

Abstract
A number of channel-forming peptides derived from the second transmembrane (TM) segment (M2) of the glycine receptor α1 subunit (M2GlyR), including the 22-residue sequence NK4-M2GlyR p22 wild type (WT) (KKKKPARVGLGITTVLTMTTQS), induce anion permeation across epithelial cell monolayers. In vitro assays suggest that this peptide or related sequences might function as a candidate for ion channel replacement therapy in treating channelopathies such as cystic fibrosis (CF). The wild-type sequence forms soluble associations in water that diminish its efficacy. Introduction of a single substitution S22W at the C-terminus, NK4-M2GlyR p22 S22W, eliminates the formation of higher molecular weight associations in solution. The S22W peptide also reduces the concentration of peptide required for half-maximal anion transport induced across Madin-Darby canine kidney cells (MDCK) monolayers. A combination of 2D double quantum filtered correlation spectroscopy (DQF-COSY), total correlation spectroscopy (TOCSY), nuclear Overhauser effect spectroscopy (NOESY), and rotating frame nuclear Overhauser effect spectroscopy (ROESY) data were recorded for both the associating WT and nonassociating S22W peptides and used to compare the primary structures and to assign the secondary structures. High-resolution structural studies were recorded in the solvent system (40% 2,2,2-Trifluoroethanol (TFE)/water), which gave the largest structural difference between the two peptides. Nuclear Overhauser effect crosspeak intensity provided interproton distances and the torsion angles were measured by spin-spin coupling constants. These constraints were put into the DYANA modeling program to generate a group of structures. These studies yielded energy-minimized structures for this mixed solvent environment. Structure for both peptides is confined to the 15-residue transmembrane segments. The energy-minimized structure for the WT peptide shows a partially helical extended structure. The S22W peptide adopts a bent conformation forming a hydrophobic pocket by hydrophobic interactions.
INTRODUCTION
Peptide-based channel replacement therapy has been proposed as a novel treatment modality for correcting genetically based diseases caused by defective channel proteins. Defective ion channels are becoming increasingly implicated in a variety of human diseases, such as episodic ataxia, diabetes, epilepsy, cystic fibrosis (CF), and Alzheimer's dementia. Numerous channel-forming peptides have been designed, synthesized, and tested to generate a peptide sequence that is deliverable from aqueous solution, binds quantitatively with membranes, and forms a de novo anion conductive pathway.
Peptides derived from the second transmembrane (TM) segment of the brain glycine receptor (M2GlyR, PARVGLGITTVLTMTTQSGSRA) have been studied extensively for their ability to form anion channels in lipid bilayers, single cells, and epithelial monolayers. The wild-type (WT) sequence, shown above, displays poor aqueous solubility and the propensity to aggregate in solution leading to reduced bioavailability and inconsistent channel forming potentials (Reddy et al., 1993; Wallace et al., 1997). It was found that by adding at least three lysines to either termini of the peptide (CKx-M2GlyR and NKx-M2GlyR) solubility was increased (Tomich et al., 1998). Nuclear magnetic resonance (NMR) data showed some soluble peptide-peptide associations remained for those sequences, indicating that there was a transition from highly associated forms to less associated forms including monomer (Tomich et al., 1998). Electrophysiological analyses were performed on a series of these synthesized peptides by studying the increase in ISC (short-circuit current, an indicator of anion secretion). The results indicated that both the CK4- and NK4-modified M2GlyR peptides did, in fact, induce a de novo anion-select current in lipid bilayers and whole cell membrane patches, as well as in epithelial monolayers (Mitchell et al., 2000; Broughman et al., 2001).
Subsequent truncation studies on both the CK4- and NK4-modified M2GlyR peptides were performed to find the minimal, fully active, peptide length (Broughman et al., 2002a). The truncations were performed on the nonlysine adducted ends of the CK4- and NK4-M2GlyR peptides. The N-terminal modified peptides showed anion transport activity (ISC) even after as many as 11 C-terminal residues were deleted. The 22-residue NK4-M2GlyR p-22 (ΔS23→A27) displayed channel properties that were indistinguishable from the full-length NK4-M2GlyR p27. Removal of two and five residues from the N-terminus of CK4-M2GlyR resulted in reduced channel forming activity. Complete loss of channel activity was seen with removal of more than five residues in CK4-M2GlyR.
Peptide-peptide solution associations were evaluated for each of the truncated peptides using chemical cross-linking with Bis[sulfosuccinimidyl]suberate (BS3). The results of these experiments showed that peptide-peptide solution associations of the NK4-M2GlyR peptide decreased with each truncation. The CK4-M2GlyR peptide, on the other hand, showed consistent amounts of monomer, dimer, and trimer throughout all truncations (Broughman et al., 2002a). NMR structural studies on NK4-M2GlyR and CK4-M2GlyR revealed substantial differences between the interactions of the lysine adducted segments with the core M2GlyR sequence. The lysines of NK4-M2GlyR p-27 do not interact with each other and are unstructured. In CK4-M2GlyR, however, the lysines fold back on the sequence to form a capping structure that is stabilized by the formation of two hydrogen bonds. The capping structure effectively reduces the overall length of the peptide. Therefore, CK4-M2GlyR truncations with deletions greater than five residues were modeled and appeared to be too short to span the bilayer (Broughman et al., 2002b). A major conclusion from this study was that the peptide-peptide association sites that lead to 1), soluble solution associations and 2), membrane bound oligomeric channel assemblies are different and separable.
Restricting our designs to NK4-M2GlyR p22, we began substituting residues to replace the deleted arginine and add an aromatic residue to help anchor the C-terminus of the TM segment. In this article, the introduction of a tryptophan residue at the C-terminus of M2GlyR peptide, NK4-M2GlyR p22 S22W, is shown to have a dramatic effect on both solution peptide-peptide association and the kinetics of channel assembly.
To better understand the mechanism by which the introduction of a single tryptophan at the C-terminus reduces solution associations, we determined and compared the solution structure of the associating NK4-M2GlyR p22 WT (KKKKPARVGLGIT-TVLTMTTQS) and the monomeric NK4-M2GlyR p22 S22W (KKKKPARVGLGITLTMTVTTQW) peptide in 40% 2,2,2-Trifluoroethanol (TFE) in water. This environment stabilizes intermediates in the folding pathway by inducing partial structure (Roccatano et al., 2002). The TFE molecules aggregate around the peptide, which helps to displace water. This displacement prevents hydrogen bonding of the CO and NH groups of the peptide backbone to water molecules providing a low dielectric environment that encourages the formation of intramolecular hydrogen bonds.
