

Abstract
ClC chloride channels are voltage-gated transmembrane proteins that have been associated with a wide range of regulatory roles in vertebrates. To accomplish their function, they allow small inorganic anions to efficiently pass through, while blocking the passage of all other particles. Understanding the conduction mechanism of ClC has been the subject of many experimental investigations, but until now, the detailed dynamic mechanism was not known despite the availability of crystallographic structures. We investigate Cl− conduction by means of an all-atom molecular dynamics simulation of the ClC channel in a membrane environment. Based on our simulation results, we propose a king-of-the-hill mechanism for permeation, in which a lone ion bound to the center of the ClC pore is pushed out by a second ion that enters the pore and takes its place. Although the energy required to extract the single central ion from the pore is enormous, by resorting to this two-ion process, the largest free energy barrier for conduction is reduced to 4 kcal/mol. At the narrowest part of the pore, residues Tyr-445 and Ser-107 stabilize the central ion. There, the bound ion blocks the pore, disrupting the formation of a continuous water file that could leak protons, possibly preventing the passage of uncharged solutes.
INTRODUCTION
Although individual macroscopic properties of ion channels have been studied extensively (Hille, 1992), until recently, very little was known with certainty about their inner workings. The discovery of the KcsA potassium channel structure (Doyle et al., 1998) provided the first high-resolution structure of a channel that was specifically selective for small ions. This discovery sparked a round of fruitful computational experiments (Bernèche and Roux, 2000, 2001; Shrivastava and Sansom, 2000; Morais-Cabral et al., 2001; Åqvist and Luzhkov, 2000) that revisited long-standing assumptions about ion permeation through ion channels.
More recently, a second generation of ion channel structures has emerged, detailing the atomic structures of a bacterial ClC chloride channel in a closed (Dutzler et al., 2002) and in a constitutively open form (Dutzler et al., 2003), and of the calcium-gated MthK (Jiang et al., 2002) and voltage-gated KvAP (Jiang et al., 2003) potassium channels. These new structures come as much needed data points in the ion channel structure landscape, and provide the opportunity to build a new framework of simulations and studies on which to base revised microscopic theories of ion channels (Roux, 1999). This article presents a computational study of the ClC channel that reveals the main energy landscape regulating the conduction of Cl−.
In addition to being one of only two currently available types of ion channel structures, members of the ClC family (Jentsch et al., 1999; Maduke et al., 2000) are important in their own right. In vertebrates, ClCs play vital cellular functions such as the regulation of blood pressure, of cell volume, of organelle pH, and of membrane excitability. In prokaryotes, it is believed that ClC allows the cells to survive extreme acid shock (Iyer et al., 2002) by permitting excess Cl− from absorbed HCl to quickly exit the cell on demand. To fulfill these roles, ClC channels are strongly selective for small anions. However, unlike cation channels they do not discriminate strongly between such anions (many ClCs are permeant to SCN−, Br−, NO3−, and many larger anions with hydrophobic chains, to name just a few) because, aside from Cl−, few other anion species are present at significant concentrations under physiological conditions. To control the flow of ions across membranes, ClCs are all voltage-gated and, as a family, span a wide range of gating behaviors (Fahlke, 2001; Pusch et al., 1995).
Structurally, ClCs are dimers in which each of the two identical subunits contains its own independently-gated pore (these independent gates are referred to as the fast gate). The ClC monomers can themselves be subdivided into two parts exhibiting very similar structures, juxtaposed in antiparallel fashion. This architecture enables three half α-helices to have their positive ends pointing toward the pore. It has been suggested that with such an alignment, the α-helix dipoles can create a favorable environment for anions in a pore composed mostly of nonpolar and noncharged residues (Dutzler et al., 2002).
METHOD
System
Our simulations were based on a published x-ray structure of ClC from Salmonella serovar typhimurium (stClC) at 3.0 Å resolution (Protein Data Bank (PDB) accession code: 1KPL) (Dutzler et al., 2002). The protein was then placed in a POPE membrane and solvated with water. 24 Cl− and 2 Na+ ions were placed at random positions in the water to neutralize the protein charge with a total ion concentration of 100 mM. The resulting 97,000 atom system was then equilibrated for 5 ns in the NPT ensemble, with a 2-fs integration timestep, periodic boundary conditions, and particle mesh Ewald (PME) electrostatics, using the NAMD2 molecular dynamics software (Kalé et al., 1999) and the CHARMM22 force fields (MacKerell et al., 1998; Schlenkrich et al., 1996) for the energy parameters for lipids, proteins, and ions, and the TIP3P model for water molecules. During the equilibration, the membrane relaxed to a state of hydrophobic match in which it became thinner near the protein than away from it. The Cl− ions redistributed themselves near the very charged cytoplasmic side of the channel. Top and side views of the final system after equilibration are shown in Fig. 1.
FIGURE 1.
