

Abstract
Two 40 ns molecular dynamics simulations of a palmitoyl-oleoyl phosphatidylserine (POPS) lipid bilayer in the liquid crystalline phase with Na+ counterions and NaCl were carried out to investigate the structure of the negatively charged lipid bilayer and the effect of salt (NaCl) on the lipid bilayer structure. Na+ ions were found to penetrate deep into the ester region of the water/lipid interface of the bilayer. Interaction of the Na+ ions with the lipid bilayer is accompanied by a loss of water molecules around the ion and a simultaneous increase in the number of ester carbonyl oxygen atoms binding the ion, which define an octahedral and square pyramidal geometry. The amine group of the lipid molecule is involved in the formation of inter- and intramolecular hydrogen bonds with the carboxylate and the phosphodiester groups of the lipid molecule. The area per lipid of the POPS bilayer is unaffected by the presence of 0.15M NaCl. There is a small increase in the order parameter of carbon atoms in the beginning of the alkyl chain in the presence of NaCl. This is due to a greater number of Na+ ions being coordinated by the ester carbonyl oxygen atoms in the water/lipid interface region of the POPS bilayer.
INTRODUCTION
Various biological processes mediated by biological membranes show a dependence on the lipid composition. For example, antimicrobial peptides can specifically target bacterial cells driven by the electrostatic interactions between the cationic peptide and negatively charged lipids in the bacterial cell membrane (Matsuzaki, 1998, 1999); anionic phospholipids are one of the determinants of membrane protein topology and their interactions with positively charged amino acids contribute to a specific orientation of membrane proteins (van Klompenburg et al., 1997); and variation in the phospholipid composition of the cell membrane is known to influence functional processes such as membrane fusion, cell division, and transbilayer movement of lipids and proteins (Epand, 1998; Luzzati, 1997).
To understand a wide variety of membrane-mediated processes, knowledge of the membrane structure and dynamics is required. Molecular dynamics (MD) computer simulation is a powerful technique to obtain detailed information on the structure and dynamics of phospholipid bilayers, which are often used as a simplified representation of the complex biological membrane. Advances in processing power and simulation methodologies have resulted in an increase in the quality of lipid bilayer simulations (Saiz and Klein, 2002; Scott, 2002; Tieleman et al., 1997). MD simulation has been used to study the structural and the dynamical properties of lipid bilayers. Examples include the water/lipid interface structure (Pandit et al., 2003a), the effects of polyunsaturation on the physical properties of bilayers (Saiz and Klein, 2001), the effects of salt and different levels of hydration on lipid bilayer structure (Böckmann et al., 2003; Mashl et al., 2001; Pandit et al., 2003b), structural fluctuations present in the lipid bilayer (Lindahl and Edholm, 2000), and interactions of cations (Gambu and Roux, 1997), and anions with lipids (Sachs and Woolf, 2003). Self-assembly of a lipid bilayer from an initial random dispersion of lipid molecules has also been simulated (Marrink et al., 2001). Thus MD simulations provide useful atomic-level insights into a growing variety of lipid bilayer systems of increasing size and complexity.
MD simulation studies of numerous homogeneous bilayers consisting of zwitterionic phospholipids have been carried out, but only a few such studies of negatively charged lipid bilayers have been done (Cascales et al., 1996; Pandit and Berkowitz, 2002). In this article we present MD computer simulations of a palmitoyl-oleoyl phosphatidylserine (POPS) lipid bilayer in the liquid crystalline phase with Na+ counterions and with both counterions and additional sodium chloride. The negatively charged phosphatidylserine (PS) lipid molecule contributes ∼1–17% of the lipid composition in mammalian cell membranes (Hauser and Poupart, 1992). A number of experimental studies on cation binding to model membranes of PS with controlled fatty acyl chain composition have been performed to understand the interactions of the negatively charged PS with cations, which are involved in membrane-mediated processes such as phase separation, fusion events, blood clotting factor binding, and enzyme regulation (Mattai et al., 1989). We investigated binding of the sodium ions to the lipid bilayer, defined by their location within the bilayer and their coordination geometry. This work further provides an additional computer model of a negatively charged lipid bilayer that will be useful for simulation studies of mixed lipid bilayers and in understanding specific process like the interaction of cationic antimicrobial peptides with negatively charged lipid bilayers.