MATERIALS AND METHODS
Peptide synthesis and purification
For this study the following sequences were chemically synthesized: NK4-M2GlyR-p22 WT (NH2-KKKKPARVGLGITTVLTMTTQS-CONH; molecular weight (MW) = 2356.9) and NK4-M2GlyR-p22 S22W (NH2-KKKKPARVGLGITTVLTMTTQW-CONH; MW = 2456.0). The peptides were prepared by solid-phase peptide synthesis by an Applied Biosystems model 431A peptide synthesizer (Foster City, CA) using 9-fluorenylmethoxycarbonyl (Fmoc) chemistries. CLEAR amide resin (0.3 mmol/g) was purchased from Peptides International (Louisville, KY) and Nα-9-fluorenylmethoxycarbonyl amino acids were purchased from Anaspec (San Jose, CA), Bachem (Torrance, CA), and Peptides International. All peptides were purified using a Phenomenex (Torrance, CA) reversed-phase C-18 column on a System Gold high performance liquid chromatography (HPLC) system (Beckman Instruments, Fullerton, CA). Peptides were eluted from the column using a linear gradient of 3.0% min−1 of 1–90% acetonitrile containing 0.1% trifluoroacetic acid (TFA) at 2 ml/min. High performance liquid chromatography-purified peptides were characterized by matrix assisted-laser desorption time-of-flight mass spectroscopy (MALDI-TOF). After characterization, peptides were lyophilized and stored as dry powders until use.
Cell culture
Madin-Darby canine kidney cells (MDCK) cells were a generous gift of Dr. Lawrence Sullivan (KUMC, Kansas City, KS) and were cultured as described previously (Wallace et al., 1997). 1-Ethyl-2-Benzimidazolinone (1-EBIO) (Acros, Morris Plains, NJ) was prepared as a 1-M stock solution in dimethyl sulfoxide (DMSO).
Transepithelial ion transport measurements
Transepithelial ion transport was evaluated in a modified Ussing chamber (model DCV9, Navicyte, San Diego, CA). For electrical measurements, MDCK cells were bathed in a modified Ringer solution (120 mM NaCl, 25 mM NaHCO3, 3.3 mM KH2PO4, 0.8 mM K2HPO4, 1.2 mM MgCl2, and 1.2 mM CaCl2 prepared fresh daily) (Broughman et al., 2001) maintained at 37°C and continuously bubbled with 5% CO2/ 95% O2 to maintain pH, provide aeration, and mix fluid in the chambers. The transepithelial membrane potential (VTE) was clamped to zero and the ISC was measured continuously with a voltage clamp apparatus (model 558C, University of Iowa, Dept. of Bioengineering, Iowa City, IA). Data acquisition was performed at 1 Hz with a Macintosh computer (Apple Computer, Cuppertino, CA) using Aqknowledge software (ver. 3.2.6, BIOPAC Systems, Santa Barbara, CA) with an MP100A-CE interface. ISC data are presented at steady-state flux levels.
Ion transport data analysis
The data points represent the mean ISC stimulated by the peptides at concentrations of 10, 30, 100, 200, 300, and 500 μM, and the error bars indicate the SE of the mean. The Differences between control and treatment data were analyzed using analysis of variance (ANOVA) and Student's t-test. The probability of making a type I error less than 0.05 was considered statistically significant. The lines are a best fit of a modified Hill equation to the data, 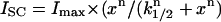 ; where k1/2 is the concentration of peptide that provides a half-maximal ISC and n represents the Hill coefficient.
; where k1/2 is the concentration of peptide that provides a half-maximal ISC and n represents the Hill coefficient.
Circular dichroism
Measurements of peptide secondary structure in water, TFE, and sodium dodecylsulfate (SDS) were performed by circular dichroism (CD). CD spectra measurements were recorded on a Jasco-720 spectropolarimeter (Jasco, Tokyo, Japan) using a 1.0-mm path length rectangular quartz cuvette (Uvonic Instruments, Plainview, NY) for the deionized (DI) H2O, TFE, and SDS studies. All spectra were recorded from 260 to 190 nm using a 1.0-nm spectral bandwidth, 0.2-nm step resolution, 20-nm/min scan speed, and 2-s response time. A baseline measurement was performed every day to check instrument stability. The peptide spectra shown are an average of eight scans with the background subtracted. Peptide stock solutions were prepared fresh daily to a concentration of 1 mM in water. Peptide stock concentrations were determined using either tryptophan fluorescence at 278 nm or the Bicinchoninic Acid protein assay (Pierce, Rockford, IL). Bovine serum albumin (BSA) served as the protein standard and all peptide samples were corrected for dye binding differences between BSA and the peptides as previously determined (Broughman et al., 2002a). Peptide concentrations in all CD studies were matched at 50 μM. DI-distilled H2O peptide samples were prepared by diluting 50 μl peptide stock solution with 950 μl DI H2O. TFE peptide samples were prepared by vortexing 50 μl peptide stock solution with appropriate volumes of 2,2,2-trifluoroethanol (Acros) and DI-distilled H2O to obtain the desired % TFE. SDS peptide samples were prepared by adding 50 μl peptide stock solution with appropriate volumes to yield 10 mM SDS, 10 mM NaCl, and 10 mM phosphate buffer at pH 7.0. The detergent containing samples were prepared using a 2-min sonication step.
BS3 chemical cross-linking reactions
The Bis[sulfosuccinimidyl]suberate cross-linking reactions were carried out as described previously (Broughman et al., 2002a). Cross-linking reactions were carried out at either 150 μM or 3 mM peptide using a 20-fold molar excess of the BS3 cross-linker. The peptide/cross-linker reaction was terminated after 30 min by the addition of 1 N HCl. Two microliters of the terminated reaction samples of 3 mM were added to 118 μL distilled water/tricine SDS sample buffer (50:50, v/v) to prepare for SDS-polyacrylamide gel electrophoresis (PAGE). A noncross-linked sample was prepared as a control. All samples were heated at 100°C in loading buffer for 5 min, then 5 μL of each SDS boiled sample was loaded on to the precast 10–20% gradient tricine gel (Novex division of Invitrogen, Carlsbad, CA). The reference well contained 1 μL of either a Mark 12 unstained standard (Novex division of Invitrogen) or a MultiMark prestained standard (Novex division of Invitrogen). The electrophoresis was carried out at a constant 110 V for 90 min. The gel was then visualized using silver stain (Invitrogen).