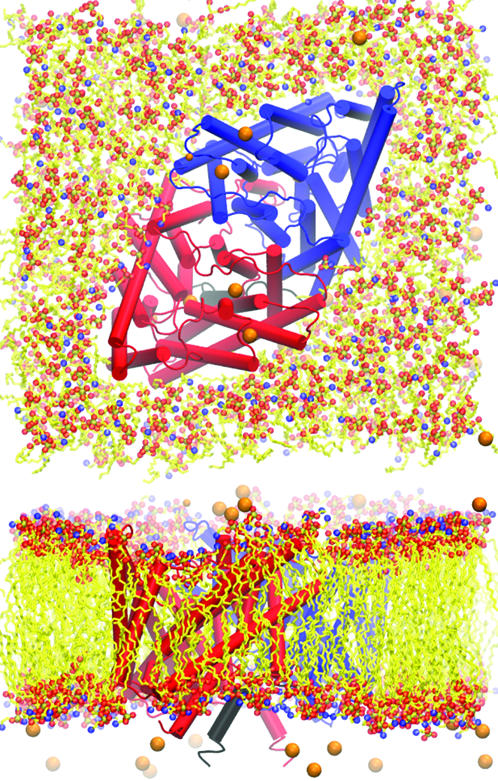
Top and side view of the ClC system showing the POPE membrane and ions. The reconstructed N-terminus is highlighted in black, and lipid tails have been simplified.
Opening the pore
Each pore of the wild-type ClC crystal structures is obstructed by a highly conserved glutamic acid residue (Glu-148), which is bent so that its negatively-charged carboxyl head is bound to a region of the pore next to the periplasmic exit. To investigate the effect of this residue, we ran interactive molecular dynamics simulations (Stone et al., 2001; Grayson et al., 2003) in which Cl− ions were pulled past Glu-148 to ascertain that the pore was indeed blocked and that the passage of Cl− across the channel was not possible without displacing the glutamate side chain. Although we can be certain that the opening of the fast gate in ClC involves a change to the conformation of Glu-148, we cannot exclude the possibility of other conformational changes. Recent indirect evidence suggests that the open ClC-0 channel exhibits additional changes at its cytoplasmic mouth, unaccounted for in the present study (Accardi and Pusch, 2003; Traverso et al., 2003). Nevertheless, we set out to modify the conformation of Glu-148 on the basis that this alteration was necessary and sufficient for unblocking ClC. Accordingly, we created an open conformation of the ClC pore by using interactive molecular dynamics to pull Glu-148's hydroxyl group out of the pore and into the channel's periplasmic vestibule.
Recent electrophysiological measurement performed on the Torpedo ray ClC-0 show that both the substitution of the pore-blocking glutamate (Glu-166 in ClC-0) with small noncharged residues (E166G, E166A, E166V, or E166Q) and the protonation of this glutamate strongly reduced the voltage dependence of the fast gate, allowing the pore to remain open for a wider range of conditions (Dutzler et al., 2003; Friedrich et al., 1999). Similar mutation studies reached identical conclusions for ClC-4 (E224A) and ClC-5 (E211A) (Friedrich et al., 1999). In addition, crystal structures of Escherichia coli ClC (electrophysiological measurements are not currently possible on the native protein) mutants (E148A, E148Q) showed them to be virtually identical to the wild-type structures except that the mutant pores were unobstructed by the Glu-148 side chain. Further supporting the validity of our assumptions about the open pore conformation and of the molecular surgery we performed is the finding that in the E148Q mutant, the neutrally charged glutamine residue, which has the same atomic geometry as glutamate, has its side chain sticking out of the pore (Dutzler et al., 2003), suggesting that Glu-148 might do the same under favorable conditions.
A superposition of the structures of the closed channel (PDB: 1KPL), of our manually opened channel after equilibration, and of the E. coli constitutively open E148Q mutant (PDB:1OTU) (Dutzler et al., 2003) are shown in Fig. 2. Inasmuch as the beginning of our study predates the publication of the constitutively open pore crystal structures, these later structures were not used as a starting point; however, our modified open-channel structure matches these new crystal structures closely.
FIGURE 2.
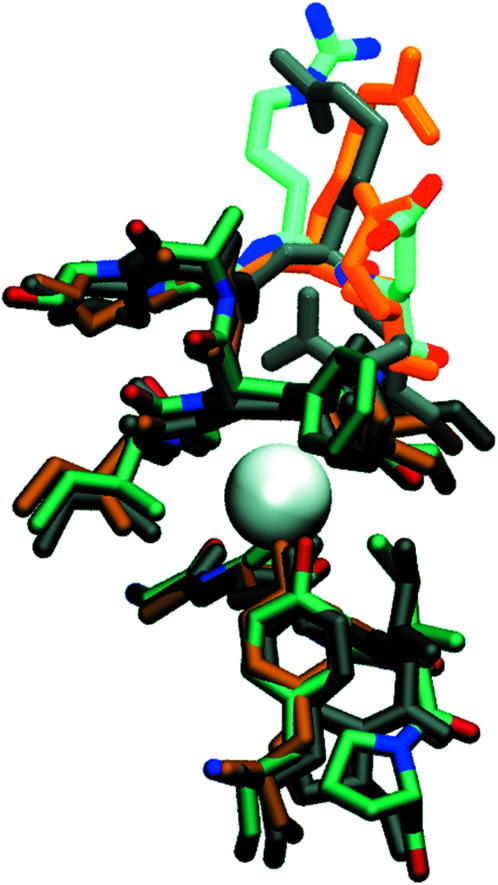
Superposition of the structures for the 1KPL closed channel (dark gray), of the 1OTU constitutively open channel E148Q mutant (orange), and of our manually opened channel after equilibration (atom-based coloring).