MATERIAL AND METHODS
Two simulations were carried out: 1), a POPS bilayer consisting of 128 lipids and 5391 water molecules with 128 Na+ ions; 2), a POPS bilayer consisting of 128 lipids and 5361 water molecules with 143 Na+ ions and 15 Cl− ions, corresponding to ∼0.15 M salt (NaCl). The single-molecule starting structure of a POPS lipid (Fig. 1) was built by modification of a palmitoyl-oleoyl phosphatidylethanolamine (POPE) lipid using the Insight II molecular modeling program (Accelrys, San Diego, CA). The initial structure of POPS lipid bilayer was obtained by arranging an 8 × 8 array of 64 lipid molecules in the x, y plane with random rotation around the z axis and random translations along the z axis with a maximum of 0.5 nm, which constituted the first layer. The second leaflet was obtained by rotation and translating this first layer. A water slab was added to solvate the headgroup region of the lipid bilayer, followed by random replacement of water by ions.
FIGURE 1.

Chemical structure of a POPS molecule with the definition of the names and structure of the groups used in the analysis of the electron density profiles in Fig. 3.
All simulations were carried out using the GROMACS 3.0 software package (Berendsen et al., 1995; Lindahl et al., 2001). In all simulations, a leapfrog integrator (Hockney et al., 1974) was used with a 2-fs time step. All bonds were constrained to their equilibrium values using the SETTLE algorithm (Miyamoto and Kollman, 1992) for water and the LINCS algorithm for all other bonds (Hess et al., 1997). A cutoff of 1.0 nm was used for calculating the Lennard-Jones interactions. The electrostatic interactions were evaluated using the particle mesh Ewald method (Darden et al., 1993; Essmann et al., 1995). The real space interactions were evaluated using a 0.9 nm cutoff and the reciprocal-space interactions were evaluated on a 0.12 nm grid with fourth-order spline interpolation. The simulations were performed with anisotropic pressure coupling, to 1 bar independently in x, y, and z directions, which allowed the area per lipid to fluctuate during the simulation. Each component of the system (i.e., lipid, ions, and water) was coupled separately to a temperature bath at 300 K, which is above the gel to liquid crystalline phase transition temperature of 287 K for the POPS lipid bilayer (Mattai et al., 1989), using the Berendsen thermostat (Berendsen et al., 1984) with a coupling constant of 0.1 ps. All the MD simulations were carried out using periodic boundary conditions. The SPC water model was used in all the simulations (Berendsen et al. 1981). The force field used was a mixture of lipid-optimized non-bonded parameters (Berger et al., 1997) and parameters based on the GROMOS87 force field (van Gunsteren et al., 1996) implemented in GROMACS as ffgmx. For the carboxylate group, the partial charges were taken from the aspartic acid side chain in ffgmx. The final structure and topology files are available for download from http://moose.bio.ucalgary.ca. Two 40 ns simulations were carried out for the POPS bilayer with and without salt. Coordinates were saved every picosecond. Analyses of the trajectories generated from the MD simulations were done with GROMACS programs. Molecular graphics were constructed using the VMD program (Humphrey et al., 1996).
RESULTS
Structure of the lipid bilayer
Fig. 2 shows the area per lipid as a function of time for the POPS lipid bilayer in the presence and absence of salt. Because the area is not known experimentally, we started at an area that is almost certainly too high and let the system equilibrate. The area per lipid in the presence and absence of NaCl averaged over the last 25 ns of the simulation time was found to be 55 ± 1 Å2. Fig. 2 shows that the area per lipid value of the POPS bilayers has stabilized between 15 ns and 40 ns of the simulation time, and therefore the last 25 ns of the trajectories were used for analysis.
FIGURE 2.

Area per lipid as a function of time for the POPS bilayer in the presence and in the absence of NaCl.
The structure and definition of the groups used for analysis of the POPS electron density profiles are shown in Fig. 1. Fig. 3 A shows the average electron density profiles for various lipid groups along the normal to the bilayer surface (z axis). Fig. 3 B shows the distribution of Na+ ions in the interface region. The Na+ ion distribution in the POPS bilayer is symmetrical with respect to the two leaflets of the bilayer. The most striking feature in the distribution profile of Na+ ions in the interface region is the appearance of two distinct peaks, which are clearly shown in Fig. 3 B. The first peak, located ∼18.5 Å from the center of the lipid bilayer, overlaps with the peak in the ester group distribution, which is located ∼18 Å from the center of the lipid bilayer. Thus the Na+ ions penetrate into the ester region of the water/lipid interface. The second peak is located ∼27 Å from the center of the lipid bilayer. The peaks in the carboxylate and phosphodiester group distributions are located ∼24 Å and ∼22 Å, respectively, from the center of the bilayer. Thus in the POPS bilayer, the Na+ ions show a strong tendency to interact with the ester group and not with the carboxylate or the phosphodiester groups. This appears to be due to the formation of inter- and intramolecular hydrogen bonds between the hydrogen atoms of the amine group (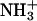 ) and the oxygen atoms of the carboxylate and phosphodiester groups in the lipids as shown in the following section.
) and the oxygen atoms of the carboxylate and phosphodiester groups in the lipids as shown in the following section.
