

Abstract
Two-photon excitation (TPE) spectra of Fura-2, -4F, -6F, -FF, and Furaptra were characterized using a tunable (750–850 nM) ultra-short pulse laser. Two-photon fluorescence of these dyes was studied in free solution and in the cytosol of isolated rabbit ventricular cardiomyocytes. The TPE spectra of the Ca2+-free and Ca2+-bound forms of the dyes were measured in free solution and expressed in terms of the two-photon fluorescence cross section (Goppert-Meyer units). The Fura dyes displayed the same Ca2+-free TPE spectrum in the intracellular volume of permeabilized and intact cardiomyocytes. Fluorescence measurements over a range of laser powers confirmed the TPE of both Ca2+-free and Ca2+-bound forms of the dyes. Single-wavelength excitation at 810 nM was used to determine the effective dissociation constants (Keff) and dynamic ranges (Rf) of Fura-2, -4F, -6F, -FF, and Furaptra dyes (Keff = 181 ± 52 nM, 1.16 ± 0.016 μM, 5.18 ± 0.3 μM, 19.2 ± 1 μM, and 58.5 ± 2 μM; and Rf = 22.4 ± 3.8, 12.2 ± 0.34, 6.3 ± 0.17, 16.1 ± 2.8, and 25.4 ± 4, respectively). Single-wavelength excitation of intracellular Fura-4F resolved diastolic and peak [Ca2+] in isolated stimulated cardiomyocytes after calibration of the intracellular signal using reversible exposure to low (100 μM) extracellular [Ca2+]. Furthermore, TPE of Fura-4F allowed continuous, long-term (5–10 min) Ca2+ imaging in ventricular cardiomyocytes using laser-scanning microscopy without significant cellular photodamage or photobleaching of the dye.
INTRODUCTION
Laser-scanning confocal fluorescence microscopy, in conjunction with the development of fluorescent Ca2+-sensitive dyes (Grynkiewicz et al., 1985) allows the imaging of transient changes of [Ca2+] in optical sections of biological samples up to a depth of ∼40 μm. These fluorescent dyes (excited by ultraviolet light) were used for epifluorescence imaging, but proved difficult to use with laser-scanning systems due to poor ultraviolet (UV) transmission characteristics of microscope optics (Bliton and Clapham, 1993) and damaging effects of short wavelength light on biological processes (Hockberger, 2002). In addition to these concerns, confocal imaging with UV excitation wavelengths can introduce chromatic aberration and focal plane offsets with reduced confocal signal collection (Bliton and Clapham, 1993; Bliton and Lechleiter, 1995). For these reasons, a series of visible excitation dyes based on the fluorescein molecule (Minta et al., 1989) are commonly used for confocal imaging. The Fluo (and Rhod) dyes have been used with laser-scanning techniques to examine Ca2+ dynamics within an optical section in a range of cells/tissues, including mammalian cardiomyocytes/myocardium. A potential problem with laser-scanning microscopy of fluorescent dyes is rapid photobleaching (Patterson and Piston, 2000), which can severely limit the timescale over which accurate estimates of intracellular [Ca2+] signals can be obtained.
With the increasing availability of reliable ultra-short pulsed near-infrared (700–1050 nM) lasers, two-photon laser-scanning microscopy is now possible on a commercial basis. Considerably lower phototoxicity can accompany the use of longer wavelength illumination, allowing long-term imaging of biological tissue (Squirrell et al., 1999). Fluorescent dyes with absorption maxima <400 nM (e.g., Fura and Indo) can be excited by near-infrared laser light. Generally the Fura dyes are preferred over the equivalent Indo versions since the latter are less fluorescent and have problems with photoisomerization and photobleaching (Grynkiewicz et al., 1985). Several Fura-based dyes are now commercially available, each with different Ca2+ (and Mg2+) sensitivities. Originally the Fura dyes were designed for dual wavelength excitation and single-wavelength band emission fluorescence (Grynkiewicz et al., 1985; Minta et al., 1989), but single-wavelength excitation may also provide an easily calibrated intracellular Ca2+ signal (Maravall et al., 2000). In this respect, Fura dyes offer additional advantages when excited at the longer (>365 nM) wavelength. Under these conditions, increases of [Ca2+] cause a decrease in fluorescence, a feature that lends itself to simple calibration procedures (Konishi et al., 1991; Ogden et al., 1995). Two-photon excitation of Fura-2 has been used to image Ca2+ within mammalian cerebral cortex (Stutzmann et al., 2003) and an epithelial cell line (Ricken et al., 1998).
Previous studies have provided two-photon excitation spectra for Fura-2 (Ca2+-bound and unbound forms; Xu et al., 1996a,b; Kuba and Nakayama, 1998). However, the multiphoton spectra of the other dyes in the Fura family have not yet been reported. The present study describes the multiphoton spectra (750–850 nM) of a range of Fura dyes (Fura-2, -4F, -6F, -FF, and Furaptra) and the Ca2+ sensitivity of these dyes when excited with light at 810 nM. This information was used to calibrate the line-scan signals of Fura-4F fluorescence in cardiac myocytes stimulated at 0.5 Hz. These measurements demonstrate for the first time that two-photon excitation of Fura-4F in isolated cardiac myocytes generates a stable Ca2+ signal during continuous line-scan imaging for prolonged periods of time (5–10 min) with no obvious phototoxicity to the cell or photobleaching of the dye.