It must be said that there is currently no certainty about the exact nature of the ClC fast gate. The mechanism suggested by Dutzler et al. (2003) is not without problems: for ClC-0, we note the conflicting studies mentioned above (Accardi and Pusch, 2003; Traverso et al., 2003); we also note that this mechanism does not account for the measured fast gate charge of 0.92 to 2.2 e for ClC (Ferroni et al., 1997; Ludewig et al., 1997; Dutzler et al., 2003); finally we note that it fails to explain the reported effect of external Cl− ions on the fast gate (Pusch et al., 1995; Chen and Miller, 1996). Nevertheless, the mentioned studies, complemented by Lin and Chen (2003), are all either indicative of, or compatible with, putative conformational changes at or outside the cytoplasmic mouth of the channel. In the present study, we seek to shed some light of our own on these matters. Our article demonstrates that anionic conduction is possible and probable across the pore of bacterial ClC as observed in current crystal structures, once the pore-blocking glutamate has been displaced. Furthermore, we find that there exists ample space for the permeating Cl− ions to move in the pore. In fact, it is probable that an open conformation of ClC that would exhibit a notably wider internal pore could lead to a dramatic loss in selectivity and is as such unlikely. It is a credible possibility, and compatible with the experimental data so far amassed, that the internal pore structure of ClC is not significantly affected by the opening and closing of the fast gate.
Molecular alterations
The crystal structure (PDB:1KPL) is missing an entire N-terminal segment for one of the two monomers (chain A). We have partially modeled this segment (residues 12–32) by duplicating it from the other monomer (chain B) and splicing it into chain A. This reconstituted segment, highlighted in black in Fig. 1, dangles underneath the monomer opposite to the one to which it belongs and binds with that monomer's C-terminus, possibly contributing to the dimer's stability.
At the end of the 5-ns equilibration, the two ClC pores, which are devoid of crystallized water molecules in the published crystal structure, did not acquire any water molecules from the bulk solution. Neither did any of the crystal structure Cl− ions budge from their binding site at the center of each ClC pore. To remedy this situation, we placed water molecules in single file across each pore. We also placed an additional Cl− at the Glu-148's binding site in each pore. This new configuration was then equilibrated for 0.5 ns and used as a starting point for all further simulations.
Mapping the potential of mean force
To reconstruct the potential of mean force (PMF) of permeation through ClC, we have employed umbrella sampling (Roux, 1995; Gullingsrud et al., 1999). In our study, the PMF describes the overall free energy profile experienced by two simultaneous Cl− ions traversing either of the ClC's two pores in the absence of an external electric field, as a function of their respective positions along the pore (Z axis), averaged over all other degrees of freedom. In each of the sampling simulations, two Cl− ions per ClC pore were separately tethered by means of virtual one-dimensional springs acting along the Z axis with spring constants of 15 kcal/mol/Å2. This was done for both pores simultaneously, which are located so far apart (40 Å) that the correlation between the configurations of the ions in each pore is negligible. The tethering points for the umbrella potentials were never distributed >1 Å apart for any ion, requiring 92 simulations of 370 ps each to sample the range of motion of the ions during their conduction through the pore. Starting configurations for the different simulations were created by translating the permeating ions to the Z-coordinate minima of the tethering (umbrella) potentials. The energy of the ions and water molecules in the pore was then minimized (keeping all the other atoms fixed) so that the ions could reposition themselves laterally in the pore. The ions were then repositioned to the correct initial Z coordinate and the pore water molecules were equilibrated for 10 ps (keeping everything else fixed), after which the system was equilibrated for an extra 70 ps. This procedure ensured that the sudden artificial displacement of the ions in the pore, at the beginning of each simulation, did not unnecessarily perturb the protein. The spatial distributions of the ions along the Z axis for each simulation were then combined using the weighted histogram analysis method (Kumar et al., 1992) applied in two dimensions to obtain the full two-ion distribution of Cl−, which was then inverted to obtain the PMF.
As is often the case with atomistic MD simulations, the studied events occur at natural timescales which cannot be directly probed using available computer power. Taking the example of ClC-0, a typical pore current of 0.5 pA (Chen and Miller, 1996) corresponds to one elementary charge exiting the pore every 200–300 ps. This includes the time for an ion to diffuse to the pore's mouth as well as the time it takes for an ion (not necessarily the same one) to subsequently exit on the other side. Since we are only interested in the permeation phase, the conservative 300-ps timescale is a high upper bound estimate of the time needed to observe one conduction event. Nevertheless, even assuming the shortest event times, an equilibrium simulation would only provide a tiny number of complete conduction events, if any at all. Umbrella sampling allows us to sample high energy states with a much higher probability than is possible with equilibrium trajectories. With sufficient sampling, this methodology permits us to accurately determine the energy barriers as well as the physical pathway taken by the ions as they permeate through the pore. The accelerated sampling does come with restrictions: nonequilibrium dynamic trajectories cannot be directly observed. And although we provide a detailed statistical description of permeation across the ClC pore, the limitation of our method is that we cannot directly resolve the concerted motions of individual residues, water molecules, and permeant ions as they occur during a conduction event.
Energetics of selectivity and conduction
In this section, we describe the energetics involved in ion conduction. First, we introduce a detailed calculation of the free energy profile that governs the coordinated conduction of two simultaneous Cl− ions in the pore in the absence of an external field. This reveals the respective motions of the two ions as they permeate through the pore, and explains the anionic binding sites in ClC. We then analyze the interaction energies between the individual ions and the major constituents of the ClC channel, providing evidence that confirms and dispels common assumptions.
