

Abstract
X-ray structures permit theoretical study of Cl− permeation along bacterial ClC Cl− pores. We determined the lowest energy curvilinear pathway, identified anion-coordinating amino acids, and calculated the electrostatic potential energy profiles. We find that all four bacterial ClC Cl− crystal structures correspond to closed states. E148 and S107 side chains form steric barriers on both sides of the crystal binding site in the StClC wild-type and EcClC wild-type crystals; both the EcClC(E148A) and EcClC(E148Q) mutants are blocked at the S107 site. We studied the effect that mutating the charge of some strongly conserved pore-lining amino acids has on the electrostatic potential energy profiles. When E148 is neutralized, it creates an electrostatic trap, binding the ion near midmembrane. This suggests a possible electrostatic mechanism for controlling anion flow: neutralize E148, displace the side chain of E148 from the pore pathway to relieve the steric barrier, then trap the anion at midmembrane, and finally either deprotonate E148 and block the pore (pore closure) or bring a second Cl− into the pore to promote anion flow (pore conductance). Side-chain displacement may arise by competition for the binding site between the oxygens of E148 and the anion moving down the electrostatic energy gradient. We also find that the charge state of E111 and E113 may electrostatically control anion conductance and occupancy of the binding site within the cytoplasmic pore.
INTRODUCTION
The ClC family of chloride channels (Jentsch et al., 1999) is present in virtually all tissues and organisms, and is widely expressed in most mammalian cells (Jentsch et al., 2002). It is now recognized that chloride channels regulate a variety of important physiological and cellular functions (see Maduke et al., 2000 and references therein). Chloride-conducting proteins are vital for regulating pH, cell volume, electrical impulses, transport of salts across cells, and voltage stabilization of excitable muscle cells (Jentsch et al., 2002). Mutations in ClC channels cause diseases such as myotonia congenita or Thomsen's disease (Steinmeyer et al., 1994), Dent's disease (Piwon et al., 2000), Bartter's syndrome (Simon et al., 1997), and an X-linked form of nephrolithiasis (Lloyd et al., 1996). In the absence of a structure, biochemical and mutagenesis analyses of these channels provided a confusing picture of the channel topology and the coupling between the architectural components of the pore; little could be said definitively about how these channels worked (Jentsch, 2002). This began to change with a cryoelectron microscopic analysis of a bacterial ClC protein (Mindell et al., 2001) and more definitively with recent x-ray structure determinations (Dutzler et al., 2002, 2003). These crystal structures confirmed many inferences from mutagenesis studies (Estévez and Jentsch, 2002): the assemblies are double-barreled (Miller, 1982; Miller and White, 1984); the two identical pores gate independently (Miller, 1982; Dutzler et al., 2003); each pore is entirely contained within a single monomer (Dutzler et al., 2002), etc. The most recent x-ray study (Dutzler et al., 2003) begins to provide detailed insight into a most intriguing property of these proteins—the coupling between conduction and fast gating (Richard and Miller, 1990; Pusch et al., 1995; Chen and Miller, 1996).
The original structures (Dutzler et al., 2002, 2003) permit theoretical study of chloride permeation, and determination of the bacterial systems' curvilinear ionic pathways. Even though the bacterial systems are transporters, not channels (Accardi and Miller, 2003), the prokaryotes share signature sequence identities with their eukaryotic relatives; consequently such atomic level analyses might help in understanding ion transport in eukaryotes and ClC channel function generally. Mutagenesis and functional studies have revealed the effect of specific amino acids on conductance and selectivity (Ludewig et al., 1996; Wollnik et al., 1997; Middleton et al., 1996), and fast and slow gating (Pusch et al., 1995; Lin et al., 1999; Lin and Chen, 2000). However, there is no evidence that the mutated residues coordinate the translocating anion. Although their mutation affects chloride conductance, they could still be located far from the pathway in various protein regions. Thus, despite a wealth of experimental mutagenesis data, major issues need to be resolved at the atomic level: What is the exact chloride pathway? Which amino acids coordinate the translocating ion and what are their roles? What is the effect of charged amino acids located within the pore mouths on the anion translocation? Identification of the role of pore-lining amino acids is important for understanding both conduction and gating, as mutations of charged residues believed to be chloride-coordinating affect both these properties simultaneously (Pusch et al., 1995). Recent experiments suggest that the electrostatic effects produced by the charged residues located in the inner pore mouth of ClC-0 play a crucial role in controlling channel conductance and occupancy of the internal Cl− binding site (Chen et al., 2003; Chen and Chen, 2003).
We focus on the translocation of Cl− ions through the pores and try to answer some of the questions just raised. We amplify on our preliminary studies (Miloshevsky and Jordan, 2003a, 2003b), provide a detailed description of chloride coordination in its translocation through the pore, and determine coordinates for the curvilinear pore through the bacterial proteins. To get an intuitive feeling for the nature of this path, we examine the effect that path-lining amino acids have on ion coordination by calculating electrostatic potential profiles. These demonstrate well-articulated electrostatic barriers and wells for Cl− translocation. The wells define positions (possible binding sites) that may be preferentially occupied by Cl−. The overall stability of the Cl− at such sites depends on entropy components, protein flexibility, gating conditions, etc., not considered in the present treatment. Even with this disclaimer, our results suggest numerous experiments that may help better characterize behavior in the ClC family generally, notwithstanding the facts that detailed electrophysiological studies are mainly limited to the ClC-0 channel, that substantial sequence differences exist between nonsignature domains in the bacterial protein and those of other family members, and that the prokaryotic assemblies are transporters, not channels.
COMPUTATIONAL MODEL
We have developed a new computational approach, the Monte Carlo ion channel proteins (MCICP) method, to simulating water-protein, ion-protein, and protein-protein interactions, for application to the study of permeation and gating in ion and water channels (Miloshevsky and Jordan, 2004). It exploits the Metropolis Monte Carlo (MMC) method (Metropolis et al., 1953) and kinetic Monte Carlo (kMC) techniques (Binder, 1992). In this implementation the computational model is based on the following assumptions. Simulations are based on the four crystallographic x-ray structures of bacterial ClC Cl− assemblies: StClC (Protein Data Bank (PDB) entry 1KPL), EcClC (PDB entry 1OTS), and two EcClC mutants (PDB entries 1OTT, i.e., the E148A mutant, and 1OTU, i.e., the E148Q mutant). Both subunits, including all explicit hydrogens (13,863 explicit protein atoms in the case of StClC), are treated in full atomic detail. Molecular details of the pore within subunit A are illustrated in Fig. 1. The coordinate origin was chosen at the contact surface between subunits A and B, defined as the center of the line connecting the subunits' central binding sites. The Z axis is oriented along the conductance pathways with the periplasmic side positive. “Missing” residues, heavy atoms, and hydrogens were added using an option of our MCICP code. We use parameters from the CHARMM22 all-hydrogen force field (MacKerell et al., 1998) and, to ensure consistency with the CHARMM22 force field (where polarization effects are not explicitly treated) immerse the protein in a low dielectric (ɛ = 1) slab. Anion translocation was analyzed for the subunit A pore (Fig. 1). This subunit's protein mouths were filled with explicit water molecules (∼2000) located in a cylinder of 25 Å radius, centered on the central binding site of subunit A. Bulk water regions abutting a membrane slab are treated as continua with ɛ = 80. The reaction field, due to dielectric differences between membrane and bulk regions, is computed by the method of images (Jackson, 1962; Dorman et al., 1996). Van der Waals (vdW) and electrostatic interactions are computed with no cutoff. The total interaction energy (electrostatic + vdW) between the chloride ion and ClC protein atoms and water molecules is calculated to determine the curvilinear conduction pathway. During simulations of chloride translocation, the protein is treated as a rigid body, similar to the Brownian dynamics (BD) approach where structure is also held fixed (Kuyucak et al., 2001). To gain a sense of the strengths of this approach, comparison with BD is informative. We treat water molecules in the pore and protein mouths explicitly. There is no abrupt dielectric boundary separating protein from the ions and the pore and mouth waters (see Fig. 1); as in standard MD these are integral parts of the functioning protein. But of course there are drawbacks as well. We cannot compute current-voltage-concentration profiles. Dielectric relaxation of the protein modulates its electrostatic influence on the mobile groups. BD treats this in a mean field sense, by assigning a “protein dielectric constant,” which is not included here. The bulk water-low ɛ-slab interfaces are chosen to be ∼32 Å from the center of the coordinate frame along the Z axis. Image planes are perpendicular to the Z axis and the outermost protein atoms, including their van der Waals radii, are included in the low ɛ-membrane slab. We examine the default charge state of the StClC and EcClC proteins (Dutzler et al., 2002, 2003) and ignore the influence of the membrane potential, reasonable since the lipid is located sufficiently far from the pore that this potential would be severely attenuated (Jordan, 1983).