NMR spectroscopy
The NMR spectroscopy was performed as described previously (Kirnarsky et al., 2000). Briefly, all high-resolution one- and two-dimensional 1H-NMR experimental data were acquired with an 11.75 T Varian UNITYplus spectrometer (Varian, Palo Alto, CA), operating at 499.96 MHz for 1H, with a 5-mm triple-resonance inverse detection probe. Using peptide concentrations of 3 mM, spectra were recorded at 10°C in 60% H2O/40% TFE-d3, and were processed and analyzed using Varian software VNMR 6.1b on a Silicon Graphics Indigo2 XZ workstation (Mountain View, CA). The invariant nature of NMR chemical shifts and line widths upon 10-fold dilution indicated that the peptide was monomeric in solution at the 3-mM concentration used for 2D-NMR analysis. Standard NMR pulse sequences were used for 2D double quantum filtered correlation spectroscopy (DQF-COSY), total correlation spectroscopy (TOCSY), nuclear Overhauser effect spectroscopy (NOESY), and rotating frame nuclear Overhauser effect spectroscopy (ROESY) experiments. Water peak suppression was obtained by low-power irradiation of the H2O resonance at 4.91-ppm peak during relaxation delay (1.2 s). A total of 512 increments of 8-K data points were collected for the DQF-COSY spectra, and a total of 256 increments of 4-K data points were recorded for all other experiments. All data sets were obtained in hypercomplex phase-sensitive mode. Proton resonance assignments were confirmed by comparison of cross peaks in a NOESY spectrum with those in a TOCSY spectrum (Wüthrich, 1986) acquired under similar experimental conditions. NOESY experiments were performed with 100-, 200-, and 400-ms mixing times. ROESY spectra were acquired with a mixing time of 300 ms. TOCSY spectra were recorded using the MLEV-17 sequence (Bax and Davis, 1985) for isotropic mixing for 90 ms at a B1 field strength of 8 KHz. The TFE peak (3.88 ppm at 10°C) was considered as reference for chemical shift assignment. The temperature dependence of amide proton chemical shifts was measured by collecting data from 5 to 35°C in steps of 5°C using a variable temperature probe. Coupling constants (3JHαNH) were measured directly from 1D- and 2D-DQF-COSY (Piantini et al., 1982) NMR spectra. All experimental data were zero-filled to 4-K data points in t1 dimension and, when necessary, spectral resolution was enhanced by Lorenzian-Gaussian apodization.
Structure calculation
The NOE spectra of mixing time 200 ms was evaluated to provide distance constraints for the structure calculations. The volume of every unambiguously identified cross peak was calculated within the VNMR 6.1b program. These volumes were converted into interproton distances by setting the volume of the peak from the two β-protons of Proline to 2.20 Å and using our program called calDist which calculates the NOE volume, which is equal to 1/R6. The program HABAS (Peter Güntert Scientific Software, Institute of Molecular Biology and Biophysics, Zurich, Switzerland) was used to convert coupling constant values to dihedral angle constraints. The torsion angle dynamics calculations were performed using the distance and dihedral angle constraints within the program DYANA (DYnamics Algorithm for NMR Applications, Peter Güntert Scientific Software) on a Silicon Graphics O2 workstation. The resulting 100 DYANA conformations were clustered with the software Sybyl version 6.7 (Tripos, St. Louis, MO). The groups were based on the pairwise root mean-square deviation (rmsd) values that were less than 1 Å deviation in the C-terminal region (from Gly-9 to Gln-21). The cluster containing the majority of the structures and lowest target function values was subjected to 100 steps of energy minimization with the software Insight II (Accelrys, San Diego, CA) utilizing the constant valence force field. The resulting structures were evaluated with the program Procheck v.3.5.4. (European Bioinformatics Institute, Cambridge, UK). The lowest-energy structure was rendered with the program MIDAS (Midas Plus Computer Graphics Laboratory, University of California, San Francisco, CA).
RESULTS
Introduction of tryptophan at C-terminus
Fig. 1 shows the concentration dependence on ISC for the full length and the truncated M2 peptides when applied to the apical membrane of MDCK cells in the presence of 1-ethyl-2-benzimidazolinone. The solid lines represent the best fit of a modified Hill equation to each data set. In paired monolayers both NK4-M2GlyR p27 and NK4-M2GlyR p22 peptides (previously characterized; Broughman et al., 2001, 2002a) display similar concentration dependency curves and induce similar anion currents at each concentration tested. The summarized ion transport data are presented in Table 1. NK4-M2GlyR p27 provides benchmark values for Imax at 26.2 ± μA/cm2, K1/2 of 270 ± 70 μM, and a Hill coefficient of 1.9, with the p22 WT displaying 23 ± 1.8 μA/cm2, K1/2 of 210 ± 70 μM, and a Hill coefficient of 2.2 ± 1.0. Introduction of the tryptophan in NK4-M2GlyR p22 S22W yields a curve with a reduced Imax (13.0 ± 1.0 μA/cm2) but more importantly a significantly reduced k1/2 (44 ± 6) with a significantly greater Hill coefficient of 5.4 ± 2.9. The sum effect of changes in the three kinetic parameters (Imax, k1/2, and Hill coefficient) is a significant reduction in the peptide concentration required to yield maximal anion secretion.
FIGURE 1.

Concentration-dependence of ISC induced by NK4-M2GlyR derived peptides on MDCK epithelial monolayers. Symbols represent the mean and SE of >6 observations for each concentration tested. Solid lines represent the best fit of a modified Hill equation to each data set. The NK4-M2GlyR derived peptides concentration dependent ISC curves are as follows: NK4-M2GlyR p27(⋄), NK4-M2GlyR p22 WT (○), and NK4-M2GlyR p22 S22W (▵).
TABLE 1.
Peptide sequences, solubility, and channel properties
| Peptide sequences | Molecular weight (Da) | Solubility (mM) | Imax (μA cm−1) | K1/2 (μM) |
|---|---|---|---|---|
| NK4-M2GlyR p27 WT KKKKPARVGLGITTVLTMTTQSSGSRA | 2817.4 | 26.2 ± 5.7 | 26.2 ± 5.7 | 271 ± 71 |
| NK4-M2GlyR p22 WT KKKKPARVGLGITTVLTMTTQS | 2358.9 | 23.7 ± 5.6 | 23.7 ± 5.6 | 210 ± 70 |
| NK4-M2GlyR p22 S22W KKKKPARVGLGITTVLTMTTQW | 2458.0 | 13.0 ± 1.0 | 13.0 ± 1.0 | 44 ± 5.9 |
Fig. 2 A represents a silver-stained gel of cross-linking reactions for the sequences presented in Table 1. Lane 1 contained a MultiMark prestained standard to provide relative molecular weights. Lanes 2 and 3 show the wild-type sequence, NK4-M2GlyR p27, in the absence and presence of a 20-fold excess of BS3, respectively. In the presence of the cross-linker, numerous higher molecular weight homooligomers are observed. Without the addition of cross-linker and after boiling the sample in SDS containing loading buffer, only monomer is observed. Lanes 4 and 5 contained NK4-M2GlyR p22 WT in the absence and presence of excess cross-linking reagent. The cross-linked lane revealed the presence of higher molecular weight oligomers formed in solution, although somewhat reduced over that seen for the full-length sequence (lane 3). Lanes 6 and 7 contain NK4-M2GlyR p22 S22W, again in the absence and presence of excess cross-linking reagent. The substituted sequence population was observed to be predominantly monomeric in solution even in the presence of the cross-linking reagent. Results indicate that NK4-M2GlyR p22 S22W (150 μM) forms few higher molecular weight associations, especially when compared to the WT M2GlyR truncated form.