Potential of mean force
Our map of the potential is shown for each pore (chains A and B) in Fig. 3. The maps describe the energetics involved in the transition between an initial state I and a final state II. In state I, a first Cl− is bound to the channel's central binding site (determined from the crystal structure) and a second Cl− is positioned in the channel's cytoplasmic entrance; in state II, the first Cl− is positioned in the periplasmic exit and the second Cl− is bound in the central binding site. This process effectively describes a conduction event since states I and II share identical pore configurations except that in state I, one Cl− is at the top of the pore, and in state II, it is at the bottom. For a new translocation to occur, one simply has to wait for a third ion to diffuse from the bulk solution into either of the pore's entrances, after which the transition I → II or II → I can repeat. In the figure, distances along the Z axis (which approximately follows the pore) are measured with respect to the center of the lipid membrane, with positive values toward the periplasm. For the naming of the ClC binding sites, we follow the nomenclature of Dutzler et al. (2003): Scen is the central binding site coordinated primarily by Ser-107 and Tyr-445, Sint is at the cytoplasmic end of the pore, and Sext is the outer binding site on the other side, for which Cl− competes with Glu-148. The location of the Cl− binding sites as well as the Z axis coordinates are indicated for reference in Fig. 6.
FIGURE 3.

Potential of mean force for both pores (chains A and B) of ClC as a function of the positions along the Z axis of the top and bottom Cl− ions. Contours represent slices of 1 kcal/mol at a spatial resolution of 0.15 Å. Red indicates a low energy whereas blue denotes high energy. The shaded background denotes areas not sampled and the solid black contour represents energies above a threshold. The position of the three Cl− binding sites from the x-ray structure of Dutzler et al. (2003) are identified by straight lines. The minimum energy path, corresponding to the most likely conduction pathway, is shown for each pore as a thick black line.
FIGURE 6.

The ClC pore, showing the positions of the permeating Cl− ions (small orange spheres), the backbone amide hydrogens involved in permeation (large green spheres), the related nonhelical backbone (red tubes), and the two pore-lining polar residues Tyr-445 and Ser-107 and the two charged residues Glu-148 and Arg-147 possibly involved in the fast gate. The location of the three crystal binding sites as well as the Z axis coordinates are displayed for reference (with z = 0 indicating the middle of the lipid bilayer).
To test the consistency and validity of our results, we have measured the PMF for conduction in both monomers of the ClC dimer. Apart from a reconstructed N-terminus (for chain A of the 1KPL PDB structure), the two monomers have the same spatial structure. We therefore expect that the variations between the PMFs calculated for the two different pores are caused, for the most part, by the different distribution of microstates being sampled, rather than by macroscopic conformational differences between the two pores. There was one exception, in which a different behavior was observed between the two monomers: in chain A, when the two ions were kept close together (≈4 Å apart) at one specific location, the top ion was temporarily pushed sideways in the pore to maintain its distances from the bottom ion, disrupting the PMF for chain A at that location (located at Ztop ≈ −1.5 and Zbottom ≈ −6 Å); this was not observed anywhere else. In describing the PMF of ClC, we will only concern ourselves with features common to both pores.
Looking at the two PMFs, we notice similar characteristics. First of all, the PMFs share a similar two-ion pathway (shown as a black line) for ion translocation across the pore. From the PMF maps, we can hypothesize a probable sequence of events describing ion permeation across the channel that proceeds in a semistepwise manner, as shown in Fig. 4. Initially, a Cl− (Cl1) is bound to Scen and a second Cl− (Cl2) enters the pore from the cytoplasm until it reaches the general area of Sint. At this point, Cl2 stays put and repels Cl1 while the latter attempts to overcome a barrier located between Scen and Sext. Once this barrier has been crossed, Cl2 can inch closer to the central binding site to a location ∼−2.5 Å below it (which we call S−). Cl1 and Cl2 then move simultaneously and gradually toward their intermediate destinations of 1.5 Å above Sext (which we call S+) and Scen, respectively. With Cl2 now tightly bound to the crystal binding site Scen, Cl1 is free to exit the pore into the periplasm.
FIGURE 4.

Sequence of steps that occur during a conduction event in which an ion is moved from the cytoplasmic side to the periplasmic side of the channel. The schematic locations of the crystal structure binding sites (defined in the text) are shown as lines.
For both pores, the second Cl− enters or exits while the first Cl− is tightly bound to Scen. We refer to this as a king-of-the-hill mechanism, in which an ion is always ultimately always left to dominate the central region of the pore. Also, in both cases, the PMF exhibits minima in regions that correspond to two of the crystal structure binding sites being simultaneously occupied (located at the intersections of the straight lines in Fig. 3): Sint and Scen can be occupied simultaneously and so can Sext and Sint. On the other hand, the simultaneous occupation of Sint and Scen is energetically unfavorable compared to the other possibilities (the appearance of a minimum of the potential of mean force at that location for chain A corresponds to the top ion being pushed to the side, as described earlier). Simultaneous occupation of these two sites had been speculated by Dutzler et al. (2003), despite the fact that the sites are only 4 Å apart, on the basis of the observation that in some crystal structures, the authors observe simultaneous occupation of a Cl− at Scen and of a charged oxygen from Glu-148's carboxylic headgroup at Sext. Instead, we observe two nearby stable intermediate states which involve alternate binding locations for either ion: one where Sext and S− are occupied, and one where Sint and S+ are occupied. It comes as no surprise that the location of S+ coincides perfectly with the location of Glu-148's other carboxylic oxygen, according to the crystal structures in which this residue is present. Occupancy of S+ and S− have not been observed to date by x-ray diffraction.