MATERIALS AND METHODS
Imaging system
A schematic of the optical system and microscope is shown in Fig. 1. The ultra-short pulsed laser source is a commercial tunable laser system with solid-state 532-nM pump laser and Titanium:sapphire oscillator in the same hermetically sealed box (Vitesse XT, Coherent Laser Group, Ely, UK). The laser wavelength (750–850 nM) was varied by adjusting the bi-refringent filter position via a stepping motor. This permitted reliable and rapid wavelength selection. The laser beam was routed through an optical isolator to minimize reflections back into the oscillator (ISO); the isolator is essential for imaging applications with this laser source. The laser beam is next routed into a commercial multiphoton excitation system (Radiance 2000MP, Bio-Rad Microscience, Hertfordshire, UK) beam combiner unit (BCU) (Dixon et al., 2001). The beam combiner unit taps off a portion of the laser beam for wavelength (λ), bandwidth measurement (Laser Spectrum Analyzer 201, IST Ltd., Alton, UK) and alignment verification with a telescoping component and phosphor site windows. The BCU also provides rapid and fine attenuation of the laser beam with a KD*P-based Pockels' cell (ω) (Model 50BK, ConOptics, Danbury, CT). The laser power is variable from 100% to 0.5%, a verified attenuation extinction range of 200:1. A separate laser-blocking shutter is located in front of the Pockels' cell. A portion of the postattenuated laser beam (10%) is tapped off and routed to the excitation dose monitors (MON). The laser beam then is expanded with a variable beam expander (Till Photonics, Grafelfing, Germany) to accommodate laser beam filling of different objective lenses. The processed laser beam leaves the BCU through two (adjustable) enhanced silver mirrors to match the optical axis of the confocal scan head (RAD). The confocal scan head was attached to the side port of an inverted microscope (TE300DV, Nikon, UK). The side port of the Nikon inverted Eclipse microscopes have been optimized for near-infrared transmission. The objective lens used for imaging investigations was the high numerical aperture (NA) Nikon Plan Apochromat 60×/1.2 NA water-immersion lens (Nikon, Surrey, UK). The BCU removed 50% of the power available from the laser system, but the power available at the sample was still sufficient to bleach the Fura dyes. Line-scan acquisitions of the confocal unit were limited to 30,000 lines/file, which amounts to 1 min of fast (2 ms/line) scanning. To circumvent this limitation a separate solid-state integration unit was directly connected to the output of the bi-alkali-cathode photon detectors (photomultiplier tube, PMT) (Dempster et al., 2002) and the signal acquired through a separate high-speed acquisition system. The bi-alkali PMT was used in all experiments. Using excitation wavelengths >800 nM, a laser-blocking filter was not necessary since the emission routing chromatic mirrors in the Radiance 2000MP do not reflect near-infrared light very well. However, a 625-nM short-pass filter (E625SP, Chroma) was used for all measurements to ensure the lower wavelengths could be used without laser source contaminating the background signal.
FIGURE 1.

System diagram: layout of modified Bio-Rad Radiance 2000MP system with non-de-scanned detectors (NDD) and custom beam monitors (MON). Laser source is a computer-controlled Coherent Vitesse XT ultra-short pulse Ti:sapphire system. Laser system includes optical isolator (ISO) and routing mirror. The multiphoton beam combiner unit (BCU) includes: optical axis adjustment mirrors, a scanning optical spectrometer (λ,B), alignment sites (circles), a Pockels' cell (PC), 10% beam tap (to monitors), and an optical telescope (ω) to change the laser beam profile at the objective lens entrance pupil. From the BCU, the main laser beam enters the Radiance scan head (RAD), which is coupled to the side port of the Nikon (TE-300DV) inverted microscope. The objective used was the Nikon Plan Apochromat 60×/1.2 water-immersion lens. The tapped-off excitation beam is routed to two photodiodes (MON) to monitor the laser beam after attenuation (PC). The average power at the sample was measured with a thermopile detector (DET). A green light-emitting diode (LED) was placed above the sample stage to serve as a timing reference signal.
System characterization
The characteristics of the laser beam were monitored with a pair of solid-state detectors (photodiodes) and correlated with the signal emerging from the high numerical aperture (NA) objective lenses used in this study. This arrangement allowed examination of the relationship between peak light intensity and fluorescence necessary to characterize the excitation mode of the dye. Furthermore, knowledge of peak and average light intensity emerging from the objective allows estimates of the fluorescence cross section of the dye to be made (Albota et al., 1998). The laser average power at the sample was measured with a thermopile detector (PM10, FieldMaster, Cascade Laser Group, Newburg, OR) and monitored and scaled with a Silicon photodiode detector (S2844K, Hamamatsu, Hertfordshire, UK). For all power measurements, the beam-blanking feature was turned off. The laser peak power was monitored with a gallium-arsenide-phosphide (GaAsP) planar diffusion photodiode (Ranka and Gaeta, 1997; Helmchen et al., 2002). The mode of excitation of the fluorophore was measured by varying the incident average power (using the Pockels' cell) and measuring the resultant fluorescence signal. As shown in an earlier publication (Wokosin et al., 1996), the number of photons required for an excitation event can be derived from the slope of the Log/Log plot of these two variables.
Measurements on solutions of fluorophore
A uniform solution of fluorophore was contained in a 200-μl chamber with a #1.5 coverslip as a base. The lens correction collar was set to the brightest fluorescence. The objective lens was focused 12 μm into the solution to employ thick sample theory (Xu et al., 1995) and avoid surface effects and movement artifacts. The generated fluorescence signal was represented by the mean of the histogram of eight accumulated (or four averaged) scans. Background counts with the laser shuttered were subtracted from all values.
Two-photon excitation fluorescence cross sections
The spectra are expressed in terms of the two-photon excitation fluorescence (2PEF) cross sections. This method has been established in previous publications as a standardized system of expressing 2PEF spectra (Albota et al., 1998). The 2PEF cross section is the product of the molecular absorption cross section and the fluorescence emission quantum efficiency and is expressed in terms of Goppert-Meyer (GM) units (1 GM = 10−50 cm4 per s per photon−1). The spectra displayed in this study were expressed in terms of 2PEF cross section by using a scaling factor representing system emission collection (SC) and a correction factor for each wavelength (CF[λ]). These values were derived using published 2PEF cross-section values for fluorescein (Albota et al., 1998), but they can also be generated without reference to a standard, but with the knowledge of a series of system parameters (see Appendix). Linear plots of the 2PEF cross sections can be found at http:www.bio-rad.com/product/multiphoton/radiance2100mp/mpspectra.htm.