FIGURE 2.

Cross-linking results using BS3 chemical reaction. (A) A 150-μM peptide solution is mixed without (c) and with (xl) BS3 cross-linker. Multiple bands in a lane with cross-linker indicate that homooligomeric forms exist in solution. The MultiMark prestained protein standard was used. (B) Cross-linking experiment showing peptide-peptide association of NK4-M2GlyR p22 S22W in both water and 40% TFE solution at a high concentration (3 mM). The Mark12 unstained protein standard was used. Note: silver staining intensities with these small peptides is somewhat variable. Loaded samples with matched concentrations can, on occasion, show different dye binding affinities particularly when cross-linked (see Fig. 2 A). Cross-linked monomer shows reduced staining intensity relative to that observed for the oligomeric assemblies that have more dye binding sites. The gels shown are representative of multiple cross-linking experiments and polyacrylamide gel electrophoresis analyses.
A second cross-linking study employed a higher concentration of NK4-M2GlyR p22 S22W (3 mM) (Fig. 2 B). The cross-linking patterns suggested that, even at higher concentration, this peptide remains monomeric in both water and in TFE (40%) solution. The NK4-M2GlyR p22 WT peptide was similarly unaffected at 3 mM and in the presence of TFE. Note that a different standard is used in this gel. Identical peptide concentrations were loaded into each lane. These data indicate that NMR can be performed on these peptides without the complication of further peptide-peptide associations affecting the spectra.
Circular dichroism analysis was performed on the two p22 peptides to make preliminary structural comparisons. Fig. 3 shows the CD spectra of M2GlyR p22 wild type and the NK4-M2GlyR p22 S22W measured in water, 40% TFE/water, and 10 mM SDS at matched peptide concentrations of 50 μM. The CD spectra of NK4-M2GlyR p22 WT (Fig. 3 A) indicated that the peptide in water was somewhat structured. The spectrum has a minimum at ∼195 nm, which is a hallmark for random coil. However it is missing the random coil's characteristic positive maximum at 215 nm. The spectra measured for the WT peptide in 40% TFE and 10 mM SDS were similar to one another and showed some helical content showing the characteristic α-helix double minima at 208 and 222 nm and a maximum at ∼192 nm. The NK4-M2GlyR p22 S22W peptide in water, however, showed considerable β- or extended structure (Fig. 3 B). The same peptide dissolved in 40% TFE showed a transition to a more helical secondary structure. Although NK4-M2GlyR p22 S22W (in 40% TFE) does not include the second minimum at 222 nm the spectrum had a similar shape to that of the p22 WT sequence indicating retention of some helical structure. The spectral data from 185 to 260 nm of the S22W peptide in TFE indicate that the sequence contained 23.4% helix and 30.3% β-structure as determined by a CD analysis program (CD Spectra Deconvolution v.2.0c, Gerald Böhm Institute für Biotechnologie, Martin-Luther-Universität, Halle-Wittenberg, Germany, http://bioinformatik.biochemtech.uni-halle.de). In SDS micelles, the peptide spectrum matches that seen for the p22 WT sequence in SDS. The S22W peptide in SDS had significantly less β-structure (10.0%) than the TFE/water sample (30.0%). These data also indicated that the helical content was slightly greater in the SDS sample (30.8% vs. 23.4% in the TFE solution by the same analysis). CD experiments were also carried out with higher peptide concentrations (3 mM) to ensure that the data was relative to our NMR samples. The results were indistinguishable from the spectra recorded at 50 μM.
FIGURE 3.

Circular dichroism spectra of NK4-M2GlyR p22 S22W and the NK4-M2GlyR p27 WT sequences. (A) NK4-M2GlyR WT (▵) in 40% TFE; (○) in H2O; and (▪) in 10 mM SDS. (B) NK4-M2GlyR S22W (▵) in 40% TFE; (○) in H2O; and (▪) in 10 mM SDS. Peptide concentrations were 50 μM. Spectra were recorded at ambient temperature. Spectra are averages of eight scans after subtraction of the reference spectra of the media.
NMR spectra
As observed by CD studies, TFE stabilized the secondary structure of the peptides, so a 3-mM solution of the peptide was made using 60% water and 40% TFE-d3. The pH of the samples was ∼2.8, which minimizes the rate of amide proton exchange.
Proton resonance assignments were made using 2D 1H-1H DQF-COSY and TOCSY for intraresidue spin systems and NOESY and ROESY spectra for interresidue connectivities by standard techniques (Wüthrich, 1986). The chemical shift assignments are summarized in Table 2. TOCSY spectra were used to identify NH-CαH, NH-CβH, NH-CγH, and NH-CδH intraresidue spin systems to determine residue assignments (Fig. 4). NOESY spectra provided dαN(i, i + 1) protons connectivity data for sequential assignments and multiple residue assignments such as lysines 1–4, leucines 10 and 16, valine 8 and 15, and threonine 13, 14, 17, 19, and 20.
TABLE 2.