Following the permeating Cl− ions along the most probable path, i.e., the path of minimum free energy, we measure similar energy profiles for both pores. Fig. 5 shows the PMF profile along this path. Between the two pore entrances, the PMF profile is relatively flat (with 1–2 kcal/mol fluctuations), permitting fast permeation. The main barrier in the profile occurs when a Cl− moves between Sint and Scen, with a height of 3–4 kcal/mol (or 4–4.5 ± 2 kcal/mol between lowest and highest energy in the pore). This barrier seems to be caused mainly by a lack of exposed backbone amides in that specific area of the pore for Cl− to interact with. The overall energy barriers are consistent with those determined by a simulation of the KcsA potassium channel (Bernèche and Roux, 2001), in which a maximum energy barrier of 2–3 kcal/mol was measured for K+ permeation (with three simultaneous ions in the pore).
FIGURE 5.

PMF profile along the minimum energy pathway for both pores (chains A and B) with the locations of the two permeating Cl− ions indicated for each local minimum. The curve follows the PMF along the solid lines of Fig. 3 and reports the minimum value of the PMF measured between this path and the two parallel paths representing the cases where the two ions are displaced toward and away from each other by 0.15 Å each. This protocol generates a fairly accurate description of the minimum energy pathway except for a small region (between 10 and 12 Å along the path) of chain A where an optimal solution could not be reliably found. The free energy is measured with respect to a base configuration in which one Cl− is bound to Scen and the other Cl− has been exchanged with a water molecule in bulk solution.
Permeation pathway
By stitching together all of our local sampling simulations along the most probable path, we obtain a picture of the physical pathway taken by Cl− as it crosses the channel. The trajectory of the topmost ion as it was moved up and down the channel joins with that of the bottom-most ion, resulting in the continuous trajectory shown in Fig. 6. Although only a full trajectory capturing entire Cl− permeation events can provide the true sequence of events during conduction, an analysis of the interaction energies between the channel and the ions, using the stitched trajectories, can reveal information about the role of the pore residues during permeation.
Fig. 7 shows the nonbonded (electrostatic and van der Waals) interaction energies between the Cl− ions and their environment as a function of their position along the pore. The energies were averaged over all four permeating ions and no appreciable variation was observed between the curves for the different ions and pores. The weakening of electrostatic interactions by the polarization of water molecules in the pore was not taken into account. The interaction energy of the ions with their surroundings can be decomposed into separate contributions from the various components of the protein, giving insight into the roles of these components in tuning channel energetics.
FIGURE 7.

Interaction energy of a permeating Cl− with the various constituents of (a) the ClC channel and (b) the pore region, calculated using a cutoff distance of 16 Å for nonbonded interactions. The standard deviation for the energy is ±5 kcal/mol for pore-lining residues, ±15 kcal/mol for water, and ±2 kcal/mol for bulk protein.
In Fig. 7 a, the energy contribution of the pore-lining residues is compared with that from the rest of the protein excluding the pore (referred to as the bulk protein). Although the pore residues dominate the interaction with Cl−, the bulk protein still contributes a significant fraction of the attractive interaction with Cl−. The bulk protein's purpose appears to be to provide a barrierless and energetically favorable background for negatively-charged particles present in the channel's pore.
Fig. 7 b shows the interaction energies between Cl− and the pore's backbone and nonpolar residues (including glycine) as well as with the pore's four polar and charged residues, as a function of position along the pore. We note that the backbone and nonpolar residues account for virtually the entirety of the total integrated interaction energy with the permeating Cl−. The backbone and nonpolar residues of the pore provide a flat basin of attraction for anions, whereas the polar and charged residues modulate the Cl− energy's position dependence. Indeed, aside from the central binding pocket in which Cl− is coordinated by polar residues and the periplasmic exit in which charged residues form a putative gate, the channel pore is lined in its entirety with nonpolar, noncharged residues.
It is intriguing to note that the pore's two conserved polar residues are present at the same location and define the pore's strongest Cl− binding site (Scen). The role of these two residues, Ser-107 and Tyr-445, is open to speculation, but it is clear that they are not by themselves responsible for the channel's anion over cation selectivity since their interaction energy with Cl− is not significant compared to the energy due to the strong electrical polarization of the protein. We believe that the most compelling reason for the existence of these residues is to keep an anion permanently in the pore to prevent such events as the formation of a proton-carrying continuous water file stretching across the channel or the passage of hydrophobic anions (Rychkov et al., 1998). Indeed, Ser-107 and Tyr-445 provide an abrupt and significant narrowing of the pore simultaneously with a very strong binding site for anions.
CLC ARCHITECTURE
Nonhelical backbone and protein polarization
It has been echoed throughout the literature on ClC that the broken-helix architecture stabilizes Cl− through α-helix electrostatic dipole interactions. Although the α-helix dipoles certainly contribute to a favorable environment for Cl−, it is not certain that their role is fundamental in determining the preferred Cl− binding sites (Åqvist et al., 1991). The interaction energy between Cl− and helices F and N (which both have their positive ends pointing toward Sext and have been credited for creating a favorable binding location), excluding the interaction with the pore-lining helix-capping residues, is shown in Fig. 7 a. This energy does not constitute a particularly prominent feature of the energy profile controlling Cl− conduction and does not explain the channel's intricate broken-helix architecture.