The correction factor (CF[λ]) represents wavelength-dependent changes in the pulse duration, i.e., the incident peak intensity at the sample. The 2PEF cross section (2PEFcs) of the dye under test was generated by
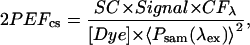 |
(1) |
where Signal is the fluorescence signal (gray levels/pixel), [Dye] is the concentration of the dye, and 〈Psam(λex)〉 is the average power measured by the silicon diode monitor.
Calibration solutions
Buffer solutions to enable the measurement of the Ca2+ sensitivity of the high affinity dyes (e.g., Fura-2 and Fura-4F) were made by mixing two principal solutions (Solutions A and B) to generate [Ca2+] values ranging from <1 nM to ∼50 μM. Both solutions A and B had the following composition: 100 mM KCl, 10 mM NaCl, 1 mM MgCl2, and 25 mM HEPES, at pH 7.0. Solution A additionally contained 10 mM K2EGTA; Solution B additionally contained 10 mM CaK2EGTA. The equilibrium [Ca2+] in the calibration solutions was calculated using a computer program with the affinity constants for H+, Ca2+, and Mg2+ for EGTA taken from Smith and Miller (1985). Corrections for ionic strength, details of pH measurement, allowance for EGTA purity, and the principles of the calculations are detailed elsewhere (Miller and Smith, 1984). Total Mg2+ was adjusted to maintain the free Mg2+ between 0.9 and 1 mM in all solutions. The sensitivity of low affinity dyes (e.g., Fura-FF) was assessed using a solution with the following composition: 100 mM KCl, 10 mM NaCl, 1 mM MgCl2, 25 mM HEPES, and 0.05 mM K2EGTA, at pH 7.0. The [Ca2+] in this solution was varied from 80 nM (no added Ca2+) to 1 mM by addition of CaCl2 from a 1 M stock solution (VWR International, Dorset, UK). Fluorescent Ca2+ indicators Fura-2, -4F, -6F, -FF (Molecular Probes, Eugene, OR) were added to the solution to give a nominal final concentration of 30 μM. All other chemicals were supplied by Sigma (Dorset, UK).
Cardiomyocyte isolation and permeabilization
Ventricular cardiomyocytes were isolated from Langendorff-perfused rabbit hearts by enzymatic digestion as previously described (McIntosh et al., 2000). Myocytes to be permeabilized were maintained in a modified Krebs solution buffered with 1 mM ethylene bis[oxyethylenenitrilo] tetra-acetic acid (EGTA) at a concentration of ∼104 cells/ml until use. The cells were allowed to settle onto the coverslip at the base of the sample chamber. β-escin (Sigma) was used to permeabilize the sarcolemma of cardiac myocytes (Loughrey et al., 2002). β-escin was added from a freshly prepared stock solution to the cell suspension to give a final concentration of 0.1 mg/ml for 0.5 to 1 min and the β-escin subsequently removed by perfusion with a mock intracellular solution with the following composition: 100 mM KCl, 5 mM Na2ATP, 10 mM Na2CrP, 5.5 mM MgCl2, 25 mM HEPES, and 0.05 mM K2EGTA, at pH 7.0 (20–21°C).
Intact myocyte measurements
Myocytes used for intact cell measurements were collected from the dissociation procedure and maintained in a modified Krebs solution with the following composition: 120 mM NaCl, 5.4 mM KCl, 3.5 mM MgCl2, 1.8 mM CaCl2, 20 mM HEPES, 11.1 mM glucose, 20 mM taurine, and 0.56 mM NaH2PO4.2H20, at pH 7.4 (20–21°C). Cells were loaded with the acetoxymethyl (AM) ester form of the dye by adding 5 μl of 1 mM dye to 0.5 ml of cell suspension. This was incubated for 10 min at 37°C. The cells were subsequently washed and resuspended in 3 ml of the above solution and left for 20 min at room temperature before use. Myocytes were placed in a shallow bath on the microscope stage and perfused with modified Krebs solution. The cardiomyocytes were field-stimulated with 2-ms voltage pulses delivered through parallel platinum wires, and the stimulation voltage was set to 1.5 times the stimulation threshold. The exact timing of electrical stimulation was marked in the confocal image by activating a light-emitting diode (LED) mounted above the cell bath for 2 ms (i.e., the duration of one line scan) 8 ms before electrical stimulation.
Statistics
Data were expressed as mean ± SD.
RESULTS
Characterization of detectors
As shown in Fig. 1, the laser power was monitored after the Pockels' cell attenuator (MON, Fig. 1). A silicon photodiode (Pave) permitted monitoring of the laser average power while the GaAsP diffusion diode (Ppk) monitored the peak intensity through two-photon absorption-induced current (Ranka and Gaeta, 1997; Helmchen et al., 2002). These diodes allow confirmation of the laser excitation energy. Fig. 2 A demonstrates that the signals from the diode monitors were reliable over three decades of laser peak intensity (limited by the 3.5 digit panel meter). The diode monitors can also be used to predict the average power and peak power at the sample plane. Fig. 2 B shows the diode signals relative to the average power measured with a thermopile detector at the sample plane (DET, Fig. 1). As expected for a linear system, the silicon diode shows a slope of 1.0 whereas the GaAsP diode retains a slope of 2.0 (Fig. 2 B). A similar principle can be used to characterize the mode of excitation for nonlinear fluorescence microscopy (Wokosin et al., 1996). As shown in Fig. 8 (see later), the mode of excitation of the dye can be derived from the slope of a Log/Log plot of the fluorescence signal versus incident average power.
FIGURE 2.

