

Abstract
The structural flexibilities of two molecular machines, myosin and Ca2+-ATPase, have been analyzed with normal mode analysis and discussed in the context of their energy conversion functions. The normal mode analysis with physical intermolecular interactions was made possible by an improved implementation of the block normal mode (BNM) approach. The BNM results clearly illustrated that the large-scale conformational transitions implicated in the functional cycles of the two motor systems can be largely captured with a small number of low-frequency normal modes. Therefore, the results support the idea that structural flexibility is an essential part of the construction principle of molecular motors through evolution. Such a feature is expected to be more prevalent in motor proteins than in simpler systems (e.g., signal transduction proteins) because in the former, large-scale conformational transitions often have to occur before the chemical events (e.g., ATP hydrolysis in myosin and ATP binding/phosphorylation in Ca2+-ATPase). This highlights the importance of Brownian motions associated with the protein domains that are involved in the functional transitions; in this sense, Brownian molecular machines is an appropriate description of molecular motors, although the normal mode results do not address the origin of the ratchet effect. The results also suggest that it might be more appropriate to describe functional transitions in some molecular motors as intrinsic elastic motions modulating local structural changes in the active site, which in turn gets stabilized by the subsequent chemical events, in contrast with the conventional idea of local changes somehow getting amplified into larger-scale motions. In the case of myosin, for example, we favor the idea that Brownian motions associated with the flexible converter propagates to the Switch I/II region, where the salt-bridge formation gets stabilized by ATP hydrolysis, in contrast with the textbook notion that ATP hydrolysis drives the converter motion. Another useful aspect of the BNM results is that selected low-frequency normal modes have been identified to form a set of collective coordinates that can be used to characterize the progress of a significant fraction of large-scale conformational transitions. Therefore, the present normal mode analysis has provided a stepping-stone toward more elaborate microscopic simulations for addressing critical issues in free energy conversions in molecular machines, such as the coupling and the causal relationship between collective motions and essential local changes at the catalytic active site where ATP hydrolysis occurs.
INTRODUCTION
Motions are essential to the function of macromolecules (Brooks III et al., 1988; Gerstein and Krebs, 1998; McCammon and Harvey, 1987). Large-scale motions are commonly found in many biological molecules such as multidomain enzymes (Bahar et al., 1999; Thomas et al., 1999), signal transduction proteins (Ma and Karplus, 1997), and the ribosome (Frank, 2003). The large structural rearrangements are essential for the functions of these systems: e.g., binding and dissociation of substrates (Joseph et al., 1990); allosteric responses to signaling events; and release of synthesized proteins (peptides). The most dramatic class of systems where large-scale motions are crucial, however, has to include molecular machines (Banting and Higgins, 2000; Blumenfeld and Tikhonov, 1994; Hill, 1977; Schliwa, 2003) that convert free energies among various forms (see below). Although the mechanism of energy conversion has been a topic of great interest some 20 years ago (Hill, 1977; Hill and Eisenberg, 1981; Jencks, 1980; Simmons and Hill, 1976), the subject has been revived recently (Blumenfeld and Tikhonov, 1994). The free energy transduction processes can now be discussed in more structural and kinetic detail (Bustamante et al., 2001; Rees and Howard, 1999; Vale and Milligan, 2000), due in part to the observation of motor functions at unprecedented resolution offered by rapidly developing single molecule techniques (Ishii et al., 2003; Jung et al., 2002; Xie, 2002). Nevertheless, due to the challenges in making concurrent measurements at both high spatial and temporal resolutions, there remain many open questions concerning the detailed mechanisms of molecular motors.
The textbook description (Alberts et al., 1994) of molecular motors is that they harness the free energy of adenosine triphosphate (ATP) hydrolysis or phosphate transfer (i.e., phosphorylation) to perform net work. For example, kinesin motors can drag vesicle cargoes along microtubules in an unidirectional manner with ATP binding and/or hydrolysis (Hirokawa and Takemura, 2003). In each functional cycle, typically one ATP molecule in solution binds to the motor protein and is hydrolyzed (or phosphate-transferred), followed by the release of products back into solution; the net free energy change associated with the entire functional cycle is equivalent to the free energy of ATP hydrolysis in solution. This is the free energy that the molecular motor can, from the thermodynamics point of view, harness to generate useful work, although the actual hydrolysis obviously occurs in the protein rather than in solution (Hill, 1977). The precise molecular mechanisms for such free energy harness or conversion process have not been fully clarified. It is clear that it is not the free energies of the chemical reactions that are directly utilized because the exothermicity of these reactions in the protein, even if significant, will most likely dissipate to the rest of the molecule in pico-/nanoseconds without strongly preferred directions (Sagnella and Straub, 2001; Wang et al., 1998), which is too short compared to the timescale of motor functions (μs-ms) to have any functional significance. Moreover, the hydrolysis reaction in the hydrolyzing state of the ATPases often has an equilibrium constant close to 1 (Boyer, 1993; Holmes and Geeves, 1999). The key to the free energy conversion, therefore, has to do with the coupling of the protein structure and the ATP molecule in its various chemical states (Yang et al., 2003); for motors with tracks, such as myosin, the protein/track interaction is also an important element. Different protein or protein-track conformations could be preferred with different ATP states (solvated ATP, bound ATP, hydrolysis transition state, and ADP+Pi), and free energy is transferred between ATP and protein/track through well-coordinated conformational changes of the protein and binding/chemical modification of ATP.
It is our long-term goal to employ various types of simulations to provide the missing link between static high-resolution crystal structures and other lower-resolution studies such as single molecule investigations and phenomenological models for molecular motors. This, in light of the above discussion, necessarily involves understanding the coupling between the conformational properties of molecular motors and the chemical events; e.g., what are the structural flexibilities of various conformational states, what are the conformational transition pathways among these states, how does ATP affect these conformational properties and, in turn, how do different conformations determine the chemical fate of ATP? In the current work, we take the initial step to discuss the structural flexibilities in two distinct types of molecular machines: myosin-II and Ca2+-ATPase. The former is a prototypical molecular motor that utilizes the free energy of ATP binding and/or hydrolysis to perform mechanical work (Bagshaw, 2000; Keike and Titus, 2003), and the latter is the first P-type ion pump that has been characterized at atomic resolution (Lee, 2002; Toyoshima et al., 2000; Toyoshima and Nomura, 2002); Ca2+-ATPase transports calcium ions across the cell membrane against a concentration gradient with the help of ATP. Both undergo substantial conformational transitions during their functional cycles (see below). We set out to explore the structural flexibility inherent in these two systems and the corresponding functional significance; i.e., we inquire whether the requirement of making specific conformational transitions poses any constraints on the construct of those systems. Another important motivation is to explore useful schemes to characterize the functional transitions in conformations and identify the key degrees of freedom (Bustamante et al., 2001) such that, ultimately, the corresponding energetics and kinetics can be probed. The particular methodology we employed is an approximate normal mode analysis approach, the block normal mode (BNM) method (Li and Cui, 2002), which is based on the rotational-translational block (RTB) method of Tama and co-workers (Durand et al., 1994; Tama et al., 2000). As discussed below, although these normal mode results are highly approximate, they provided useful insights into the intrinsic structural features of myosin and Ca2+-ATPase; the results also revealed a set of collective coordinates that can be used in more elaborate microscopic simulations that can potentially offer more complete descriptions of the conformational transitions that occur in the functional cycle of molecular motors. In a recent article (Li and Cui, 2002), the low-frequency modes of Ca2+-ATPase in the calcium-loaded state (Toyoshima et al., 2000) have been discussed with the BNM method; here we focus on the correlation between low-frequency modes and the conformational transitions involving both the calcium-loaded and empty states, which was made possible by the recently solved x-ray structure for Ca2+-ATPase in the empty state (Toyoshima and Nomura, 2002).
The article is organized as follows. We first briefly reiterate the general value and limitation of normal mode analysis in studying large-scale conformational transition problems in biomolecules. We will then concisely review the approximate normal mode approach used in the calculations. Next, we present the normal mode results of myosin-II and Ca2+-ATPase, and discuss the relation between low-frequency modes of these proteins and their functions. Finally, we conclude with a number of summarizing statements.
COMPUTATIONAL METHODS
Normal mode analysis of large-scale conformational transitions: values and limitations
Although normal mode analysis has been applied to a large number of macromolecules (Bahar et al., 1997a; Case, 1994; Kitao and Go, 1999; Ma and Karplus, 1998; Marques and Sanejouand, 1995), it is useful to reiterate the value and limitation of this simple approach for analyzing conformational transitions in biological systems. The major challenge that hampers us from directly simulating large-scale conformational transitions in macromolecules is that these processes are diffusive and occur at a timescale (microseconds to milliseconds) much longer than what can be achieved using regular atom-based molecular dynamics with standard computational facilities (Elcock, 2002; Karplus and McCammon, 2002). One solution is to employ equilibrium simulations to explore thermodynamical quantities such as potential of mean force, in which time is not explicitly involved and kinetic properties can be estimated based on various rate theories (Hanggi et al., 1990); such types of calculations proved to be valuable for exploring the folding mechanism of small peptides and proteins (Shea and Brooks III, 2001). Another possibility is to use nonequilibrium simulations in which the biomolecule is biased along certain degrees of freedom, such as the root-mean square (RMS) difference from a given conformation (Schlitter et al., 1993); this type of calculation has been used to explore qualitative features associated with conformational transitions in molecular motors (Bockmann and Grubmuller, 2002; Ma et al., 2002) or ligand dissociations/translocations (Grubmuller et al., 1996; Isralewitz et al., 2001). In both schemes, the key is to identify appropriate reaction coordinate or order-parameters that concisely characterize the slow process. A fundamental assumption in those simulations is that all orthogonal degrees of freedom are in equilibrium during the simulation, which is usually difficult to justify for macromolecules but has to be made for practical purposes.
Compared to molecular dynamics-based methods such as principal component analysis (Amadei et al., 1993; Hayward et al., 1993; Kitao et al., 1991), normal mode analysis (NMA) is a simple but powerful approach for exploring the intrinsic structural flexibility (motion) of macromolecules (Brooks et al., 1995; Case, 1994; Go et al., 1983). The most significant advantage of NMA is that it is not limited by the timescale of the process under study, which makes it a unique tool for analyzing large-scale conformational transitions (Gerstein and Krebs, 1998). For example, several previous studies (Ma and Karplus, 1998; Tama and Sanejouand, 2001; Thomas et al., 1996, 1999) have illustrated that large-scale conformational changes can often be represented concisely by a very small number of low-frequency normal modes, which makes NMA an efficient approach for defining collective coordinates (Basu et al., 1995; Hayward and Go, 1995; Kitao and Go, 1999) that can be used in either equilibrium or nonequilibrium MD simulations; this is one of the important motivations for us to carry out NMA for the molecular motors of interest before more elaborate MD studies.
