negatively charged Cys-42. In the calculation, the major reason for the predicted neutral state of the cysteine is the very large intrinsic pKa shift, due to the reduction in solvent-exposed area. In other words, because cysteine is so small, it is very hard to bury it in a charged state to the extent indicated in the crystal structure (Fig. 6). The energetic cost of burying a negative cysteine deep into the surface more than outweighs the potential energetic advantage of a hydrogen bond to the nearby arginine. The observed movement of residue Arg-82 relative to its position in the wild-type structure is thus more likely to be due to its repulsion from Arg-132 than to attraction to Cys-42.
Heuristic for analyzing pKa results
Representing protein with an elevated dielectric constant affects pKa shifts in two ways. Firstly, the use of elevated dielectric constant decreases interaction energies of a residue with the rest of the protein. This decreases the magnitude of change in free energy that comes from transferring its ionized state from water to protein. Such a change is visible as shifts in intrinsic and apparent pKa toward their respective model pKa. Secondly, use of an elevated dielectric constant decreases the Born energy of transferring a charge from bulk water to protein. In some cases, the differences in magnitudes of these shifts across a variation of PD constant would be negligible, whereas in other cases these differences would be very large.
To understand the implication of these differences, we classify all residues into four categories. Using q = +1 to represent a basic residue and q = −1 to represent an acidic residue, we define two categories of shifts in intrinsic pKa observed over model pKa and two categories of shifts in apparent pKa observed over intrinsic pKa as follows:
Type I shift
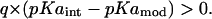 |
This represents a situation where the ionized state of a residue is stabilized when it is inserted in a neutral protein containing no other titratable sites. In the macroscopic model used in our calculations, this stabilization comes from opposite charges (−q) in the protein. Because our objective is to calculate ionization states at neutral pH, a further shift in pKa, which may come from other factors due to a residue's natural tendency to lower its free energy, will not be a concern for residue types Glu, Asp, Arg, and Lys (respective model pKa 4.4, 4.0, 12, and 10.4). Even if we had to take into account a projected per-residue stabilization and calculate a resulting intrinsic pKa, we would have to represent such sites by a lower dielectric constant (Schutz and Warshel, 2001). A calculation with an elevated dielectric constant would only decrease the magnitude and not the sign of a stabilizing pKa shift (unless solvent separates these interacting protein charges). Furthermore, if during calculation of apparent pKa, we find no other titratable residues in proximity, a stabilizing shift in intrinsic pKa for any assumed PD constant is then enough to expect such isolated residues to be charged at neutral pH. In cases of His, Cys, and Tyr, which by default are protonated at neutral pH (respective model pKa 6.3, 8.3, and 9.6), such a stabilizing shift in pKa can reverse the probability of ionization states from uncharged to charged. Thus, residues His, Cys, and Tyr belonging to this category need to be dealt with separately.
Type II shift
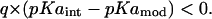 |
This represents a situation where the ionized state of a residue is destabilized when it is inserted in protein. This may be induced from two possible sources: a), proximity to q-type protein charges and b), reduction in solvent-exposed area. In either case, the magnitude of these shifts would decrease upon simulations with elevated protein dielectric constants. Because water penetration is expected to increase the PD constant, one may find that some residues that appear to be uncharged at neutral pH in simulations with lower dielectric constants may actually appear to be fully charged when represented with elevated dielectric constants. Thus, residues Glu, Asp, Arg, and Lys belonging to this category need to be dealt with separately. On the other hand, the prediction of ionization states of residues His, Cys, and Tyr belonging to this category becomes independent of the choice of PD constant. For any assumed higher or lower PD constant, these residues can be expected to be uncharged at neutral pH. It is important to note that such reasoning is valid only in the absence of other titratable sites. In the presence of other titratable sites, we shall see that resolving ionization states of many residues belonging to the former set also becomes a much easier task.