FIGURE 1.

Molecular representation of the pore in ClC Cl− subunit A. View from within the membrane along the X axis connecting the anions in the central binding sites in subunits A and B. The backbone α-helices of subunit A are individually color-coded. Atoms belonging to interhelical polypeptide loops are shown in conventional colors: carbons, gray; oxygens, red; nitrogens, cyan; and hydrogens, white. The anion in the central binding site of subunit A is a green sphere. The anionic pathway is shown as a curved green arrow. The bulk regions are continua with dielectric constant ɛ = 80; a membrane slab is a low dielectric medium (ɛ = 1). The image planes are dark-blue panels. The water molecules are shown in the protein mouths. The figure was generated using our MCICP code.
For anion motion, at each MC step all atomic positions in the model system are determined and the interaction energy is directly calculated. The difficulty is that each MC interaction energy calculation requires a time-consuming reaction field computation (e.g., with 13,755 real StClC charges, 10 generations of images yield 275,100 image charges). The total interaction energy includes interaction among all real charges in the system (partially charged protein atoms, all ions and waters) and with the image charges. To accelerate calculations we first decompose the model system into separate rigid groups: protein subunits and helices, ions and water molecules. Within each group interatomic distances are fixed; thus interaction between atoms in individual groups is invariant and need not be computed. Each rigid group has a specific number of atoms and the groups are relatively mobile. Depending on available computer power, the approach can be refined, further decomposing some groups into relatively mobile subgroups. Having excluded contributions from interactions between fixed charges inside each group, the calculations are greatly accelerated, but by itself this is insufficient. The following multistep MC technique significantly speeds up calculation. Consider various groups (mobile parts of protein, ions, or waters), each with different numbers of atoms.
Before starting MC trials the total system energy (the interaction energy among mobile groups) is calculated and stored in RAM. It is the intergroup Coulomb and van der Waals energy plus the interaction with all image charges. The interaction energy of individual groups with the rest of the model system is also stored in main arrays in RAM, a time-consuming calculation performed only once.
MC trials are begun and repeated; the mobile group is sampled randomly. Interaction energies between atoms of the sampled group and those of all other groups are rewritten into temporary arrays. The total system energy is stored as an old system energy. The interaction energy between the sampled group and all other groups is temporarily stored as an old group energy. The old group term is subtracted from the total system energy, a computationally inexpensive step.
The sampled group is then moved, changing its atoms' positions relative to those of atoms in other groups. The interaction energy between this group and the other groups (including all image charges) is calculated and stored in the main arrays. The change in the total interaction energy of this group with all the others is added to the total energy of the system, yielding a new system energy. As group energies must be calculated this is computationally intensive. However, it only involves the interaction between the mobile sampled group and the rest of the system. Recalculation of the interaction energy between immobile groups is not required.
The MMC procedure (Metropolis et al., 1953) is now performed. Old and new total system energies are compared. If the new configuration is accepted, we return to step 2. Otherwise, energies of the sampled group are returned from temporary arrays to the main arrays. The total energy of the system is assigned to an old system energy and we return to step 2. This step is also computationally cheap.
The procedure is fast and highly efficient. Computations for large protein systems can be performed on an ordinary personal computer. Protein structures may be decomposed into mobile groups of specific interest or treated as totally rigid. A big advantage is that the strong interactions between charged species, including long-range terms, are directly calculated. Van der Waals and electrostatic interactions are computed without cutoffs. The MCICP code, based on the principles outlined, is written in C++ and designed to allow three-dimensional graphical output of results during the simulation process using OpenGL three-dimensional graphics (Woo et al., 1999).
Determination of the ion-protein interaction energy along the curved paths is not straightforward, as a priori coordinates for the low energy paths are unknown. To overcome the steep energy barriers found along the ClC pathways we combine a constrained MMC method with the kMC approach, developing a bootstrap technique for determining reaction coordinates: kinetic Monte Carlo reaction-path following (kMCRPF). It is known that the MMC method permits moves to states of higher energy with a Boltzmann probability. The smaller the energy difference the greater the probability accepting an uphill move. Using this property we constrain the anionic Z coordinate (the obvious choice for reaction coordinate) and allow only its unidirectional increment (constrained MMC). Contrary to familiar techniques (Torrie and Valleau, 1974, 1977; Carter et al., 1989; Rosso et al., 2002), that also constrain the selected coordinate or decouple it from the other degrees of freedom, in the kMCRPF approach the reaction coordinate step is not fixed in advance; it is sampled randomly and corresponds to accepted configurations with a Boltzmann probability. All other monitored degrees of freedom (e.g., anionic X and Y coordinates, positions and orientations of explicit waters) are unconstrained. It is important that water rearrangement is included. The reaction coordinate evolves slowly relative to these other degrees of freedom. For accepted configurations many MC trials are used to relax the anion, fixing the new value of the reaction coordinate. The remaining degrees of freedom then relax fully, responding to small movements of the anion along the reaction coordinate; this is most important as the anion evolves downhill, since any new lower energy configuration will always be accepted. This describes a major aspect of the anionic evolution along the reaction pathway. However, it is incomplete since the energy fluctuates within the lowest-energy groove. Thermal fluctuations will, in general, spread the set of transition states of the system. A large set of closely related paths may be sampled within a minimum energy pathway. By tracking numerous MC reaction paths we can establish the most favorable one(s), i.e., those that connect very distinct states at the lowest cost in energy. Preferential sampling techniques (Owicki and Scheraga, 1977; Mehrotra et al., 1983) were incorporated to move the water molecules near the anion more frequently than those further away.
