

Abstract
The reincorporation of lipids into monolayers at the air-water interface after collapse is important to the maintenance of low surface tensions on subsequent expansion and compression cycles. For single component, anionic dipalmitoylphosphatidylglycerol monolayers, the fraction of recovered lipid is proportional to the subphase ionic strength. The collapse mechanism and structure of the collapsed materials appear unchanged with ionic strength. A simple electrostatic barrier model shows that the fractional recovery depends exponentially on the Debye length; this is verified by experiment. This simple model suggests possible catalytic roles for the cationic lung surfactant specific proteins SP-B and SP-C that induce structural changes in the monolayer that may act as charge-neutralizing docking sites for surfactant in the subphase, leading to faster and more efficient recovery.
INTRODUCTION
Our understanding of the organization of lipid and other insoluble surfactant monolayers at the air-water interface under equilibrium conditions is quite advanced, as the subject has been of great interest in biology, chemistry, and physics for nearly a century (Kaganer et al., 1999; Knobler and Desai, 1992; McConnell, 1991; Schwartz, 1997; Zasadzinski et al., 2001). Less is known about the nonequilibrium aspects of monolayer structure and function, especially monolayer collapse (which ultimately limits the surface tension reduction possible by a given monolayer) (Diamant et al., 2000; Ding et al., 2001; Gopal and Lee, 2001; Guitter et al., 1988; Kampf et al., 1999; Lipp et al., 1996, 1998; Longo et al., 1993; Risdale et al., 2001; Schief et al., 2000; Tchoreloff et al., 1991; Warriner et al., 2002; Ybert et al., 2002). Even less is known about the subsequent recovery of surfactant removed from the interface into the subphase that may or may not return into the monolayer on reexpansion (Ding et al., 2001; Lipp et al., 1998; Lu et al., 2002; Walters et al., 2000).
The collapse and recovery of surfactant monolayers at the air-water interface is especially important to the function of lung surfactants, a lipid-protein layer that coats the inside of the lung alveoli. Human lung surfactant is a complex mixture of lipids and proteins that coats the alveolar liquid-air interface. This film modulates the surface tension of the lung, lowering the normal air-water surface tension of ∼70 mN/m to near zero on expiration, thereby stabilizing alveoli against collapse during expiration and minimizing the work of expanding the alveolar surface during inhalation (Goerke, 1998; Notter, 2000). Lack of effective surfactant in premature infants results in neonatal respiratory distress syndrome (NRDS), a potentially fatal disorder characterized by reduced lung compliance and oxygenation (Notter, 2000). Replacement lung surfactants for treatment of NRDS consist primarily (>98% by weight) of dipalmitoylphosphatidylcholine (DPPC), unsaturated phosphatidylcholines and phosphatidylglycerols, fatty acids, and cholesterol. There are small fractions (∼2 wt%) of two surfactant specific proteins, SP-B and SP-C (Ding et al., 2001, 2003; Mizuno et al., 1995; Notter, 2000; Tanaka et al., 1986). There is a wide variation in lipid and protein content between the various replacement and native surfactants; the optimal lung surfactant composition has not yet been established, nor is it clear that there is a universal surfactant composition for treatment of NRDS (Bernhard et al., 2000).
DPPC, the main lipid component of native lung surfactant, forms a rigid monolayer capable of surface tensions near zero when fully compressed (Lee et al., 1999). However, DPPC fails as a lung surfactant (Poulain and Clements, 1995; Robertson and Halliday, 1998) as it is slow to adsorb from solution and respreads slowly when compression is relieved. This helps explain the significant fraction of unsaturated phospholipids and hydrophobic proteins in native surfactant (Bernhard et al., 2000). Although unsaturated lipids and proteins likely facilitate surfactant adsorption and spreading, they collapse at relatively high surface tensions via the ejection of material from the monolayer (Ding et al., 2001; Lipp et al., 1996, 1998; Takamoto et al., 2001). Although the individual components of lung surfactant are either good at lowering surface tension (DPPC) or fluidizing the monolayer (unsaturated PG and PC; proteins), no single lipid or protein exhibits both properties. This dichotomy of necessary material properties has led to the “squeeze-out” theory of lung surfactant function (Notter, 2000). This theory states that the unsaturated lipids and proteins in lung surfactant are selectively removed, or “squeezed out,” from the monolayer during compression, leading to a DPPC-enriched monolayer capable of low surface tension. However, in vitro studies of captive (Schürch et al., 1989, 1998) and pulsating air bubbles in contact with aqueous surfactant show that the necessary mass transfer requires that the squeezed-out surfactant remain within a few nanometers of the interface. Hence, current thought is that the lipids and proteins “squeezed out” from the monolayer occupy a “surface associated reservoir” near the interface (Schürch et al., 1995). However, the mechanisms and kinetics of readsorption of this surface associated reservoir into the monolayer are essentially unexplored.