Type III shift
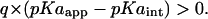 |
This indicates a stabilization of the ionized state of a residue from proximal −q-type titratable residues. Such a shift is very likely to be observed along with a Type II shift. If two titratable sites were close to each other, then the proton donor (acceptor) of one residue would also be close to the proton acceptor (donor) of the other residue. This proximity of proton acceptor and donor would result in a Type II shift. A combination of shifts Type II and Type III observed for a residue may thus indicate a strong possibility of salt-bridge or hydrogen-bond formation at neutral pH, the strength of which may be judged from the magnitude of this shift. It is easy to project that the use of an elevated protein dielectric would decrease the strength of interaction energy W between two titratable sites and thereby decrease the magnitude of such a shift (following Eq. 7) unless of course, solvent separates these interacting residues. Thus, residues showing a combination of Type II and Type III shifts that appear ionized at neutral pH in calculations assuming lower PD constants may be predicted uncharged for higher assumed PD constants making it imperative to calculate their ionization states at multiple PD constants. Only if the ionization state of such a residue comes out independent of an assumed PD constant, one can safely predict its ionization state. Using the same set of arguments we may also say that residues Asp, Glu, Arg, and Lys showing a combination of Type I and Type III shifts would be predicted charged regardless of an assumption of PD constant.
Type IV shift
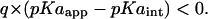 |
This indicates a destabilization of the ionized state of a residue from proximal q-type titratable residues. For a basic (acidic) residue, such a shift is observed in a scenario where multiple strongly interacting basic (acidic) residues are competing for proton binding (dissociation) and that this residue has an intrinsic pKa relatively lower (higher) than the other residues. Calculations with increased dielectric constants would not only decrease the interaction energy W between such titratable sites but would also alter the relative differences between their respective intrinsic pKa. Thus, in such cases, calculations with different PD constants may predict completely different ionization state sets. This makes it imperative to calculate pKa shifts for multiple assumptions of PD constants before assigning the ionization state of residues belonging to this category.
Statement of the heuristic
This gives us a clear strategy, or heuristic, to analyze results from multiple assumed dielectric constants:
We first consider results from a calculation assuming a low PD constant of 4. A PD constant of 4 is chosen because representing protein with this dielectric constant does not include any stabilizing factors that may be implicitly absorbed in pKa calculations.
From this set, we extract residues DERK that show a combination of Type I and Type III shifts and have a higher probability of being charged at neutral pH. These residues would be predicted charged regardless of the assumption of PD constant.
We then extract residues CYH that show a combination of Type II and Type IV shifts and have a higher probability of being uncharged at neutral pH. A Type II shift lowers the pKa of histidines or increases that of cysteines and tyrosines. Given that these residues show a Type IV shift implies that they are least favored in a race condition for proton binding (His) or proton dissociation (Cys and Tyr) and that their uncharged ionization state is more probable at neutral pH. A calculation assuming a higher PD would not only decrease the magnitudes of their shifts but also of their competitors indicating a possibility that these residues may still have a preferred neutral (uncharged) ionization state even in results from calculations assuming higher PD constants. Nevertheless, if calculations over a range of PD constants also predict these residues to have a higher probability of being uncharged, then they can be expected to be neutral.
We then consider residues DERK showing a combination of Type II and Type III shifts and have a higher probability of being charged at neutral pH. For a PD constant of 4, these residues indicate a possibility of a salt-bridge or H-bond formation, the strength of which may be evident from the magnitude of the Type III shift. If calculations for a given range of PD constants predict them charged, these residues are set charged otherwise their ionization states would depend upon an assumption of PD constant.
The remaining residues then need to be addressed individually.
SUMMARY AND CONCLUSIONS
An important methodological conclusion from this article is that for the special case of computing ionization states in the narrow lumen in a protein channel, it appears justified to formally neglect the effects of electrolyte shielding, rendering the calculations simpler in that respect. We draw this conclusion from the result that our results for the ionization states at neutral pH are unchanged whether the calculations are done with physiological ionic strength taking all the nonlinear terms in the PBE into account, or done with the linearized PBE at physiological ionic strength, or done with essentially zero ionic strength.
On the other hand, it does seem necessary to consider all the combinatorial possibilities for protonation states of residues in the lumen that are close enough to each other to have significant electrostatic interactions. In this article we have presented a strategy for considering the combinatorial possibilities. In the first stage of the strategy, we identified residues whose ionization states were sensitive to a choice of titration sites on residues. For these residues, we used a pairwise interaction energy cutoff of 1 kcal/mol to identify titratable residue clusters in the protein where a choice of H-bond network would also play a significant contribution in ionization state prediction. In the second stage, we identified residues whose ionization states were sensitive to a choice of PD constant. In the third stage of our strategy, all possible combinations of ionization states were considered at varying dielectric constants to create alternative scenarios for combination of ionization states. These combinations of ionization states were then evaluated in the context of experiment to decide on the most appropriate ionization state for model simulations. This procedure seems to us sufficiently comprehensive that, for a given structure, it must arrive at either the correct protonation state or the set of possible protonation states. (With the caveat that possible effects of motional fluctuations are not taken into account, nor the possible effects of permeant ions in the channel interior.)