The most serious limitation of NMA is the harmonic approximation around a single conformational minima (Brooks et al., 1995), which seems to be a tremendously oversimplified model for macromolecules considering the rugged energy landscape associated with the high-dimensionality of these systems (Frauenfelder et al., 1991); another practical constraint is that NMA is usually carried out without the explicit representation of solvent molecules, which are well known to play important roles in determining the equilibrium and dynamical properties of macromolecules. Application of normal mode analysis to a large number of medium-size proteins, however, have shown that the characters of collective motions that correspond to the very low-frequency modes (Kitao and Go, 1999) are rather robust and NMA often gives similar results compared to molecular dynamics simulations (e.g., quasiharmonic analysis, Doruker et al., 2000; or singular value decomposition, Kundu et al., 2002). Physically, these results indicate that the motions associated with the low-frequency modes of macromolecules are most sensitive to the packing density of atoms; indeed, it has been shown that fairly robust results can be obtained for the low-frequency modes and patterns in the thermal atomic fluctuations by considering simplified models that contain only the packing distributions of atoms (Atilgan et al., 2002; Bahar et al., 1997b; Halle, 2002; Tirion, 1996). We emphasize that what is conserved in NMA is the direction of the collective motions, but not their actual timescales or thermal amplitudes; after all, these motions were found to be typically overdamped by the solvent molecules, which are usually neglected in NMA calculations. Moreover, for an analysis of conformational transitions, the directions of low-frequency modes are more relevant than their thermal amplitude (see below). Another important effect of the solvent molecules is that their presence increases the number of local minima on the potential energy surface, thus there are frequent transitions among various minima in solution. However, at least for small systems such as melittin, calculations indicated that the envelope of the potential energy surface in solution can be predicted correctly by normal mode analysis in vacuum (Kitao et al., 1991). In other words, although there may not be 1:1 correspondence between normal modes and the more realistic principal components that include anharmonic motions of the system, the subspace that dominates the atomic fluctuations can be captured by a linear combination of the low-frequency, collective normal modes (Kitao and Go, 1999; Kitao et al., 1998).
Approximate normal mode analysis: the block normal mode (BNM) approach
Despite the observed success of simplified elastic models in describing low-frequency modes of macromolecules (Doruker et al., 2000; Kundu et al., 2002; Tama and Brooks III, 2002), it is still desirable to be able to explore these motions with atomic level details and a physical representation of intermolecular interactions to ensure robustness of the calculations. This is especially true when the molecular system involves heterogeneous distributions of different chemical groups, such as in ribosome and RNA polymerase, which involves both protein and nucleic acids. To make connections between the normal modes of biomolecules and their mechanical properties (e.g., elastic modulus), a model with physical interactions is also more useful.
The computational expense associated with normal mode analysis in an all-atom representation, unfortunately, scales rather steeply with the size of the system (N atom); nominally, the Hessian matrix scales as N2, and the diagonalization of this Hessian scales as N3 with the most standard diagonalization procedures. To reduce the computational cost, we have recently made an efficient implementation of the BNM approach (Li and Cui, 2002), which was based on the RTB method originally proposed by Tama and co-workers (Durand et al., 1994; Tama et al., 2000). In RTB, the macromolecule is first partitioned into blocks, where each block may range from a single residue to a secondary structure element depending on the problem in hand. The full atomic Hessian matrix is then projected onto a subspace spanned by the translation and rotation vectors associated with these blocks, which results in a much smaller matrix to be diagonalized. BNM is more general than RTB in that other eigenvectors of the blocks can also be included, which is appropriate when blocks are large in size. The merit of the RTB/BNM approach is that the cost of calculation is dramatically reduced with the full account of intermolecular interactions; the coarse-graining procedure does not compromise the accuracy of results when only the low-frequency, collective modes of the macromolecule are of interest (Li and Cui, 2002; Tama et al., 2000). Test calculations using small protein systems (<200 residues) and one residue per block indicated that a very good correlation (R2 ∼ 0.997) between the BNM frequencies and standard NMA frequencies was observed up to at least 40 cm−1; although the overlap between the BNM eigenvectors and standard NMA eigenvectors become <0.5 after the frequency reaches 10 cm−1, the subspace spanned by the NMA eigenvectors can be represented >90% by the BNM eigenvectors for frequencies up to 25 cm−1.
Compared to the original work of Tama and co-workers (Durand et al., 1994; Tama et al., 2000), our BNM implementation has the essential advantage that the projected Hessian matrix is constructed in a direct fashion (i.e., without the construction of the entire atomic Hessian), which makes the application to fairly large systems (N < 1000 residues) very straightforward. Recently, we have substantially increased the applicability of BNM by taking advantage of a sparse matrix diagonalization procedure (Cullum and Willoughby, 1985; Lanczos, 1950) implemented in the PARPACK package (Maschhoff and Sorensen, 1996) and parallel computations. These refinements made it possible to analyze collective motions of supermolecular assemblies such as the ribosome at atomic resolution with modest computational cost; BNM calculations for a 50S ribosome (∼128,000 atoms with a polar hydrogen set of force field, Brooks et al., 1983) with one residue per block took <17 h on a single node of a 2.0-GHz Athlon. The details of the new implementation and application to large molecular assemblies will be given separately.
Computational models and protocols
The newly implemented BNM method in CHARMM (Brooks et al., 1983) was used to analyze the structural flexibilities in two molecular motors where large-scale motions are critical to their functions: Ca2+-ATPase (Lee, 2002) and myosin (Holmes and Geeves, 1999; Steffen et al., 2003). For myosin, calculations were done for the prehydrolyzing state (see Scheme 1) with bound Mg2+·ATP (PDB code 1FMW, Bauer et al., 2000); the structures for the motor domain without ATP or ADP were found to be very similar to 1FMW (Bauer et al., 2000; Gulick et al., 1997) and therefore have not been calculated. Substantial conformational transition occurs in the ATP hydrolyzing state (PDB code 1VOM, Smith and Rayment, 1996), and therefore the normal modes for the prehydrolysis state was analyzed in terms of the conformational difference with the hydrolyzing state. Due to the many missing residues in the 1VOM structure, no normal mode analysis has been done for that structure.
SCHEME 1.

The functional cycle of myosin-II-actin (only a myosin-monomer is shown).
The Ca2+-ATPase consists of a transmembrane domain (TMD) that includes 10 helices, and a cytoplasmic piece that includes three domains that have been termed N (nucleotide binding), P (phosphorylation), and A (actuator) domains (Toyoshima et al., 2000). In the popular proposal (Lee and East, 2001), the functional cycle of Ca2+-ATPase adopts at least two conformational states, generally referred to as E1 and E2 (Scheme 2); it was proposed that the x-ray structures with and without Ca2+ ions correspond to the E1·Ca2+ (Toyoshima et al., 2000) and E2 states (Toyoshima and Nomura, 2002), respectively, although the E2 state was crystallized with the inhibitor thapsigargin. The low frequency modes for the E1·Ca2+ state have been briefly analyzed in a previous publication (Li and Cui, 2002); the availability of the E2 x-ray structure (Toyoshima and Nomura, 2002) allowed us to characterize the conformational transitions in terms of the low-frequency modes, which is the major focus of the current work.
SCHEME 2.

The functional cycle of Ca2+-ATPase in the E1-E2 model.
For all the systems studied here, the protein atoms were described with the united atom representation associated with the CHARMM 19 force field (Neria et al., 1996). The solvation effect was approximated for myosin with an effective solvent model EEF1 (Lazaridis and Karplus, 2000), which contains screened electrostatic interactions and a Gaussian term that represents hydrophobic interactions. For Ca2+-ATPase, which is a membrane-bound system, vacuum calculations were performed; the partial charges associated with charged residues were scaled by 0.3 to approximately account for the dielectric shielding due to the environment. Although such a treatment does not accurately account for the heterogeneous lipid-solvent environment, it is expected to be sufficient for exploring global structural features associated with the Ca2+-ATPase. This is also supported by the fact that MD simulations with a similar setup maintained the system close (RMS deviation from the x-ray structures are <3 Å for all heavy atoms) to the x-ray structures (for both calcium-loaded and calcium-free states) for 10 ns. Moreover, such a protocol leads to a salt-bridge interaction on the order of 1 to 3 kcal/mol, which is in accord with the results of molecular dynamics simulations with explicit solvents as well as mutation experiments involving charged groups (Northrup et al., 1980; Simonson et al., 1997). In both EEF1 and scaled-partial charge setups, a distance-dependent dielectric constant was used. Previous comparisons of vacuum normal mode analysis and solution quasiharmonic analysis indicate that the two approaches in general gave similar descriptions for the directions of collective motions of proteins (Hayward and Go, 1995; Hayward et al., 1993; Kitao et al., 1991), although the explicit solvents lead to the overdamp of large-scale motions (i.e., substantially longer relaxation times) and increase of number of minima on the potential energy surface.
Starting from the x-ray structures (see Table 1 for a summary), the systems were first energy-minimized before normal mode analysis. The minimization protocol started with 500 steps of steepest descent followed by ∼5000 steps of adapted basis Newton-Rhapson (Brooks et al., 1983). To prevent undesired structural distortions, gradually decreasing harmonic constraints with respect to the x-ray structure as the reference was used in the minimization. Another 3000 adapted-basis Newton-Rhapson minimization steps were finally carried out without constraints, which produces the final structure with a typical RMS gradient on the order of 0.01 kcal/(mol × Å). Such a threshold is sufficient for the current purpose, as reflected by the fact that the normal mode analysis does not produce imaginary modes. Moreover, the translation and rotation modes have frequencies on the order of 0.05 to 0.10 cm−1. The RMS deviations between x-ray and minimized structure are typically 1.5 Å (see Table 1).
TABLE 1.
Systems studied using the BNM method in this work
| System | PDB code | Res. | No. of residues* | No. of atoms | Dimension (Å) | RMSD†(Å) | Sparse ratio‡ | No. of computed modes | Wall time‡;§ |
|---|---|---|---|---|---|---|---|---|---|
| Ca2+-ATPase, E1·Ca2+ | 1EUL | 2.6 | 994/0 | 9307 | 86 × 99 × 117 | 1.30 | 40 | 200 | 2 |
| Ca2+-ATPase, E2 | 1IWO | 3.1 | 994/0 | 9305 | 84 × 81 × 135 | 1.60 | 40 | 200 | 2 |
| Myosin-II | 1FMW | 2.15 | 738/451 | 8688 | 68 × 93 × 69 | 1.59 | 31 | 100 | 0.75 |
The numbers before the slash are the number of amino acids or nucleotides; the numbers after the slash are the number of x-ray-resolved water molecules.
The RMSD is between the minimized structures relative to the x-ray data for all backbone atoms. For the calculation setups, see subsection Approximate Normal Mode Analysis: the Block Normal Mode (BNM) Approach. For myosin, an implicit solvent model (EEF1) was used, and the corresponding group-based cutoff scheme is 11.0/9.0/7.0 Å; for Ca2+-ATPase, which is a membrane bound protein, no implicit solvent model was used, and the corresponding atom-based cutoff scheme is 8.0/7.5/6.5 Å. In the cutoff scheme, the first number is the distance cutoff in generating the list of nonbond pairs; the second number is the distance at which the switching function eliminates all contributions from a pair in calculating nonbond energies; and the third number is the distance at which the switching function begins to reduce a pair's nonbond contribution.