FIGURE 3.

Electron density profile of the various groups (as defined in Fig. 1) along the bilayer normal. The density profiles were averaged over the last 25 ns of the trajectories.
The electron density profiles (Fig. 3) were used to calculate the component volumes of specific lipid atoms in the simulation without NaCl. The lipid volume (VL) was calculated using
 |
(1) |
where A is the average area per lipid molecule (55 Å2), D is the average height of the simulation box (90.1 Å), Nw (42.12) is the number of water molecules per lipid, and Vw is the volume of a water molecule (30.5 Å3) (Petrache et al., 1997). Thus using Eq. 1, we obtain the volume of lipid as 1194 Å3. The volumes of methyl, methylene, double bond, and headgroup (which contains all lipid atoms, including the carbonyl carbon and oxygen of the lipid chains but not the rest of the chains) were found to be 70.3 Å3, 26.5 Å3, 41.8 Å3, and 264 Å3, respectively.
Hydrogen bonding in the water/lipid interface region
Fig. 4 shows the radial distribution function (RDF) for various oxygen atoms from water and the lipid in the system, with hydrogen atoms of the 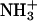 group of the POPS lipid. The first minima in the RDF for the carboxylate and phosphodiester oxygen atoms occur at ∼2.4 Å, indicating hydrogen bonding with the amine hydrogen atoms. To further analyze the hydrogen bonding between various polar headgroups, we calculated the average number of inter- and intramolecular hydrogen bonds between the various oxygen atoms in the system and hydrogen atoms of the amine group (Table 1). A cutoff distance of 2.4 Å between the donor and acceptor atoms (corresponding to the first minima in the RDF, Fig. 4), and a cutoff angle of 35° between the H donor and acceptor atoms were used as hydrogen bond criteria. Table 1 shows that the number of intramolecular hydrogen bonds formed by the oxygen atoms of the carboxylate and phosphodiester groups is larger than the number of intermolecular hydrogen bonds. More intramolecular hydrogen bonds are formed by the amine hydrogen atoms with the phosphodiester oxygen atoms than with the carboxylate oxygen atoms. The observed intra- and intermolecular hydrogen bonds are thus responsible for the low number of direct interactions between the Na+ ions and these oxygen atoms.
group of the POPS lipid. The first minima in the RDF for the carboxylate and phosphodiester oxygen atoms occur at ∼2.4 Å, indicating hydrogen bonding with the amine hydrogen atoms. To further analyze the hydrogen bonding between various polar headgroups, we calculated the average number of inter- and intramolecular hydrogen bonds between the various oxygen atoms in the system and hydrogen atoms of the amine group (Table 1). A cutoff distance of 2.4 Å between the donor and acceptor atoms (corresponding to the first minima in the RDF, Fig. 4), and a cutoff angle of 35° between the H donor and acceptor atoms were used as hydrogen bond criteria. Table 1 shows that the number of intramolecular hydrogen bonds formed by the oxygen atoms of the carboxylate and phosphodiester groups is larger than the number of intermolecular hydrogen bonds. More intramolecular hydrogen bonds are formed by the amine hydrogen atoms with the phosphodiester oxygen atoms than with the carboxylate oxygen atoms. The observed intra- and intermolecular hydrogen bonds are thus responsible for the low number of direct interactions between the Na+ ions and these oxygen atoms.
FIGURE 4.

Radial distribution functions of various oxygen atoms with the lipid amine group (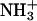 ) hydrogen atoms.
) hydrogen atoms.
TABLE 1.
Average number of hydrogen bonds between various oxygen atoms in the system and the hydrogen atom in the amine group (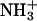 ) of the lipid molecule
) of the lipid molecule
| Oxygen atoms in | Intermolecular hydrogen bond/lipid | Intramolecular hydrogen bond/lipid |
|---|---|---|
| Carboxylate group | 0.25 ± 0.04 (0.26)* | 0.5 ± 0.05 (0.5) |
| Phosphodiester group | 0.25 ± 0.02 (0.26) | 0.8 ± 0.03 (0.84) |
| Ester carbonyl group | 0.065 ± 0.001 (0.064) | ∼0 |
Numbers in parentheses correspond to the POPS bilayers with NaCl.