Native LS extracts adsorb to air/water interfaces rapidly to form monolayers both in vivo and in vitro. The LS monolayer is initially fluid-like at large areas per molecule. On compression, LS monolayers achieve near-zero surface tension (or surface pressures in excess of 70 mN/m, where the surface pressure, π, is defined as the surface tension of pure water minus the surface tension in the presence of a monolayer). LS can maintain these low tensions or high surface pressures past collapse of the monolayer and is capable of respreading rapidly and reversibly upon reexpansion from the collapsed state. These properties lead to a stable and reversible hysteresis in cyclic compression and expansion isotherms, and are believed to be key to reducing the work of breathing and mechanically stabilizing the lungs in vivo (Notter, 2000). Thus, to properly account for the behavior of functional lung surfactant, a reasonable model must account for: 1), rapid adsorption, 2), low surface tensions upon compression, and 3), rapid and reversible respreading on expansion.
Monolayer collapse occurs via several mechanisms, including large-scale folding into the subphase (Diamant et al., 2000; Ding et al., 2001; Gopal and Lee, 2001; Lipp et al., 1996, 1998; Risdale et al., 2001; Schürch et al., 1998; Warriner et al., 2002); fracture of the monolayer (Lipp et al., 1996, 1998; Longo et al., 1993); and squeeze-out of bilayer vesicles or other small bilayer aggregates (Risdale et al., 2001; Schief et al., 2000; Schürch et al., 1998; Takamoto et al., 2001; Ybert et al., 2002). It has been suggested that a high recovery necessitates that the monolayer collapse by a folding mechanism and the collapse material remain attached to the monolayer (Diamant et al., 2000; Gopal and Lee, 2001; Lipp et al., 1998; Warriner et al., 2002). For this mechanism to occur the monolayer must have areas of differing spontaneous curvature just before collapse (Diamant et al., 2000). For monolayers containing the SP-B and SP-C proteins, some of the collapsed material forms three-dimensional structures that store protein and lipids in multilayer patches until expansion (Ding et al., 2003; Takamoto et al., 2001; von Nahmen et al., 1997).
Surfactant recovery is defined experimentally as the amount of material that returns to the monolayer after collapse during reexpansion of the monolayer area, divided by the amount of material initially removed from the monolayer (Notter, 2000). This is generally determined from cyclic Langmuir isotherms. Little is known about recovery mechanisms, or the conditions necessary for complete recovery in single component monolayers, much less the multicomponent lipid-protein monolayers common to lung surfactants. Here we show that for simple, single component, anionic dipalmitoylphosphatidylglycerol (DPPG) monolayer, surfactant recovery is proportional to the subphase ionic strength. We use various mono- and divalent salt concentrations in the subphase to control the interactions between the collapse structures and the monolayer. The recovery is consistent with an electrostatic barrier, the height of which depends on the Debye length, or equivalently, the square root of the ionic strength of the subphase. The collapse structures appear to be bilayer aggregates or vesicles regardless of the salt concentration. However, how these aggregates interact with the monolayer changes with the salt concentration. A simple model of the probability of a vesicle rejoining the monolayer depends on the vesicle concentration and the height of the barrier. At low salt, the vesicles diffuse away from the interface due to the barrier and do not reincorporate their material into the monolayer on expansion. At high salt, the net interaction appears attractive as the vesicles stay near the air-water interface allowing easy respreading back into the monolayer. This simple model suggests possible catalytic roles for the cationic lung surfactant specific proteins SP-B and SP-C in respreading. Both SP-B and SP-C have multiple excess positively charged residues and preferentially locate in fluid and anionic monolayer domains. Both proteins induce extended three-dimensional structures into the subphase that may act as overall positively charged sites for docking surfactant in the subphase onto the monolayer (Ding et al., 2001, 2003), thereby bypassing the electrostatic barrier to surfactant recovery.
MATERIALS AND METHODS
A modified commercial Langmuir trough (NIMA, Coventry, England) with a stainless steel ribbon barrier was used to measure compression-expansion-compression cyclic surfactant isotherms. The ribbon barrier minimizes leakage of surfactant around the barriers at low surface tensions (high surface pressures), which can complicate measures of the fractional recovery. Temperature control of the subphase is achieved through recirculating water. The trough can be operated over a temperature range of 10–50°C. A simple feedback loop allows for measurement and control of the subphase temperature; all experiments were done at 30°C, which is well above the triple point for DPPG. Expansion and compression speeds ranged from quasistatic (∼30–60 min per expansion/compression cycle) to the maximum speed available in our trough (∼30 s/cycle); no significant variations in the isotherms were observed over these cycle times. A Wilhelmy plate pressure sensor with a filter paper plate was calibrated before each experiment using the liquid-expanded-liquid-condensed (LE-LC) kink of palmitic acid at 25°C (Peterson et al., 1992; Warriner et al., 2002).
DPPG (Avanti Polar Lipids, Alabaster, AL; >99% stated purity) was deposited from a 2-mg/ml chloroform solution onto subphases composed of 0.2 mM sodium bicarbonate at pH 7.0 ± 0.2 at 30°C with varying NaCl or other salt concentrations. After allowing the solvent to evaporate for 10 min, the DPPG monolayer was compressed beyond collapse, expanded, and then compressed again. The degree of overlap in the isotherms of the collapse region was used to determine the fractional recovery (Notter, 2000; Warriner et al., 2002).