In those calculations, each block consists of one residue (amino acid, nucleotide, water molecule, or a Magnesium ion). The sparse ratio is defined as the number of elements larger than a given threshold (10−8 kcal/(mol × Å2)) divided by the total number of projected Hessian elements.
The wall time (in h) was for one 1.2-GHz Athlon machine with 1-GB memory.
In the BNM analysis, each block included only one residue; each residue could be an amino acid, nucleotide, water molecule, or metal-ion. Due to the use of a sparse matrix diagonalization procedure, the BNM calculations can be readily carried out for all the systems on a single CPU in <2 h.
The results of the BNM calculations were analyzed in terms of the root-mean square atomic fluctuations (RMSF, 〈Δr2〉), participation ratios (Leitner, 2001; Sagnella and Straub, 1999), and visual inspections. The RMSF and participation ratios associated with the kth normal mode are defined by
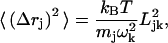 |
(1) |
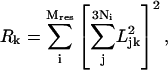 |
(2) |
where Mres is the number of residues in the system, mj is the mass of the jth atom, and Ni is the number of atoms in the ith residue; Ljk and ωk are the jth components and vibrational frequency associated with the kth eigenvector, respectively. As discussed extensively in previous work (Leitner, 2001; Sagnella and Straub, 1999), the reciprocal of Rk corresponds approximately to the number of residues involved in the kth normal mode. To further characterize the motions involved in the normal modes, the Dyndom package (Hayward et al., 1997) was used to divide the structure into dynamical domains (to be distinguished from the structural domain identified based on static geometry) and identify hinge-bending residues associated with the relative displacements of these dynamical domains; for each normal mode analyzed, two structures were generated by following the corresponding eigenvector with phase angles of 90° and 270°, respectively, which were then used as inputs for Dyndom.
For myosin and Ca2+-ATPase, where multiconformations are known from x-ray crystallography, the individual involvement coefficients were also computed to reveal the connection between the normal modes and the conformational transitions (Tama and Sanejouand, 2001),
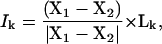 |
(3) |
where X1–X2 is the displacement vector between two conformations. A large involvement coefficient indicates that the motion along this normal mode is highly relevant to the conformational transition being examined. A related quantity that indicates the weight of a set of normal modes in a conformational transition is the cumulative involvement coefficients,
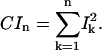 |
(4) |
Since the normal mode eigenvectors of a macromolecule form a complete orthonormal set for the 3N-dimensional space, CI3N is expected to be 1; in other words, 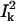 measures the maximal percentile contribution of exciting motions along the kth mode to the conformational difference between the two states.
measures the maximal percentile contribution of exciting motions along the kth mode to the conformational difference between the two states.
For an analysis of conformational transitions, the involvement coefficients from normal mode analysis are of great interest because they measure the relative importance of motions along a given normal mode. If the involvement coefficients are dominated by low-frequency normal modes, it is reasonable to expect that Brownian motions along these directions can accomplish large-scale conformational transitions with significant probability/rate. We note that thermal amplitude of a normal mode, which is an equilibrium quantity defined in terms of the mean-square displacement of the oscillation, is less relevant (compared to the direction of the mode) in conformational transition problems; therefore, the fact that normal mode analysis with simplified solvent treatment does not give quantitative thermal amplitudes is of little concern for our purpose. Take a simple example of gauche-trans isomerization of butane. The isomerization is dominated by a single torsional mode, which implies an involvement coefficient close to 1 for that mode. Although a normal mode analysis cannot directly predict any rate constant, the fact that the isomerization motion is dominated by a very low-frequency torsional mode implies that the barrier is likely to be low, relative to other type of processes such as breaking of the C–C bond. The thermal amplitude of the torsional mode is small at low temperatures, but this is obviously not contradictory to the importance of the torsional mode in the isomerization.
STRUCTURAL FLEXIBILITY AND FUNCTION OF BROWNIAN MOLECULAR MACHINES: MYOSIN AND CA2+-ATPASE
In this section, the structural flexibilities of myosin and Ca2+-ATPase are first discussed in terms of their low-frequency modes from BNM calculations. The functional relevance of these motions is illustrated with involvement coefficients, which provide a semiquantitative scheme for characterizing large-scale conformation transitions that are valuable for more elaborate MD simulations in the future. In the last subsection, we put the results into a more general framework and discuss the value of low-frequency modes in understanding motor functions.
Low-frequency modes of the motor domain in myosin
As shown in Fig. 1 a, the majority of conformational transitions in the myosin motor domain occurs in the C-terminal region, which is known as the converter; during the functional cycle of myosin, it is the large-scale displacement of this converter region that guides the motion of the longer level-arm (absent from the structure used here). When the two states of myosin (ATP prehydrolysis state and ATP hydrolyzing state, which will be referred to by their PDB code names, 1FMW, Bauer et al., 2000; and 1VOM, Smith and Rayment, 1996, respectively) are best fit with the first 650 amino acids, the total root-mean square difference (RMSD) for these residues is only 2.4 Å; the rest of the residues in the C-terminal region after such alignment, by contrast, have a much larger RMSD of 20.8 Å (Fig. 1 a). The displacement of the C-terminal region can be approximately described as a rigid body motion because residues in this region have a small RMSD of 2.5 Å after they are superimposed in the two states. In addition to the C-terminal part, two nearby regions, the N-terminal region (∼Asp-23) and a long α-helical region (Ser-465 to Glu-497, the “relay” helix) adjacent to the ATP binding site also undergo substantial changes (Fig. 2 a). Among the three structural motifs essential to ATP binding and hydrolysis (P-loop, Switch I, and Switch II), the Switch II region undergoes a substantially larger change compared to the other two (Fig. 1 b); it corresponds to the formation and break of a salt-bridge (Arg-238 and Glu-459), which was proposed to be important for ATP hydrolysis based on mutation (Onishi et al., 2002) and computational studies (G. Li and Q. Cui, unpublished). An essential issue for the mechanochemical coupling in myosin concerns the relation between the larger-scale C-terminal motion and the more localized changes in the ATP binding site, and to what extent these motions are reflected by the intrinsic structural flexibility of the motor domain (i.e., low-frequency modes).
FIGURE 1.

The difference between the prehydrolysis (1FMW) and hydrolyzing (1VOM) state of the Dictyostelium discoideum myosin-II motor domain. (Left) The superposition of the motor domain in the two states based on the backbone atoms in residues 1–650; red and blue corresponds to 1FMW and green and yellow indicates the 1VOM state. As illustrated by the arrow, the majority of the conformational difference occurs in the C-terminal converter region. (Right) The structural difference between the active sites of the two states, with the same color coding; the ATP molecule is shown in van der Waals representation, and the three important motifs for ATP hydrolysis are shown in ribbon-type representation.
FIGURE 2.

The correspondence between structural difference and low-frequency normal modes in myosin-II motor domain. (a) The displacements of Cα atoms between the prehydrolysis (1FMW) and hydrolyzing (1VOM) structures, with the essential active site motifs highlighted; (b) the root-mean square fluctuations (RMSF) of Cα atoms in the prehydrolysis state calculated using the 100 lowest-frequency modes at 300 K (Eq. 1); (c) the individual involvement coefficients (Eq. 3) for the first 100 modes; and (d) the cumulative involvement coefficients (Eq. 4). In c and d, the overall translational and rotational modes are shown with negative mode indices; they contribute because the structures are superimposed with residues 1–650 (see text).
Based on the root-mean square fluctuations (RMSF) from the first 100 modes (Fig. 2 b), the most mobile regions include the C-terminal converter and a loop region in the upper 50-K domain (Fig. 1 a), which form the opposite ends of the structure (Fig. 1 a). The two regions that are close to the C-terminal converter (the N-terminal and the relay helix), which exhibit substantial differences between the two states (Fig. 2 a), also have significant fluctuations (Fig. 2 b). Residues in other regions, including those at the ATP binding site (P-loop, Switch I, and Switch II), have much smaller fluctuations (see below). Therefore, the general picture is that the low-frequency motions in myosin mainly involve the floppy ends of the structure (C-terminal, upper 50 K) and little displacement in the active site. The intrinsic flexibility in the C-terminal converter region is consistent with previous small-angle x-ray solution scattering (Sugimoto et al., 1995) and an approximate normal mode analysis (Higo et al., 2001). It is also consistent with the observation that the motor domain can adopt either the open (1FMW) or the closed states (1VOM) with ADP·BeF3 as the ligand (Fisher et al., 1995; Holmes and Geeves, 1999; Houdusse et al., 2000).
The relevance of the low-frequency modes in myosin to the conformational transition is more quantitatively illustrated by the involvement coefficients (Fig. 2, c and d). The individual involvement coefficients (Ik) are dominated by a number of low-frequency modes; e.g., only two intramolecular modes have Ik > 0.20. Since the two states (1FMW and 1VOM) were superimposed based on the first 650 residues, the overall translation and rotation also have notable contributions, although superimposing the two structures based on all heavy atoms gave very similar results in terms of involvement coefficients of internal normal modes. For example, one rotational mode has a large involvement coefficient ∼0.30 (Fig. 2 c), which highlights the fact that the conformational transition substantially changes the center of mass and moments of inertia of the system (note that intramolecular normal modes conserve the center of mass properties). The cumulative involvement coefficients (Fig. 2 d) indicate that nearly 50% of the overall conformational changes can be described in terms of motions in the direction of the 10 lowest-frequency modes. After the 10th mode, the increase of the cumulative involvement coefficients becomes substantially slower (Fig. 2 d); including all 100 modes, nearly 60% of the conformational changes can be accounted for.
The lowest-frequency internal mode (ω1 = 1.8 cm−1) has the largest involvement coefficient (I1 = 0.32), and it mainly involves the displacement of the C-terminal converter region and the upper 50-K domain (Fig. 3 a). As shown in Fig. 4 a, the converter region undergoes a rigid-body rotation, which is precisely the motion that corresponds to the essential conformational transition between 1VOM and 1FMW. Dynamical domain analysis (Hayward et al., 1997) using the two end-structures found the hinge axis, which is also shown in Fig. 4 a; the hinge regions (see Table 2) include residues Gln-112–Thr-117, Phe-482, Ile-514, and Gly-680, which are shown as the dark green region in the Dyndom plot (also see Supporting Materials for the positions of the hinge residues in the structure); Phe-482 and Gly-680 are in close contact in the crystal structure, with Phe-482 on the relay helix whereas Gly-680 is on the so-called SH1–SH2 helix (Fig. 1 a). To conserve the center of mass, a loop and the long α-helix in the upper 50-K domain rotate in the opposite direction. Although the difference in the converter is substantially larger than that in the upper 50-K domain between the two conformational states (Fig. 1 a), the magnitude of their motion is comparable in the normal modes, which is imposed by the conservation of the center-of-mass position. The magnitude of rotation in the converter domain predicted from the normal mode analysis at 300 K is much smaller than the difference between the actual x-ray structures of the two states (Fig. 1), which reflects the limitation of the harmonic assumption inherent in the normal mode analysis. As mentioned above, the active site has relatively small degree of motion in the low-frequency modes. In the first mode, for example, the observed displacement in Switch II (Fig. 4 b) has little overlap with the functional transition between the prehydrolysis to the hydrolyzing states (Fig. 1 b).