Interaction of the ions with the water/lipid bilayer interface
The Na+ ion distribution profiles (Fig. 3 B) show that these ions penetrate deep into the ester region of the water/lipid interface. To further analyze the interactions between Na+ ions and the water/lipid interface, we calculated the RDF of all oxygen atoms with the Na+ ions, which is shown in Fig. 5 A. The RDFs show that the oxygen atoms form a first coordination shell within 3.3 Å of the Na+ ion. From the integral of the first peak, from zero to the first minima at 3.3 Å, we obtain average coordination numbers of 4.3, 0.9, 0.04, and 0.07 for water, ester carbonyl, phosphodiester, and carboxylate oxygen atoms, respectively, with the Na+ ion. These coordination numbers, however, are obtained by averaging over all the Na+ ions in the system regardless of their position in the lipid bilayer. The broad distribution of Na+ ions shows that they occupy different positions along the water/lipid interface (Fig. 3 B), and the coordination number of the Na+ ion with the various oxygen atoms will vary along the width of the water/lipid interface. Fig. 5, B and C, show the RDFs of Na+ ions in bulk water and the ester region of the water/lipid interface, respectively, throughout the last 25 ns of the POPS bilayer simulation, with water and ester carbonyl oxygen atoms. As shown by the integral of the first peak, from zero to 3.3 Å, there are ∼5.7 water molecules in the first coordination shell of the Na+ ions in bulk water (Fig. 5 B), and ∼2.5 ester carbonyl and water oxygen atoms each in the first coordination shell of Na+ ions in the ester region of the water/lipid interface (Fig. 5 C). Fig. 5 D further illustrates the variation in the coordination number of Na+ ions with various oxygen atoms in the first coordination shell as function of distance, Z, from the bilayer center, averaged over the two leaflets of the lipid bilayer. The simulation cell was divided into 1 Å slabs, and the average coordination number of various oxygen atoms with the Na+ ion was calculated for each slab. Fig. 5 D also shows that the interaction of Na+ ions with the lipid bilayer is accompanied by a decrease in the number of water molecules and an increase in the number of ester carbonyl oxygen atoms around the ion. At a distance of ∼17.5 Å from the center of the bilayer (corresponding to the region around the ester group), the Na+ ion on average has ∼2.5 water and ester carbonyl oxygen atoms each in the first coordination shell. This corresponds to a situation where a Na+ ion has either three water and two ester carbonyl or two water and three ester carbonyl oxygen atoms in the first coordination shell. Fig. 6 shows the coordination of a Na+ ion with water and ester carbonyl oxygen atoms. As the Na+ ion penetrates deeper in the water/lipid interface region (distance <17.5 Å), the average number of water oxygen atom increases to 2.75 and the number of ester carbonyl oxygen atom decreases to 2 (Fig. 5 D). However, the number of Na+ ions at a distance <17.5 Å is significantly less than the number of ions at a distance ≥17.5 Å from the center of the lipid bilayer as can be seen in the Na+ ion electron density profiles in Fig. 3 B.
FIGURE 5.

Radial distribution functions of various oxygen atoms with all the Na+ ions in the POPS bilayer (A), the Na+ ions in bulk water (B), and the Na+ ions in the ester region (C) of the water/lipid interface. Coordination number of various oxygen atoms around the Na+ ion as a function of distance Z from the bilayer center (D).
FIGURE 6.

Snapshot of the Na+ ion coordination by lipid oxygen atoms.
To define the coordination geometry of oxygen atoms around the Na+ ion, we calculated the distribution of oxygen-Na+-oxygen angle θ, for the oxygen atoms within the first coordination shell, and the percentages of tetra, penta, hexa, and other lower and higher coordinated Na+ ions present in the simulation. As shown in Table 2, the percentage of tetra- and the other lower coordinated Na+ ions are significantly less than that of penta- and hexa-coordinated Na+ ions. Fig. 7 shows the distribution of the angle θ around the hexa- and the penta-coordinated Na+ ions. The peaks at ∼90° and 180° in the distribution profile suggest that the Na+ ions prefer to adopt an octahedral and a square pyramidal geometry in the hexa- and the penta-coordinated species, respectively. The structure of the hydration shell around the Cl− ion in the POPS bilayer system with NaCl was not as well defined, which is consistent with previous results (Grossfield et al., 2003; Sachs and Woolf, 2003). We also studied the distribution of the angle θ between the P-N vector and the outward bilayer normal in the POPS bilayer with and without NaCl (Fig. 8). This distribution is slightly broader in the bilayer without NaCl (Fig. 8 A), although the average value of the angle θ was found to be ∼75° in both systems.
TABLE 2.
Percentage of different types of Na+-oxygen atom coordinated species present in the POPS bilayer with and without NaCl
| Percentage of | POPS bilayer with NaCl | POPS bilayer without NaCl |
|---|---|---|
| Tetra | 4 | 3 |
| Penta | 45 | 51 |
| Hexa | 50 | 46 |
| Lower | 1 | 0 |
| Higher | <1 | <1 |
FIGURE 7.

Distribution of the oxygen-Na+-oxygen angle θ for the oxygen atoms in the first coordination shell of the hexa- and penta-coordinated species in the POPS bilayer with NaCl.