Atomic force microscopy
To visualize the DPPG collapse structures, the collapsed monolayers at high surface pressure were deposited onto freshly cleaved mica discs using conventional Langmuir-Blodgett deposition at 30°C. The mica disc was placed in the subphase before spreading the DPPG. After the monolayer was compressed past collapse, the mica disc was pulled through the monolayer by a motorized dipping mechanism (NIMA). A modified Nanoscope III AFM (Veeco Instruments, Santa Barbara, CA) was used for imaging in air at ambient temperature. The samples were glued or taped to magnetic stainless steel discs, which were then attached to the piezoelectric tube scanner via an internal magnet on the scanner. Atomic force microscopy (AFM) imaging was done with a 150-μm × 150-μm (J) scanner in contact mode. Silicon nitride tips with a spring constant of 0.12 N/m were used. Exerting large forces on the sample was a concern during imaging, so samples were checked often for deformation. This was done by imaging for a few minutes on a smaller region (∼20 μm) and then zooming out to check whether damage had been done to the scanned region.
Brewster angle microscopy
A 7–30 mW 686-nm diode laser was used as light source. A Glen-Thompson polarizer (Melles-Griot, Sunnyvale, CA) placed between laser and trough provided p-polarized light at the Brewster angle (53.1° from vertical for a pure water surface). A 70XL zoom lens (Optem International, Fairport, NY) with magnification ranging from 2.25 to 15.75× was used to focus the light onto a Sony XC-E150 near-infrared camera. An additional polarizer was used at the entrance to the 70XL lens to improve contrast in the images, which were recorded to a JBC super VHS VCR (Elmwood Park, NJ). The Brewster angle microscope (BAM) was located over a homebuilt Langmuir trough equipped with a Wilhelmy-type pressure-measuring device and two computer-controlled barriers that provided a symmetric compression. DPPG was deposited onto various subphases as described above.
RESULTS AND DISCUSSION
Isotherms
Representative isotherms at 30°C (displaced by 20 A2/molecule) of DPPG at three different salt concentrations: buffer only, buffer plus physiological NaCl (150 mM), buffer plus 1 M NaCl, are shown in Fig. 1. Isotherms are determined by decreasing (or increasing) the area available to a monolayer, A, by imposing an external surface pressure, π, which lowers the normal air-water surface tension, γo, to γ. π = γo − γ. Above the “triple-point” temperature for a particular lipid, compression induces the formation of the liquid-expanded phase from the gaseous phase at low surface pressure. The triple-point temperature of DPPG is ∼23°C, and decreases with increasing salt concentration in the subphase (Takamoto et al., 2001). The 30°C temperature of the experiments was thus well above the triple point for DPPG. In the LE phase, the hydrophobic parts of the molecules contact each other and lift from the water surface, but remain largely disordered and fluid. Further compression leads to a first-order transition to the “liquid-condensed” phase, marked by a plateau in the isotherm beginning at ∼20 mN/m and ending at ∼30 mN/m corresponding to LE-LC coexistence (Fig. 1). The LC phase is characterized by longer-ranged molecular order and lower compressibility than the LE phase (Bringezu et al., 2001; Lee et al., 2002).
FIGURE 1.

First compression (1); expansion (E); second compression (2). Langmuir isotherms (offset by 20 A2/mol) of dipalmitoylphosphatidylglycerol at pH 7 at 30°C, well above the triple point for DPPG. The isotherm to the left is for a DPPG monolayer on a subphase of 0.2 mM sodium bicarbonate, the center isotherm is for DPPG on 0.2 mM sodium bicarbonate with 150 mM sodium chloride, and the isotherm to the right is for DPPG on 0.2 mM sodium bicarbonate with 1 M NaCl. The same amount of DPPG was spread from a chloroform solution for each isotherm. The main difference between the isotherms is the second compression cycle (labeled 2). For the 1-M isotherm, the second compression almost retraces the first (labeled 1), indicating little material lost and the fractional recovery is 0.95. For the isotherm on buffer only, the second compression is shifted toward smaller areas per molecule in comparison to the first compression, indicating a significant loss of material from the monolayer. The fractional recovery was only ∼0.30. The expansion isotherms were nearly identical, suggesting that the surface pressure at which material was reincorporated (plateau at ∼25 mN/m) was independent of the ionic strength.
The initial compression (labeled 1 in Fig. 1) and expansion (labeled E in Fig. 1) cycles are similar between all three isotherms; it is only on the second compression cycle (labeled 2 in Fig. 1) that differences are manifested. Collapse of each monolayer occurs at ∼70 mN/m, the LE-LC transition occurs at a similar range of 20–30 mN/m on compression, and the expansion cycle shows a distinct break at ∼20–30 mN/m independent of ionic strength. This suggests that the reincorporation of material occurs at the same surface pressure for all ionic strengths tested. However, in each case, the second compression cycle is offset toward smaller areas per molecule, consistent with some loss of monolayer material to the subphase. This offset did not occur when DPPG monolayers were cycled to a maximum pressure of 40 mN/m, confirming that the offset was due to material lost during monolayer collapse (Takamoto et al., 2001). The degree of overlap of the consecutive compressions determines the percent recovery of this lost material (Notter, 2000; Warriner et al., 2002).