Proton chemical shifts (ppm) for NK4-M2GlyR p22 (WT) and NK4-M2GlyR p22 (S22W) in 40% TFE in water
| Residues | NH | CαH | CβH | CγH | Others | ||||
|---|---|---|---|---|---|---|---|---|---|
| WT | |||||||||
| Lys-1 | 4.00 | ||||||||
| Lys-2 | 8.59 | 4.29 | 1.72* | 1.63 | δ 1.28 | ɛ 2.92 | |||
| Lys-3 | 8.31 | 4.26 | 1.71* | 1.62 | δ 1.26 | ɛ 2.92 | |||
| Lys-4 | 8.21 | 4.50 | 1.71* | 1.63 | δ 1.28 | ɛ 2.92 | |||
| Pro-5 | 4.34 | 1.83* | 2.23 | 1.95 | δ 3.51, 3.75 | ||||
| Ala-6 | 8.15 | 4.22 | 1.33 | 3.62 | |||||
| Arg-7 | 8.08 | 4.22 | 1.72 | 1.78* | 1.55 | δ 3.10 | NɛH 7.12 | ||
| Val-8 | 7.80 | 3.97 | 2.00* | 0.76 | 0.87 | ||||
| Gly-9 | 8.08 | 3.86 | 3.86 | ||||||
| Leu-10 | 7.91 | 4.21 | 1.56* | δ 0.82 | |||||
| Gly-11 | 8.28 | 3.87 | 3.87 | ||||||
| Ile-12 | 8.08 | 3.86 | 1.80 | 1.49* | 1.16 | γ 0.87 | δ 0.82 | ||
| Thr-13 | 7.91 | 3.87 | 4.18 | 1.18 | |||||
| Thr-14 | 7.85 | 3.97 | 4.32 | 1.17 | |||||
| Val-15 | 7.78 | 3.60 | 2.11 | 0.84 | .94* | ||||
| Leu-16 | 8.55 | 3.87 | 1.44* | 1.75 | δ 0.75 | ||||
| Thr-17 | 8.03 | 3.87 | 4.27 | 1.15 | |||||
| Met-18 | 8.25 | 4.22 | 2.08* | 2.13 | 2.64 | 2.50 | |||
| Thr-19 | 8.11 | 4.09 | 4.24 | 1.21 | |||||
| Thr-20 | 7.76 | 4.21 | 4.26 | 1.22 | |||||
| Gln-21 | 7.95 | 4.25 | 2.03 | 2.37 | 2.15 | ||||
| Ser-22 | 7.85 | 4.32 | 3.86 | ||||||
| S22W | |||||||||
| Lys-1 | 7.92 | 3.93 | 1.67* | δ 1.40 | ɛ 3.02 | ||||
| Lys-2 | 8.59 | 4.29 | 1.72* | 1.63 | δ 1.38 | ɛ 2.90 | |||
| Lys-3 | 8.31 | 4.25 | 1.71* | 1.63 | δ 1.36 | ɛ 2.91 | |||
| Lys-4 | 8.21 | 4.49 | 1.71* | 1.61 | δ 1.38 | ɛ 2.90 | |||
| Pro-5 | 4.33 | 1.82* | 2.20 | 1.93 | δ 3.72, 3.49 | ||||
| Ala-6 | 8.15 | 4.21 | 3.72 | ||||||
| Arg-7 | 8.07 | 4.22 | 1.73* | 1.53 | δ 3.08 | NɛH 7.09 | |||
| Val-8 | 7.81 | 3.98 | 1.98* | 0.86 | |||||
| Gly-9 | 8.08 | 3.84 | 3.84 | ||||||
| Leu-10 | 7.89 | 4.21 | 1.55* | 1.79 | δ 0.81 | ||||
| Gly-11 | 8.28 | 3.86 | 3.86 | ||||||
| Ile-12 | 8.05 | 3.88 | 1.79 | 1.45* | 1.16 | γ 0.85 | δ 0.78 | ||
| Thr-13 | 7.89 | 3.88 | 1.16 | ||||||
| Thr-14 | 7.82 | 3.88 | 1.15 | ||||||
| Val-15 | 7.73 | 3.61 | 2.08 | 0.93* | 0.83 | ||||
| Leu-16 | 8.40 | 4.00 | 1.40* | 1.70 | δ 0.73 | ||||
| Thr-17 | 7.87 | 4.19 | 1.15 | ||||||
| Met-18 | 8.10 | 4.24 | 2.06* | 2.09 | 2.48* | 2.59 | |||
| Thr-19 | 7.81 | 4.12 | 1.15 | ||||||
| Thr-20 | 7.53 | 4.25 | 1.08 | ||||||
| Gln-21 | 7.82 | 4.25 | 2.17 | 1.85 | |||||
| Trp-22 | 7.75 | 4.63 | 3.20* | 3.29 | Indole-HN | 9.73 | |||
| 2H 7.20 | 4H 7.54 | ||||||||
| 5H 7.01 | 6H 7.08 | ||||||||
| 7H 7.33 | |||||||||
An asterisk corresponds to the hydrogen atom in the stereo chemical position 2, as defined by the IUPAC-IUB Commission on Biochemical Nomenclature (1970).
FIGURE 4.

Two-dimensional TOCSY spectra of NK4-M2GlyR p22 WT (A) and S22W (B). Individual spin systems are connected with a vertical line and labeled with the residue number. The chemical shift values of all the residue spin systems were established from these identifications. As seen by the spectra there are several differences in residue chemical shifts. The experiments were performed at 10°C and pH 2.8.
The chemical shift dispersion for the NH and CαH proton resonances supported the presence of a folded structure as predicted from the CD spectra recorded in TFE. An estimate of both peptides' secondary structures was made based on the α-proton chemical shift values as compared with the random coil values in TFE, termed the chemical shift index (CSI). It has been shown that these values can provide a valuable approximation of secondary structure (Wishart et al., 1992; Merutka et al., 1995). The deviation, in ppm, of each residue from the expected random coil values observed in TFE are shown in Fig. 5. A negative value (<−0.10 ppm, upfield shift) suggested a turn in the structure and several consecutive residues with negative values can mean a helical conformation exists, whereas a comparatively positive value (downfield shift) suggested a β-sheet type conformation. As visible in the figure, the deviation of α-proton chemical shift values of residues 12–16 and 18–19 of both sequences were, in fact, heavily shifted upfield. Residues 5–11 and 20–21 were shifted slightly upfield. These shifts suggest that a large percentage of the peptides could be made up of helical regions or a series of turns. The main difference between the two sequences is that the chemical shift of the Thr-17 Cα-proton resides in the heavily negative region in the WT sequence but is only slightly negative in the S22W sequence. This chemical shift difference suggests a continuous helix from Ile-12 to Thr-19 in the WT sequence but a split in the helical structure in the S22W peptide, which could result in separated helical portions.
FIGURE 5.

Chemical shift value differences between experimental values and the random coil values for Cα protons. Random coil values correspond to the values given for peptides in TFE solution (Merutka et al., 1995). A large negative deviation (<−0.10) suggests a turn in the structure and several consecutive residues with negative values suggests a helical conformation exists.
The amide regions (NH-NH) of the NOESY spectra are overlayed in Fig. 6 to display the differences. Other connectivities found including dNN(i, i + 1), dαN(i, i + 1), dNN(i, i + 2), and dαN(i, i + 3) by the NOE spectra provided some structural information about the peptides. The observed NOEs were summarized graphically in Fig. 7. The observation of sequential dNN(i, i + 1) proton NOEs for the Gly-(9)–Thr-(17) strongly indicated a continuous stretch of backbone φ- and ψ-values in the helical regions of conformational space (Wüthrich,1986). In addition, the observation of dβN(i, i + 1) proton connectivities and particularly the presence of dαN(i, i + 3) protons NOE between Gly-9 and Ile-12, Gly-11 and Thr-14, Ile-12 and Val-15, Thr-14 and Thr-17, and Met-18, Leu-16 and Thr-19, and Thr-17 and Thr-20 confirmed that this region of the peptide was in α-helical conformation. The S22W sequence displayed many of these same interactions but lacked the Ile-12–Val-15 and Thr-14–Thr-17 connectivities. This would suggest a difference in the backbone conformation between the two peptides in this region.