Source monitoring. (A) Laser monitoring: Log/Log plot demonstrating the range of two-photon absorption current for the diodes (MON) monitoring the laser after attenuation. The peak power is plotted against average power and the slope of the Log/Log plot is ∼2.0. Valid two-photon excitation monitoring covers three orders of magnitude. (B) Sample scaling: Log/Log plot showing the signal of the diode monitors relative to average power measured with a thermopile detector at the sample plane after the objective lens. The silicon diode voltage scaled linearly with the sample average power (slope ∼ 1) and the GaAsP diode voltage scaled with the square of the average power (slope ∼ 2). The resistor values were set to match the tapped-off laser beam average power to the 200-mV to 100-μV range of the DVM.
FIGURE 8.

Signal quantification. The excitation mode was measured at 780 nM, 810 nM, and 850 nM; Fura-2 signals were represented by photons per pixel counted with fast photon counting in eight accumulated scans, 2.6 μs dwell-time, 0.4 μm pixels. Values are acquired from a focal plane 12 μm inside a homogeneous sample. Log/Log plots showing fluorescence signal of Fura dyes relative to the average power at the sample. (A) Determination of excitation mode: values plotted for Fura-2 (circles) at 800 nM (open symbols) and 850 nM (solid symbols). Two-photon excitation is verified at all three wavelengths from the slope of the relationship. (B) Fluorophore bleaching: Log/Log plot showing fluorescence signal of the Ca2+-bound form of Fura-2 relative to the peak intensity at the sample. Two-photon excitation of the Ca2+-bound form is verified when the incident peak intensities are <1 arbitrary unit (equivalent to ∼1 × 1030 photons/cm2 per s) for all three wavelengths. Similar bleaching thresholds at these wavelengths were noted for Fura-FF and Furaptra (results not shown).
Two-photon fluorescence excitation spectra
Fig. 3 A shows the two-photon excitation fluorescence spectrum of the Ca2+-free form of the Fura-2, -4F, -6F, -FF, and Furaptra from 750 nM to 850 nM. The spread of data points is representative of the system measurement errors. Fura-5F produced similar spectra (data not shown). Fig. 3 B indicates typical spectra for the Ca2+-bound forms of the dyes. Note lower fluorescence values compared to Fig. 3 A. Spectra of both Ca2+-free and Ca2+-bound forms of Fura-4F are shown in Fig. 3 C to illustrate the approximately parallel decrease in fluorescence that occurs between 780 and 850 nM. Also plotted is the typical background fluorescence signal demonstrating the magnitude of the signal over the background value for the Ca2+-bound form of Fura-4F.
FIGURE 3.

Action cross-section spectra. (A) Ca2+-free form of indicator: high fluorescence (fmax) signal, acquired in photon counting of eight accumulated scans, 2.6 μs dwell-time, 0.4 μm pixels from focal plane 12 μm inside a homogeneous sample. The acquired values are scaled by a constant at each wavelength to obtain GM units for action cross section. Values are shown for Fura-2 (○), Fura-4F (□), Fura-6F (▵), Fura-FF (⋄) and Furaptra (▿). (B) Ca2+-bound form of indicator: low fluorescence (fmin) signal, acquired in same manner as A. 2PEF cross-section spectra are shown for Fura-2 (•), Fura-4F (▪), Fura-6F (▴), Fura-FF (♦), and Furaptra (▾). (C) Fura 4F: Log plot of Fura-4F 2PEF cross-section spectra; indicator unbound (□) and indicator bound to calcium (▪). The lowest plot (shaded square and shaded line) is a typical background signal recorded in the absence of laser light, and scaled for GM units.
Fura fluorescence in permeabilized cardiomyocytes
Fig. 4 A,i shows a representative pseudocolored image of a permeabilized cardiac myocyte superfused with a mock intracellular solution containing <1 nM Ca2+ (30 μM Fura-4F) excited with 810 nM. The 2PEF cross-section spectra for various regions of the image are shown in Fig. 4 A,ii. The spectra are scaled for GM units, but these values only strictly apply to “outside” and “cuvette” signals. The higher signal recorded in the “cytoplasm” and “nucleus” is probably due to changes in local dye concentration, but changes in absorptive and radiative efficiencies cannot be excluded (see Appendix). Fura-2, Fura-6F, Fura-FF, and Furaptra produced similar differences in intensity between the nuclear region and cytoplasm, as well as between the cytoplasm and outside the cell. Measurements from both cytoplasm and nucleus produced effectively the same shape of spectrum as that recorded from free solution (outside the cell) when normalized to the fluorescence at 780 nM (inset, Fig. 4 A,ii).
FIGURE 4.

Cardiomyocytes. (A,i) Permeabilized cells: a representative image (810 nM excitation) of a cardiac myocyte stained with Fura-4F. Scale bar = 10 μm. The pseudocolored images were scaled in terms of GM units as indicated by the adjacent lookup table. (A,ii) Cellular 2PFE spectra: data acquired as per Fig. 3 A. Region pixel boxes were analyzed from the nucleus (○), cytoplasm (□) and values outside the cell (×). The cuvette data (0.1 nM Ca2+ from Fig. 3 A) is also shown for comparison (+). (Inset) The cellular data normalized to values at 780 nM (relative CS) to highlight the similarity of spectral response in the different regions of the cell, and compared to the cuvette samples. (B,i) AM-loaded cardiomyocyte: a representative image (810-nM excitation) of a cell loaded with Fura-4F-AM. Scale bar = 10 μm. (B,ii) 2PEF spectra: data acquired as per Fig. 3 A, but with 40% of average power used previously. Signals recorded from the cytoplasm (▪) and from the cytoplasm of an unloaded cell (□). The internal dye concentration is unknown (or nominally zero in the unloaded cell), therefore the signal is expressed as 2PEFcs × [Dye]/30 μM. (B,iii) Normalized 2PFE spectra: normalized 2PEF cross-section spectra (to 780 nM value) are plotted versus laser center wavelength. Region pixel boxes were analyzed from the nucleus (open circles), cytoplasm (open squares). The cuvette data is also shown (+).