The recovery of surfactant after collapse steadily increased with increased subphase ionic strength, from a low of ∼30% at 0.2 mM buffer concentration to almost complete recovery at 1 M NaCl. Fig. 2 shows that the log (recovery) is proportional to the Debye length (Israelachvili, 1992) in the subphase for a number of 1:1 and 2:1 monovalent and divalent salts. The Debye length, 1/κ is given by:
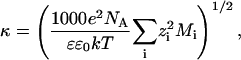 |
(1) |
in which e is the electron charge, NA is Avogadro's number, k is Boltzmann's constant, Mi is the molar concentration of electrolyte of valence zi, ɛ0 is the permittivity of vacuum, and ɛ is the dielectric constant of the subphase. At 30°C, κ−1 = 0.43(2I)−1/2 nm, in which I is the ionic strength of the subphase: 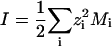 (Israelachvili, 1992). The ionic strength of the subphase determines both the range and magnitude of the electrostatic interactions between the charged DPPG monolayer and the charged DPPG bilayer aggregates squeezed out from the monolayer (Israelachvili, 1992). There is some variability in the fractional recovery with the nature of the ion, but the overall trend is clear. The line is the best fit to the NaCl data as a function of ionic strength.
(Israelachvili, 1992). The ionic strength of the subphase determines both the range and magnitude of the electrostatic interactions between the charged DPPG monolayer and the charged DPPG bilayer aggregates squeezed out from the monolayer (Israelachvili, 1992). There is some variability in the fractional recovery with the nature of the ion, but the overall trend is clear. The line is the best fit to the NaCl data as a function of ionic strength.
FIGURE 2.

Log (fractional recovery) versus inverse Debye length showing the exponential increase in recovery with increased ionic strength of the subphase for a variety of monovalent and divalent salts. The line is the best fit to the NaCl data. This trend is consistent with an electrostatic barrier to vesicle fusion with the monolayer as described in Eq. 2.
In Fig. 1, the plateau upon expansion around 25 dyne/cm corresponds closely to the LC-LE phase transition pressure (Mansour et al., 2001; Takamoto et al., 2001) and is independent of ionic strength. From the relatively rapid change in molecular area with decreasing surface pressure (below ∼25 mN/m), it appears that the DPPG expelled to the subphase begins to respread when the surface pressure is lower than the LC-LE phase transition pressure. When the surface pressure drops below this pressure, the surface pressure within the vesicles is likely similar or higher than in the monolayer and the vesicle can readsorb into the monolayer. An alternate explanation is that the monolayer must be sufficiently fluid to make room for additional material at the interface. Thus, reincorporation only occurs when a significant fraction of the monolayer is in the fluid state and has a low surface viscosity (Ding et al., 2002). Hence, in addition to the repulsive electrostatic interaction discussed above, an attractive interaction at lower surface pressures may cause the surfactant to readsorb. However, the isotherms do not reveal anything about the organization of the collapse structures.
Brewster angle microscopy
BAM images confirm our earlier fluorescence microscopy (Takamoto et al., 2001) that shows the material lost from the monolayer forms bilayer aggregates or vesicles in the subphase at all salt concentrations (Fig. 3). At collapse, these bilayer aggregates appears as bright spots, smaller than the resolution of the BAM. The number of bright spots depends on how many of these aggregates stay within the depth of field of the BAM. In the buffer-only case, a few large bright streaks of aggregated material are evident in images recorded at collapse, indicating that little collapse material remains near the monolayer (Fig. 3 A). This is to be compared with both the 150-mM and 1-M salt samples (Fig. 3, B and C), which have numerous bright spots in the BAM images. The 1-M sample (Fig. 3 C) has significantly more bright spots at collapse than the 150-mM sample (Fig. 3 B), likely due to more collapsed material remaining near the monolayer. This correlates directly to the higher fractional recovery of the monolayer on expansion (Figs. 1 and 2).
FIGURE 3.

Brewster angle microscope images of DPPG monolayers on the different ionic strength buffers at a surface pressure of ∼70 mN/m immediately after monolayer collapse. (A) For the DPPG monolayer on a subphase of 0.2 mM sodium bicarbonate, only large-scale folds or cracks in the monolayer are visible (central feature in the image). (B) For DPPG on 0.2 mM sodium bicarbonate with 150 mM sodium chloride, there are still folds and cracks in the monolayer (not shown), but there are also small, circular bright spots consistent with bilayer aggregates in the subphase near the monolayer. (C) For DPPG on 0.2 mM sodium bicarbonate with 1 M sodium chloride, the number of bright spots increases, suggesting that there are more bilayer aggregates near the monolayer.