FIGURE 3.

The RMSF of Cα atoms in selected normal modes of the prehydrolysis state myosin-II motor domain (1FMW) that have large individual involvement coefficients. For each mode, the three numbers above each plot are the vibrational frequency (in cm−1), reciprocal participation ratio (Eq. 2), and the individual involvement coefficient (Eq. 3).
FIGURE 4.


Superposition of end-structures (in cartoon format) in selected normal modes of the prehydrolysis state myosin-II motor domain (1FMW) that have large individual involvement coefficients. The end-structures are generated based on the corresponding equation of motion, and they correspond to a phase angle of 90° and 270°, respectively; to clearly illustrate the structural variations, a high temperature of 1500 K was used. In each mode, red and blue indicate one structure; green and yellow correspond to the other. The arrows indicate qualitative in-plane motion of the modes, whereas “⊕” indicates out-of-plane rocking motion. Similar to Fig. 3, the three numbers in each plot are the vibrational frequency (in cm−1), reciprocal participation ratio (Eq. 2), and the individual involvement coefficient (Eq. 3). Also shown are the results (in Trace format) of dynamical domain analysis using the Dyndom software with these end-structures. Different domains are colored differently, with dark green indicating regions undergoing significant bending going from one end-structure to the other; for the hinge residues, see Table 2 and Supporting Materials. The arrows indicate the hinge-bending axis, where the colors of the axis and arrowhead indicate the two dynamical domains involved in the relative motion. In b, displacements in the ATP (in vander Waals representation) binding site are shown (see text).
TABLE 2.
Hinge regions identified for the normal modes have large involvement coefficients based on block normal mode analysis of myosin-II
| Mode | Hinge index | Hinge residues |
|---|---|---|
| Q1 | 1 | Q112-T117, I514, F482, G680 |
| Q2 | 1 | L222, S237, R238, S260 |
| 2 | L354, A378, S379, S417, R418 | |
| Q6 | 1 | L27, T28, F692, P693 |
| 2 | L421-L425, L529, | |
| Q8 | 1 | F25-T28, N81 |
| 2 | D509, S510 |
For each normal mode examined here, end-structures were first generated by following the corresponding eigenvectors at 1500 K; the two end-structures have phase angles of 90° and 270°, respectively. These end-structures were then analyzed with the Dyndom package, to identify the dynamical domains involved in the normal mode motion and the corresponding hinges for the relative displacements of these dynamical domains. See Supporting Materials for figures that illustrate the position of the hinge residues in the structure.
In the other internal modes that have large individual involvement coefficients (the second, sixth, and eighth modes), a common feature is the simultaneous motion in the converter region and the upper 50-K domain, with the majority part of the system staying rigid. The fluctuations of other regions are generally smaller in these modes compared to the first mode, which is also reflected by the participation ratios that are smaller by almost 100 (Fig. 3). Due to the orthogonality to the first mode, however, the patterns in the two regions (Fig. 4) are different from those in the lowest-frequency mode. For example, in the second mode (Fig. 4 c), the displacements in the converter and upper 50-K domain are nearly perpendicular to those in the first mode. In the eighth mode (Fig. 4 e), it is interesting that the actin binding motif (shown as violet in Fig. 4 e, also see Fig. 1 a) was also found to have substantial mobility. The hinge residues of these modes (see Table 2) are mainly focused at the upper 50-K domain (e.g., Leu-354, Ser-379) and the converter domain (e.g., Phe-692) as well as the N-terminal residues (e.g., Phe-25–Thr-28). It is interesting to note that the Switch I motif (Ser-237, Arg-238) is involved as the hinge region in the second mode.
Low-frequency modes of the Ca2+-ATPase in different functional states
As shown in Fig. 5, large changes are observed in the TMD and cytoplasmic domains between E1·Ca2+ and E2, and the magnitude of conformational change depends on the scheme used to superimpose the two structures. If best fitted based on TMD, the N, P, and A domains in the two states have very large RMSDs of 39.9, 14.3, and 20.2 Å, respectively; the TMDs have a RMSD of 5.8 Å. If best fitted based on all heavy atoms, the RMSDs for N, P, and A are 17.0, 9.4, and 19.3 Å, respectively, and that for TMDs is 10.3 Å. Therefore, aligning the structures based on TMD significantly amplifies the displacement of the N domain (Fig. 5). It is not immediately clear which is the better way to characterize the conformational transition: on one hand, the TMD is held by the lipid and thus it appears to be more physical to minimize the displacements of TMD during the best-fit procedure; on the other hand, normal mode analysis indicates that the low-frequency modes contain significant components in TMD that correspond to the transition between the two states when best fitted based on all heavy atoms. Qualitative trends for the correlation between low-frequency modes and conformational transition are the same using either scheme for alignment. Therefore, in the following discussion, we focus on results obtained by fitting the E1·Ca2+ and E2 structures based on all heavy atoms. It should be noted that the internal structures of the N, P, and A domains change little in the two states, especially for N and P; the internal RMSD in the three domains are 1.5, 1.2, and 3.6 Å, respectively. Therefore, the dominant components in the conformational transition between E1·Ca2+ and E2 involve rigid domain-motions of N, P, and A relative to TMD, and to a somewhat smaller extent, the internal structural changes in TMD.
FIGURE 5.

The superposition of calcium-loaded and calcium-free Ca2+-ATPase structures when they are aligned based on (a) backbone atoms in the transmembrane domain (TMD) and (b) all backbone atoms. In c and d, the corresponding displacements in Cα are shown respectively. In a and b, brown-red-purple-dark blue corresponds to the E1·Ca2+ state, and gray-green-blue-yellow corresponds to E2. In c and d, pale blue indicates the A domain, yellow indicates the N domain, pink indicates the P domain, and the rest corresponds to the TMD.
Overall, the BNM calculations indicate that the conformational difference between the E1·Ca2+ and E2 states can be characterized by a small number of modes in either state, although the number of modes with large contributions is smaller and more concentrated on the lowest-frequency modes in E1·Ca2+ (Fig. 6). After the two states are superimposed based on all heavy atoms, only five E1·Ca2+ modes have Ik > 0.20 (Fig. 6 a), and the first mode has a dominating value of 0.53 (i.e., 25% of the conformational difference between E1·Ca2+ and E2 can be accounted for by motions in the direction of the first mode). More strikingly, the cumulative involvement coefficient is almost 0.7 when considering only the 10 lowest-frequency E1·Ca2+ modes (Fig. 6 c). For modes in the E2 structure (Fig. 6 b), the individual involvement coefficients for the low-frequency modes are also generally larger than those of high frequencies, although the dominance is less obvious and shifted to higher frequency compared to the E1·Ca2+ case. For example, the E2 mode with the largest individual involvement coefficient is the fourth mode, which has a frequency of 1.1 cm−1 and an Ik of 0.40; the 18th mode has a frequency of 3.16 cm−1 and an Ik of 0.36. A few other modes have Ik ∼ 0.20, including, for instance, the lowest-frequency E2 modes and the 16th mode (2.98 cm−1). As a result, the cumulative involvement coefficient for the first 10 E2 modes is only 0.3, compared to the value of 0.7 with E1·Ca2+ modes; in other words, although 70% of the conformational difference between E1·Ca2+ and E2 can be described by motions in the direction of the 10 lowest-frequency modes in E1·Ca2+, only 30% of the conformational difference can be accounted for with motions in the direction of the 10 lowest-frequency modes of E2. Since motions along modes of higher frequencies are required in going from E2 to E1·Ca2+, the results imply that energy rises more rapidly around E2 than around E1·Ca2+ during the functional transitions.
FIGURE 6.

Individual (a, b) and cumulative (c) involvement coefficients calculated when the calcium-loaded/-free forms are aligned based on all backbone atoms. In a and b, the eigenvectors associated with the calcium-loaded and calcium-free structures are used, respectively. The overall translational and rotational modes are shown with negative mode indices, although they do not contribute significantly (see text).
Next, it is instructive to examine and compare the characters of low-frequency modes in the two functional states that are relevant to the conformational transition (Figs. 7–11). From a structural point of view, E2 is expected to be less flexible than E1·Ca2+ considering the fact that the cytoplasmic domains (N, P, and A) form more close interactions compared to the E1·Ca2+ structure where the N and A domains are far apart. This is clearly reflected in the difference between the low-frequency modes of E2 and E1·Ca2+.
FIGURE 7.

The RMSF of Cα atoms in selected normal modes of the calcium-loaded form (E1·Ca2+) of Ca2+-ATPase that have large individual involvement coefficients. For each mode, the three numbers above each plot are the vibrational frequency (in cm−1), reciprocal participation ratio (Eq. 2), and the individual involvement coefficient (Eq. 3).
FIGURE 11.

Similar to Fig. 10, but for the normal modes of the calcium-free form (E2) of Ca2+-ATPase.
In E1·Ca2+, the four modes with the largest individual involvement coefficients are the first, second, fourth, and ninth modes (Fig. 6 a). In the first two modes (Fig. 7, a and b; and Fig. 10, a and b), it is essentially the rocking of the N domain relative to the rest of the proteins; the directions for the rocking are perpendicular in the two modes. To conserve the center of mass, the rest of the protein also has substantial displacements, although the relative arrangements of P, A, and TMD remain essentially the same. Both modes have hinge residues (see Table 3) in the range of Thr-357–Gln-360 and Asp-601–Arg-604, which are at the interface between the N and P domains (see Supporting Materials for illustrations). In the E1·Ca2+ structure, the ATP binding site in the N domain (Lys-492) is very far (>25 Å) from the phosphorylation site (Asp-351) in the P domain, and therefore the high structural elasticity is critical such that the conformational transition to the phosphorylation state can occur in a diffusive manner without the requirement of much external driving force (Xu et al., 2002). This is clearly demonstrated by the fact that the two lowest-frequency modes in E1·Ca2+ have essentially pure rocking of the N domain relative to the rest of the system, and they both have large individual involvement coefficients; motions in the direction of two modes constitute 45% of the conformational difference between the two states! The second mode also involves motion of the P domain relative to the A/TMD domains (Fig. 10 b), for which the hinge residues are those at the P/A interface, such as Ile-348, Cys-349, and Ala-699–Thr-701 (Table 3).
FIGURE 10.