The kMC methodologies widely used in theoretical condensed matter physics to study phase transitions, polymer systems, and critical phenomena in alloys and magnets (Binder, 1992; Landau and Binder, 2000) can be modified, elaborated upon, and profitably adapted to treat permeation and gating. In traditional kMC simulations all transitions and transition probabilities must be known in advance of simulation (Binder, 1992; Landau and Binder, 2000). They are usually used for lattice systems (crystals) with fixed atomic sites. However, ions, water molecules, and protein atoms are not at fixed locations. All possible permeation or gating pathways cannot be cataloged and specified in advance. The relevant transition probabilities must be found on the fly during simulation. The point is to propagate the anion slowly along the lowest energy path (without a lattice approximation, with no predefined probability table). We focus on applications where only the initial state of the path (binding site, closed or open protein conformation) is known. The kMC method is ideal for simulating kinetic evolution processes that are characterized by atomic jumps to one of the empty nearest neighbor sites. Our kMC algorithm includes the following steps: 1), sample a random direction for anion transport, which is constrained within a forward or backward hemisphere (unidirectional motion along the Z coordinate); 2), determine the nearest water or protein atom in this direction, and calculate the distance 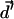 between the anion and this putative collision partner; 3), calculate a new anion coordinate,
between the anion and this putative collision partner; 3), calculate a new anion coordinate, 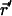 , using
, using 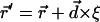 , where ξ is a random number in the interval from 0 to 1; and 4), move the anion to this new location. The constrained MMC technique monitors motion along the lowest energy pathway. Thus, evolution proceeds via small anionic jumps toward the nearest lowest energy uphill or downhill states (kMC technique) and jumps are thermally activated (constrained MMC). The result is a new kMCRPF method, with transition state search on the fly. As system evolution is slow, energy barriers need not be known in advance. Atoms need not be mapped onto a lattice and all possible states of the system need not be known. The kMCRPF technique allows the anion to overcome steep electrostatic and even steric barriers in following the lowest energy path. The energy function is not biased; sampling is Boltzmann-weighted. A limitation, to be lifted in the future, is that kMCRPF sampling relies on a good initial guess of reaction coordinate. The approach was tested by comparison with grid search (Leach, 2001) used to determine ion-protein interaction energy at points on a cubic lattice superimposed onto the ClC pores with a three-dimensional grid of 0.1 × 0.1 × 0.2 Å. The grid points corresponding to the lowest energies were identified with the paths of interest. Both approaches yield identical energy profiles over the whole Z range, with good agreement in both uphill and downhill directions. Strikingly, even along downhill paths, kMCRPF does not stray from the “exact” reaction coordinate; naturally it does not agree exactly, as the energy exhibits some thermal fluctuations within the reaction pathway.
, where ξ is a random number in the interval from 0 to 1; and 4), move the anion to this new location. The constrained MMC technique monitors motion along the lowest energy pathway. Thus, evolution proceeds via small anionic jumps toward the nearest lowest energy uphill or downhill states (kMC technique) and jumps are thermally activated (constrained MMC). The result is a new kMCRPF method, with transition state search on the fly. As system evolution is slow, energy barriers need not be known in advance. Atoms need not be mapped onto a lattice and all possible states of the system need not be known. The kMCRPF technique allows the anion to overcome steep electrostatic and even steric barriers in following the lowest energy path. The energy function is not biased; sampling is Boltzmann-weighted. A limitation, to be lifted in the future, is that kMCRPF sampling relies on a good initial guess of reaction coordinate. The approach was tested by comparison with grid search (Leach, 2001) used to determine ion-protein interaction energy at points on a cubic lattice superimposed onto the ClC pores with a three-dimensional grid of 0.1 × 0.1 × 0.2 Å. The grid points corresponding to the lowest energies were identified with the paths of interest. Both approaches yield identical energy profiles over the whole Z range, with good agreement in both uphill and downhill directions. Strikingly, even along downhill paths, kMCRPF does not stray from the “exact” reaction coordinate; naturally it does not agree exactly, as the energy exhibits some thermal fluctuations within the reaction pathway.
RESULTS
Chloride-coordinating amino acids
A Cl− ion was placed at the central crystallographic site of the four original structures (Dutzler et al., 2002, 2003) in pore A (Fig. 1) and simulations carried out at 300 K based on the kMCRPF technique. Explicit water molecules were equilibrated using the MMC method. The anionic pathway was followed by moving the anion away from the central binding site and toward the cytoplasm and periplasm, thus determining the lowest energy pathway, the coordinating residues, and the corresponding anion interaction energy profiles.
Before describing the particulars of ionic coordination, a notational comment on atomic identification is needed. We use standard computational conventions for realizations of PDB notation: an HN hydrogen is bound to a backbone amide, an HA to an α-carbon, etc. The anion conduction pathway through pore A in the StClC wild-type (WT) structure determined using the kMCRPF technique is illustrated in Fig. 2. The pore length is ∼16 Å. Pore-lining residues and α-helices are labeled. In the cytoplasmic region the pore is lined by helices R and D. At the extracellular entrance and in midmembrane the ion pathway is coordinated by helices F and N. Water molecules are illustrated in the mouth cavities on the cytoplasmic (∼−6 Å) and extracellular (∼10 Å) sides of the pore. There is no room for water molecules along the ∼16 Å length of the pore region of the StClC crystal structure. In the cytoplasm the pore entrance is formed by I448, S107, P110, F348, and Y445. In all four crystal structures (StClC, EcClC, and its two mutants) the side chains of S107 and Y445 sterically restrict anion entrance into the pore from the cytoplasmic region. At the central binding site the anion (green sphere) is coordinated by backbone HNs of I356 and F357 (helix N) and by hydroxyl HOs of Y445 and S107. In the periplasm the pore entrance is formed by G146, R147, E148, F190, and A358. The side chain of R147 is easily accessible to water molecules. Then the E148 side chain, which forms bonds with numerous protein hydrogens, sterically blocks the pathway in WT StClC and EcClC.
FIGURE 2.

Anion pathway along pore A in StClC. The Cl− trajectory is represented by the green curve. The anion in the central binding site is illustrated as a green sphere. Dashed lines connect the anion with the coordinating atoms from S107, Y445, I356, and F357. The symbol Θ indicates trajectory sites where the pore is sterically blocked by S107 and E148 side chains. Individual protein atoms are conventionally colored as indicated in Fig. 1. Important residues lining the pore are labeled. The α-helices to which they belong are indicated in brackets. Water molecules in the ClC protein mouths are illustrated. The figure was generated using our MCICP code.
Fig. 3 details the anion's pathway in the cytoplasmic part of pore A in StClC. At ∼−6 Å (the internal binding site; Dutzler et al., 2003) the anion faces the intracellular water molecules (Fig. 2). Side chains of P110, I448, and F348 (Fig. 3 a) coordinate the anion's entrance into the pore. The aryl ring of F348 points toward the pore pathway, aligned roughly perpendicular to the ion trajectory, permitting favorable anion interaction with an aryl hydrogen and shielding the chloride from the ring's π-electrons. The anion is coordinated by backbone amide hydrogens from the end of helix D (G108 and S107) on the other side of the pore (Fig. 3 a). In WT StClC and EcClC crystals and in both EcClC mutants the S107 side chain sterically restricts ion entry to the central binding site (Fig. 3 b). The steric block is due to CA, CB, and OG of S107 and CE2 and OH of Y445. The anion's trajectory follows the contour of the S107 side chain. It should be noted that the residues adjacent to S107 are strictly conserved glycines, which may permit the flexibility needed to reorient the side chains of S107 and/or Y445 and open the pore. At the central binding site (Fig. 3 c) the anion is coordinated by backbone HNs of I356 and F357 and hydroxyl HOs of Y445 and S107 (Dutzler et al., 2002). Backbone HNs of I109 and G149 also contribute to anion coordination.