FIGURE 8.

Distribution of the angle between the vector joining phosphorous and nitrogen in POPS and the outward normal to the bilayer for the POPS bilayer (A) in the presence of NaCl and (B) without NaCl.
Order parameters for the tails
Fig. 9 shows the SCD order parameter of the palmitoyl (A) and oleoyl chains (B) of the POPS lipid in the presence and absence of NaCl. The SCD order parameters were calculated using
 |
(2) |
where θ is the angle between the CD bond and the bilayer normal. Deuterium positions were constructed from the neighboring carbons assuming ideal geometries. The brackets imply averaging over time and molecules. Fig. 9 shows a small increase in the order parameter in the beginning of the alkyl chains in the presence of NaCl. Since the area per lipid of the POPS bilayers in the presence and absence of NaCl was found to be the same (see “Structure of the lipid bilayer” section), one would expect the order parameters of the alkyl chains of POPS lipids to be the same in the presence and absence of NaCl (Nagle and Tristram-Nagle, 2000; Petrache et al., 1999). The discrepancy between the area per lipid and the order parameter can be explained in terms of the Na+ ion coordination with the ester carbonyl oxygen atoms in the water/lipid interface region of the lipid bilayer.
FIGURE 9.

Comparison of the deuterium order parameter (SCD) of the alkyl chains in the POPS bilayer in the presence and in the absence of NaCl (A, palmitoyl; B, oleoyl).
Fig. 10 shows the number density of Na+ ions averaged over the two leaflets of the lipid bilayer as a function of distance, Z, from the bilayer center. The simulation cell was divided into 1 Å slabs and the number density of Na+ ions was calculated for each slab. The number of Na+ ions that penetrate deeper into the ester region of the water/lipid interface is higher in the POPS bilayer system with NaCl compared to the POPS bilayer system without NaCl as shown by a larger peak in the Na+ ion distribution at 17.5 Å, which corresponds to the ester region. This coordination of Na+ ion by the ester carbonyl oxygen atoms results in an increase in the order parameter of the carbon atoms in the beginning of the alkyl chains of the POPS bilayer in the presence of NaCl (see discussion below). Comparison of the fraction of the chain dihedral angles in the trans conformation in the presence and absence of NaCl shows that there is no significant difference in the number of trans conformations in the two systems (data not shown).
FIGURE 10.

Comparison of the number of Na+ ions averaged over the two leaflets of the lipid bilayer as a function of distance Z from the bilayer center in the POPS bilayer in the presence and in the absence of NaCl.
DISCUSSION
In this article we study the structure of a POPS lipid bilayer in the presence and absence of NaCl using MD computer simulation. Earlier MD simulations of negatively charged lipid bilayers include the dipalmitoylphosphatidylserine (DPPS) lipid bilayer simulations by Cascales et al. (1996) and Pandit and Berkowitz (2002). As the structure of the POPS and DPPS lipid molecules differs only by the presence of a double bond in the POPS lipid, one would expect comparable structures of DPPS and POPS lipid bilayers. We compare the DPPS bilayer simulation results from Cascales et al. and Pandit and Berkowitz with the present POPS bilayer simulations.
The average value of the area per lipid of the POPS bilayer in the presence and absence of NaCl was found to be the same. MD simulation studies of the effect of NaCl on the structure of zwitterionic phospholipid bilayer of DPPC show that the area per lipid of the lipid bilayer decreases in the presence of NaCl (Pandit et al., 2003b). This might be due to the coordination of Na+ ions with different DPPC molecules, which further restricts the motion of the lipid molecules in the bilayer compared to the lipid molecules in a bilayer without salt. In the case of the negatively charged POPS bilayer, the Na+ ions coordinate with different POPS molecules (Fig. 6), similar to what is found in the DPPC bilayer with NaCl. Further addition of 0.15M NaCl does not have a significant effect on the area per lipid (Fig. 2) because the movement of the lipid molecules already appears to be restricted by the coordination of the Na+ ion by lipids.
MD simulation studies of a DPPS bilayer gave an area per lipid of DPPS of ∼54 Å2 (Cascales et al., 1996; Pandit and Berkowitz, 2002), which is comparable to the area per lipid of ∼55 Å2 for the POPS bilayer obtained in this study. One would expect the area per lipid to increase in the presence of a double bond in the alkyl chain (Murzyn et al., 2001). The DPPS bilayer simulations, however, were done at a higher temperature of 350 K compared to 300 K for the POPS bilayer simulations in this study. The difference in temperature between the two simulations may be responsible for an unexpected similar area per lipid values of POPS and DPPS. The earlier results obtained for the DPPS bilayers are from rather short simulations. A recent study on the methodological issues in lipid bilayer simulation shows that of the order of 10–20 ns equilibration time is required for MD studies of DPPC bilayers (Anézo et al., 2003).