We believe that the broken-helix architecture stems from nature's desire to expose its backbone's amide groups to the permeant ions. Whereas the conventional picture of a membrane protein is that of a bundle of parallel α-helices, other structurally known channel proteins exhibit a broken-helix conformation. Notable examples are the potassium channels, the only other ion channels of known structure, and the aquaporin family of water channels (Fu et al., 2000; Tajkhorshid et al., 2002 ). In these other channels, it is the protein's backbone carbonyl groups which are exposed to the pore, as opposed to backbone amide groups for the case of ClC. It must be noted that the amide-to-Cl− interaction is by itself electrostatically unfavorable, but that the interaction between Cl− and the total dipole moment of an amino acid's backbone, when its amide group points toward Cl−, is favorable over a region of a few Å. In all cases, the proteins seem to favor the presence of a nonhelical secondary structure in the pore region, making the backbone available for interaction with solutes. The lack of a stable secondary structure would presumably require that the nonhelical segments be held in place at their ends by α-helices solidly anchored inside the protein. A cursory look at the location of the nonhelical segments in ClC reveals that, with few exceptions, these are all either at the surface of the protein and act as connecting loops, or are concentrated near the pore. Further examination of the conserved genetic sequence shows that all three pore-lining five-peptide segments end with a helix-breaking proline and are rich in small flexible hydrophobic residues (Dutzler et al., 2002).
Given the small size of the pore region compared to the great size and complexity of the scaffold that supports its nonhelical structure (if one can consider the ClC protein as a scaffold), there must be a strong incentive for channel proteins to expose their naked backbone. We believe that there is. The discoveries of the first ion channel structures elicited surprise because of the absence of significant charge in their pore regions, leading to the suggestion that strong charges would be problematic because, although the channels would be very selective, the strong electrostatic interactions could prevent the solute from unbinding from the pore. With this in mind, if we consider that a channel's idealized function in the absence of any bias is to provide a flat free energy potential for solutes, mimicking the bulk solution, then the ideal channel (from a permeation point of view) would have a continuous line of charge along which ions could glide. The important idea here is that of a flat potential energy surface: the ion should not get particularly attached to any location in the pore. In this respect, the backbone dipole moments provide a ladder of closely-spaced, but weak, identical interactions. Such a configuration confers a much higher mobility to Cl− than would a few isolated strong charges.
The role of the protein's electrical polarization is not to be neglected, however. As we have seen, most of the pore-to-ion attraction is caused by either backbone atoms or by nonpolar residues. The same is true of the interactions between the permeating Cl− ions and the channel as a whole (except at the cytoplasmic end of the pore, where the interactions between Cl− and the positively-charged (+28e) cytoplasmic side is important). At first, the role of the backbone atoms and nonpolar residues might seem strange. However, if the protein relied on polar residues to create favorable basins of attraction for anions, there would always be the danger that if the side chains of these residues were mobile enough, they would reorient themselves to be attractive to cations. Furthermore, polar residues interact strongly with external electrical fields, and their collective polarization might disrupt the field experienced by the permeating ions. Conversely, an external field would disrupt their interactions with the permeating ions, affecting the selective behavior of the channel. On the other hand, backbone atoms and nonpolar residues do not react strongly to external electrical fields or the presence of ions. In that sense, they provide a reliable frozen interaction with the permeating ions, since their polarization is dependent more on the channel architecture and on local interactions with neighboring residues, and less on external electrical fields. These weak dipolar interactions become quite substantial when one accounts for the sheer number of nonpolar residues involved.
Interrupted water file geometry
The water geometry that we observed in the ClC pore did not conform to the single water file picture expected of narrow channels. After equilibration, we found that the ClC pore instead encouraged an interrupted double-file geometry of water molecules around the Cl− ions. In Fig. 8, we have superimposed the locations of the water oxygens in and around the ClC pore for all local sampling simulations along the permeation pathway. One sees clear evidence that, on average, the water molecules are localized into two distinct files. One of these files is always present and also carries the permeating Cl−. The second file, only observed in the immediate proximity of Cl−, reflects the observed fact that in most of the pore, permeating Cl− ions remained partially hydrated (one water molecule above and below and up to three on the outer side).
FIGURE 8.

Overlay of the positions of the water molecules (blue) and Cl− (orange) for all the local sampling simulations along the permeation pathway showing the water double-file.
This partial hydration shell did not follow the permeating anions all the way, however. A continuous water file would create many problems such as loss of selectivity against larger anions (if the pore were wide throughout) and the conduction of protons if a bridge of connected water molecules were to span the pore. Instead, the double-file is broken at the constriction at Scen, to which a Cl− is quasipermanently bound. An obvious advantage of the broken double-file geometry is that the pore can offer an environment for permeating anions that is similar to that of the bulk solution outside the pore, thereby reducing the free energy cost of removing the anion's water shell as it enters the pore from either end. The only exception is at the central binding site Scen, where the ion-to-pore attraction is very strong, but there the ion can more easily part with its solvation shell.
Since the double water file is so advantageous, one may wonder why it is not observed in the potassium channel. Theoretical studies on generalized channels suggest that although electrostatic effects contribute to selectivity between ions of different charge, the discrimination between ions of same charge and valence can be controlled by the pore's radius and size fluctuations (Laio and Torre, 1999), energetic contributions that arise in addition to the dehydration energies at the pore entrance. These considerations are important for cation channels where strong selectivity is crucial, but in ClC there is little need to discriminate between different inorganic anions. Therefore one may imagine that, unlike for the potassium channel, the ClC pore geometry attempts to optimize permeation rather than interanion selectivity.