During the expansion of the 1-M salt monolayer, some of the bright spots corresponding to the collapse material persist down to a surface pressure of 5 mN/m. However, the number of bright spots begins to decrease at <30 mN/m, which corresponds to the kink in the isotherm (Fig. 1). The collapse material appears to spread out heterogeneously, suggesting that the readsorption depends on local conditions and local interactions with the monolayer. The expansion of the 150-mM salt monolayer is quite different. By 50 mN/m, the bright spots have disappeared, only to reappear at 20 mN/m. This is due to the collapse material staying near the monolayer, but outside of the depth of field of the microscope, until the material starts to respread at ∼20 dynes/cm. This suggests that the interactions between the monolayer and the aggregate grow more attractive as the surface pressure is lowered. The expansion of the monolayer on buffer is quite different. The only visible features are sparsely distributed bright streaks present at collapse (Fig. 3 A) that disappear by 40 dyne/cm. Very little contrast is seen throughout the expansion. No bright spots are seen near the monolayer at any surface pressure, consistent with the small fraction of material recovered in this monolayer (Figs. 1 and 2).
Atomic force microscopy
The BAM images suggest a quite different arrangement of the collapsed material depending on the subphase ionic strength. AFM images of monolayers transferred by Langmuir-Blodgett deposition on mica substrates after collapse (Zasadzinski et al., 1994) also showed significantly different organization of the collapsed material depending on the subphase ionic strength. For the monolayer transferred from a subphase containing only buffer, little material remained near the monolayer (Fig. 4 A). The transferred monolayer was smooth, with few bright spots in the AFM image and an occasional multilayer patch, which was ∼5 nm high, consistent with the expected thickness of a DPPG bilayer (Fig. 4 A, inset). This suggested a monolayer folding or buckling mechanism, consistent with the appearance of such folds and cracks in the BAM images (Fig. 3 A).
FIGURE 4.

AFM images (50 × 50 μm) of DPPG monolayers transferred via Langmuir-Blodgett deposition to mica substrates just after collapse at a surface pressure near 70 mN/m. (A) DPPG monolayer deposited from a subphase of 0.2 mM sodium bicarbonate. The AFM image shows a generally smooth background with a single fold. The inset shows that the fold is ∼5-nm high, consistent with a bilayer structure. No other collapse material remains with the monolayer. (B) DPPG monolayer deposited from subphase with 0.2 mM sodium bicarbonate with 150 mM sodium chloride. The monolayer is decorated with 50–100-nm bright spots (inset) that are consistent with small bilayer aggregates or vesicles (see Fig. 5). The arrows indicate a ring of collapse material surrounding a solid monolayer domain. More collapsed material remains near the interface than in A, but much less than C. (C) DPPG monolayer deposited from subphase with 0.2 mM sodium bicarbonate with 1 M sodium chloride. The vesicles collect into distinct rings around solid phase domains. Individual vesicles are difficult to see, as there is so much collapsed material near the monolayer. The bright areas have increased to ∼200 nm in height (inset).
The sample transferred from the 150-mM salt subphase had a quite different appearance. There were numerous, distinct bright spots corresponding to 50–100 nm high structures, presumably the same bilayer aggregates present in the BAM images (Fig. 4 B), scattered across the surface. Higher resolution images reveal small spherical structures 50–100 nm in diameter scattered beneath the monolayer (Fig. 5), consistent in size and shape with small bilayer vesicles. These spherical structures aggregate and preferentially decorate the borders of the solid phase domains of the monolayer (Fig. 4 B). This indicates the collapsed material likely was ejected from the edge of the solid domains and remains near its ejection point. The LE phase, if any remains, is most likely to be found near the boundaries of the solid phase domains.
FIGURE 5.

Higher magnification AFM image (1 × 1 μm) of cluster of spherical collapse structures formed on DPPG monolayer transferred from a 150-mM salt buffer. Each sphere is 50–100 nm in diameter. The bright rings in Fig. 4 C are also formed by aggregated spheres similar to these.
The monolayer transferred after collapse on a 1-M salt subphase monolayer retains significantly more collapse material near the monolayer. The network structure of the bright areas is obvious in the AFM image and is similar to the distribution of the solid phase domains in the uncollapsed monolayer (Fig. 4 C). The bright network is ∼200 nm in height; higher resolution images (not shown) of the 1-M monolayer show the same vesicle aggregates as in Fig. 5. In the 1-M monolayer, mounds of the vesicle aggregates pile up to account for the increased height of the network, again preferentially decorating the borders of the solid phase domains. The fractional recovery of each monolayer correlates with the amount of material seen near the monolayer in the AFM images.
CONCLUSIONS
BAM and AFM images reveal that the materials squeezed out from DPPG monolayers at collapse organize as bilayer aggregates in the subphase at all ionic strengths. At low-ionic strength, these aggregates apparently diffuse away from the monolayer and remain in the subphase, whereas at high-ionic strength; the vesicles remain attached to the monolayer (or at least in the immediate vicinity of the monolayer) and rapidly reincorporate into the monolayer on expansion of the film. This readsorption behavior suggests that for charged monolayers (or charged domains of the multicomponent monolayers common to lung surfactants (Lipp et al., 1996, 1998; Takamoto et al., 2001) and large Debye length (or low subphase ionic strength), there is an electrostatic energy barrier to readsorption of the charged vesicles to the charged interface. In opposition to this electrostatic repulsion are a complicated set of hydrophobic, van der Waals, and other attractive interactions that promote readsorption of the bilayer aggregates to the interface. The surface pressures and phase behavior of DPPG in the monolayer relative to the bilayer likely determines the strength of these attractive interactions; recovery of surfactant into the monolayer only occurs at surface pressures below the LE-LC phase transition pressure of ∼30 mM/m. A balance of the attractive and repulsive forces gives the net interaction schematically shown in Fig. 6.