Comparison of the structural parameters of dioleoyl phosphatidylserine (DOPS) bilayers with those of dioleoyl phosphatidylcholine (DOPC) bilayers in the fluid state at the same temperature from x-ray diffraction and NMR spectroscopy shows that the volume and area per lipid of DOPC are ∼6% and ∼10% larger than the volume and area per lipid of DOPS (Petrache et al., 2004). In this work, a similar comparison from MD simulations of POPS and POPC bilayers under the same conditions gives a volume and area per lipid of POPC of ∼6% and ∼12% larger than that of POPS respectively. Similar differences in area per lipid were also obtained by comparing simulations results of DPPS and DPPC lipid bilayers (Cascales et al., 1996; Pandit and Berkowitz, 2002). The smaller area per lipid for DOPS compared to DOPC, a “condensing effect”, is thought to be due to the presence of hydrogen bonds (Petrache et al., 2004). In the present simulations, we find a significant fraction of intramolecular hydrogen bonds among POPS lipids, which appear responsible for its smaller area per lipid compared to POPC. X-ray diffraction and NMR spectroscopy also show similarity between PS and phosphatidylethanolamine (PE) lipids; for example, PS and PE have very similar volumes (Petrache et al., 2004). Volumes of POPS and POPE lipids from MD simulations under similar conditions are found to be 1194 Å3 and 1199 Å3, respectively, which are quite similar and in agreement with the experimental observation (Mukhopadhyay et al., 2004). Thus, lipid force field parameters can reproduce, at least qualitatively, the variation in bilayer structural parameters with different types of lipids.
The electron density profiles of the POPS bilayer (Fig. 3) show that the peak in the Na+ ion density profile overlaps with the peak in the ester group density profile and thus the Na+ ions are found mostly next to the ester group. This is contrary to the results obtained from the DPPS bilayer simulations, where Pandit and Berkowitz (2002) and Cascales et al. (1996) found the Na+ ions next to the carboxylate and the phosphodiester groups respectively. Comparison of the density profiles of the POPS bilayer averaged over 0–10 ns and 30–40 ns time intervals shows that the peaks in the Na+ ion distribution profile are closer to the peaks in the carboxylate and the phosphodiester group distribution profiles for the first 10 ns of the trajectory. Thus in our simulation we do see interactions between the Na+ ions and the carboxylate and the phosphodiester groups over the first 10 ns of the trajectory; by the time the system reaches an equilibrium state, which is between 15 and 40 ns time interval as seen by a stable area per lipid (Fig. 2), however, these interactions decrease and the Na+ ion-carbonyl oxygen atom interactions become dominant. Mattai et al. (1989), using proton-decoupled 31P NMR studies, showed that the phosphodiester group stays mobile in a Na+ ion-POPS bilayer system, indicating that the Na+ ions interact weakly with the phosphodiester oxygen atoms. However, based on spin-label electron spin resonance techniques and differential scanning calorimetry studies on the thermotropic behavior of POPS in the presence of Na+ ions, they also concluded that the Na+ ions display only weak interactions with POPS bilayers. Interactions between Na+ ions and ester carbonyl oxygen atoms in lipid molecules are also found in a POPC lipid bilayer simulation with NaCl (Böckmann et al., 2003). The coordination of Na+ ions by the ester carbonyl oxygen atoms of lipid molecules is clearly shown in Fig. 6.
Interactions of the Na+ ion with the lipid bilayer are accompanied by a loss of water molecules around the ion and a simultaneous increase in the number of ion-carbonyl oxygen coordination (Fig. 5 D). We also show that the coordination geometry of the oxygen atoms in the first coordination shell of the Na+ ion is well defined (Fig. 7). The hexa- and the penta-coordinated Na+ ions, which are the predominant species present in the system (Table 2), have an octahedral and a square pyramidal arrangement of oxygen atoms around the Na+ ion, respectively. The high charge density of the Na+ ion results in a well-defined geometry of water molecules around a solvated Na+ ion and hence the kosmotropic nature of the ion (Collins, 1997). Thus Na+ ions interacting with the lipid bilayer impose spatial restrictions on the water and ester carbonyl oxygen atoms with which they coordinate in the water/lipid interface region.