Superposition of end-structures in selected normal modes of the calcium-loaded form (E1·Ca2+) of Ca2+-ATPase that have large individual involvement coefficients. The end-structures are generated based on the corresponding equation of motion, and they correspond to a phase angle of 90° and 270°, respectively; to clearly illustrate the structural variations, a high temperature of 1500 K was used. The dotted circles indicate that the enclosed domains move almost like a rigid body. The brown-red-purple-dark blue corresponds to one end-structure, and gray-green-blue-yellow corresponds to the other. Also shown are the results (in Trace format) of dynamical domain analysis using the Dyndom software with these end-structures. Different domains are colored differently, with dark green indicating regions undergoing significant bending going from one end-structure to the other; for the hinge residues, see Table 3 and Supporting Materials. The arrows indicate the hinge-bending axis, where the colors of the axis and arrowhead indicate the two dynamical domains involved in the relative motion.
TABLE 3.
Hinge regions identified for the normal modes have large involvement coefficients based on block normal mode analysis of Ca2+-ATPase
| System | Mode | Hinge index | Hinge residues (regions) |
|---|---|---|---|
| E1·Ca2+ | Q1 | 1 | T357-Q360 (N/P); D601-R604 (N/P) |
| 1EUL | Q2 | 1 | K352, L356, T357, C364 (N/P); L600-P603 (N/P) |
| 2 | I348, C349, A699-T701, A710 (P) | ||
| Q4 | 1 | Q360-S362 (N/P); M599-D601 (N/P) | |
| 2 | V747, R751, Y754 (P/M5); P821, R822, L828 (L67) | ||
| 3 | W50, I54 (A/M1); T247, L249, Q250, K252, L253 (A/M3) | ||
| Q9 | 1 | L356-S362 (N/P); M599-D601 (N/P) | |
| 2 | E340, A746, V747 (P/M5); S823, P824 (L67) | ||
| E2 | Q1 | 1 | I743-A746 (M5) |
| 1IWO | 2 | I315 (M4); K758-I761 (P/M5) | |
| Q2 | 1 | E40-P42 (A/M1); L119 (A/M2); I232, G233, I235, R236 (A/M3) | |
| 2 | K246, T247 (M3) | ||
| 3 | L356, W552, T558, L600 (N/P) | ||
| 4 | E575, M576, S581-Y587 (N/A) | ||
| 5 | Y763, L764 (M5) | ||
| Q4 | 1 | L249, Q250 (M3); P312-T316 (M4) | |
| 2 | K572, E575, S581-Y587 (N/A) | ||
| 3 | E231 (A/M3) | ||
| 4 | D601 (N/P) | ||
| Q8 | 1 | F256, G257 (M3); A305-P308 (M4), S767-E771 (M5); L787-L795 (M6); M897-L904 (M8) | |
| 2 | L119, K120 (A/M2) | ||
| 3 | V734 (M5) |
See the legend of Table 2 for the procedures used to identify these hinges. The notations in the parentheses indicate the structural region to which the hinge residues belong. See Supporting Materials for figures that illustrate the position of the hinge residues in the structure.
In the fourth and ninth modes, the motions are very different from the first two modes. In the fourth mode (Figs. 7 c and 10 c), the N and P domains have very small displacements; the A domain rocks with an axis nearly parallel to the TMD, whereas the entire TMD twists around the norm of the membrane. The internal structure of either A or TMD remain rather rigid during the rocking and twisting motions. The hinge residues for the rocking of the A domain include those at the A/TMD interface, such as Trp-50 (A/M1) and Thr-247–Leu-253 (A/M3); the twist of TMD relative to the N/P domains mainly involve hinge residues on the M5 TMD helix, such as Arg-751 and Tyr-754. It is interesting to note that several residues in the loop connecting M6 and M7, Pro-821, Arg-822, and Leu-828, also serve as hinges for the motions involved; this loop is termed L67, and is believed to be essential for the signal transduction between the calcium-binding motifs in the TMD and the N/P domains (Lee and East, 2001; Zhang et al., 2001). In the ninth mode (Figs. 7 d and 10 d), the displacements of the P domain show up clearly; the P and A domains rock relative to each other, whereas the TMD twists as a rather rigid body around the norm of the membrane. The N/P interface (Leu-356–Ser-362; Met-599–Asp-601) is the hinge for the relative N/P motion and those on M5 (e.g., Val-747) as well as on L67 (e.g., Ser-823, Pro-824) are the hinges for the TMD twists.
In E2, the modes with large involvement coefficients are the first, second, fourth, and eighth modes, and the 14th, 16th, 17th, and 18th modes (Fig. 6 b); the involvement coefficients are the largest for the fourth and 18th modes, in contrast to the situation in E1·Ca2+ (Fig. 6 a). Due to the more compact structures, especially in terms of the orientation of the N domain and its interaction with the A domain, the normal modes in E2 are more complex and usually involve multiple hinges. Moreover, the modes in E2 exhibit evident internal motions of the TMD, with many hinge residues in the middle of transmembrane helices (see Table 3 and Supporting Materials). In the first mode, the P and A domains move approximately as a group, and the motion involves the rocking of this group, the N domain, and the twisting of the TMD (Figs. 8 a and 11 a). The hinges for the P-A motions are at the interface between P and M5 (Ile-743–Ala-746), whereas the hinges for the TMD twist involves a number of residues on M4 (Ile 315) and M5 (Lys-758–Ile-761). The motion of the second mode is rather complex and involves a number of relative displacements of various groups (Figs. 8 b and 11 b); these include the rocking of the N and A domains in directions perpendicular to those in mode 1, and twists of the TMD (for the displacement axis, see Fig. 11 b). The hinge residues exist on the interface between N/P, A/M1, A/M3, and A/P; two residues on M5, Tyr-763, and Leu-764 were found as hinges for the internal motion of the TMD. In the fourth mode (Figs. 8 c and 11 c), the N, P, and A domains and TMD move almost like rigid bodies, and the hinge residues occur at the interfaces between them (Table 3). In the eighth mode (Fig. 8 d and 11 d), the motion is rather distinct: it involves the relative opening and closing of the N and A domains, and complex motion of the TMD; there are many hinge residues that occur in different parts of TMD, including M3, M4, M5, and M8 (Table 3). The few higher-frequency modes (∼3 cm−1) that have large involvement coefficients (Fig. 9) are more complex in terms of atomic motions, and include various relative displacements of the N, P, and A domains and TMD; the TMD also undergo notable intradomain rearrangement (see the figure in Supporting Materials).
FIGURE 8.

The RMSF of Cα atoms in selected normal modes of the calcium-free form (E2) of Ca2+-ATPase that have large individual involvement coefficients. For each mode, the three numbers above each plot are the vibrational frequency (in cm−1), reciprocal participation ratio (Eq. 2), and the individual involvement coefficient (Eq. 3).
FIGURE 9.

Similar to Fig. 8, but for a few modes of substantially higher frequencies that have large involvement coefficients for E2.
DISCUSSION
Low-frequency modes and functional transitions in Brownian molecular machines
Given the x-ray structures for the various conformational states, what new insights can be obtained into the structure-function relationship of the system by performing normal mode analysis? Before the present study, similar normal mode studies (Ma and Karplus, 1998; Marques and Sanejouand, 1995; Tama and Sanejouand, 2001; Thomas et al., 1999) for a number of protein systems that involve large-scale conformational transitions found that a major component of these transitions can often be captured by a small number of low-frequency modes. The trend has been rationalized by the simple idea that such a design will ensure that the functional conformational transitions proceed without a significant energy penalty; i.e., the conformational transition is largely a diffusive rather than a thermally activated process. Here we explore this idea in the context of two representative molecular machines: myosin and Ca2+-ATPase, and investigate to what extent the functional transitions in these systems can be characterized as collective motions that are represented by low-frequency modes; the latter is an important objective because the results will impact on the design of more elaborate MD simulations for probing the energetics and kinetics of the conformational transitions.
For the two systems studied, which represent two distinct classes of molecular machines that convert free energy among various forms (i.e., chemical → mechanical in myosin, chemical → ion gradient in Ca2+-ATPase), it was found that the various conformational states indeed have the flexibilities that are highly correlated with the conformational transitions required by the function. The direct numerical evidence is that the individual involvement coefficients are significant for only a small number of low-frequency modes. In myosin, 45% of the conformational change that converts the system from the prehydrolysis to the hydrolyzing state can be described with motions in the direction of the first 10 normal modes. In the E1·Ca2+ state of Ca2+-ATPase, strikingly, 70% of the conformational difference from the E2 state can be described with only the 10 lowest-frequency modes. With the 10 lowest-frequency normal modes of the E2 state, however, <40% of the expected conformational transition can be accounted for, which implies that energy rises more rapidly around E2 compared to E1·Ca2+ during the isomerization (i.e., the initial transitions involves motions along modes of higher frequencies).
Intrinsic structural flexibilities in proteins that perform functions through conformational transitions have been probed with NMR for signal transduction proteins (Volkman et al., 2001). For several receiver domains involved in two component systems, for example, it was argued based on NMR relaxation and chemical shifts measurements (Volkman et al., 2001) that these proteins have the intrinsic structural flexibility in regions that undergo large-scale conformational transitions. Therefore, in contrast to the traditional mechanism in which conformational transition is triggered by chemical events (e.g., phosphorylation), it was argued that the protein has certain population in the active conformation even without the chemical activation; the role of the chemical event is simply to shift more populations toward the active conformation. In light of the normal mode results discussed here, we speculate that many molecular motors work through such a population-shift mechanism; i.e., the motor has intrinsic structural flexibility such that Brownian motions can lead to significant population of specific conformations where chemistry (e.g., ATP hydrolysis) occurs. For example, myosin has to switch from the open to the closed state to allow ATP hydrolysis, whereas Ca2+-ATPase has to swing the N (by >25 Å) and A domains to allow ATP binding and the subsequent phosphorylation. For myosin, this has been demonstrated with bulk tryptophan fluorescence studies for a single tryptophan-containing mutant of Dictyostelium myosin (Malnasi-Csizmadia et al., 2001a,b, 2000), which revealed that the open (1FMW)-closed (1VOM) transition does not require ATP hydrolysis per se and can be induced by a nonhydrolyzable ATP analog. The flexible constructs of myosin and Ca2+-ATPase is an important component of the Brownian ratchet model, which has been proposed to rationalize the unidirectional character of molecular motors (Astumian and Hanggi, 2002; Julicher et al., 1997). The ratchet effect, however, cannot be addressed directly with the normal mode results. In the literature (Astumian and Hanggi, 2002; Julicher et al., 1997), it is often loosely stated that the ratchet effect arises because ATP hydrolysis breaks detailed balance by driving the system locally out of equilibrium. We emphasize that the term out-of-equilibrium should not refer to the hydrolysis reaction per se, because the equilibrium constant associated with the ATP hydrolysis step is usually measured at close to 1 in motor systems (Boyer, 1993; Holmes and Geeves, 1999). In other words, ATP and ADP·Pi are in constant equilibrium, which has been illustrated by 18O isotope exchange results in systems such as F1-ATPase (Bagshaw et al., 1975) and myosin (Dale and Hackney, 1987). As already mentioned in the Introduction, this makes sense, because otherwise the energy released will be dissipated to the environment rather rapidly, which compromises the efficiency of the motor. Therefore, it is likely that the ratchet effect is related to other features of the functional cycle; e.g., it might be that products of the chemical events stabilize the new conformation over the initial one, or further events can occur with the new conformation; in the case of myosin, these correspond to the possibilities that ADP·Pi stabilize the closed conformation over the open (G. Li and Q. Cui, unpublished), and that the ADP·Pi state favors rebinding of myosin to actin, which is practically irreversible.