Calculations using the above procedure for native OmpF lead us to suggest that at neutral pH, all residues near the permeation pathway except Asp-127 and Glu-296 have ionization states characteristic of their bulk value. This result was in fact quite surprising. The titratable residues lining the permeation pathway of the channel are either buried in a cavity surrounded by protein or are in the vicinity of other charged residues. Despite such large perturbations from the default conditions, almost all the residues wound up in their default ionization states. We found Glu-296 to be neutral (rather than negatively charged) because of its proximity to Asp-312. The ambiguity regarding the ionization state of Asp-312 was resolved by comparing the differences in experimental values of ionic selectivity of wild-type OmpF with one of its mutants, E117Q. The only residue for which we do not make an unambiguous prediction is Asp-127, because its ionization state is irreducibly dependent on assumed PD constant. If PD is assumed lower than 12, Asp-127 is computed to be neutral; if assumed >12, it is computed to be negatively charged. The “correct” PD may depend on the extent to which the region where Asp-127 is located (between loop 3 and the barrel) is penetrated by water.
We found several differences between our results and those previously proposed in literature and used in molecular simulations (Karshikoff et al., 1994; Watanabe et al., 1997; Tieleman and Berendsen, 1998; Schirmer and Phale, 1999; Im and Roux, 2002a,b; Van Der Straaten et al., 2002). The most notable differences are in the ionization states of residues Lys-80, Asp-121, Asp-127, Arg-167, Arg-168, and Asp-312. These residues had been considered either fully or partially uncharged whereas our calculations predict them to be fully ionized at neutral pH. Because there are three basic and three acidic residues in this group, setting the ionization states of all these residues from uncharged to charged will have no effect on the net charge in the lumen of the protein. However local fields in the vicinity of these residues will be quite different with our charge assignment. We note that in previous molecular dynamics simulations on this channel (including from our laboratory), the structure of the narrow part of the channel lumen deviated significantly from the crystal structure, becoming significantly more narrow (Tieleman and Berendsen, 1998; Im and Roux, 2002b; Van Der Straaten et al., 2002). Our laboratory has undertaken a new set of molecular dynamics simulations with the charge assignments indicated in this article, specifically setting Asp-312 to be fully charged and using the H-bond network shown in Fig. 4 c as the starting condition. Preliminary analysis based on 2 ns of simulation indicates that the width of the lumen is remaining at the value indicated by the crystal structure. These simulations will be the subject of a future publication.
We then extended the application of our strategy to elucidate the effect of several mutations on the ionization states of wild-type residues in the constriction zone. We found that although these mutations did cause shifts in pKa to other titratable residues in the constriction zone, these shifts were not large enough (even at low PD constants) to cause any changes in ionization states at neutral pH. There was, however, one interesting observation. In the mutant R42C, where residue Arg-42 is substituted with a cysteine, cysteine was earlier postulated to be fully ionized and forming a salt bridge with a neighboring arginine Arg-132. We found Cys-42 to be uncharged for all assumed PD constants ranging from 4 to 80. In calculations assuming a low PD constant, the energetic cost of inserting a charged cysteine into the protein was prohibitive and in calculations assuming a high PD constant, the interaction between Cys-42 and Arg-82 was too attenuated to result in charging the cysteine.
In this paper, we have not attempted to calculate the ionization states of point mutants E71A and E71Q. The x-ray crystal structures of these two mutants (Phale et al., 1998) were resolved with a Cl− ion in a position previously occupied by Glu-71 indicating that the stabilization of the ionized states of nearby arginines may be brought about by the presence of this discrete ion. Thus, to correctly resolve ionization states of nearby residues, an accounting of this discrete ion may be essential. We have thus considered the resolution of the ionization states of these two mutants to be beyond the scope of this paper. We also have not presented the ionization states of titratable residues buried in the surrounding hydrophobic part of the lipid.
Acknowledgments
Computational time from the National Computational Science Alliance is gratefully acknowledged. Simulations were carried out on the Origin 2000 at the National Center for Supercomputing Applications at the University of Illinois Urbana-Champaign. We also thank Shreedhar Natarajan and all other lab members for their helpful comments and discussions.