FIGURE 3.

Residues defining the Cl− conduction pathway in the cytoplasmic region of StClC's pore A. Stereo view is along the pore from the cytoplasmic side. Individual atoms are conventionally colored. The residues coordinating the anion are labeled. Cl− is represented as a green sphere. (a) Cl− located at Z = −6 Å (the internal binding site). (b) Cl− located at Z = −2.7 Å (steric barrier due to the S107 side chain). (c) Cl− located at its central binding site, Z = 0 Å. The figures were generated using our MCICP code.
Fig. 4 illustrates residues on the pathway in the extracellular part of pore A in StClC. At the entrance (∼10 Å) the anion is coordinated by the side chain of R147 and by backbone HNs from G315, G316, and F317 (Fig. 4 a). This site is accessible to water molecules and the anion also interacts with three or four waters (see Fig. 2). The E148 side chain belonging to helix F forms hydrogen bonds with its own polypeptide loop (helix F) and residues belonging to another polypeptide chain (helix N) (Fig. 4 b). The OE1 oxygen of E148 forms hydrogen bonds with three helix F residues: the HNs of E148 (oxygen-hydrogen separation, d =1.9 Å) and R147 (d = 3.0 Å) and the HA of G146 (d = 2.6 Å). The OE2 oxygen of E148 forms hydrogen bonds with two helix F residues: HN of G149 (d = 2.5 Å) and HA of G146 (d = 2.4 Å); it also hydrogen-bonds with helix N residues: the HNs of A358 (d = 2.3 Å) and F357 (d = 3.3 Å) and HA of G355 (d = 2.4 Å). Thus, the E148 oxygens, which form bonds with many protein hydrogens, sterically block the pathway.
FIGURE 4.

Residues defining the Cl− conduction pathway in the periplasmic region of StClC's pore A. Stereo view is along the pore from the cytoplasmic side. Individual atoms are conventionally colored. The residues coordinating the anion are labeled. Cl− is represented as a green sphere. (a) Cl− located at Z = 10 Å (the entrance from the periplasm). (b) Cl− located at Z = 4.8 Å. The E148 side chain sterically blocks the pore. The figures were generated using our MCICP code.
Pore size
The effective radius of pore A along the pathway in WT StClC is illustrated in Fig. 5. The pore radius was determined using the MCICP code by squeezing a variable radius sphere along the pathway. At Z ∼ 6 Å the extracellular pore is narrowest, ∼0.46 Å, forming the constriction region leading to the binding site from the periplasmic pore entrance. Here the E148 oxygens block the pore. At the binding site (Z ∼ 0 Å) the pore is not constricted (pore radius ∼ 1.88 Å and Cl− fits perfectly). On the cytoplasmic side the constriction region is located at Z ∼ −2.5 Å. Here the pore narrows again; its radius drops to ∼0.92 Å due to the S107 and Y445 side chains which prohibit anion escape from the binding site and access to the cytoplasmic side of the pore. At approximately Z = −6 Å the pore again opens out and its radius increases to ∼2 Å. These results clearly demonstrate that the crystal structure has captured the bacterial system in a closed state, with access forbidden from either side of the central binding site.
FIGURE 5.

The effective pore radius as a function of Z along subunit A's Cl− pathway in WT StClC.
Anionic electrostatic potential energy profiles
As the pore is sterically constricted, the total anion nonbonded interaction energy (electrostatic + vdW) has no meaning. However, the electrostatic potential energy profile along the pathway may provide clues to the energy barriers and wells of the open pore. Our rationale is that the blocking residues must rearrange themselves slightly (Dutzler et al., 2002, 2003) for the pore to open and the anion to move unimpeded. These local processes most likely do not require global structural alterations. We present electrostatic potential energy profiles for WT EcClC, as this is the more accurately resolved ClC Cl− structure (2.5 Å resolution). Energies are measured relative to anionic energy in the pore mouths, where anionic solvation is bulk-like.
Fig. 6 A illustrates the electrostatic potential energy profiles along the pathway in pore A of the EcClC WT structure. As the kMCRPF technique describes the kinetics of anion permeation, the electrostatic potential energy profiles exhibit energy fluctuations. Depending on the height of the energy barriers typically 8000–10,000 small MC steps are needed for full translocation. The amplitude of potential energy noise is larger in the protein mouths than in the pore region. In the cytoplasmic and extracellular mouths the anion is solvated by the water molecules and less constricted. Minima and maxima in the energy profiles reflect the influence of protein structure. In the pore region there are large electrostatic barriers at the E148 and S107 sites (black trace, Z ± 4 Å). These barriers are clearly overestimated as the anion passes very close to the glutamate and serine oxygens (due to the pore's local steric constriction). We focus on the charge state of the E148 and R147 residues. The mutation E148A or the neutralization of E148 by lowering pH (Iyer et al., 2002, Miller, 2003) creates a deep electrostatic trap. The barrier near 4 Å is converted into a deep well (red trace). The electrical potential energy profiles for both the E148A and E148Q mutants are very close to that of a neutralized E148 with R147 in its native state. The energy minimum at Z ∼ 4 Å is then located precisely at the position previously occupied by the OE2 oxygen of E148 and the Cl− is sixfold coordinate, on one side of the pore by three HNs from helix F (G149, R147, and E148) and on the other side by HNs from helix N (A358, F357, and I356). This locus (Z ∼ 4 Å) has been found experimentally to be the external binding site in the open ClC structure (Dutzler et al., 2003). The anion's energy is less than in the periplasmic mouth, a favorable condition for anion entrance into the pore from the periplasm. When R147 is also neutralized by proton transfer to E148 (the OE2(E148)-HH12(R147) distance is 6.4 Å) an energy barrier appears at ∼10 Å (green trace). This illustrates the importance of the strictly conserved positive charge at the periplasmic pore entrance (Fahlke et al., 1997a; Lin and Chen, 2000). This charge may facilitate the smooth anionic flow from the periplasmic mouth into the pore. It should be noted that the charge of E148 and R147 affects the electrostatic potential energy at the central binding site and even in the S107 region, demonstrating long-range electrostatic effects.
FIGURE 6.

Effect of the charge state of selected charged residues on anionic electrostatic potential energy profiles in the periplasmic pore of WT EcClC. (A) Charge mutations of E148 and R147, with the locations of S107, E148, and R147 labeled. The black trace is for the native state of WT EcClC (with both E148 and R147 charged). The red trace illustrates the effect of the mutation E148A or of neutralizing E148 by pH decrease. The green trace describes the case where R147 is also neutralized by proton transfer to E148. (B) Charge mutations of E111 and E113, with the locations of S107, E111, and E113 labeled. The black trace is for the double mutation, S107G and E148A, introduced to artificially create an open permeation pathway. The other traces illustrate effects of further mutations. Red and blue traces demonstrate the effect that neutralizing E111 (red trace) or E113 (blue trace) has on the electrostatic energy profile in the cytoplasmic pore region. The green trace corresponds to the case with both E111 and E113 neutralized.