To understand the effect of NaCl on the POPS lipid bilayer structure, we calculated the SCD order parameters of the alkyl chains in the presence and absence of NaCl. Comparison of these SCD order parameters shows that there is a small increase in the order parameter in the beginning of the alkyl chains in the presence of NaCl (Fig. 9). This increase is due to the greater number of Na+ ions coordinated by the ester carbonyl oxygen atoms with well-defined coordination geometry in the water/lipid interface region of the POPS bilayer with NaCl. For the carbon-carbon bonds adjacent to the ester group, which are in a region of high tail density and low free volume (Tieleman et al., 1997), coordination of the Na+ ion with the ester carbonyl oxygen atoms further restricts their movement. For the carbon-carbon bonds near the center of the bilayer, this coordination does not affect the movement of the bonds due to the greater spatial and orientational flexibility available to these bonds. Thus an increase in the order parameter of the carbon atoms in the beginning of the alkyl chains in the POPS bilayer in the presence of NaCl is because of the restricted motion of the carbon-carbon bonds adjacent to the ester carbonyl group.
CONCLUSIONS
Molecular dynamics computer simulations of palmitoyl-oleoyl phosphatidylserine bilayers in the liquid crystalline phase with Na+ ions show that the Na+ ion penetrates deep into the ester region of the water/lipid interface of the lipid bilayer. The amine group (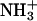 ) of the lipid molecule is involved in the formation of inter- and intra-molecular hydrogen bonds with the carboxylate and the phosphodiester groups of the lipid molecule. The interaction of Na+ ions with the lipid bilayer is accompanied by a loss of water molecules around the ion and a simultaneous increase in coordination of the ion by ester carbonyl oxygen atoms. The coordination geometry in the first coordination shell around the Na+ ions is well defined as is expected from the kosmotropic nature of the Na+ ion. The area per lipid of the POPS bilayer is very similar in the presence and in the absence of NaCl, 55 Å2. A small increase in the chain order parameter of the carbon atoms in the beginning of the alkyl chains in the presence of NaCl is observed, which might be due to the greater number of Na+-carbonyl oxygen atoms interacting in the water/lipid interface region of the POPS bilayer with NaCl compared to the POPS bilayer without NaCl.
) of the lipid molecule is involved in the formation of inter- and intra-molecular hydrogen bonds with the carboxylate and the phosphodiester groups of the lipid molecule. The interaction of Na+ ions with the lipid bilayer is accompanied by a loss of water molecules around the ion and a simultaneous increase in coordination of the ion by ester carbonyl oxygen atoms. The coordination geometry in the first coordination shell around the Na+ ions is well defined as is expected from the kosmotropic nature of the Na+ ion. The area per lipid of the POPS bilayer is very similar in the presence and in the absence of NaCl, 55 Å2. A small increase in the chain order parameter of the carbon atoms in the beginning of the alkyl chains in the presence of NaCl is observed, which might be due to the greater number of Na+-carbonyl oxygen atoms interacting in the water/lipid interface region of the POPS bilayer with NaCl compared to the POPS bilayer without NaCl.
Acknowledgments
We thank Mr. Tariq Rajan for initial steps in this project. We thank Dr. H. Petrache for providing comments and data on DOPS before publishing.
This work was supported by the Natural Science and Engineering Research Council of Canada. D.P.T. is a Scholar of the Alberta Heritage Foundation for Medical Research.
References
- Anézo, C., A. H. de Vries, H.-D. Höltje, D. P. Tieleman, and S. J. Marrink. 2003. Methodological issues in lipid bilayer simulations. J. Phys. Chem. B. 107:9424–9433. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht. 331–342.
- Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS—a message-passing parallel molecular-dynamics implementation. Comput. Phys. Commun. 91:43–56. [Google Scholar]
- Berger, O., O. Edholm, and F. Jähnig. 1997. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 72:2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böckmann, R. A., A. Hac, T. Heimburg, and H. Grubmüller. 2003. Effect of sodium chloride on a lipid bilayer. Biophys. J. 85:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales, J. J. L., J. G. de la Torre, S. J. Marrink, and H. J. C. Berendsen. 1996. Molecular dynamics simulation of a charged biological membrane. J. Chem. Phys. 104:2713–2720. [Google Scholar]
- Collins, K. D. 1997. Charge density-dependent strength of hydration and biological structure. Biophys. J. 72:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- Epand, R. M. 1998. Lipid polymorphism and protein-lipid interactions. Biochim. Biophys. Acta. 1376:353–368. [DOI] [PubMed] [Google Scholar]
- Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Gambu, I., and B. Roux. 1997. Interaction of K+ with a phospholipid bilayer: a molecular dynamics study. J. Phys. Chem. B. 101:6066–6072. [Google Scholar]
- Grossfield, A., R. Pengyu, and J. W. Ponder. 2003. Ion solvation thermodynamics from simulation with a polarizable force field. J. Am. Chem. Soc. 125:15671–15682. [DOI] [PubMed] [Google Scholar]
- Hauser, H., and G. Poupart. 1992. Lipid structure. In The structure of biological membranes. P. Yeagle, editor. CRC Press, Boca Raton, FL. 3–71.