This work was supported by DARPA, National Science Foundation, and National Institutes of Health.
References
- Alexov, E. G., and M. R. Gunner. 1997. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys. J. 72:2075–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antosiewicz, J., J. M. Briggs, A. H. Elcock, M. K. Gilson, and J. A. McCammon. 1996a. Computing the ionization states of proteins with a detailed charge model. J. Comput. Chem. 17:1633–1644. [Google Scholar]
- Antosiewicz, J., A. J. McCammon, and M. K. Gilson. 1994. Prediction of pH-dependent properties of proteins. J. Mol. Biol. 238:415–436. [DOI] [PubMed] [Google Scholar]
- Antosiewicz, J., A. J. McCammon, and M. K. Gilson. 1996b. The determinants of pKa's in proteins. Biochemistry. 35:7819–7833. [DOI] [PubMed] [Google Scholar]
- Bashford, D., and M. Karplus. 1990. pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 29:10219–10225. [DOI] [PubMed] [Google Scholar]
- Bashford, D., and M. Karplus. 1991. Multiple-site titration curves of proteins: an analysis of exact and approximate methods for their calculation. J. Phys. Chem. 95:9556–9561. [Google Scholar]
- Beroza, P., D. R. Fredkin, M. Y. Okamura, and G. Feher. 1991. Protonation of interacting residues in a protein by a Monte Carlo method: application to lysozyme and the photosynthetic reaction center of Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA. 88:5804–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher, C. J. F. 1973. Theory of Electric Polarization. Elsevier Press, Amsterdam.
- Cowan, S. W., T. Schirmer, G. Rummel, M. Steiert, R. Ghosh, R. A. Pauptit, J. N. Jansonius, and J. P. Rosenbusch. 1992. Crystal structures explain functional properties of two E. coli porins. Nature. 358:727–733. [DOI] [PubMed] [Google Scholar]
- Delcour, A. H. 1997. MiniReview: function and modulation of bacterial porins: insights from electrophysiology. FEMS Microbiol. Lett. 151:115–123. [DOI] [PubMed] [Google Scholar]
- Demchuk, E., and R. C. Wade. 1996. Improving the continuum dilectric approach to calculating pka's of ionizable groups in proteins. J. Phys. Chem. 100:17373–17387. [Google Scholar]
- Dwyer, J. J., A. G. Gittis, D. A. Karp, E. E. Lattman, D. S. Spencer, W. E. Stites, and E. B. Garcia-Moreno. 2000. High apparent dielectric constants in the interior of a protein reflect water penetration. Biophys. J. 79:1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth, W. R., M. K. Gilson, J. Antosiewicz, O. R. Jaren, and A. D. Robertson. 1998. Theoretical and experimental analysis of ionization equilibria in ovomucoid third domain. Biochemistry. 37:8643–8652. [DOI] [PubMed] [Google Scholar]
- Georgescu, R. E., E. G. Alexov, and M. R. Gunner. 2002. Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophys. J. 83:1731–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibas, C. J. 1996. Computer simulation of titration behavior in proteins. PhD thesis. University of Illinois at Urbana-Champaign, Urbana, IL. 275.