For most important conformational transitions, one expects a combination of collective displacements and local rearrangements in the active site. In myosin, the collective displacement mainly concerns the converter region, whereas the local rearrangement involves the salt-bridge region between Switch I and Switch II (Fig. 1 b), which is essential for the chemistry in the mechanochemical cycle (Holmes and Geeves, 1999; G. Li and Q. Cui, unpublished; Rayment, 1996). In calcium pump, the collective motion mainly involves displacements of the N and A domains, as well as some adjustments in the TMD, whereas local changes involve the reorientation of the side chains in Glu-58, Glu-771, and Glu-908 (Lee, 2002; Toyoshima and Nomura, 2002), which are essential for calcium binding. A key issue in the mechanochemical coupling of molecular motors concerns, therefore, the relationship between the collective and local motions. In the normal mode analysis for all the structures studied here, no local motions were evident in the low-frequency modes that have large involvement coefficients, although certain active site residues function as hinges for the essential normal modes (e.g., Arg-238 in myosin and Glu-361 in Ca2+-ATPase; see Tables 2 and 3). A possible scenario is that the local arrangement is a thermally activated process and thus corresponds to high energy/frequency motions, irrespective of the collective motions. Alternatively, it is possible that the local motions are gated by the collective motion (Berlin et al., 2001), and are also diffusive in nature (or a very fast thermally activated process) after the collective motions have occurred up to a certain degree; a remotely relevant classical example concerns the ring flip of a Tyr in the core of protein BPTI (McCammon and Karplus, 1980), which is gated by the breathing motion of the entire protein. Considerations along those lines suggest that functional transitions in some molecular motors might be more appropriately described as global elastic motions regulating essential local structural changes, which is in contrast with the conventional description of local structural perturbations somehow getting amplified into large-scale motions. In the case of myosin, for example, we favor that the critical issue is how Brownian motions associated with the converter domain get transmitted to the active site (Switch I/II regions), rather than the amplification of conformational transitions in the active site to the converter, as usually discussed in the literature. Nevertheless, it is clear that molecular dynamics simulations need to be carried out to examine the coupling and causal relationship between collective motions and local structural changes. In this regard, another important value of the BNM results is that they provide a set of collective coordinates that can be used to characterize the progress of large-scale conformational transitions in more detailed MD simulations.
It is of interest to relate the BNM analysis with available experimental results on the identity of residues important to the functional transitions. The general idea is that residues that act as hinge residues are expected to be important. We note that performing hinge analysis (Hayward et al., 1997) with important normal modes allows one to examine potentially important residues in more detail, compared to using only the x-ray structures as the two ending-points; with Ca2+-ATPase, for example, the only hinge regions identified with the x-ray structures are those at the N/P and A/TMD interfaces, whereas analysis using structures generated with normal modes identified many other hinges at the P/TMD interfaces and within the TMD (see subsection Computational Models and Protocol).
For myosin, we noted a very recent experiment by Ito et al. (2003), who showed that mutating Phe-482 into a smaller residue such as Ala abolished the motility of myosin in vivo. The double mutant F482A/G680F, by contrast, was functional both in vivo and in vitro. These experimental observations indicated the importance of F482/G680 in the functional transition, which is consistent with the fact these residues were found to be hinge residues for the first mode in the current BNM analysis (see Table 2). Another mutagenesis study (Sasaki et al., 2003) that also appeared recently demonstrated that mutations I499A and F692A completely abolished the motor function of myosin II in vivo and in vitro, although the ATPase activity was not altered. In the BNM calculations, Phe-692 was found to be a hinge residue in the sixth mode (Table 2), which has a large involvement coefficient. Therefore, the BNM analysis generated encouraging insights into the role of specific residues in the functional transitions, although more detailed analysis with molecular dynamics is clearly required for more thorough understanding. We hope that the current study can stimulate further mutations studies on residues that we predict to be important in the functional transitions.
On calcium pump, a large number of mutation studies have been carried out (Andersen, 1995; Andersen and Sorensen, 1996; Krupka, 1994), although many of these are targeted toward calcium binding pathways or inhibitor binding sites (Andersen and Vilsen, 1995). Among the mutants constructed, the most relevant to the current study are those unable to phosphorylate with ATP/Pi as substrate. Most of these phosphorylation-negative mutants involve mutations at the N-P interface (Asp-351–Thr-357, and Asp-601, Pro-603) (Andersen and Vilsen, 1995). Other than Asp-351, which is chemically involved in the phosphorylation, it is likely that other residues are critical because they regulate the structural flexibility of the N domain; this is consistent with our finding that they are hinge residues in the critical low-frequency modes of E1·Ca2+ (Table 3). The loop connecting M6 and M7 (L67) (Andersen et al., 2003; Zhang et al., 2001) was also found to be important to the communication between calcium binding and the ATPase activity (Lee and East, 2001). Mutating the conserved residues Asp-813, Asp-815, and Asp-818 to Asn was found to interfere strongly with ATPase activation by Ca2+ binding (Zhang et al., 2001). Kinetic measurements also demonstrated that the hydrogen bond network involving Asn-813 and Lys-758 is important for the E2 → E1·Ca2+ transition (Andersen et al., 2003). These mutation results are qualitatively consistent with the current results that residues in L67 and Lys-758 are hinges in the low-frequency modes of E1·Ca2+ and E2, although a more concrete understanding requires further simulation studies. There are also many mutations that block the transitions between E1P and E2P states (Andersen and Vilsen, 1995); they are more difficult to interpret with the current results since calculations were only performed at the E1·Ca2+ and E2 states.
Before closing, we note that not all the low-frequency modes are important to the conformational transitions that are directly relevant to the function. Indeed, many low-frequency modes have vanishingly small involvement coefficients in myosin and Ca2+-ATPase studied here. One explanation is that the x-ray structures represent only a subset of conformations available to the motor system. Alternatively, it is also possible that only a subset of structural flexibility is taken advantage of by the motor protein to perform relevant functions. The superfluous flexibility could be simply a byproduct of evolution (e.g., an inevitability of packing amino acids in a certain way) or could be utilized for other purposes. Therefore, for systems with detailed structural information for only one conformational state, it remains difficult to predict unambiguously other relevant functional states with normal mode analysis alone. Combined with other information, however, normal mode analysis is a powerful approach for stimulating new experiments to test various hypotheses that ultimately will lead to an improved understanding of the relevant mechanism. For example, it should be highly interesting to use the normal mode eigenvectors from a high-resolution structure as the basis set to carry out structure reconstruction for other functional states with low-resolution data (e.g., electron microscopy); i.e., one optimizes the linear combination coefficients of the eigenvectors such that the produced high-resolution model reproduces the low-resolution data for the new functional state. Compared to other flexible docking algorithms (Wriggers and Chacon, 2001), the advantage of a normal mode-based approach is that the number of variables need to be optimized is significantly smaller.
CONCLUSIONS
Myosin-II and Ca2+-ATPase are two prototypical molecular motors that carry out bioenergy transductions using the chemical energy in ATP: the former performs mechanical work whereas the latter carries out vectorial transport of calcium ions against a concentration gradient. As an attempt to understand the principles behind the construction of these molecular machines, we analyzed the structural flexibility of myosin-II and Ca2+-ATPase in terms of their low-frequency normal modes and explored the connection between the normal modes with the large-scale conformational transitions implicated in their functional cycles. The present analysis was made possible by a further improvement of a coarse-grained normal mode analysis method, the block normal mode (BNM) approach (Li and Cui, 2002; Tama et al., 2000), that has been recently implemented into the molecular simulation package CHARMM.
It was found that for all the structures analyzed here, there is a good correspondence between the low-frequency modes and the large-scale conformational transitions in the functional cycles of these systems. Although it is well recognized that atomic fluctuations in macromolecules are dominated by low-frequency modes (Case, 1994; Kitao and Go, 1999), it is not necessarily obvious that a majority of the conformational transition is dominated by a small number of low-frequency modes, as found in the current analysis of molecular motors as well as previous studies on several other protein assemblies whose functions rely on conformational flexibilities (Marques and Sanejouand, 1995; Tama and Sanejouand, 2001; Thomas et al., 1996, 1999). We believe that such a feature is more prevalent in molecular motors than in other simpler protein systems that also undergo conformational transitions for their functions (e.g., signal transduction proteins) because, in motor systems, the conformational transition often has to occur spontaneously and other events (such as ATP hydrolysis in myosin) only occur subsequently. Therefore, it is critical that the motor structures have intrinsic flexibilities such that significant conformational changes can occur with Brownian motions. In this sense, the name Brownian molecular machine is a well-deserved choice for molecular motor systems, although much remains to be explored on the mechanism of the ratchet effect implicated in various phenomenological models (Astumian and Hanggi, 2002; Julicher et al., 1997). On the other hand, perhaps not surprisingly, the low-frequency modes involve little local structural rearrangement in the ATP or calcium-binding site, which suggests that these local changes are either high in energy barrier or, very likely, gated by collective low-frequency modes. These arguments suggest that it might be more appropriate to describe functional transitions in some molecular motors as collective Brownian motions regulating local structural changes, which is in contrast with the conventional description of small changes in the active site somehow amplified into large-scale motions.
The current work represents a useful step toward a microscopic understanding of the free energy conversion mechanism in molecular motors. A valuable output from the normal mode analysis is a set of collective coordinates that can be used to characterize the progress of global conformational transitions. These coordinates would be very useful in molecular dynamics simulations for further exploring the coupling and causal relationship between collective motions and local structural changes that are essential to the subsequent chemical steps.
SUPPORTING MATERIALS
Brief discussions on the BNM results compared to Gaussian elastic network models and x-ray B-factors are included; the effect of using a minimized structure instead of the x-ray structure in normal mode analysis is also discussed. The hinge residues in the normal modes with large involvement coefficients are shown in the structures to illustrate their positions. Finally, a figure that illustrates the motions of several higher frequency modes with large involvement coefficients in E2-Ca2+-ATPase is included. MPEG movies that illustrate the motions of the various modes discussed in the text can be obtained from the author (cui@chem.wisc.edu) by request.
Supplementary Material
Acknowledgments
We thank Mr. A. Van Wynsberghe for discussion and a critical reading of the manuscript.
Q.C.'s group is partially supported by the starting-up fund from the Department of Chemistry and College of Letters and Science at University of Wisconsin, Madison, a PRF-G grant administered by the American Chemical Society, a Research Innovation Award from the Research Corporation, and a National Science Foundation grant.
References
- Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. D. Watson. 1994. Molecular Biology of the Cell, 3rd Ed. Garland Publishing, New York and London, UK.
- Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Prot. Struct. Funct. Gen. 17:412–425. [DOI] [PubMed] [Google Scholar]
- Andersen, J. P. 1995. Dissection of the functional domains of the sarcoplasmic reticulum Ca2+-ATPase by site-directed mutagenesis. Biosci. Rep. 15:243–261. [DOI] [PubMed] [Google Scholar]
- Andersen, J. P., J. D. Clausen, A. P. Einholm, and B. Vilsen. 2003. Mutagenesis of residues involved in control of the Ca2+ entry pathway and conformational changes associated with Ca2+ binding in the SR Ca2+-ATPase. Ann. NY Acad. Sci. 986:72–81. [DOI] [PubMed] [Google Scholar]
- Andersen, J. P., and T. Sorensen. 1996. Site-directed mutagenesis studies of energy coupling in the sarcoplasmic reticulum Ca2+-ATPase. Biochim. Biophys. Acta Bioenerg. 1275:118–122. [DOI] [PubMed] [Google Scholar]
- Andersen, J. P., and B. Vilsen. 1995. Structure-function relationships of cation translocation by Ca2+- and Na+, K+-ATPases studied by site-directed mutagenesis. FEBS Lett. 359:101–106. [DOI] [PubMed] [Google Scholar]
- Astumian, R. D., and P. Hanggi. 2002. Brownian motors. Phys. Today. 55:33–39. [Google Scholar]
- Atilgan, A. R., S. R. Durell, R. L. Jernigan, M. C. Demirel, O. Keskin, and I. Bahar. 2002. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagshaw, C. R. 2000. Motors in muscle: the function of conventional myosin II. In Molecular Motors. S. J. Higgins, editor. Portland Press, London, UK. 19–31. [DOI] [PubMed]
- Bagshaw, C. R., D. R. Trentham, R. G. Wolcott, and P. D. Boyer. 1975. Oxygen exchange in the γ-phosphoryl group of protein-bound ATP during Mg2+-dependent adenosine triphosphatase activity of myosin. Proc. Natl. Acad. Sci. USA. 72:2592–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar, I., A. R. Atilgan, M. C. Demirel, and B. Erman. 1997a. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 80:2733–2736. [Google Scholar]
- Bahar, I., A. R. Atilgan, and B. Erman. 1997b. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Folding Des. 2:173–181. [DOI] [PubMed] [Google Scholar]
- Bahar, I., B. Erman, R. L. Jernigan, A. R. Atilgan, and D. G. Covell. 1999. Collective motions in HIV-1 reverse transcriptase: examination of flexibility and enzyme function. J. Mol. Biol. 285:1023–1037. [DOI] [PubMed] [Google Scholar]
- Banting, G., and S. J. Higgins, editors. 2000. Molecular Motors. Portland Press, London, UK.
- Basu, G., A. Kitao, F. Hirata, and N. Go. 1995. A collective motion description of the 3(10-)/α-helix transition—implications for a natural reaction coordinate. J. Am. Chem. Soc. 116:6307–6315. [Google Scholar]
- Bauer, C. B., H. W. Holden, J. B. Thoden, R. Smith, and I. Rayment. 2000. X-ray structures of the apo and MgATP-bound states of Dictyostelium discoideum myosin motor domain. J. Biol. Chem. 275:38494–38499. [DOI] [PubMed] [Google Scholar]
- Berlin, Y. A., A. L. Burin, L. D. A. Siebbeles, and M. A. Ratner. 2001. Conformationally gated rate processes in biological macromolecules. J. Phys. Chem. A. 105:5666–5678. [Google Scholar]
- Blumenfeld, L. A., and A. N. Tikhonov. 1994. Biophysical Thermodynamics of Intracellular Processes. Springer-Verlag, New York.
- Bockmann, R. A., and H. Grubmuller. 2002. Nanoseconds molecular dynamics simulation of primary mechanical energy transfer steps in F1-ATP synthase. Nat. Struct. Biol. 9:198–202. [DOI] [PubMed] [Google Scholar]
- Boyer, P. D. 1993. The binding change mechanism for ATP synthase—some probabilities and possibilities. Biochim. Biophys. Acta. 1140:215–250. [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comp. Chem. 4:187–217. [Google Scholar]
- Brooks, B. R., D. Janezic, and M. Karplus. 1995. Harmonic analysis of large systems. I. Methodology. J. Comp. Chem.16:1522–1542. [Google Scholar]
- Brooks III, C. L., M. Karplus, and B. M. Pettitt.1988. Proteins: A Theoretical Perspective of Dynamics, Structure, and Thermodynamics. John Wiley & Sons, New York.
- Bustamante, C., D. Keller, and G. Oster. 2001. The physics of molecular motors. Acc. Chem. Res. 34:412–420. [DOI] [PubMed] [Google Scholar]
- Case, D. A. 1994. Normal-mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 4:285–290. [Google Scholar]
- Cullum, J. K., and R. A. Willoughby. 1985. Lanczos Algorithms for Large Symmetric Eigenvalue Computations. Birkhauser, Boston, MA.
- Dale, M. P., and D. D. Hackney. 1987. Analysis of positional isotope exchange in ATP by cleavage of the β-P-O-γ-P bond—demonstration of negligible positional isotope exchange by myosin. Biochemistry. 26:8365–8372. [DOI] [PubMed] [Google Scholar]
- Doruker, P., A. R. Atilgan, and I. Bahar. 2000. Dynamics of proteins predicted by molecular dynamics simulations and analytical approaches: application to a-Amylase inhibitor. Prot. Struct. Funct. Gen. 40:512–524. [PubMed] [Google Scholar]
- Durand, P. G., Y. Trinquier, and Y. Sanejouand. 1994. New approach for determining low-frequency normal-modes in macromolecules. Biopolymers. 34:759–771. [DOI] [PubMed] [Google Scholar]
- Elcock, A. H. 2002. Modeling supramolecular assemblages. Curr. Opin. Struct. Biol. 12:154–160. [DOI] [PubMed] [Google Scholar]
- Fisher, A. J., C. A. Smith, J. B. Thoden, R. Smith, K. Sutoh, H. W. Holden, and I. Rayment. 1995. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP·BeFx and MgADP·AlF4-. Biochemistry. 34:8960–8972. [DOI] [PubMed] [Google Scholar]
- Frank, J. 2003. Electron microscopy of functional ribosome complexes. Biopolymers. 68:223–233. [DOI] [PubMed] [Google Scholar]
- Frauenfelder, H., S. G. Sligar, and P. G. Wolynes. 1991. The energy landscape and motions of proteins. Science. 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- Gerstein, M., and W. Krebs. 1998. A database of macromolecular motions. Nuc. Acids Res. 26:4280–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go, N., T. Noguti, and T. Nishkawa. 1983. Dynamics of a small globular protein in terms of low-frequency vibrational modes. Proc. Natl. Acad. Sci. USA. 80:3696–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmuller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the streptavidin biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- Gulick, A. M., C. B. Bauer, J. B. Thoden, and I. Rayment. 1997. X-ray structure of the MgADP, MgATPgS, and MgAMPPNP complexes of the Dictyostelium discoideum myosin motor domain. Biochemistry. 36:11619–11628. [DOI] [PubMed] [Google Scholar]
- Halle, B. 2002. Flexibility and packing in proteins. Proc. Natl. Acad. Sci. USA. 99:1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanggi, P., P. Talkner, and M. Borkovec. 1990. Reaction-rate theory: fifty years after Kramers. Rev. Mod. Phys. 62:251–341. [Google Scholar]
- Hayward, S., and N. Go. 1995. Collective variable description of native protein dynamics. Annu. Rev. Phys. Chem. 46:223–250. [DOI] [PubMed] [Google Scholar]
- Hayward, S., A. Kitao, and H. J. C. Berendsen. 1997. Model-free methods of analyzing domain motions in proteins from simulation: a comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Prot. Struct. Funct. Gen. 27:425–437. [DOI] [PubMed] [Google Scholar]
- Hayward, S., A. Kitao, F. Hirata, and N. Go. 1993. Effect of solvent on collective motions in globular protein. J. Mol. Biol. 234:1207–1217. [DOI] [PubMed] [Google Scholar]
- Higo, J., Y. Sugimoto, K. Wakabayashi, and H. Nakamura. 2001. Collective motions of myosin head derived from backbone molecular dynamics and combination with x-ray solution scattering data. J. Comp. Chem. 22:1983–1994. [Google Scholar]
- Hill, T. L. 1977. Free Energy Transduction in Biology. Academic Press, New York.
- Hill, T. L., and E. Eisenberg. 1981. Can free energy transduction be localized at some crucial part of the enzymatic cycle? Q. Rev. Biophys. 14:463–511. [DOI] [PubMed] [Google Scholar]
- Hirokawa, N., and R. Takemura. 2003. Kinesin superfamily proteins. In Molecular Motors. M. Schliwa, editor. Wiley-VCH, Munich, Germany.
- Holmes, K. C., and M. A. Geeves. 1999. Structural mechanism of muscle contraction. Annu. Rev. Biochem. 68:687–728. [DOI] [PubMed] [Google Scholar]
- Houdusse, A., A. G. Szent-Gyorgyi, and C. Cohen. 2000. Three conformational states of scallop myosin S1. Proc. Natl. Acad. Sci. USA. 97:11238–11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii, Y., K. Kitamura, H. Tanaka, and T. Yanagida. 2003. Molecular motors and single-molecule enzymology. Methods Enzymol. 361:228–245. [DOI] [PubMed] [Google Scholar]
- Isralewitz, B., M. Gao, and K. Schulten. 2001. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11:224–230. [DOI] [PubMed] [Google Scholar]
- Ito, K., T. Q. P. Uyeda, Y. Suzuki, K. Sutoh, and K. Yamamoto. 2003. Requirement of domain-domain interaction for conformational change and functional ATP hydrolysis in myosin. J. Biol. Chem. 278:31049–31057. [DOI] [PubMed] [Google Scholar]
- Jencks, W. P. 1980. The utilization of binding energy in coupled vectorial processes. Adv. Enzymol. 51:75–106. [DOI] [PubMed] [Google Scholar]
- Joseph, D., G. A. Petsko, and M. Karplus. 1990. Anatomy of a protein conformational change: hinged “lid” motion of the triosephosphate isomerase. Science. 249:1425–1428. [DOI] [PubMed] [Google Scholar]
- Julicher, F., A. Ajdari, and J. Prost. 1997. Modeling molecular motors. Rev. Mod. Phys. 69:1269–1281. [Google Scholar]
- Jung, Y. J., E. Barkai, and R. J. Silbey. 2002. Current status of single-molecule spectroscopy: theoretical aspects. J. Chem. Phys. 117:10980–10995. [Google Scholar]
- Karplus, M., and J. A. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9:646–652. [DOI] [PubMed] [Google Scholar]
- Keike, M. C., and M. A. Titus. 2003. The myosin superfamily: an overview. In Molecular Motors. M. Schliwa, editor. Wiley-VCH, Munich, Germany.