Fura fluorescence measurements from intact cardiomyocytes
Fig. 4 B,i shows a representative image of an intact cardiac myocyte after loading with Fura-4F by incubation with the –AM form of the dye. The fluorescence signal from the cytoplasm and an autofluorescence signal from a different cell are shown in Fig. 4 B,ii. On the basis of these measurements, the signal/background ratio (S/B) increased from ∼50 at 750 nM to ∼150 at 810 nM before decreasing to a value of ∼50 at 850 nM. The signal values for nuclear region and cytoplasm are all normalized to their values at 780 nM because the relative amount of dye and intracellular [Ca2+] within different regions of the cell are not known. The spectra are shown in Fig. 4 B,iii. Fura-2, Fura-6F, Fura-FF, and Furaptra produced similar 2PEF cross sections (data not shown). Again, all locations within the cell produced effectively the same shape of 2PEF cross section and the cellular spectra agree with the spectra of the dyes in free solution (Fig. 4 B,ii). Using standardized –AM loading procedures (see above), the average cytoplasmic Fura-4F fluorescence in intact cells in the presence of low extracellular Ca2+ (100 μM) was 114 ± 66 (n = 10) times higher than the typical background fluorescence measured in unloaded cardiomyocytes. Typically the Fura-4F fluorescence excited at 810 nM varied on a cell-to-cell basis from 35 to 150 times the background autofluorescence value. The effect of this uncertainty of signal over background has implications for the estimation of intracellular [Ca2+], and is discussed later (Fig. 9).
FIGURE 9.

[Ca2+] calibration errors. (A) Error due to changes in Fbg (ΔFbg): the fluorescence signal of Fura-4F (Fig. 6 A, 810-nM excitation) is plotted with varying Ca2+ values normalized by the indicator Keff (1.15 μM). A typical cellular Ca2+ concentration range is highlighted in shaded box (115 nM to 1.15 μM). The left axis (thick line) highlights the relative fluorescence where the full Rf is available at low [Ca2+]. The right axis (thin lines) represents the percentage of error in [Ca2+] due to variations in the intracellular background (0.5, 1.5, 2, and 3 times measured background). The dashed lines denote zero error level. (B) Error due to changes in background Rf (ΔRf): as per A, but now the right axis (thin lines) represents the percentage of error in [Ca2+] due to a lower intracellular Rf (0.6, 0.8, 0.9, and 1.1 × Rf). The curves indicate that below the indicator Keff /[Ca2+] of 2.0, the estimation of [Ca2+] should have reasonable accuracy (error < 20%). (C) Error analysis due to Rf for Fura-2 and Furaptra. The left axis (thick line) highlights the relative fluorescence where the full Rf is available at low [Ca2+]. The right axis (thin lines) represents the percentage of error in [Ca2+] due to a lower intracellular Rf (0.6, 0.8, 0.9, and 1.1 × Rf). The right-hand shaded rectangle represents the range of [Ca2+]/Keff expected for the 0.1–1 μM range of intracellular [Ca2+] for the dye Fura-2. The left-hand shaded rectangle is the typical range of [Ca2+]/Keff values appropriate for the dye Furaptra.
Variation of fluorescence ratio (Rf) with wavelength
The dynamic range of the fluorescence indicator is determined by the ratio of the fluorescence recorded from the Ca2+-free form (fmax) divided by the fluorescence from the Ca2+-bound form (fmin). The variation of this ratio (Rf) over the range of wavelengths is shown in Fig. 5. The Rf increased progressively from 750 nM to ∼790 nM; beyond this wavelength, the value remained constant for the majority of the Fura dyes. As shown in Fig. 3, the value of fmax decreases progressively beyond ∼810 nM. For these reasons, the optimal wavelength for two-photon excitation was chosen as 810 nM, yielding the highest dynamic range and highest fmax value for a range of Fura dyes. The mean Rf values for the Fura dyes studied are shown in Table 1. Fura-2 and Furaptra exhibited the highest Rf values, Fura-FF and Fura-4F the next largest, and Fura-6F displayed the lowest Rf value. Preliminary work with Fura-5F indicated an even lower Rf value (∼3, data not shown).
FIGURE 5.

Fluorescence ratio (Rf). Unbound indicator signal (Fig. 3 A) relative to bound indicator signal (Fig. 3 B) as a function of excitation center wavelength. Values highlight the dynamic range (Rf = fmax/fmin) for the [Ca2+] ratio imaging. Image background counts have been subtracted from each signal before the ratio. Values plotted for Fura-2 (○), Fura-4F (□), Fura-6F (▵), Fura-FF (⋄), and Furaptra (▿).
TABLE 1.
Ca2+ sensitivity and dynamic range of Fura fluorescence on excitation at 810 nM
| Dye | Keff | Rf |
|---|---|---|
| Fura-2 | 181 ± 52 nM (n = 14) | 22.4 ± 3.8 (n = 9) |
| Fura-4F | 1.16 ± 0.016 μM (n = 8) | 12.2 ± 0.34 (n = 8) |
| Fura-6F | 5.18 ± 0.3 μM (n = 12) | 6.3 ± 0.17 (n = 8) |
| Fura-FF | 19.2 ± 1.0 μM (n = 9) | 16.1 ± 2.8 (n = 13) |
| Furaptra | 58.5 ± 2.0 μM (n = 4) | 25.4 ± 4 (n = 4) |
The dissociation constant (Keff) was obtained from best-fit logistic curve to fluorescence measured at a range of [Ca2+]. The dynamic range (Rf) is the relative change in fluorescence from the Ca2+-bound form (fmin) to the Ca2+-free form (fmax). Estimates of fmin and fmax were obtained from parameters of the logistic curve fit to the calibration data. (Individual assays are shown as mean ± SD.)