FIGURE 6.

An overlay of the expanded NH-NH region of the NOE spectra of NK4-M2GlyR p22 WT and S22W. The expanded NH-NH region of the two dimensional NOESY spectra. The overlay of WT (blue) and S22W (red) indicates more backbone interactions in the WT peptide, a result of more secondary structure.
FIGURE 7.

Summary of the 3JHNHα coupling constants and NOE connectivities of NK4-M2GlyR p22 WT (A) and S22W (B) in 40% TFE. The solid circles, just below the amino acid residues, represent the coupling constants that were <6.5 Hz and open circles represent the coupling constants >6.5 Hz. The rest of the coupling constants were not measurable due to the overlapping of signals. Unambiguous sequential and short-range connectivities are shown by lines. The thicknesses of the lines indicate the observed intensity. Dashed lines indicate ambiguity, as a result of overlapping.
Structural calculation
Using the NOE cross peaks, 136 for WT and 141 for S22W, distance-constraints were calculated. Due to a large number of overlapping peaks in CαH regions of the DQF-COSY spectra, only a limited number of angle constraints could be calculated for these 22 amino-acid peptides. The distance constraints and the 3JαHNH coupling constants were submitted to HABAS for a local conformational analysis, which resulted in dihedral angle constraints on five residues and four residues for WT and S22W peptides, respectively. With the distance constraints and the dihedral angle constraints, 100 conformations were obtained by torsion angle dynamics calculations using SYBYL/DYANA. The resulting structures had no significant violations of the input constraints (<0.66 Å), and they had a target function value of 1.15 ± 0.78 Å2 for the WT sequence and 0.45 ± 0.42 Å2 for the S22W peptide. All one hundred structures were clustered into groups based on rmsd values by SYBYL. The WT peptide structures were clustered into 20 groups. A group containing 11 conformations was examined based on its target function values. The structure of these conformers was limited to the C-terminal region. This structure included a continuous stretch of helix from Ile-12 to Thr-19. The rmsd values for the backbones of the 11 structures was 1.95 ± 0.60. The S22W peptide structures were clustered into 11 groups and only one major group was observed, containing 30 conformations.
These structures were energy-minimized in Insight II with all the constraints, and the resulting structures' ψ- and φ-angles were evaluated against the Ramachandran Plot values by the program PROCHECK (Table 3). The plot of the WT peptide cluster showed that >99.9% of the ψ- and φ-angles of the peptide backbone reside within the allowed regions. The analysis of S22W showed that 91.7% of residues were in the favored region, 8.3% were in other allowed regions, and no residues were in disallowed region.
TABLE 3.
Structure statistics
| WT | S22W | |
|---|---|---|
| Target function | 1.15 ± 0.78 Å | 0.45 ± 0.42 Å |
| Experimental NMR constraints | ||
| NOE distance constraints | ||
| Intraresidue | 47 | 97 |
| Sequential | 67 | 40 |
| Medium range | 15 | 4 |
| Long range | 7 | 0 |
| Angle constraints | 10 | 8 |
| NMR constraint violations | ||
| NOE constraint violations (Å) | ||
| Sum | 2.68 ± 1.06 | 1.25 ± 0.95 |
| Maximum | 0.43 ± 0.20 | 0.39 ± 0.28 |
| Angle constraint violations | ||
| Sum | 0.00 | 0.20 |
| Maximum | 0.00 | 0.20 |
| Energy (kcal/mol−1) | ||
| Evan der Waals | −39.61 ± 8.28 | 101.07 ± 30.85 |
| Eelectrostatic | −219.67 ± 59.51 | 69.86 ± 18.28 |
| Etotal | −259.28 ± 54.39 | 171.71 ± 32.00 |
| Rmsd from the mean structure | ||
| Residues 9–20 backbone atoms | 0.00 ± 0.00 Å | 0.40 ± 0.20 Å |
| Ramachandran statistics | ||
| Residues in allowed regions | >99.9% | 91.7% |
| Residues in generously allowed regions | <0% | 8.3% |
| Residues disallowed in regions | <0% | 0% |
The structures of the S22W peptide, like the WT sequence, could be broken up into two conformationally distinct parts. The first eight residues of the N-terminal, including the four capping lysines were highly variable in conformation. Even with angle and NOE constraints pertaining to this area, it was not limited to a strict conformation by structure simulation. The results indicate this portion does not have a strict structure and can be free in solution. Throughout the group of structures, Gly-9–Gly-11 were found to create a turn that led into the α-helix-like C-terminal region. The rmsd values for the backbones in the region from Gly-9 to Thr-20 between these 30 conformations and the corresponding mean coordinates of this group is 0.40 ± 0.20 Å (Table 3).
The structures with the lowest total energies are shown in Fig. 8 created in Qmol (J. D. Ganes, Shalloway Labs, Cornell University, http://www.mbg.cornell.edu/shalloway/shalloway.html). Fig. 9 shows a representative modeled structure of the two sequences presented as an aid to this discussion.
FIGURE 8.

Clustered DYANA conformers of NK4-M2GlyR p22 WT and S22W. The superimposed structures represent the cluster with the lowest target function for each peptide. A structured portion closer to the N-terminal can be identified in the clusters of both peptides.
FIGURE 9.

Calculated structure models of NK4-M2GlyR p22 WT and S22W. A representative conformation modeled for the solution structures. The backbones are shown as a tube with side chains visible in a wire frame. The models illustrate the difference in the structures of the two peptides.
The WT peptide structure is linear with a continuous helix in the C-terminus. The backbone of this region agrees with the NOE data. The helix structure allows the α- and amide protons to come in close contact with other protons on the same side of the helix. Although the S22W peptide, like the WT sequence, has a flexible, extended region from Lys-1 to Val-8, a turn stabilized by the presence of hydrogen bonding between the amide group of Thr-14 and Val-15 to the carbonyls of Gly-11 and Ile-12, respectively, redirects the C-terminal portion of the chain forming a turn that looks like a pocket. The three segments, from Val-8 to Gly-10, from Gly-11 to Leu-16, and from Thr-17 to Trp-22, formed the three sides of the pocket. Gly-9 and Thr-19 came close at the opening of the pocket with NH of Gly-9 and the γ-oxygen of Thr-19 forming a hydrogen bond. PROCHECK reported that the C-terminal region contained two α-helix domains. Residues Ile-12, Thr-13, Thr-14, and Val-15 formed a one-cycle α-helix in that Thr-13 and Thr-14 extruded outside of the pocket. The seven residues at the C-terminal end formed a two-cycle α-helix. It was observed that all the residues facing out of the pocket are hydrophilic residues, and most of the residues facing inward are hydrophobic.