- Hess, B., H. Bekker, H. J. C. Berendsen, and J. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- Hockney, R. W., S. P. J. Goel, and J. W. Eastwood. 1974. Quite high resolution computer models of a plasma. J. Comput. Phys. 14:148–158. [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Lindahl, E., and O. Edholm. 2000. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 79:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- Luzzati, V. 1997. Biological significance of lipid polymorphism: the cubic phases. Curr. Opin. Struct. Biol. 7:661–668. [DOI] [PubMed] [Google Scholar]
- Marrink, S. J., E. Lindahl, O. Edholm, and A. E. Mark. 2001. Simulation of the spontaneous aggregation of phospholipids into bilayers. J. Am. Chem. Soc. 123:8638–8639. [DOI] [PubMed] [Google Scholar]
- Mashl, R. J., H. L. Scott, S. Subramaniam, and E. Jakobsson. 2001. Molecular simulation of dioleoylphosphatidylcholine lipid bilayers at differing levels of hydration. Biophys. J. 81:3005–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki, K. 1998. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta. 1376:391–400. [DOI] [PubMed] [Google Scholar]
- Matsuzaki, K. 1999. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta. 1462:1–10. [DOI] [PubMed] [Google Scholar]
- Mattai, J., H. Hauser, R. A. Demel, and G. G. Shipley. 1989. Interactions of metal ions with phosphatidylserine bilayer membrane: effect of hydrocarbon chain unsaturation. Biochemistry. 28:2322–2330. [DOI] [PubMed] [Google Scholar]
- Miyamoto, S., and P. A. Kollman. 1992. SETTLE—an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13:952–962. [Google Scholar]
- Mukhopadhyay, P., H. J. Vogel, and D. P. Tieleman. 2004. Distribution of pentachlorophenol in phospholipid bilayers: A molecular dynamics study. Biophys. J. 86:337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzyn, K., T. Rog, G. Jezierski, Y. Takaoka, and M. Pasenkiewicz-Gierula. 2001. Effects of phospholipid unsaturation on the membrane/water interface: a molecular simulation study. Biophys. J. 81:170–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, S. A., and M. L. Berkowitz. 2002. Molecular dynamics simulation of dipalmitoylphosphatidylserine bilayer with Na+ counterions. Biophys. J. 82:1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit, S. A., D. Bostick, and M. L. Berkowitz. 2003a. An algorithm to describe molecular scale surface and its application to the study of a water/lipid bilayer interface. J. Chem. Phys. 119:2199–2205. [Google Scholar]
- Pandit, S. A., D. Bostick, and M. L. Berkowitz. 2003b. Molecular dynamics simulation of a dipalmitoylphosphatidylcholine bilayer with NaCl. Biophys. J. 84:3743–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache, H. I., K. C. Tu, and J. F. Nagle. 1999. Analysis of simulated NMR order parameters for lipid bilayer structure determination. Biophys. J. 76:2479–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache, H. I., S. E. Feller, and J. F. Nagle. 1997. Determination of component volumes of lipid bilayers from simulations. Biophys. J. 70:2237–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrache, H. I., S. Tristram-Nagle, K. Gawrisch, D. Harries, V. A. Parsegian, and J. F. Nagle. 2004. Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 86:1574–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs, J. N., and T. B. Woolf. 2003. Understanding the Hofmeister effect in interactions between chaotropic anions and lipid bilayers: molecular dynamics simulation. J. Am. Chem. Soc. 125:8742–8743. [DOI] [PubMed] [Google Scholar]
- Saiz, L., and M. L. Klein. 2001. Structural properties of a highly polyunsaturated lipid bilayer from molecular dynamics simulations. Biophys. J. 81:204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiz, L., and M. L. Klein. 2002. Computer simulation studies of model biological membranes. Acc. Chem. Res. 35:482–489. [DOI] [PubMed] [Google Scholar]
- Scott, H. L. 2002. Modeling the lipid component of membranes. Curr. Opin. Struct. Biol. 12:495–502. [DOI] [PubMed] [Google Scholar]
- Tieleman, D. P., S. J. Marrink, and H. J. C. Berendsen. 1997. A computer perspective of membranes: molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta. 1331:235–270. [DOI] [PubMed] [Google Scholar]
- van Gunsteren, W. F., P. Kruger, S. R. Billeter, A. E. Mark, A. A. Eising, W. R. P. Scott, P. H. Huneberg, and I. G. Tironi. 1996. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Biomos Hochschulverlag AG an der ETH Zurich, Groningen, Germany.
- van Klompenburg, W., I. Nilsson, G. von Heijne, and B. de Kruijff. 1997. Anionic phospholipids are determinants of membrane protein topology. EMBO J. 16:4261–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]