Multiion conduction
The pore's central binding site binds to Cl− very tightly through the side-chain hydroxyl groups of Tyr-445 and Ser-107 as well as through the backbone dipole moments of Phe-357 and Ile-356, such that a pore configuration devoid of the presence of anions is not possible in ClC. To dislodge the central Cl−, its binding energy needs to be considerably reduced. The presence of additional anions in the pore is thus required so that the channel-to-Cl− attraction and the Cl−-to-Cl− repulsion can balance each other. Our results confirm what had been previously suggested for ClC (Pusch et al., 1995) and has already been established for the K+-channel (Bernèche and Roux, 2001): conduction across the channel requires more than one ion in the pore. In addition, our PMF describes a permeation process for two simultaneous Cl− ions in the ClC pore, establishing that this number may be sufficient for the Cl− conduction in ClC.
With both ion channels of known structure (ClC and KcsA) exhibiting multiion permeation, we can ask whether multiion permeation is an ion-channel necessity or merely a coincidence. On one hand, if the aim is to ensure that permeation can occur along a path with a flat energy profile, then increasing the dimensionality of the PMF (i.e., adding essential degrees of freedom) makes it easier to get around barriers. On the other hand, it is conceivable that one could engineer an ion channel with single ion occupancy by relegating the degrees of freedom of the extra ions to internal components of the channel. Multiple occupancy would, in that case, not be a necessity. There is, however, a problem with single occupancy pores: if the single ion is allowed to exit the channel, then the possibility arises that a continuous file of water across the channel connects both sides of the cell membrane, possibly allowing for proton conduction to the detriment of the cell.
CONCLUSION
We have mapped the energetics involved in the conduction of a pair of Cl− ions across the ClC channel. The result suggests that ion dynamics in the pore follows a king-of-the-hill mechanism, in which two Cl− ions compete over an energetically favorable central location in the pore. This strategy would ensure that an ion is always left inside the pore to block it. During a conduction event, the ion configurations in the pore appear to evolve through a succession of four stable states. The positions of the ions in these states coincide with the locations of three binding sites observed by x-ray crystallography. In addition, we observe stable intermediate states in which the ions are located at two novel locations, S− and S+.
Inspection of the interaction energies between the ClC channel and the permeating Cl− ions reveals the importance of the protein's overall polarization in making the pore attractive to anions. Indeed, backbone and nonpolar residues account for a large majority of the attraction between Cl− and the channel. Our calculations do not, a priori, support the common assumption that polar residues and specific α-helix dipole interactions play a pivotal role in assuring the pore's anion over cation selectivity. Instead, we suggest that the main role for the ClC's broken-helix structure is to provide the pore region with a nonhelical backbone structure, allowing anions to interact favorably with the exposed backbone's electric dipole moment. This type of interaction has the advantage that it is rather evenly distributed along the pore and prevents Cl− from getting stuck in deep energy wells. We also suggest a novel role for the polar residues Ser-107 and Tyr-445 that is consistent with the nature of their measured interaction with Cl−: they ensure that the pore remains blocked by an anion at all times, preventing the formation of a continuous water file. These residues likely also contribute to the size-selectivity of the pore and prevent the passage of hydrophobic particles, and this will likely be resolved by further experimental and theoretical work investigating selectivity (Fahlke et al., 1997; Shrivastava et al., 2002; Bernèche and Roux, 2001) in ClCs.
Compared to the other ion channel family of known structure, the K+ channels, the ClC narrow-pore region is longer and wider. Whereas KcsA has a very symmetrical pore and tight interactions with the permeating K+ ions, the ClC pore is irregular and accommodates partially solvated anions through most of its length. The consequence of these two diverging architectures is that ClC is a lot less selective between ion species of same charge than is KcsA, in harmony with a lack of evolutionary pressure in that direction. On the other hand, both channels share a similar peculiar feature: they both exhibit a broken-helix architecture resulting in their pores being lined almost exclusively with a nonhelical backbone structure. This pore architecture has been previously shown to be a crucial ingredient for the efficient conduction and selectivity of ions in K+ channels and here we observe the same for ClC. Based on these discoveries, one should expect to observe the exposed backbone architecture in the pores of the many ion channel structures yet to come.
Acknowledgments
This work was supported by the National Institutes of Health Research grants PHS5P41RR05969 and 1R01GM60946-01, as well as the National Science Foundation supercomputer time grant NRAC MCA93S028.