- Kitao, A., and N. Go. 1999. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 9:164–169. [DOI] [PubMed] [Google Scholar]
- Kitao, A., S. Hayward, and N. Go. 1998. Energy landscape of a native protein: jumping-among-minima model. Prot. Struct. Funct. Gen. 33:496–517. [DOI] [PubMed] [Google Scholar]
- Kitao, A., F. Hirata, and N. Go. 1991. The effects of solvent on the conformation and the collective motions of a protein: normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem. Phys. 158:447–472. [Google Scholar]
- Krupka, R. M. 1994. Interpreting the effects of site-directed mutagenesis on active transport systems. Biochim. Biophys. Acta Biomem. 1193:165–178. [DOI] [PubMed] [Google Scholar]
- Kundu, S., J. S. Melton, D. C. Sorensen, and J. G. N. Phillips. 2002. Dynamics of proteins in crystals: comparison of experiment with simple models. Biophys. J. 83:723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanczos, C. 1950. An iteration method for the solution of the eigenvalue problem of linear differential and integral operators. J. Res. Natl. Bur. Stand. 45:255–282. [Google Scholar]
- Lazaridis, T., and M. Karplus. 2000. Effective energy functions for protein structure prediction. Curr. Opin. Struct. Biol. 10:139–145. [DOI] [PubMed] [Google Scholar]
- Lee, A. G. 2002. A calcium pump made visible. Curr. Opin. Struct. Biol. 12:547–554. [DOI] [PubMed] [Google Scholar]
- Lee, A. G., and J. M. East. 2001. What the structure of a calcium pump tells us about its mechanism. Biochem. J. 356:665–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner, D. 2001. Vibrational energy transfer in helices. Phys. Rev. Lett. 87:188102. [Google Scholar]
- Li, G., and Q. Cui. 2002. A coarse-grained normal mode approach for macromolecules: an efficient implementation and application to Ca2+-ATPase. Biophys. J. 83:2457–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, G., and Q. Cui. 2003. Mechanochemical coupling in myosin: a theoretical analysis with molecular dynamics and combined QM/MM reaction path calculations. J. Phys. Chem. B. In press.
- Ma, J., T. C. Flynn, Q. Cui, A. G. W. Leslie, J. E. Walker, and M. Karplus. 2002. A dynamic analysis of the rotation mechanism for conformational change in F1-ATPase. Structure. 10:921–931. [DOI] [PubMed] [Google Scholar]
- Ma, J., and M. Karplus. 1997. Ligand induced conformational changes in ras p21: a normal mode and energy minimization study. J. Mol. Biol. 274:114–131. [DOI] [PubMed] [Google Scholar]
- Ma, J., and M. Karplus. 1998. The allosteric mechanism of the chaperonin GroEL: a dynamic analysis. Proc. Natl. Acad. Sci. USA. 95:8502–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malnasi-Csizmadia, A., M. Kovacs, R. J. Woolley, S. W. Botchway, and C. R. Bagshaw. 2001a. The dynamics of the relay loop tryptophan residue in the Dictyostelium myosin motor domain and the origin of spectroscopic signals. J. Biol. Chem. 276:19483–19490. [DOI] [PubMed] [Google Scholar]
- Malnasi-Csizmadia, A., D. S. Pearson, M. Kovacs, R. J. Woolley, M. A. Geeves, and C. R. Bagshaw. 2001b. Kinetic resolution of a conformational transition and the ATP hydrolysis step using relaxation methods with a Dictyostelium myosin II mutant containing a single tryptophan residue. Biochemistry. 40:12727–12737. [DOI] [PubMed] [Google Scholar]
- Malnasi-Csizmadia, A., R. J. Woolley, and C. R. Bagshaw. 2000. Resolution of conformational state of Dictyostelium myosin II motor domain using tryptophan (W501) mutants: implications for the open-closed transition identified by crystallography. Biochemistry. 39:16135–16146. [DOI] [PubMed] [Google Scholar]
- Marques, O., and Y. Sanejouand. 1995. Hinge-bending motion in citrate synthase arising from normal mode calculations. Prot. Struct. Funct. Gen. 23:557–560. [DOI] [PubMed] [Google Scholar]
- Maschhoff, K. J., and D. C. Sorensen. 1996. A portable implementation of ARPACK for distributed memory parallel computers. Prelim. Proc. Copper Mountain Conf. Iterat. Meth.
- McCammon, J. A., and S. C. Harvey. 1987. Dynamics of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK.
- McCammon, J. A., and M. Karplus. 1980. Dynamics of Tyrosine ring rotation in a globular protein. Biopolymers. 19:1375–1405. [Google Scholar]
- Neria, E., S. Fischer, and M. Karplus. 1996. Simulation of activation free energies in molecular systems. J. Chem. Phys. 105:1902–1921. [Google Scholar]
- Northrup, S. H., M. R. Pear, J. A. McCammon, and M. Karplus. 1980. Molecular dynamics of ferrocytochrome c. Nature. 286:304–305. [DOI] [PubMed] [Google Scholar]
- Onishi, H., T. Ohki, N. Mochizuki, and M. F. Morales. 2002. Early stages of energy transduction by myosin: roles of Arg in Switch I, of Glu in Switch II, and of the salt-bridge between them. Proc. Natl. Acad. Sci. USA. 99:15339–15344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment, I. 1996. The structural basis of the myosin ATPase activity. J. Biol. Chem. 271:15850–15853. [DOI] [PubMed] [Google Scholar]
- Rees, D. C., and J. B. Howard. 1999. Structural bioenergetics and energy transduction mechanisms. J. Mol. Biol. 293:343–350. [DOI] [PubMed] [Google Scholar]
- Sagnella, D. E., and J. E. Straub. 1999. A study of vibrational relaxation of B-state carbon monoxide in the heme pocket of photolyzed carboxymyoglobin. Biophys. J. 77:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagnella, D. E., and J. E. Straub. 2001. Directed energy “funneling” mechanism for heme cooling following ligand photolysis or direct excitation in solvated carbonmonoxy myoglobin. J. Phys. Chem. B. 105:7057–7063. [Google Scholar]
- Sasaki, N., R. Ohkura, and K. Sutoh. 2003. Dictyostelium myosin II mutations that uncouples the converter swing and ATP hydrolysis cycle. Biochemistry. 42:90–95. [DOI] [PubMed] [Google Scholar]
- Schlitter, J., M. Engels, P. Kruger, E. Jacoby, and A. Wollmer. 1993. Targeted molecular-dynamics simulation of conformational change-application to the T/R transition in insulin. Mol. Simul. 10:291–308. [Google Scholar]
- Schliwa, M., editor. 2003. Molecular Motors. Wiley-VCH, Munich, Germany.
- Shea, J. E., and C. L. Brooks III. 2001. From folding theories to folding proteins: a review and assessment of simulation studies of protein folding and unfolding. Annu. Rev. Phys. Chem. 52:499–535. [DOI] [PubMed] [Google Scholar]
- Simmons, R. M., and T. L. Hill. 1976. Definitions of free energy levels in biochemical reactions. Nature. 263:615–618. [DOI] [PubMed] [Google Scholar]
- Simonson, T., G. Archontis, and M. Karplus. 1997. Continuum treatment of long-range interactions in free energy calculations: application to protein-ligand binding. J. Phys. Chem. B. 41:8349–8362. [Google Scholar]
- Smith, C. A., and I. Rayment. 1996. X-ray structure of the magnesium(II)·ADP·vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 Å resolution. Biochemistry. 35:5404–5417. [DOI] [PubMed] [Google Scholar]
- Steffen, W., D. Smith, and J. Sleep. 2003. The working stroke upon myosin-nucleotide complexes binding to actin. Proc. Natl. Acad. Sci. USA. 100:6434–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto, Y., M. Tokunaga, Y. Takezawa, M. Ikebe, and K. Wakabayashi. 1995. Conformational changes of the myosin heads during hydrolysis of ATP as analyzed by x-ray solution scattering. Biophys. J. 68:29S–34S. [PMC free article] [PubMed] [Google Scholar]
- Tama, F., and C. L. Brooks III. 2002. The mechanism and pathway of pH-induced swelling in cowpea chlorotic mottle virus. J. Mol. Biol. 318:733–747. [DOI] [PubMed] [Google Scholar]
- Tama, F., F. X. Gadea, O. Marques, and Y. Sanejouand. 2000. Building block approach for determining low-frequency normal modes of macromolecules. Prot. Struct. Funct. Gen. 41:1–7. [DOI] [PubMed] [Google Scholar]
- Tama, F., and Y. Sanejouand. 2001. Conformational change of proteins arising from normal mode calculations. Prot. Eng. 14:1–6. [DOI] [PubMed] [Google Scholar]
- Thomas, A., M. J. Field, and D. Perahia. 1996. Analysis of the low-frequency normal modes of the R state of aspartate transcarbamylase and a comparison with the T state modes. J. Mol. Biol. 261:490–506. [DOI] [PubMed] [Google Scholar]
- Thomas, A., K. Hinsen, M. J. Field, and D. Perahia. 1999. Tertiary and quaternary conformational changes in aspartate transcarbamylase: a normal mode study. Prot. Struct. Funct. Gen. 34:96–112. [DOI] [PubMed] [Google Scholar]
- Tirion, M. M. 1996. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77:1905–1908. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., M. Nakasako, H. Nomura, and H. Ogawa. 2000. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 405:647–655. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., and H. Nomura. 2002. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 418:605–611. [DOI] [PubMed] [Google Scholar]
- Vale, R. D., and R. A. Milligan. 2000. The way things move: looking under the hood of molecular motor proteins. Science. 288:88–95. [DOI] [PubMed] [Google Scholar]
- Volkman, B. F., D. Lipson, D. E. Wemmer, and D. Kern. 2001. Two-state allosteric behavior in a single domain signaling protein. Science. 291:2429–2433. [DOI] [PubMed] [Google Scholar]
- Wang, Q., C. F. Wong, and H. Rabitz. 1998. Simulating energy flow in biomolecules: application to tuna cytochrome c. Biophys. J. 75:60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wriggers, W., and P. Chacon. 2001. Modeling tricks and fitting techniques for multiresolution structures. Structure. 9:779–788. [DOI] [PubMed] [Google Scholar]
- Xie, X. S. 2002. Single-molecule approach to dispersed kinetics and dynamic disorder: probing conformational fluctuation and enzymatic dynamics. J. Chem. Phys. 117:11024–11032. [Google Scholar]
- Xu, C., W. J. Rice, W. He, and D. L. Stokes. 2002. A structural model for the catalytic cycle of Ca2+-ATPase. J. Mol. Biol. 316:201–211. [DOI] [PubMed] [Google Scholar]
- Yang, W., Y. Q. Gao, Q. Cui, J. Ma, and M. Karplus. 2003. The missing link between thermodynamics and structure in F1-ATPase. Proc. Natl. Acad. Sci. USA. 100:874–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z., D. Lewis, C. Sumbilla, G. Inesi, and C. Toyoshima. 2001. The role of the M6–M7 loop (L67) in stabilization of the phosphorylation and Ca2+ binding domains of the sarcoplasmic reticulum Ca2+-ATPase (SERCA). J. Biol. Chem. 276:15232–15239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.