Measurement of the effective dissociation constant for Ca2+ (Keff)
As illustrated in Fig. 6 A, the Keff was estimated by fitting a binding isotherm to the relationship between fluorescence and [Ca2+]. All dyes generated satisfactory sigmoidal relationships with slopes close to unity. The average Keff values are shown in Table 1. The relationship between the Keff measured at 810 nM and those observed on one-photon excitation of the dye are shown in Fig. 6 B. UV light at 380 nM (10 nM bandwidth) was provided by a Cairn Optoscan (Cairn Research, Faversham, UK) spectrophotometer. Emitted light was measured at >500 nM. In all dyes studied, the Keff of the fluorescence change was similar for excitation at 380 nM and at 810 nM.
FIGURE 6.

Effective Ca2+ binding constants, Keff. Values are plotted for Fura-2 (○), Fura-4F (□), Fura-6F (▵), Fura-FF (⋄), and Furaptra (▿). (A) Ca2+ calibration: fluorescence signal (810 nM excitation) of Fura dyes measured for various buffered free-calcium concentrations. Signal represents shaded levels per pixel of 8 or 25 accumulated scans in photon counting mode, 2.6 μs dwell-time, 0.4 μm pixels. Values are from a focal-plane 12 μm inside uniform solution samples. Ca2+ concentration axis plotted in Log scale (0.1 nM to 10 mM). (B) One- and two-photon excitation: the same solution samples used in Fig. 6 A were also measured with an epifluorescence monochromator system to determine the Keff values at 340 nM and 380 nM.
Line-scan imaging of Ca2+ transients in isolated intact cardiomyocytes
Long-term line-scan imaging of intracellular Ca2+ was achieved using Fura-4F (1.15 μM Keff) loaded into the cell by incubation with the –AM form of the dye. As shown in Fig. 7, the myocytes were continuously scanned (2 ms/scan) with light at 810 nM (∼2.3 mW average power, ∼395 fs, 80 MHz) for 10 min while the cell was stimulated at 0.5 Hz using field electrodes. No noticeable bleaching or photodamage was evident as shown by samples of the 1-min acquisitions in Fig. 7 A. The average fluorescence from the central 20 pixels of the line scan are shown in Fig. 7 B. Each stimulus event was marked by a 2-ms flash from a green LED. This was followed by a transient fall in fluorescence as a result of a transient rise in intracellular [Ca2+]. The fmax value was obtained by initially superfusing the cell with a low Ca2+ solution (100 μM) to enable the intracellular [Ca2+] to fall to levels that would generate an fmax (intracellular [Ca2+] < 10 nM). Using this fmax value with the Rf and Keff values from Table 1, the signal was converted to intracellular [Ca2+]. As shown in Fig. 7 B,ii, this procedure indicated a resting [Ca2+] of ∼150 nM and peak systolic of ∼800 nM, comparable to values gained from other indicators.
FIGURE 7.

Long-term line scans. (A) Records of Fura-4F fluorescence recorded at 1, 2, 4, and 5 min from a single rabbit ventricular cardiomyocyte field-stimulated at 1 Hz. The fluorescence signals are derived from a continuous line-scan image. The mean fluorescence signal from an intracellular section 20 pixels (6 μm) wide is shown. The stimulus is marked by a 2-ms flash by a green light-emitting diode (LED) placed above the cell (see Fig. 1). The [Ca2+] in the superfusion solution was increased from 0.1 mM to 1.8 mM at the time indicated above the trace. (B,i) Average of eight transient signals of fluorescence, the flash due to the photodiode is marked at stimulus above the trace. (B,ii) The fluorescence signals shown in B,i converted to [Ca2+] using the Keff and Rf values shown in Table 1.
DISCUSSION
Verification of two-photon excitation
The incorporation of detectors into the light path allowed the fluorescence of the Fura dyes to be studied at a range of peak powers. As shown in Fig. 8 A, the normalized Log/Log plot of peak intensity versus fluorescence signal for the Ca2+-free form of Fura-2 was assessed at three different wavelengths. The slopes of these relationships were very close to 2.0 when measured at low peak intensities confirming two-photon excitation (mean slope at 810 nM = 1.98 ± 0.06, n = 4). Similar relationships were exhibited by the Ca2+-bound forms (Fig. 8 B). As peak excitation intensity is increased, the slopes deviated from 2.0 at all wavelengths, behavior that can be explained by bleaching of the dye at higher light intensities. Measurements of the average power and pulse duration at the sample plane indicate an upper limit of peak excitation intensity of ∼0.5 × 1030 photons/cm2 per s, above which bleaching occurred. Bleaching was highlighted as the most likely cause since increased dwell times decreased the signal further and calculations show the peak intensities are far from saturation (Xu et al., 1996b). Similar bleaching characteristics were obtained with all Fura dyes studied.
A previous study has shown a slope >2 for the Ca2+-bound form of Fura-2 with excitation wavelengths beyond 820 nM (Xu et al., 1996b). A slope of >2 would suggest that part of the fluorescence signal detected is generated by three-photon excitation accessing the 340-nM arm of the Fura excitation spectrum. However, nonlinear excitation modes beyond two are quite sensitive to the peak intensity, the objective lens NA, and the pulse duration at the sample (Xu et al., 1996b; Hopt and Neher, 2001). The peak intensity (∼0.5 × 1030 photons/cm2 per s) and NA (1.2) used in the present study were similar to the previous study, but measurements of the laser pulse duration at the sample plane (456 fs at 850 nM) indicate longer pulse durations in the present study than the ∼75 fs most likely used for three-photon imaging with a pulse-precompensated system. The longer pulse durations would yield considerably less (∼37 times) three-photon excitation for the same incident peak intensity and may explain the lack of three-photon events observed in this study.