Fig. 6 B illustrates how putative changes in the charge state of conserved acidic residues (E111 and E113), located close to the cytoplasmic pore entrance, alter the electrostatic potential energy profile. We focus on WT EcClC, introducing mutations S107G and E148A to artificially create an open permeation pathway. The S107G mutation has reduced the electrostatic barrier at the S107 site (compare black trace of Fig. 6 B with red trace of Fig. 6 A). It is known that mutation of this strictly conserved serine to threonine in ClC-0 influences selectivity and fast gating, and dramatically reduces anion conductance (Ludewig et al., 1996). The gating role, if any, of S107 is unclear. A recent study of ClC-0 found no evidence of a physical gate cytoplasmic to the central binding site, i.e., no moving structure on the intracellular side of the selectivity filter that participates in fast gating (Lin and Chen, 2003). Fig. 6 B shows that E111 and E113 residues influence the energy in the region from Z ∼ −15 to Z ∼ 5 Å. Neutralizing E111 impacts the electrostatic barrier at the S107 site (red trace). The internal binding site at Z ∼ −6 Å becomes pronounced. The energy drops to the level of that in the cytoplasmic mouth. Neutralization of E113 has the same effect as neutralizing E111. The electrostatic effect of E113 is even more pronounced in the range from Z ∼ 0 to Z ∼ 5 Å than that of E111 (blue trace) because E113 is closer to the central binding site. When both E111 and E113 are neutralized, there is the clear energy well in the range from Z ∼ −15 to Z ∼ 10 Å (green trace). The center of this energy well is somewhat shifted toward the cytoplasm. Altering the charge of conserved residues also affects the configuration of the water molecules and the anion trajectory within the cytoplasmic mouth.
DISCUSSION
We have described a Monte Carlo study of chloride permeation through prokaryotic ClC Cl− pores based on recent x-ray structures (Dutzler, et al., 2002, 2003), finding the lowest energy curved anionic pathway through the constricted pores, and determining the anion-coordinating amino acids. Also, we studied the effect that charge mutation of some important amino acids, highly conserved throughout the ClC family and located close to the periplasmic and cytoplasmic pore entrances, has on electrostatic potential energy profiles. Our kMCRPF technique allowed us to determine the lowest energy anionic pathway even in the sterically constricted pore. We did not distort the native protein structure trying to open the pore and severely disrupting and rearranging the flexible side chains. In general our results agree well with the recent experimental study of selectivity filter gating (Dutzler, et al., 2003). Our model determinations of the coordinating amino acids and the location of the two outer binding sites (at Z ∼ 4 Å and Z ∼ 0 Å) as well as the inner site (Z ∼ −6 Å) are in excellent agreement with experimental findings, which confirms the validity of our kMCRPF approach and the feasibility of using it to establish the anionic pathway.
The x-ray structures of the bacterial ClCs correspond to closed states. The anion is locked in the central binding site. In addition to the E148 oxygens the side chains of S107 and Y445 sterically impede anionic passage from the central binding site into the cytoplasm in all four structures. Unlike the E148 side chain, which is perpendicular to the pore, the S107 side chain lies along the anionic pathway coordinating the translocating anion; its hydroxyl hydrogen is at the central binding site and its backbone amide hydrogen at the inner binding site (Z ∼ −6 Å). The steric block extends the length of the S107 side chain. In both the EcClC E148A and EcClC E148Q mutants there is still a small steric barrier near E148. This suggests that the anion fits snugly over the whole length of the pore and that protein flexibility is crucial for anion permeation, since anionic coordination is mainly with flexible side chains, not backbone hydrogens.
When the E148 is neutralized at low pH (Iyer et al., 2002), it creates an electrostatic trap, binding the anion near midmembrane (Z ∼ 4 Å, see Fig. 6 A). Both E148G and E148A mutations exhibit nearly the same electrical effect. Since the potential energy drops steadily along the pathway, conditions are favorable for anion motion down the electrical gradient from the periplasmic mouth (∼25 Å) to the pore entrance (at ∼10 Å). This suggests the possibility of an electrostatic mechanism for controlling anion flow through the periplasmic pore: neutralize E148, displace the side chain of E148 from the pore pathway to relieve the steric barrier, then trap the anion at midmembrane, and finally either deprotonate E148 and block the pore (pore closure) or bring a second Cl− into the pore to promote the anion flow (pore conductance). This mechanism may explain the observation that the gating of ClC-0 is tightly coupled to anion permeation (Pusch et al., 1995; Chen and Miller, 1996). It has been suggested that the movement of the anion from a site at the extracellular pore mouth to a site well within the pore promotes the opening of the ClC Cl− channel (Pusch et al., 1995; Chen and Miller, 1996), i.e., Cl− acts as a gating charge. This idea is also supported by a recent experiment (Dutzler, et al., 2003) on the role of the E148 side chain as a gate. In the E148Q mutant, where the glutamate residue is neutralized but retains its polar character, the side chain moved away from its original location, opening the pore and permitting an anion to occupy the 4 Å locus; the side chain was displaced from the path and directed toward the extracellular mouth (Dutzler, et al., 2003). The Cl− can be driven from the central binding site at midmembrane toward the cytoplasmic pore exit either by moving the negative charge of E148 or by moving another Cl− ion into the E148 locus (Dutzler et al., 2003). This negative charge raises the potential energy at midmembrane expelling Cl− from the central binding site toward the cytoplasm. But, insertion of the E148 side chain into the pore blocks it sterically, thus interrupting anion flow. To maintain the anion flux another Cl− ion is needed at Z ∼ 4 Å to push Cl− out of the central binding site toward the cytoplasm, a cooperative unblocking mechanism reminiscent of behavior in potassium channels (Chung et al., 2002; Jordan, 2002). When the pore is open conduction occurs via repulsive interaction of adjacent Cl− ions. The molecular details of this mechanism, by which the side chain of E148 opens or blocks the pore and interrupts anion flow, are unclear. The specific interactions that lead to breaking and restoring the hydrogens bonds between E148 oxygens and the many surrounding protein hydrogens are also obscure. Our analysis of Fig. 6 A suggests that these may be due to competition between the carboxylate oxygens of neutralized E148, with their reduced hydrogen-bonding ability, and the anion moving from the periplasm down the electrical gradient. Our results demonstrate that there may be two binding sites in the pore (one at midmembrane and the other at the OE2 oxygen locus of E148). As these sites are only ∼4 Å apart it is very unlikely that both would be simultaneously occupied by chloride; for this to happen the ions would have to be in contact, there not being enough room to intercalate a water molecule. It would seem more likely that the second anion plays the part of a trigger, promoting movement of the Cl− away from the midmembrane site. In any event, for conduction to occur there must be negative charge at the position of the E148 oxygens. It is known that the anion conductance in the periplasmic pore is also affected by the charge state of R147 (Fahlke et al., 1997a; Lin and Chen, 2000). Mutations of the homologous residues (K231 in ClC-1 and K165 in ClC-0) were found, respectively, to alter the anionic selectivity sequence and significantly increase cation permeability (Fahlke et al. (1997a) and to render the channel nonfunctional (Lin and Chen, 2000). If R147 is neutralized by proton transfer to E148 (energetically plausible since the pK difference of 8.5, equivalent to ∼500 mV, can be more than compensated for by Coulombic attraction, ∼2.3 V at the 6.4 Å separation) an energy barrier appears at Z ∼10 Å that could lower anion conductance. Thus, Fig. 6 A demonstrates that the strictly conserved positive charge at this location has an important role in guaranteeing smooth communication between the periplasmic mouth and the pore. In addition, we also examined the influence of D54. This strictly charge-conserved helix B residue (E54 in StClC) forms hydrogen bonds with residues from helices F (HE atom of R147 and T151) and G (A189). R147 screens the pore from the carboxylate oxygens of D54. We found the negative charge of D54 has little electrostatic influence on the anionic energy profile. Neutralizing D54 yields a small energy well in the 10–15 Å region (results not shown). It seems that this residue plays a different role, possibly by participating in an important salt-bridge linking helices B, F, and G, thus orienting the charged end of the R147 side chain toward the pore.