DISCUSSION
The results presented in this study suggest important properties about the solution structure of transmembrane peptides derived from the M2GlyR sequence in aqueous environments. The sequences are derived from an anion selective glycine receptor channel found in the spinal cord. These sequences have been proposed as possible therapeutic agents for treating disease conditions that are the result of an absent or defective channel (Wallace et al., 1997). When applied to membranes the peptides insert and then assemble into helical bundles to give ion transport activities that can be measured in synthetic bilayers or cells. Although this peptide displays its biological activity in a membrane environment, delivery from an aqueous solution is of paramount importance. The self-assembling native M2GlyR sequence has been shortened and modified through an iterative process to improve water-solubility and reduce solution associations of the peptide. Based on aggregation studies (Tomich et al., 1998) it appears that only the lower molecular weight forms of the peptide are active. In solution, the hydrophobic surfaces of these amphipathic peptides apparently self-associate to form soluble oligomers. With the hydrophobic portions of the molecules sequestered from and the hydrophilic surfaces exposed to solvent, there is little driving force for the soluble peptide oligomers to associate with membranes. Therefore we hypothesize that monomer is the most bioactive species. Increasing the amount of monomer in solution should enhance membrane affinity thereby increasing efficacy.
The profiles for the ion transport data in Fig. 1 appear similar except for the lowest concentrations tested for the full-length NK4-M2GlyR p27 WT and the truncated NK4-M2GlyR p22 WT sequences. As judged by cross-linking patterns shown in Fig. 2 A, the two peptides show different propensities to form higher molecular weight associations and NK4-M2GlyR p22 WT appears to have more monomer present.
Replacement of the C-terminal serine with a tryptophan affects both the kinetics of assembly of the functional channel pore and the free peptide's solution properties. The peptide NK4-M2GlyR p22 S22W displayed anion conductance activity (Imax = 13.0 ± 1.0 μA/cm−1) with increased affinity for channel assembly (K1/2 = 45 μM) relative to the WT sequences. The observed reduction in maximal anion current is postulated to be the result of an alteration in the pore structure imposed by the introduction of the tryptophan.
The cross-linking studies reveal an absence of higher soluble molecular weight peptide-peptide associations for the S22W peptide. In fact, most of the soluble material appears as monomer. Increasing the amount of bioactive material could explain the improved efficacy of the compound, which is important in the design of membrane active drugs that need to be delivered from buffered aqueous solution. The abundance of soluble monomer in the presence of cross-linking agent clearly indicates that the tryptophan containing peptide must reside primarily in conformations that favors only intramolecular interactions, whereas the WT sequences adopts multiple conformations that stabilize monomer, dimer, and the other oligomers by allowing both intra- and intermolecular interaction.
CD studies showed that both peptides displayed different secondary structures in water and 40% TFE. NK4-M2GlyR p22 S22W appears to adopt a structure that was intermediate to those observed in water or SDS micelles. On the other hand, the structures for NK4-M2GlyR p22 WT in 40% TFE and SDS micelles were quite similar. Since NK4-M2GlyR p22 WT had little structure in water and the NK4-M2GlyR p22 WT and NK4-M2GlyR p22 S22W had similar CD spectra in SDS micelles, high-resolution structural studies were undertaken in the solvent system that gave the largest structural difference, preserved some of the soluble characteristics of the peptide, and provided samples that could be analyzed by NMR. CD studies and cross-linking studies performed at the higher concentrations required for NMR analyses showed no concentration-induced structural or aggregation effects.
Chemical shift values of CαH protons indicative of secondary structure existed predominately in the C-terminal end and were found to be accurate predictions in the final structure. The overall shapes of the two molecules are substantially different. The overlapping of the minimized structures showed that the region spanning from residue 11 to 22 was highly structured (KKKPARVGLGITTVLTMTTQW). All of the 11 structured residues (underlined) are located in the putative transmembrane segment (bolded).
Both peptides were found to have an unstructured N-terminus and a structured C-terminal portion. The wild-type peptide is made up of a continuous helix with an extended, linear segment at the N-terminus. The S22W peptide, unlike the WT peptide, adopts a more closed structure due to a turn in the region separating the structured and unstructured sections. The two glycines in both peptides may limit inter-residue interaction ensuring that the segment containing Val-8–Gly 11 can be flexible enough to extend the N-terminal of the peptide into a linear structure or to fold the backbone so that a closed structure like the S22W peptide is formed. Although the proline residue in both of these sequences lies within the unstructured portion of the peptide, the absence of α- to α-proton connectivity in the NOESY spectra between the preceeding lysine and the proline of both peptides rules out the presence of a cis isoform.
A measurement of the linear WT peptide structure shows that the length of the entire TM segment (from valine 8 to the anchoring tryptophan 22) could be greater than 32 Å, more than enough to span the hydrophobic core of the membrane. This structure is similar to the results shown in micelle studies done on the wild-type glycine receptor α1 subunit (Tang et al., 2002; Yushmanov et al., 2003). Their sequences are identical to our WT p22 sequence except for the inclusion of other WT residues at the termini. An identical region was found to be helical when incorporated in SDS micelles. The rest of the peptide was found to be unstructured and flexible, including the longer C-terminus. The results of this article suggest that the NMR solution structure of the WT peptide in 40% TFE is only slightly different, if at all, from the structure the peptide assumes in SDS micelles. These findings also indicate that the structured portion of the peptide is restricted to residues 8–22 regardless of the length of the peptide. The WT structure resembles other TM segments observed in the x-ray crystal structure of a CLC chloride channel. In that structure, the Cl− binding TM segments are made up of shorter helices that do not completely span the width of the membrane (Dutzler et al., 2002; Estévez and Jentsch, 2002).
The structured C-terminus of the S22W peptide was made up of a single-turn helix (residues 11–14), a stretched β-like turn (residues 14–17), and then another two-turn helix (residues 15–21). The observed structure indicated a helical content of ∼40%, which was in good agreement with CD data for the tryptophan containing 22-residue peptide. These backbone structures allow the peptide to loop over into a closed structure. This fold gathers the hydrophobic residues such that the acyl side chains have a reduced exposure to water. Clearly, this structure has little relevance to a TM segment where the aliphatic side chains would be fully exposed and interacting with the lipid acyl chains in the membrane bilayer. On the other hand, this may explain why the peptide remains in the monomeric form in solution, since the hydrophobic groups are not exposed and able to associate with other hydrophobic groups from other peptides. Based on the CD data obtained using 10-mM SDS micelles (Fig. 3 B), the peptide is able to adopt a more helical structure, much like the WT peptide.