- Gibas, C. J., and S. Subramaniam. 1996. Explicit solvent models in protein pKa calculations. Biophys. J. 71:138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson, M. K. 1993. Multiple-site titration and molecular modeling: two rapid methods for computing energies and forces for ionizable groups in proteins. Proteins. 15:266–282. [DOI] [PubMed] [Google Scholar]
- Gilson, M. K., and B. H. Honig. 1986. The dielectric constant of a folded protein. Biopolymers. 25:2097–2119. [DOI] [PubMed] [Google Scholar]
- Gilson, M. K., K. Sharp, and B. H. Honig. 1988. Calulating the electrostatic potentials of molecules in solution: method and error assessment. J. Comput. Chem. 9:327–335. [Google Scholar]
- Holst, M., R. E. Kozack, F. Saied, and S. Subramaniam. 1994. Treatment of electrostatic effects in proteins: multigrid-based Newton iterative method for solution of full nonlinear Poisson-Boltzmann equation. Proteins. 18:231–245. [DOI] [PubMed] [Google Scholar]
- Im, W., and B. Roux. 2002a. Ion permeation and selectivity of OmpF porin: a theoretical study based on molecular dynamics, Brownian dynamics, and continuum electrodiffusion theory. J. Mol. Biol. 322:851–869. [DOI] [PubMed] [Google Scholar]
- Im, W., and B. Roux. 2002b. Ions and counterions in a biological channel: a molecular dynamics simulation of OmpF porin from Escherichia coli in an explicit membrane with 1 M KCl aqueous salt solution. J. Mol. Biol. 319:1177–1197. [DOI] [PubMed] [Google Scholar]
- Jap, B. K., and P. J. Walian. 1990. Biophysics of the structure and function of porins. Q. Rev. Biophys. 23:367–403. [DOI] [PubMed] [Google Scholar]
- Jorgensen, W. L., D. S. Maxwell, and J. Tirado-Rives. 1996. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. JACS. 118:11225–11236. [Google Scholar]
- Jorgensen, W. L., and J. Tirado-Rives. 1988. The OPLS potential functions for proteins: energy minimizations for crystals for cyclic peptides and crambin. JACS. 110:1657–1666. [DOI] [PubMed] [Google Scholar]
- Karshikoff, A. 1995. A simple algorithm for the calculation of multiple site titration curves. Protein Eng. 8:243–248. [DOI] [PubMed] [Google Scholar]
- Karshikoff, A., V. Spassov, S. W. Cowan, R. Ladenstein, and T. Schirmer. 1994. Electrostatic properties of two porin channels from Escherichia coli. J. Mol. Biol. 240:372–384. [DOI] [PubMed] [Google Scholar]
- King, G., F. S. Lee, and A. Warshel. 1991. Microscopic simulations of macroscopic dielectric constants of solvated proteins. J. Chem. Phys. 95:4366–4377. [Google Scholar]
- Kozack, R. E., M. J. d'Mello, and S. Subramaniam. 1995. Computer modeling of electrostatic steering and orientational effects in antibody-antigen association. Biophys. J. 68:807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou, K.-L., N. Saint, A. Prilipov, G. Rummel, S. A. Benson, J. P. Rosenbusch, and T. Schirmer. 1996. Structure and functional characterization OmpF porin mutants selected for larger pore size. I. Crystallographic analysis. J. Biol. Chem. 271:20669–20675. [PubMed] [Google Scholar]
- Madura, J. D., J. M. Briggs, R. C. Wade, M. E. Davis, B. A. Luty, A. Ilin, J. Antosiewicz, M. K. Gilson, B. Bagheri, L. R. Scott, and A. J. McCammon. 1995. Electrostatics and diffusion of molecules in solution: simulations with the University of Houston Brownian dynamics program. Program. Comp. Phys. Comm. 91:57–95. [Google Scholar]
- Mohan, V., M. E. Davis, A. J. McCammon, and B. M. Pettitt. 1992. Continuum model calculations of solvation free energies: accurate evaluation of electrostatic contributions. J. Phys. Chem. 96:6428–6431. [Google Scholar]
- Nielsen, J. E., and G. Vriend. 2001. Optimizing the hydrogen-bond network in Poisson-boltzmann equation-based pKa calculation. Proteins. 43:403–412. [DOI] [PubMed] [Google Scholar]
- Nikaido, H. 1993. Transport across the bacterial outer membrane. J. Bioenerg. Biomembr. 25:581–589. [DOI] [PubMed] [Google Scholar]
- Oberoi, H., and N. M. Allewell. 1993. Multigrid solution of the nonlinear Poisson-Boltzmann equation and calculation of titration curves. Biophys. J. 65:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethig, R. 1992. Protein-water interactions determined by dielectric methods. Annu. Rev. Phys. Chem. 43:177–205. [DOI] [PubMed] [Google Scholar]