Recently the highly conserved E111 (E127 in ClC-0) was shown to electrostatically control anion conductance and occupancy of the internal binding site in the cytoplasmic pore (Chen and Chen, 2003). In ClC-0 this negatively charged residue is part of a salt bridge linking helices R and D. Here the E111 carboxylate oxygens (helix D) interact directly with the hydroxyl of T452 (K519 in ClC-0) (helix R) and the R120 guanidinium group hydrogens. R120 is a nonconserved residue from the polypeptide loop connecting helices D and E. The distance between the E111 carboxylate oxygens and the R120 guanidinium group hydrogens is ∼3 to 4 Å. Nearby a nonconserved E103 from the polypeptide loop connecting helices C and D also participates in this complex bridge forming hydrogen bonds with T452 and R120. The distance between OE1 atoms of E103 and E111 is 4.5 Å. Thus, helix D is linked by means of E111 and E103 to the polypeptide loop between helices D and E (residue R120) and to helix R (residue T452). Possibly the positive charge of R120 or of R451 (adjacent to T452) may play the role of K519 in ClC-0. The effect of K519 charge on channel conductance has been demonstrated experimentally (Pusch et al., 1995; Middleton et al., 1996; Chen and Chen, 2003). Our results show that neutralizing E111 affects energetics in the cytoplasmic mouth (see Fig. 6 B, red trace). Energy wells become pronounced at ∼−6 Å (the internal binding site) and at ∼−11 Å. When both E111 and R120 or both E111 and R451 are neutralized the energy profile is located between the black and red traces, closer to the black trace (results not shown). If only R120 or R451 is neutralized, the energy barrier moves above the black trace by roughly the energy difference between black and red traces (results not shown). This demonstrates the importance of positive charge at the cytoplasmic pore entrance (Chen and Chen, 2003). Taken together these results suggest that charged residues in the cytoplasmic mouth influence Cl− permeation by an electrostatic mechanism. The replacement of E111 by a neutral residue decreases the energy barrier, which should increase conductance. Occupancy of the internal Cl− binding site is also affected by a negative charge at position 111.
In the bacterial system E113 (helix D) forms a bridge with helix R (residue S446). The side chain of Y445 is also coordinated by one of the carboxylate oxygens from E113. In eukaryotic channels this bridge is stronger and has a reversed polarity, E113(D) and S446(R) being strictly replaced by lysine and aspartic acid respectively (in ClC-0, K129(D), and D513(R)). Thus, the negative charge and the bridge between helices D and R near the central binding site is conserved throughout the ClC family. We examined the effect that the negative charge at residue 113 of helix D (shifted to residue 513 of helix R in ClC-0) has on the anionic potential energy profile. The electrostatic effect is very close to that due to residue 111 (Fig. 6 B, blue trace). This suggests that E113(D) (the charge equivalent to the strictly conserved aspartic acid of helix R in eukaryotic channels) may also regulate pore conductance electrostatically. When both E111 and E113 are neutralized the charge effect nearly doubles (Fig. 6 B, green trace). The absence of negative charge on the E111 and E113 side chains leads to a deep energy well in the region from Z ∼ −15 to Z ∼ 10 Å, which would favor anion entry into the protein pore from both the periplasmic and cytoplasmic mouths. Energy fluctuations in the well reflect the influence of the protein structure. Finally, Fig. 6 shows that anionic energy in EcClC (a feature common to all four bacterial systems) is higher in the periplasmic mouth than in the cytoplasmic mouth.
The positively-charged amino groups R98 and K455 (Z ∼ −11 Å) of the bacterial structures (strictly nonconserved in the eukaryotic channels, although their role may be played by the lysine pairs K519 and K520 and K585 and K586 in ClC-0 and ClC-1, respectively, which are shifted one residue toward the N-terminus of helix R relative to the bacterial K455 residue, but are not found in other eukaryotes) attract Cl− ions into the cytoplasmic mouth. In the bacterial systems these residues directly coordinate the translocating anion deep in the cytoplasmic mouth; their positive charges are pointed toward the pore. If these are discharged, in homology to ClC-0 or ClC-1, where the corresponding residues are neutral, Q114 and P522 and H180 and P588 respectively, the cytoplasmic mouth profile is drastically altered. There is then a large energy barrier from the cytoplasmic mouth to Z ∼ 0 Å (results not shown), suggesting that anion permeation energetics may be quite different in ClC-0 and ClC-1. There are substantial sequence differences that can significantly alter channel properties. An obvious case in point is that conduction in ClC-0 is ohmic (Miller, 1982), whereas that in ClC-1 is inwardly rectifying (Fahlke et al., 1995, 1997b). Thus, extrapolations to the behavior of other members of the ClC family must proceed cautiously. Even though the resolved bacterial structures are transporters (Accardi and Miller, 2003), not channels, they still may serve as templates for understanding eukaryotes.
The electrostatic potential energy profiles shown in Fig. 6 were calculated choosing the dielectric constant (ɛ) of the membrane protein to be unity, consistent with the CHARMM22 force field. Our experience with ɛ-modification has shown that increasing ɛ simply scales the electrostatic potential energy by a factor 1/ɛ (Miloshevsky and Jordan, 2004). The structure of the energy profiles (including all minima and maxima) is unaffected. Thus, it is possible to adjust the value of ɛ to obtain the “right” energy values and make quantitative comparisons with experimental data. However, this is not our aim. We focus on qualitative electrostatic effects that strongly conserved charged residues have on the anionic potential energy in the ClC Cl− pore.
Our simulation results suggest a wealth of interesting possibilities for mutations of amino acids lining the chloride pathway, which can guide further experimental investigation of the chloride-permeation mechanism of ClC Cl− channels. This is somewhat problematical since much of the coordination is with amide hydrogen atoms of the peptide backbone. Mutations of many polar and nonpolar residues and their effect on permeation, selectivity, and gating are discussed in recent reviews (Estévez and Jentsch, 2002; Jentsch et al., 2002). Conceivably the glycine residues in the signature sequences (G106, G108, G146, G149, and G355) are required to permit a wider sampling of Ramachandran space than is available to optically active residues. For instance, mutation G230E in ClC-1 (G146 in EcClC) greatly distorts permeation properties of the channel (Fahlke et al., 1997b) causing Thomsen's disease (George et al., 1993). Of nonsignature residues, those for which the side chains most closely coordinate the anion on its travels through the pore are I448, F348, and Q277, corresponding to I515, V409, and I308 in ClC-0; if the pore structures are the same, their modification could well affect eukaryotic conductance. In the vestibules, nonsignature residues affecting the electrical energy profile are all cytoplasmic: R98, E111, E113, T452, and K455. Of the corresponding ClC-0 residues, Q114, E127, K129 (or D513), K519, and P522, the importance of E127 and K519 has already been demonstrated (Chen and Chen, 2003). Modifications of the others could be profitable speculations, with one caveat. K129 or D513 appear to be helix-bridging residues; mutating them may have structural consequences. In addition to the transformations suggested, the mutation R147K in the bacterial pore would provide insight into possible effects that allotropism rather than charge have on the electrostatic potential energy profile.