Two-photon excitation spectra
The measured values for the 2PEF cross section of the Ca2+-free form of the indicator (Fig. 3 A) for all Fura dyes studied agree very well with previously published Fura-2 spectra (Xu et al., 1996a,b), except for the outgoing “bump” feature from 800 nM to 820 nM in previously published spectra. No anomalies were observed with measurements of fluorescein and Fluo-3 spectra (the same dye concentrations normalized by average power and only scaled at one wavelength, 780 nM, SC, see Eq. 1); therefore the reasons for the minor differences in the Fura spectra are not clear. It is worth noting that our measured values from 750 nM to 850 nM (7.5-fold drop) also agree well with the one-photon values from 375 nM to 425 nM (eightfold drop) (Haugland, 2002). Similarly, the fall in signal with increasing wavelength (750–850 nM) described by the 2PEF spectrum for the Ca2+-bound form of Fura-2 (Fig. 3 B) agrees well with the one-photon fluorescence cross section (375–425 nM) (Reynolds, 2003).
Fortuitously, it appears that the useful portions of the Fura excitation spectrum for single-wavelength excitation are contained within the narrow spectral range presented in this study. Excitation wavelengths <750 nM suffer reduced dynamic range (Rf) and increased autofluorescence contribution, whereas excitation wavelengths >850 nM will produce a dramatically lower S/B when imaging cardiac myocytes. Previously published work has indicated the 2P isosbestic wavelength for Fura-2 is 710 nM (Xu et al., 1996b) and suggests that the excitation peak of Fura-2 unbound to calcium may be 700 nM. The absorption peak will be difficult to confirm with excitation wavelengths available on Ti:sapphire lasers with broad-band mirror sets (700-nM minimum). As assumed in the original description of the Fura dye (Grynkiewicz et al., 1985), the Keff of the dye is independent of the excitation wavelength, as now demonstrated with the two-photon wavelength (Fig. 6 B).
While investigating the spectral characteristics of the Ca2+-bound form of the various Fura dyes, it became apparent that excessively high Ca2+ levels (>1 mM) caused a reduction in the fluorescence signal beyond the value expected from the standard sigmoid relationship (Fig. 6 A). Fluorophore quenching by high [Ca2+] seems the most obvious reason for this effect. To avoid this effect leading to an overestimation of Rf, the values listed in Table 1 were derived from the estimates of minimum fluorescence (fmin) generated by the calibration curves.
A standard wavelength of 810 nM was chosen as a suitable compromise between maximizing the Rf value and the S/B. The 2PEF cross section (i.e., normalized signal) for a given dye decreases with increasing wavelength, whereas myocyte cellular autofluorescence (background) exhibits a local minimum ∼800 nM. As shown in Table 1, Rf values measured at 810 nM varied considerably across the range of Fura dyes studied. As shown in Fig. 6 B, the values of Keff measured at 810 nM are approximate to those measured at 340 nM and 380 nM. The unusual relationship between wavelength and Rf observed with Furaptra was consistent between different batches of the dye and was also observed with single-photon excitation. One possible explanation is the progressive quenching of the fluorescence signal by Ca2+, a phenomenon observed only at Ca2+ levels required to achieve fmin (see above).
The present study has emphasized the two-photon excitation characteristics of Fura dyes, but the single-excitation wavelength approach may also be applied to confocal imaging. Based on preliminary measurements using Fura-4F, excitation with a 405-nM laser (Gallium Nitride, Coherent Violet laser) may provide a feasible approach. Further work is required to compare the two optical sectioning techniques (confocal versus two-photon).
Autofluorescence
Nonloaded cardiac myocytes have detectable autofluorescence in the wavelength range investigated (Fig. 4 B,ii). The level of autofluorescence is comparable to the fmin values obtained at the excitation wavelengths >825 nM, Fig. 3 C. The spectral dependence of the autofluorescence 2PEFcs × [Dye] product does follow the shape of NAD(P)H and flavoprotein. The excitation and emission spectra are compatible with previous measurements on isolated cardiomyocytes (Huang et al., 2002). Based on these autofluorescence spectra and that for the dye Fura-4, 800–810 nM provides the best S/B values within the excitation range studied. The signal/background ratio at 810-nM excitation can be improved 150% by restricting the emission window to 500–550 nM (compared to 420–625 nM), but at the expense of one-half the Fura signal photons.
Measurement of intracellular [Ca2+]
The bright intracellular fluorescence signals with basal and low intracellular [Ca2+] provides an opportunity to examine intracellular loading uniformity, a procedure more difficult with the standard Fluo-based dyes due to the low fluorescence levels in low intracellular [Ca2+]. Commonly, anomalous spectra of Ca2+-sensitive dyes are observed using wide-field UV excitation after –AM loading; the remarkably similar Fura-4F spectra observed in 1), free solution, 2), permeabilized cell cytoplasm, and 3), –AM-loaded dye intact cells may be based on the use of the near-infrared excitation wavelength. These wavelengths avoid major absorption and scattering concerns common with UV excitation. Furthermore, the optical sectioning technique should remove optical path differences of the acquired signal volume. The simple calibration step used in this study (superfusion with 0.1 mM extracellular [Ca2+]) can be used a number of times without adverse effects on the cell.
The attribution of the free [Ca2+] value to a fluorescence signal is critically dependent on the value of the Keff of the dye. In this study, the assumption was made that the Keff of Fura-4F within the cytosol is similar to that measured in buffer solutions. The Keff values of a range of Ca2+-sensitive dyes have been shown to increase on interaction with the protein aldolase (Baylor and Hollingworth, 2000). However, calibration of an intracellular Fura-2 signal in smooth muscle cells indicated only a small increase in Keff (Williams and Fay, 1990).