References
- Accardi, A., and M. Pusch. 2003. Conformal changes in the pore of CLC-0. J. Gen. Physiol. 122:277–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åqvist, J., H. Luecke, F. A. Quiocho, and A. Warshel. 1991. Dipoles localized at helix termini of protein stabilize charges. Proc. Natl. Acad. Sci. USA. 88:2026–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Åqvist, J., and V. Luzhkov. 2000. Ion permeation mechanism of the potassium channel. Nature. 404:881–884. [DOI] [PubMed] [Google Scholar]
- Bernèche, S., and B. Roux. 2000. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 78:2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernèche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- Chen, T.-Y., and C. Miller. 1996. Nonequilibrium gating and voltage-dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, D. A., J. M. Cabral, R. A. Pfuetzer, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, and R. MacKinnon. 2003. Gating the selectivity filter in ClC chloride channels. Science. 300:108–112. [DOI] [PubMed] [Google Scholar]
- Fahlke, C. 2001. Ion permeation and selectivity in ClC-type chloride channels. Am. J. Physiol. Ren. Physiol. 280:F748–F757. [DOI] [PubMed] [Google Scholar]
- Fahlke, C., H. Yu, C. L. Beck, T. R. Rhodes, and J. A. L. George. 1997. Pore-forming segments in voltage-gated chloride channels. Nature. 390:529–532. [DOI] [PubMed] [Google Scholar]
- Ferroni, S., C. Marchini, M. Nobile, and C. Rapisarda. 1997. Characterization of an inwardly rectifying chloride conductance expressed by cultured rat cortical astrocytes. Glia. 21:217–227. [DOI] [PubMed] [Google Scholar]
- Friedrich, T., T. Breiderhoff, and T. J. Jentsch. 1999. Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J. Biol. Chem. 274:896–902. [DOI] [PubMed] [Google Scholar]
- Fu, D., A. Libson, L. J. W. Miercke, C. Weitzman, P. Nollert, J. Krucinski, and R. M. Stroud. 2000. Structure of a glycerol conducting channel and the basis for its selectivity. Science. 290:481–486. [DOI] [PubMed] [Google Scholar]
- Grayson, P., E. Tajkhorshid, and K. Schulten. 2003. Mechanisms of selectivity in channels and enzymes studied with interactive molecular dynamics. Biophys. J. 85:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullingsrud, J., R. Braun, and K. Schulten. 1999. Reconstructing potentials of mean force through time series analysis of steered molecular dynamics simulations. J. Comp. Phys. 151:190–211. [Google Scholar]
- Hille, B. 1992. Ionic Channels of Excitable Membranes, 2nd Ed. Sinauer Associates, Sunderland, MA.
- Iyer, R., T. M. Iverson, A. Accardi, and C. Miller. 2002. A biological role for prokaryotic ClC chloride channels. Nature. 419:715–718. [DOI] [PubMed] [Google Scholar]
- Jentsch, T. J., T. Friedrich, A. Schriever, and H. Yamada. 1999. The ClC chloride channel family. Pflug. Arch. Eur. J. Physiol. 437:783–795. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Kalé, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comp. Phys. 151:283–312. [Google Scholar]
- Kumar, S., D. Bouzida, R. H. Swendsen, P. A. Kollman, and J. M. Rosenberg. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comp. Chem. 13:1011–1021. [Google Scholar]
- Laio, A., and V. Torre. 1999. Physical origin of selectivity in ionic channels of biological membranes. Biophys. J. 76:129–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C.-W., and T.-Y. Chen. 2003. Probing the pore of ClC-0 by substituted cysteine accessibility method using methane thiosulfonate reagents. J. Gen. Physiol. 122:147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig, U., T. J. Jentsch, and M. Pusch. 1997. Inward rectification in ClC-0 chloride channels caused by mutations in several protein regions. J. Gen. Physiol. 110:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell Jr., A. D., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, I. W. E. Reiher, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-hydrogen empirical potential for molecular modeling and dynamics studies of proteins using the CHARMM22 force field. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Maduke, M., C. Miller, and J. A. Mindell. 2000. A decade of ClC chloride channels: structure, mechanism, and many unsettled questions. Annu. Rev. Biophys. Biomol. Struct. 29:411–438. [DOI] [PubMed] [Google Scholar]
- Morais-Cabral, J. H., Y. Zhou, and R. MacKinnon. 2001. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 414:37–41. [DOI] [PubMed] [Google Scholar]
- Pusch, M., U. Ludewig, A. Rehfeldt, and T. J. Jentsch. 1995. Gating of the voltage-dependent chloride channel ClC-0 by the permeant anion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- Roux, B. 1995. The calculation of the potential of mean force using computer simulations. Comp. Phys. Comm. 91:275–282. [Google Scholar]
- Roux, B. 1999. Statistical mechanical equilibrium theory of selective ion channels. Biophys. J. 77:139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkov, G. Y., M. Pusch, M. L. Roberts, T. J. Jentsch, and A. H. Bretag. 1998. Permeation and block of the skeletal muscle chloride channel, ClC-1, by foreign anions. J. Gen. Physiol. 111:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlenkrich, M., J. Brickmann, A. D. MacKerell, Jr., and M. Karplus. 1996. Empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz and B. Roux, editors. Birkhauser, Boston, MA. 31–81.
- Shrivastava, I. H., and M. S. P. Sansom. 2000. Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys. J. 78:557–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava, I. H., D. P. Tielemann, P. C. Biggin, and M. S. P. Sansom. 2002. K+ versus Na+ in a K channel selectivity filter: a simulation study. Biophys. J. 83:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, J., J. Gullingsrud, P. Grayson, and K. Schulten. 2001. A system for interactive molecular dynamics simulation. In 2001 ACM Symposium on Interactive 3D Graphics. J. F. Hughes and C. H. Séquin, editors. ACM SIGGRAPH, New York. 191–194.
- Tajkhorshid, E., P. Nollert, M. Ø. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud, and K. Schulten. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 296:525–530. [DOI] [PubMed] [Google Scholar]
- Traverso, S., L. Elia, and M. Pusch. 2003. Gating competence of constitutively open ClC-0 mutants revealed by the interaction with a small organic inhibitor. J. Gen. Physiol. 122:295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]