There is one limitation to our MC simulations that, in our view, is not crucial. In simulating chloride translocation, the protein is treated as rigid, so possible effects of peptide flexibility on the reaction pathway cannot be taken into consideration. This is a temporary constraint and will be lifted in future development. Fixing the protein structure in our MC model shares features with the BD approach where structure is also held fixed (Kuyucak et al., 2001). A composite continuum theory for calculating ion current through a protein channel, incorporating information about channel dynamics, demonstrated that no substantial changes in protein geometry occur during ion translocation (Mamonov et al., 2003). Protein flexibility, an important contributor to the “protein dielectric constant” (Schutz and Warshel, 2001), will certainly perturb the lowest energy pathways and the corresponding energy profiles. However, in the absence of a gating rearrangement, we do not expect this to crucially alter anion behavior or the pore pathway coordinates, although it will modulate energies. Here we rely on numerous MD simulations of KcsA (Bernèche and Roux, 2000; Shrivastava and Sansom, 2000; Shrivastava et al., 2000). When compared with high-resolution protein structures, simulational studies demonstrate overall protein stability, suggesting that these crystal structures are reasonably representative of the equilibrium state. Comparing MD simulations with the KcsA crystal structure indicates that root-mean square fluctuations of various structural elements are in a range of 0.2 to 2 Å (Shrivastava and Sansom, 2000; Shrivastava et al., 2000). In no instance does MD suggest that the equilibrium conformation of a channel differs drastically from the high-resolution crystal structure. This is quite understandable chemically, as the forces bonding the different groups (subunits, helices, amino groups, etc.) that give rise to the crystal structure are always clearly delineated. However, to determine how flexibility affects ion energetics, future developments will incorporate protein flexibility by combining the scaled collective variables method, introduced by Noguti and Go (1985) to simulate thermal conformational fluctuations in proteins, with normal mode analysis techniques (Tirion, 1996; Tama et al., 2000; Li and Cui, 2002).
In summary, we found that the charge state of the strictly conserved E148 and the strongly conserved E111 and E113 (the salt-bridge equivalent of D513 in ClC-0) may electrostatically control anion conductance and occupancy of the binding sites in the ClC Cl− pore. Although our model identifies numerous amino acids lining the bacterial conduction pathway, the specific role of many of them remains unclear. An intriguing open question is the role of S107, the residue blocking the pathway at the cytoplasmic side. Further developments will introduce protein flexibility and will mutate important amino acids lining the pathway to study their effect on the chloride-conduction mechanism.
Acknowledgments
We thank Chris Miller and Joe Mindell for their comments on the manuscript.
Work was supported by National Institutes of Health grant GM-28643.
References
- Accardi, A., and C. Miller. 2003. pH activation of a prokaryotic chloride channel. J. Gen. Physiol. 122:37A. [Google Scholar]
- Bernèche, S., and B. Roux. 2000. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 78:2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder, K. 1992. The Monte Carlo Method in Condensed Matter Physics. Springer-Verlag, Berlin, Germany.
- Carter, E. A., G. Ciccotti, J. T. Hynes, and R. Kapral. 1989. Constrained reaction coordinate dynamics for the simulation of rare events. Chem. Phys. Lett. 156:472–477. [Google Scholar]
- Chen, T.-Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. F., C. W. Lin, and T.-Y. Chen. 2003. Conductance determinants in the pore of ClC-0 chloride channel. Biophys. J. 84:2358a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M.-F., and T.-Y. Chen. 2003. Side-chain charge effects and conduction determinants in the pore of ClC-0 chloride channels. J. Gen. Physiol. 122:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, S. H., T. W. Allen, and S. Kuyucak. 2002. Modeling diverse range of potassium channels with Brownian dynamics. Biophys. J. 83:263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman, V., M. B. Partenskii, and P. C. Jordan. 1996. A semi-microscopic Monte Carlo study of permeation energetics in a gramicidin-like channel: the origin of cation selectivity. Biophys. J. 70:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Estévez, R., and T. J. Jentsch. 2002. ClC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 12:531–539. [DOI] [PubMed] [Google Scholar]
- George, A. L., M. A. Crackower, J. A. Abdalla, A. J. Hudson, and G. C. Ebers. 1993. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita). Nat. Genet. 3:305–310. [DOI] [PubMed] [Google Scholar]
- Fahlke, C., R. Rüdel, N. Mitrovic, and M. Zhou. 1995. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron. 15:463–472. [DOI] [PubMed] [Google Scholar]
- Fahlke, C., H. T. Yu, C. L. Beck, T. H. Rhodes, and A. L. George, Jr. 1997a. Pore-forming segments in voltage-gated chloride channels. Nature. 390:529–532. [DOI] [PubMed] [Google Scholar]
- Fahlke, C., C. L. Beck, and A. L. George. 1997b. A mutation in autosomal dominant myotonia congenita affects pore properties of the muscle chloride channel. Proc. Natl. Acad. Sci. USA. 94:2729–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, R., T. M. Iverson, A. Accardi, and C. Miller. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. [DOI] [PubMed] [Google Scholar]
- Jackson, J. D. 1962. Classical Electrodynamics. John Wiley, New York.
- Jentsch, T. J., T. Friedrich, A. Schriever, and H. Yamada. 1999. The ClC chloride channel family. Pflugers Arch. 437:783–795. [DOI] [PubMed] [Google Scholar]
- Jentsch, T. J., V. Stein, F. Weinreich, and A. A. Zdebik. 2002. Molecular structure and physiological function of chloride channels. Physiol. Rev. 82:503–568. [DOI] [PubMed] [Google Scholar]
- Jentsch, T. J. 2002. Chloride channels are different. Nature. 415:276–277. [DOI] [PubMed] [Google Scholar]
- Jordan, P. C. 1983. Electrostatic modeling of ion pores. II. Effects attributable to the membrane dipole potential. Biophys. J. 41:189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, P. C. 2002. Unclogging a pipe: potassium channel pinball. Biophys. J. 83:2–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyucak, S., O. S. Andersen, and S. H. Chung. 2001. Models of permeation in ion channels. Rep. Prog. Phys. 64:1427–1472. [Google Scholar]
- Landau, D. P., and K. Binder. 2000. A Guide to Monte Carlo Simulations in Statistical Physics. University Press, Cambridge, UK.
- Leach, A. R. 2001. Molecular Modelling: Principles and Applications. Prentice Hall, Harlow, England; New York.