FIGURE 6.

Schematic of interaction potential between charged bilayer aggregates and charged monolayer. Bilayer structures in solution are either attracted or repelled from the monolayer depending on the magnitude of the electrostatic repulsion, which scales with the Debye length of the subphase. At low salt concentrations, the Debye length is large, and the vesicles diffuse away from the monolayer. The lipid in the vesicles is not recovered on expansion of the monolayer and the recovery is low. At higher salt concentrations, the Debye length is small, and the interaction can even become attractive. The vesicles in solution remain near the monolayer and can reincorporate into the monolayer during expansion, leading to a high recovery of lipid.
The rate of readsorption to the interface should be given by a Boltzmann factor reflecting the probability of the vesicle approaching the monolayer, times the local bilayer aggregate concentration, Cves. The height of the barrier, Emax, depends primarily on the strength of the repulsive electrostatic interactions, as the attractive interactions between bilayers are much less dependent on ionic strength (Israelachvili, 1992):
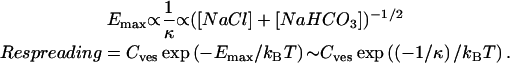 |
(2) |
In these experiments, the monolayer was compressed to the same limiting area on each cycle, so roughly the same amount of material is likely squeezed out at all salt concentrations, so Cves should be roughly the same in all experiments. When the electrostatic barrier is high, the fraction of vesicles returning to the monolayer is low, and most vesicles are lost to the subphase by simply diffusing away. If the barrier is such that the attractive interactions dominate, the rate or readsorption is enhanced, and more vesicles readsorb before they diffuse away into the subphase. Fig. 2 shows that the log (respreading) of the monolayer is proportional to the Debye length for a given cycle speed as suggested by the above equations.
Although DPPG monolayers are simple in comparison to the multicomponent lipid and protein lung surfactant monolayers present in vivo, these experiments do suggest a general framework that may help explain other factors that enhance or inhibit adsorption and respreading. For example, recent work has shown that a broad range of hydrophilic, nonionic polymers, including polyethylene glycols (Lu et al., 1999, 2000; Taeusch et al., 1999), dextrans (Kobayashi et al., 2001), and hyaluronan (W. Taeusch, private communication) of widely varying molecular weights enable surfactants to better resist inhibition. Such polymers are known to dehydrate multilamellar lamellar phases (Kuhl et al., 1998b; Parsegian et al., 2000), or induce a depletion attraction between aggregates and a surface (Kaplan et al., 1994; Kuhl et al., 1998a,b), thereby making bilayer aggregates less stable relative to the monolayer, and/or changing the height of the energy barrier to readsorption. Altering the LE-LC transition pressure of the monolayer may have similar effects on readsorption by increasing the attractive interactions. The cationic surfactant specific proteins SP-B and SP-C might provide ways around the energy barrier to readsorption as they form multilayer patches that may provide locally, net positive charged docking sites for anionic surfactant adsorption (Ding et al., 2003; Takamoto et al., 2001; von Nahmen et al., 1997). SP-B and SP-C locate preferentially in anionic and/or fluid bilayers as well (Ding et al., 2003; Takamoto et al., 2001). These docking sites for readsorption due to SP-B and SP-C suggests that it is the multiple cationic residues of these proteins that lead to faster and more efficient monolayer recovery after collapse.
Acknowledgments
The authors acknowledge financial support from the National Institutes of Health (grant HL-51177) and the University of California Tobacco Related Disease Research Program (grant 11RT-0222). H.E.W. was supported by an NIH postdoctoral fellowship.
References
- Bernhard, W., J. Mottaghian, A. Gebert, G. A. Rau, H. von der Hardt, and C. F. Poets. 2000. Commerical versus Native Surfactants. American J. of Critical Care Medicine. 162:1524–1533. [DOI] [PubMed] [Google Scholar]
- Bringezu, F., J. Q. Ding, G. Brezesinski, and J. A. Zasadzinski. 2001. Changes in model lung surfactant monolayers induced by palmitic acid. Langmuir. 17:4641–4648. [Google Scholar]
- Diamant, H., T. A. Witten, A. Gopal, and K. Y. C. Lee. 2000. Unstable topography of biphasic surfactant monolayers. Europhys. Lett. 52:171–177. [Google Scholar]
- Ding, J., I. Doudevski, H. Warriner, T. Alig, J. Zasadzinski, A. J. Waring, and M. A. Sherman. 2003. Nanostructure changes in lung surfactant monolayers induced by interactions between POPG and SP-B. Langmuir. 19:1539–1550. [Google Scholar]
- Ding, J., D. Y. Takamoto, A. Von Nahmen, M. M. Lipp, K. Y. C. Lee, A. J. Waring, and J. A. Zasadzinski. 2001. Effects of lung surfactant proteins, SP-B and SP-C, and palmitic acid on monolayer stability. Biophys. J. 80:2262–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, J. Q., H. E. Warriner, J. A. Zasadzinski, and D. K. Schwartz. 2002. Magnetic needle viscometer for Langmuir monolayers. Langmuir. 18:2800–2806. [Google Scholar]
- Goerke, J. 1998. Pulmonary surfactant: functions and molecular composition. Biochim. Biophys. Acta. 1408:79–89. [DOI] [PubMed] [Google Scholar]
- Gopal, A., and K. Y. C. Lee. 2001. Morphology and collapse transitions in binary phospholipid monolayers. J. Phys. Chem. B. 105:10348–10354. [Google Scholar]
- Guitter, E., F. David, S. Leibler, and L. Peliti. 1988. Crumpling and buckling transitions in polymerized membranes. Phys. Rev. Lett. 61:2949–2952. [DOI] [PubMed] [Google Scholar]
- Israelachvili, J. N. 1992. Intermolecular and Surface Forces. Academic Press, London, UK.