After closer examination of the side chains of the structures it was determined that the polar threonines are exposed on a single side of the structure. This suggested that this side of the structure might be involved in the pore lining face of the assembled channel. Once assembled in the membrane these side chains would face toward the lumen of the pore and would be available to interact with either water or ions passing across the membrane.
In this study we report the structural consequences of incorporating a tryptophan at the C-terminus of an amphipathic channel-forming peptide. The tryptophan reduces solution aggregation thereby increasing the concentration of monomer. Realizing that producing detailed structures of TM sequences in a TFE/water solvent system is not ideal, nonetheless, the structural findings have provided remarkable insight into the possible membrane structure of the membrane-bound forms of the peptides. The structural data presented here has helped to explain how a 15-residue TM segment can and does span the bilayer to form functional channels. Aggregation is reduced due to the peptides ability to adopt a solution conformation in water that shields the hydrophobic residues from solvent. Future experiments are planned for further structural determinations of this peptide in micelles and oriented bilayers using both solution and solid-state NMR techniques.
The two structures in water containing 40% TFE show differing degrees of helical, extended β-like, and random coil structures. The segments displaying secondary structure are confined to those residues believed to form the transmembrane segments for the two sequences. The WT sequence is linear in 40% TFE, whereas the S22W peptide shows a fold that is stabilized by hydrophobic interactions. The structure of the S22W peptide in SDS micelles shows an increase in helical content as well as a dramatic decrease in β-structure. This suggests a significant alteration in secondary structure upon entering a membrane. A structural transition upon membrane insertion would reposition the hydrophobic residues to maximize interaction with the fatty acid acyl chains.
Acknowledgments
The authors are grateful to Gary Radke and Robert Brandt for technical support.
The work is supported by the National Institute of General Medical Sciences (NIGMS) grant GM43617 (J.M.T.) and a structural supplement for the same grant. This work is contribution 03-179-J from the Kansas Agricultural Experiment Station (J.M.T., B.D.S., and O.P.).
References
- Bax, A., and D. G. Davis. 1985. MLEV-17 based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65:355–360. [Google Scholar]
- Broughman, J. R., K. E. Mitchell, R. L. Sedlacek, T. Iwamoto, J. M. Tomich, and B. D. Schultz. 2001. NH2-terminal modification of a channel-forming peptide increases capacity for epithelial anion secretion. Am. J. Physiol. Cell Physiol. 280:C451–C458. [DOI] [PubMed] [Google Scholar]
- Broughman, J. R., L. P. Shank, W. Takeguchi, T. Iwamoto, K. E. Mitchell, B. D. Schultz, and J. M. Tomich. 2002a. Distinct structural elements that direct solution aggregation and membrane assembly in the channel forming peptide M2GlyR. Biochemistry. 41:7350–7358. [DOI] [PubMed] [Google Scholar]
- Broughman, J. R., L. P. Shank, T. Iwamoto, O. Prakash, B. D. Schultz, J. M. Tomich, and K. E. Mitchell. 2002b. Structural implications of placing cationic residues at either the NH2- or COOH-terminus in a pore-forming synthetic peptide. J. Membr. Biol. 190. 93–103. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Cambell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Estévez, R., and T. Jentsch. 2002. CLC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 12:531–539. [DOI] [PubMed] [Google Scholar]
- IUPAC-IUB Commission on Biochemical Nomenclature. 1970. Abbreviations and symbols for the description of the conformation of polypeptide chains. J. Mol. Biol. 52:1–17. [PubMed] [Google Scholar]
- Kirnarsky, L., O. Prakash, S. M. Vogan, M. Nomoto, M. A. Hollingsworth, and S. Sherman. 2000. Structural effects of O-glycosylation on a 15-residue peptide from the mucin (MUC1) core protein. Biochemistry. 39:12076–12082. [DOI] [PubMed] [Google Scholar]
- Merutka, G., H. J. Dyson, and P. E. Wright. 1995. ‘Random coil’ 1H chemical shifts obtained as a function of temperature and trifluoroethanol concentration for the peptide series GGXGG. J. Biomol. NMR. 5:14–24. [DOI] [PubMed] [Google Scholar]
- Mitchell, K. E., T. Iwamoto, J. M. Tomich, and L. C. Freeman. 2000. A synthetic peptide based on a glycine-gated chloride channel induces a novel chloride conductase in isolated epithelial cells. Biochim. Biophys. Acta. 1466:47–60. [DOI] [PubMed] [Google Scholar]
- Piantini, U., O. W. Sorensen, and R. R. Ernst. 1982. Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 104:6800–6801. [Google Scholar]
- Reddy, L. G., T. Iwamoto, J. M. Tomich, and M. Montal. 1993. Synthetic peptides and four-helical bundle proteins as model systems for the pore-forming structure of channel proteins. III. Transmembrane segment M2 of the brain glycine receptor channel is a plausible candidate for the pore-lining structure. J. Biol. Chem. 268:14608–14615. [PubMed] [Google Scholar]
- Roccatano, D., G. Colombo, M. Fioroni, and A. E. Mark. 2002. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: a molecular dynamics study. Proc. Natl. Acad. Sci. USA. 99:12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, P., P. V. Mandal, and Y. Xu. 2002. NMR Structures of the second transmembrane domain of the human glycine receptor α1 subunit: model of pore architecture and channel gating. Biophys. J. 83:252–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomich, J. M., D. Wallace, K. Henderson, K. E. Mitchell, G. Radke, R. Brandt, C. A. Ambler, A. J. Scott, J. Grantham, and T. Iwamoto. 1998. Aqueous solubilization of transmembrane peptide sequences with retention of membrane insertion and function. Biophys. J. 74:256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, D. P., J. M. Tomich, T. Iwamoto, K. Henderson, J. J. Grantham, and L. P. Sullivan. 1997. A synthetic peptide derived from glycine-gated Cl− channel induces transepithelial Cl− and fluid secretion. Am. J. Physiol. 272:C1672–C1679. [DOI] [PubMed] [Google Scholar]
- Wishart, D. S., B. D. Sykes, and F. M. Richards. 1992. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 31:1647–1651. [DOI] [PubMed] [Google Scholar]
- Wüthrich, K. 1986. NMR of Proteins and Nucleic Acids. Wiley Inc., New York.
- Yushmanov, V. E., P. K. Mandal, Z. Liu, P. Tang, and Y. Xu. 2003. NMR structure and backbone dynamics of the extended second transmembrane domain of the human neuronal glycine receptor α1 subunit. Biochemistry. 42:3989–3995. [DOI] [PubMed] [Google Scholar]