- Phale, P. S., A. Philippsen, T. Kiefhaber, R. Koebnik, V. P. Phale, T. Schirmer, and J. P. Rosenbusch. 1998. Stability of trimeric OmpF porin: the contributions of the latching loop L2. Biochemistry. 37:15663–15670. [DOI] [PubMed] [Google Scholar]
- Phale, P. S., A. Philippsen, C. Widmer, V. P. Phale, J. P. Rosenbusch, and T. Schirmer. 2001. Role of charged residues at the OmpF porin channel constriction probed by mutagenesis and simulation. Biochemistry. 40:6319–6325. [DOI] [PubMed] [Google Scholar]
- Richards, F. M. 1977. Areas, volumes, packing and protein structure. Annu. Rev. Biophys. Bioeng. 6:151–176. [DOI] [PubMed] [Google Scholar]
- Schabert, F. A., C. Henn, and A. Engel. 1995. Native Escherichia coli OmpF porin surfaces probed by atomic force microscopy. Science. 268:92–94. [DOI] [PubMed] [Google Scholar]
- Schirmer, T. 1998. General and specific porins from bacterial outer membranes. J. Struct. Biol. 121:101–109. [DOI] [PubMed] [Google Scholar]
- Schirmer, T., and P. S. Phale. 1999. Brownian dynamics simulation of ion flow through porin channels. J. Mol. Biol. 294:1159–1167. [DOI] [PubMed] [Google Scholar]
- Schulz, G. E. 1996. Porins: general to specific, native to engineered passive pores. Curr. Opin. Struct. Biol. 6:485–490. [DOI] [PubMed] [Google Scholar]
- Schutz, C. N., and A. Warshel. 2001. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins. 44:400–417. [DOI] [PubMed] [Google Scholar]
- Sham, Y. Y., Z. T. Chu, and A. Warshel. 1997. Consistent calcuations of pKa's of ionizable residues in proteins: semi-microscopic and microscopic approaches. J. Phys. Chem. 101:4458–4472. [Google Scholar]
- Sham, Y. Y., I. Muegge, and A. Warshel. 1998. The effect of protein relaxation on charge-charge interactions and dielectric constants of proteins. Biophys. J. 74:1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonson, T. 1998. Dielectric constant of cytochrome c from simulations in a water droplet including all electrostatic interactions. JACS. 120:4875–4876. [Google Scholar]
- Simonson, T., D. Perahia, and G. Bricogne. 1991. Intramolecular dielectric screening in proteins. J. Mol. Biol. 218:859–886. [DOI] [PubMed] [Google Scholar]
- Sitkoff, D., K. Sharp, and B. H. Honig. 1994. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98:1978–1988. [Google Scholar]
- Smith, P. E., R. M. Brunne, A. E. Mark, and W. Van Gunsteren. 1993. Dielectric properties of trypsin inhibitor and lysozyme calculated from molecular dynamics simulations. J. Phys. Chem. 97:2009–2014. [Google Scholar]
- Sternberg, M. J., F. R. Hayes, A. J. Russell, P. G. Thomas, and A. R. Fersht. 1987. Prediction of electrostatic effects of engineering of protein charges. Nature. 330:86–88. [DOI] [PubMed] [Google Scholar]
- Takashima, S., and H. P. Schwan. 1965. Dielectric dispersion of crystalline powders of amino acids, peptides, and proteins. J. Phys. Chem. 69:4176–4182. [DOI] [PubMed] [Google Scholar]
- Tanford, C., and J. G. Kirkwood. 1957. Theory of the protein titration curves. I. General equations for impenetrable spheres. JACS. 79:5333–5339. [Google Scholar]
- Tanford, C., and R. Roxby. 1972. Interpretation of protein titration curves. Application to lysozyme. Biochemistry. 11:2192–2198. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., and H. J. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. (See comments) Biophys. J. 74:2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Straaten, T., S. Varma, S. W. Chiu, J. Tang, N. R. Aluru, R. S. Eisenberg, U. Ravaioli, and E. Jakobsson. 2002. Combining Computational Chemistry and Computational Electronics to Understand Protein Ion Channels. ICCN. Puerto Rico, USA. 60–63.
- Warshel, A., S. T. Russell, and A. K. Churg. 1984. Macroscopic models for studies of electrostatic interactions in proteins: limitations and applicability. Proc. Natl. Acad. Sci. USA. 81:4785–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, M., J. Rosenbusch, T. Schirmer, and M. Karplus. 1997. Computer simulations of the OmpF porin from the outer membrane of Escherichia coli. Biophys. J. 72:2094–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, M. S., and G. E. Schulz. 1992. Structure of porin refined at 1.8 A resolution. J. Mol. Biol. 227:493–509. [DOI] [PubMed] [Google Scholar]
- Yang, A.-S., M. R. Gunner, R. Samponga, K. Sharp, and B. Honig. 1993. On the calculation of pka's in proteins. Proteins. 15:252–265. [DOI] [PubMed] [Google Scholar]