- Li, G., and Q. Cui. 2002. A coarse-grained normal mode approach for macromolecules: an efficient implementation and application to Ca2+-ATPase. Biophys. J. 83:2457–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. W., C. W. Lin, and T.-Y. Chen. 1999. Elimination of the slow gating of ClC-0 chloride channel by a point mutation. J. Gen. Physiol. 114:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. W., and T.-Y. Chen. 2000. Cysteine modification of a putative pore residue in ClC-0. Implication for the pore stoichiometry of ClC chloride channels. J. Gen. Physiol. 116:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. W., and T.-Y. Chen. 2003. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfinate reagents. J. Gen. Physiol. 122:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, S. E., S. H. Pearce, S. E. Fisher, K. Steinmeyer, B. Schwappach, S. J. Scheinman, B. Harding, A. Bolino, M. Devoto, P. Goodyear, S. P. Rigden, O. Wrong, T. J. Jentsch, I. W. Craig, and R. V. Thakker. 1996. A common molecular basis for three inherited kidney stone diseases. Nature. 379:445–449. [DOI] [PubMed] [Google Scholar]
- Ludewig, U., M. Pusch, and T. J. Jentsch. 1996. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 383:340–343. [DOI] [PubMed] [Google Scholar]
- MacKerell, A. D. Jr., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Maduke, M., C. Miller, and J. A. Mindell. 2000. A decade of ClC chloride channels: structure, mechanism, and many unsettled questions. Annu. Rev. Biophys. Biomol. Struct. 29:411–438. [DOI] [PubMed] [Google Scholar]
- Mamonov, A. B., R. D. Coalson, A. Nitzan, and M. G. Kurnikova. 2003. The role of the dielectric barrier in narrow biological channels: a novel composite approach to modeling single-channel currents. Biophys. J. 84:3646–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrotra, P. K., M. Mezei, and D. L. Beveridge. 1983. Convergence acceleration in Monte Carlo computer simulation on water and aqueous solutions. J. Chem. Phys. 78:3156–3166. [Google Scholar]
- Metropolis, N., A. Rosenbluth, M. Rosenbluth, A. Teller, and E. Teller. 1953. Equations of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1091. [Google Scholar]
- Middleton, R. E., D. J. Pheasant, and C. Miller. 1996. Homodimeric architecture of a CLC-type chloride ion channel. Nature. 383:337–340. [DOI] [PubMed] [Google Scholar]
- Miller, C. 1982. Open-state substructure of single chloride channels from Torpedo electroplax. Philos. Trans. R. Soc. Lond. B Biol. Sci. 299:401–411. [DOI] [PubMed] [Google Scholar]
- Miller, C., and M. M. White. 1984. Dimeric structure of single chloride channels from Torpedo electroplax. Proc. Natl. Acad. Sci. USA. 81:2772–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, C. 2003. ClC channels: reading eukaryotic function through prokaryotic spectacles. J. Gen. Physiol. 122:129–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miloshevsky, G. V., and P. C. Jordan. 2003a. Details of ion translocation through a ClC chloride channel. Biophys. J. 84:412a. [Google Scholar]
- Miloshevsky, G. V., and P. C. Jordan. 2003b. Theoretical study of the passage of chloride ions through a bacterial ClC chloride channel. J. Gen. Physiol. 122:32a. [Google Scholar]
- Miloshevsky, G. V., and P. C. Jordan. 2004. Gating gramicidin channels in lipid bilayers: reaction coordinates and the mechanism of dissociation. Biophys. J. 86:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell, J. A., M. Maduke, C. Miller, and N. Grigorieff. 2001. Projection structure of a ClC-type chloride channel at 6.5 Å resolution. Nature. 409:219–223. [DOI] [PubMed] [Google Scholar]
- Noguti, T., and N. Go. 1985. Efficient Monte Carlo method for simulation of fluctuating conformations of native proteins. Biopolymers. 24:527–546. [DOI] [PubMed] [Google Scholar]
- Owicki, J. C., and H. A. Scheraga. 1977. Preferential sampling near solutes in Monte Carlo calculations on dilute solutions. Chem. Phys. Lett. 47:600–602. [Google Scholar]
- Piwon, N., W. Grunther, M. Schwake, M. R. Bosl, and T. J. Jentsch. 2000. ClC-5 Cl− channel disruption impairs endocytosis in a mouse model for Dent's disease. Nature. 408:369–373. [DOI] [PubMed] [Google Scholar]
- Pusch, M., U. Ludewig, A. Rehfeldt, and T. J. Jentsch. 1995. Gating of the voltage-dependent chloride channel CIC-0 by the permeant anion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- Richard, E. A., and C. Miller. 1990. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 247:1208–1210. [DOI] [PubMed] [Google Scholar]
- Rosso, L., P. Minary, Z. W. Zhu, and M. E. Tuckerman. 2002. On the use of the adiabatic molecular dynamics technique in the calculation of free energy profiles. J. Chem. Phys. 16:4389–4402. [Google Scholar]
- Schutz, C. N., and A. Warshel. 2001. What are the dielectric “constants” of proteins and how to validate electrostatic models. Prot. Struct. Funct. Gen. 44:400–417. [DOI] [PubMed] [Google Scholar]
- Shrivastava, I. H., C. E. Capener, L. R. Forrest, and M. S. P. Sansom. 2000. Structure and dynamics of K channel pore-lining helices: a comparative simulation study. Biophys. J. 78:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava, I. H., and M. S. P. Sansom. 2000. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J. 78:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, B. D., R. S. Bindra, T. A. Mansfield, C. Nelson-Williams, E. Mendonca, R. Stone, S. Schurman, A. Nayir, H. Alpay, A. Bakkaloglu, J. Rodri-Guez-Soriano, J. M. Morales, S. A. Sanjad, C. M. Taylor, D. Pilz, A. Brem, H. Trachtman, W. Griswold, G. A. Richard, E. John, and R. P. Lifton. 1997. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat. Genet. 17:171–178. [DOI] [PubMed] [Google Scholar]
- Steinmeyer, K., C. Lorenz, M. Pusch, M. Koch, and T. J. Jentsch. 1994. Multimeric structure of ClC-1 chloride channel revealed by mutations in dominant myotonia congenita (Thomsen). EMBO J. 13:737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tama, F., F. X. Gadea, O. Marques, and Y.-H. Sanejouand. 2000. Building-block approach for determining low-frequency normal modes of macromolecules. Prot. Struct. Funct. Gen. 41:1–7. [DOI] [PubMed] [Google Scholar]
- Tirion, M. M. 1996. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys. Rev. Lett. 77:1905–1908. [DOI] [PubMed] [Google Scholar]
- Torrie, G. M., and J. P. Valleau. 1974. Monte Carlo free-energy estimates using non-Boltzmann sampling—application to subcritical Lennard-Jones fluid. Chem. Phys. Lett. 28:578–581. [Google Scholar]
- Torrie, G. M., and J. P. Valleau. 1977. Non-physical sampling distributions in Monte Carlo free-energy estimation—umbrella sampling. J. Comp. Phys. 23:187–199. [Google Scholar]
- Wollnik, B., C. Kubisch, K. Steinmeyer, and M. Pusch. 1997. Identification of functionally important regions of the muscular chloride channel CIC-1 by analysis of recessive and dominant myotonic mutations. Hum. Mol. Genet. 6:805–811. [DOI] [PubMed] [Google Scholar]
- Woo, M., J. Neider, T. Davis, and D. Shreiner. 1999. OpenGL(r) 1.2 Programming Guide, 3rd Ed: The Official Guide to Learning OpenGL, Version 1.2. OpenGL Architecture Review Board, Addison-Wesley, Boston, MA.