- Kaganer, V. M., H. Mohwald, and P. Dutta. 1999. Structure and phase transitions in Langmuir monolayers. Rev. Mod. Phys. 71:779–819. [Google Scholar]
- Kampf, J. P., C. W. Frank, E. E. Malmstrom, and C. J. Hawker. 1999. Adaptation of bulk constitutive equations to insoluble monolayer collapse at the air-water interface. Science. 283:1730–1733. [DOI] [PubMed] [Google Scholar]
- Kaplan, P. D., J. L. Rourke, A. G. Yodh, and D. J. Pine. 1994. Entropically driven surface phase separation in binary colloidal mixtures. Phys. Rev. Lett. 72:582–585. [DOI] [PubMed] [Google Scholar]
- Knobler, C. M., and R. C. Desai. 1992. Phase transitions in monolayers. Annu. Rev. Phys. Chem. 43:207–236. [Google Scholar]
- Kobayashi, T., K. Tashiro, X. Cui, T. Konzaki, Y. Xu, C. Kabata, and K. Yamamoto. 2001. Experimental models of acute respiratory distress syndrome: clinical relevance and response to surfactant therapy. Biol. Neonate. 80:26–28. [DOI] [PubMed] [Google Scholar]
- Kuhl, T. L., A. D. Berman, S. W. Hui, and J. N. Israelachvili. 1998a. Direct measurement of depletion attraction and thin-film viscosity between lipid bilayers in aqueous polyethylene oxide solutions. Macromolecules. 31:8250–8257. [Google Scholar]
- Kuhl, T. Y., A. D. Berman, S.-W. Hui, and J. N. Israelachvili. 1998b. Cross-over from depletion attraction to adsorption: PEG induced electrostatic repulsion between lipid bilayers. Macromolecules. 31:8258–8263. [Google Scholar]
- Lee, K. Y. C., A. Gopal, A. Von Nahmen, J. A. Zasadzinski, J. Majewski, G. S. Smith, P. B. Howes, and K. Kjaer. 2002. Influence of palmitic acid and hexadecanol on the phase transition temperature and molecular packing of dipalmitoylphophatidyl-choline monolayers at the air-water interface. J. Chem. Phys. 116:774–783. [Google Scholar]
- Lee, K. Y. C., J. Majewski, K. Kjaer, P. Howes, M. M. Lipp, A. J. Waring, and J. A. Zasadzinski. 1999. Incorporation of lung surfactant specific protein SP-B into lipid monolayers at the air-fluid interface: a synchrotron x-ray study. Biophys. J. 76:216a. (Abstr.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp, M. M., K. Y. C. Lee, D. Y. Takamoto, J. A. Zasadzinski, and A. J. Waring. 1998. Coexistence of buckled and flat monolayers. Phys. Rev. Lett. 81:1650–1653. [Google Scholar]
- Lipp, M. M., K. Y. C. Lee, J. A. Zasadzinski, and A. J. Waring. 1996. Phase and morphology changes in lipid monolayers induced by SP-B protein and its amino-terminal peptide. Science. 273:1196–1199. [DOI] [PubMed] [Google Scholar]
- Longo, M., A. Bisagno, J. Zasadzinski, R. Bruni, and A. Waring. 1993. A function of lung surfactant protein SP-B. Science. 261:453–456. [DOI] [PubMed] [Google Scholar]
- Lu, K. W., H. W. Taeusch, J. Goerke, and J. A. Clements. 1999. Hydrophilic polymers prevent surfactant inactivation. J. Investig. Med. 47:32a. (Abstr.). [Google Scholar]
- Lu, K. W., H. William Taeusch, B. Robertson, J. Goerke, and J. A. Clements. 2000. Polymer-surfactant treatment of meconium-induced acute lung injury. Am. J. Respir. Crit. Care Med. 162:623–628. [DOI] [PubMed] [Google Scholar]
- Lu, W. X., C. M. Knobler, R. F. Bruinsma, M. Twardos, and M. Dennin. 2002. Folding Langmuir Monolayers. Phys. Rev. Lett.89:6107–6110. [DOI] [PubMed] [Google Scholar]
- Mansour, H., D. S. Wang, C. S. Chen, and G. Zografi. 2001. Comparison of bilayer and monolayer properties of phospholipid systems containing dipalmitoylphosphatidylglycerol and dipalmitoylphosphatidylinositol. Langmuir. 17:6622–6632. [Google Scholar]
- McConnell, H. 1991. Structures and transitions in lipid monolayers at the air-water interface. Annu. Rev. Phys. Chem. 42:171–195. [Google Scholar]
- Mizuno, K., M. Ikegami, C. M. Chen, T. Ueda, and A. H. Jobe. 1995. Surfactant protein B supplementation improves in vivo function of a modified natural surfactant. Pediatr. Res. 37:271–276. [DOI] [PubMed] [Google Scholar]
- Notter, R. H. 2000. Lung surfactant: Basic Science and Clinical Applications. Marcel Dekker, Inc., New York, Basel, Switzerland.
- Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: a comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson, I. R., V. Brzezinski, R. M. Kenn, and R. Steitz. 1992. Equivalent states of amphiphilic lamellae. Langmuir. 8:2995–3002. [Google Scholar]
- Poulain, F. R., and J. A. Clements. 1995. Pulmonary surfactant therapy. West. J. Med.162:43–50. [PMC free article] [PubMed] [Google Scholar]
- Risdale, R. A., N. Palaniyar, F. Possmayer, and G. Harauz. 2001. Formation of folds and vesicles by dipalmitoylphosphatidylcholine monolayers spread in excess. J. Membr. Biol. 180:21–32. [DOI] [PubMed] [Google Scholar]
- Robertson, B., and H. L. Halliday. 1998. Principles of surfactant replacement. Biochim. Biophys. Acta. 1408:346–361. [DOI] [PubMed] [Google Scholar]
- Schief, W. R., L. Touryan, S. B. Hall, and V. Vogel. 2000. Nanoscale topographic instabilities of a phospholipid monolayer. J. Phys. Chem. B. 104:7388–7393. [Google Scholar]
- Schürch, S., H. Bachofen, J. Goerke, and F. Possmayer. 1989. A captive bubble method reproduces the in situ behavior of lung surfactant monolayers. J. Appl. Physiol. 67:2389–2396. [DOI] [PubMed] [Google Scholar]
- Schürch, S., F. H. Y. Green, and H. Bachofen. 1998. Formation and structure of surface films: captive bubble surfactometry. Biochim. Biophys. Acta. 1408:180–202. [DOI] [PubMed] [Google Scholar]
- Schürch, S., R. Qanbar, H. Bachofen, and F. Possmayer. 1995. The surface-associated surfactant reservoir in the alveolar lining. Biol. Neonate. 67:61–76. [DOI] [PubMed] [Google Scholar]
- Schwartz, D. K. 1997. Langmuir-Blodgett film structure. Surface Science Reports. 27:241–334. [Google Scholar]
- Taeusch, H. W., K. W. Lu, J. Goerke, and J. A. Clements. 1999. Hydrophilic polymers added to surfactant enhance treatment of lung injury by reducing surfactant inactivation. Am. J. Respir. Crit. Care Med. 159:595a. (Abstr.).
- Takamoto, D. Y., M. M. Lipp, A. Von Nahmen, K. Y. C. Lee, A. J. Waring, and J. A. Zasadzinski. 2001. Interaction of lung surfactant proteins with anionic phospholipids. Biophys. J. 81:153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, Y., T. Takei, T. Aiba, K. Masuda, A. Kiuchi, and T. Fujiwara. 1986. Development of synthetic lung surfactants. J. Lipid Res. 27:475–485. [PubMed] [Google Scholar]
- Tchoreloff, P., A. Gulik, B. Denizot, J. E. Proust, and F. Puisieux. 1991. A structural study of interfacial phospholipid and lung surfactant layers by transmission electron microscopy after Blodgett sampling: influence of surface pressure and temperature. Chem. Phys. Lipids. 59:151–165. [DOI] [PubMed] [Google Scholar]
- von Nahmen, A., M. Schenk, M. Sieber, and M. Amrein. 1997. The structure of a model pulmonary surfactant as revealed by scanning force microscopy. Biophys. J. 72:463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters, R. W., R. R. Jenq, and S. B. Hall. 2000. Distinct steps in the adsorption of pulmonary surfactant to an air-liquid interface. Biophys. J. 78:257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warriner, H. E., J. Ding, A. J. Waring, and J. A. Zasadzinski. 2002. A concentration-dependent mechanism by which serum albumin inactivates replacement lung surfactants. Biophys. J. 82:835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ybert, C., W. Lu, G. Moller, and C. M. Knobler. 2002. Kinetics of phase transitions in monolayers: collapse. J. Phys.: Condensed Matter. 14:4753–4762. [Google Scholar]
- Zasadzinski, J. A., J. Ding, H. E. Warriner, F. Bringezu, and A. J. Waring. 2001. The physics and physiology of lung surfactants. Curr. Opinion Coll. Int. Sci. 6:506–513. [Google Scholar]
- Zasadzinski, J. A., R. Viswanathan, L. Madsen, J. Garnaes, and D. K. Schwartz. 1994. Langmuir-Blodgett films. Science. 263:1726–1733. [DOI] [PubMed] [Google Scholar]