Error analysis for estimation of intracellular [Ca2+]
The use of single-excitation wavelength techniques to measure intracellular [Ca2+] is more susceptible to error as a result of potential changes in excitation light levels or sample movement. All-solid-state laser systems can have long-term stability on the order of 3% for the two-photon signal, i.e., much higher stability than the equivalent gas lasers commonly used in laser-scanning confocal microscopy. As proposed earlier, continuous monitoring of laser source intensity can effectively remove these concerns. However, for isolated cardiac myocytes, cell movement/contraction is still a potential source of error. Furthermore, the spectral properties of the dye may be altered by the intracellular environment, the most prominent effect being the reduction in dynamic range of fluorescence signal within the cell (Maravall et al., 2000). To assess the ability of Fura fluorescence to accurately represent intracellular [Ca2+], computations were made to estimate the errors resulting from changes in the standard parameters necessary for calibration. As shown in Fig. 9 B, the error resulting from change under- or overestimation in Rf is dependent on the relative Keff of the dye. Up to a [Ca2+]/Keff value of 2.0, a large overestimation of Rf (real value 40% less than estimate) causes an overestimation of intracellular [Ca2+] of <20%. For the dye Fura-4F, this limits the maximum measurable [Ca2+] to ∼2.3 μM, a value seldom achieved physiologically in mammalian cells. As shown in Fig. 9 C, for the commonly used dye Fura-2 this upper limit is reduced to ∼400 nM. This restricts the accurate use of this dye to measurements of resting [Ca2+] (50–400 nM). For illustrative purposes, Fig. 9 C indicates the range of fluorescence changes anticipated for Furaptra in cardiac muscle cells. The high Keff of this indicator precludes its use for accurate measurements of cytosolic [Ca2+].
Conversion of the fluorescence signal to [Ca2+] requires an estimate of the background fluorescence, i.e., systematic background (Fig. 3 C) plus cellular autofluorescence (Fig. 4 B,ii). In this study, the protocol used to load Fura-4F into intact cells using the –AM form generated a range of maximum fluorescence values ranging from 35 to 150 (mean 115) times the background level. Another source of uncertainty is the variation in background fluorescence within the cell. Both considerations result in an uncertainty over the background fluorescence, but as shown in Fig. 9 A, the calculation of intracellular [Ca2+] is relatively insensitive to errors in this quantity if the [Ca2+]/Keff value of the dye remains <2.0. This analysis suggests the best dye to cover the range of physiologically [Ca2+] is Fura-4F. Assuming the Keff of the dye is unchanged within the cytoplasm, this dye has the best dynamic range and the lowest number of errors of all the Fura dyes tested.
Viability
The ability to acquire continuous line scans over a long time period (>5 min) permits [Ca2+] calibration before and after experimental intervention. The bracketed calibration coupled with the lack of fluorophore bleaching (Fig. 7 A) allows accurate calibration of an intracellular signal (Fig. 7 C). The marked resistance to bleaching compared to similar short wavelength dyes (e.g., Indo-1) and long wavelength indicators (e.g., Fluo dyes) is most likely due to the inherent stability of Fura fluorescence (Grynkiewicz et al., 1985). These characteristics mean that the duration of the recording is limited only by the ability of the cell to tolerate continuous exposure to intense laser light and the rate of dye leakage from the cell. A previous study (Hopt and Neher, 2001) has examined phototoxicity in cultured bovine chromaffin cells, using excitation of Fura-2 with light at 840 nM and peak laser power levels ∼10 times higher than that of the present study. Extrapolating from these measurements, based on the peak intensity used (see Appendix), the toxic effects of the intense light would be anticipated after 90,000 scans. In contrast, cardiac myocytes could be scanned continuously for up to 10 min (300,000 scans) without noticeable detrimental effects. The basis for this difference between predicted and observed sensitivity is not known, but this may reflect the threshold nature of the damage process. Furthermore, freshly isolated cardiac myocytes (i.e., primary culture) may have a higher potential to resist oxidative damage (Keelan et al., 2001). Previous publications suggest that lower dose phototoxicity is an indirect two-photon excitation phenomenon, i.e., not related to local heating or UVB effects due to three-photon excitation (Koester et al., 1999). Instead phototoxic effects may result from absorption by NAD(P)H and endogenous flavonoids (Hockberger et al., 1999; Hockberger, 2002). From the endogenous 2PEF cross-section spectra provided by Huang et al. (2002), there is an effective local absorption minimum at ∼800 nM. This agrees well with the measured background autofluorescence from unlabeled myocytes (Fig. 4 B,ii). This may contribute to the long-term viability demonstrated by scanning at 810 nM in cardiac myocytes.
CONCLUSION
Excitation wavelengths of ∼810 nM have been shown to be optimal for Fura dyes in terms of indicator dynamic range and S/B for cardiac myocytes. The single-wavelength ratio method demonstrated a robust method to image optically sectioned Ca2+ dynamics in cardiomyocytes while minimizing the entire sample's exposure to UVA/violet/blue-induced effects. Long-term (5–10 min), continuous line-scan imaging of Ca2+ dynamics was possible with Fura dyes excited via two-photon excitation. The single-wavelength excitation scheme has been shown to provide accurate assessment of the [Ca2+] inside living cells (and tissues). Arrays of Keff values (182 nM, 1.15 μM, 5.18 μM, 19.2 μM, and 58.5 μM) are available to fine-tune the expected [Ca2+] to the dynamic range of the indicator. Fura-4F (1.15 μM Keff) appears to be the best indicator to study cytoplasmic [Ca2+] in cardiac myocytes.
Acknowledgments
The authors thank Dr. John Dempster (Strathclyde University) and Prof. Phil Hockberger (Northwestern University) for commenting on earlier forms of this manuscript.
The Wellcome Trust and the British Heart Foundation and Scottish Higher Education Funding Council financially supported this research. An Engineering and Physical Sciences Research Council studentship and the School of Veterinary Medicine, University of Glasgow, financially supported C.L.
APPENDIX
The equation for the acquired two-photon excitation signal in laser-scanning systems was taken from Xu and Webb (1996) and is reproduced here (Eq. 2),
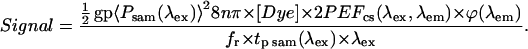 |
(2) |
The following assumptions are made to arrive at this equation:
The excitation mode is truly two-photon excitation.
The paraxial approximation is assumed to derive the illumination intensity.
There is negligible fluorophore bleaching.
There is negligible fluorophore saturation (ground-state depletion).
The slope of the Log/Log plots validates the first assumption (Fig. 8 A) and safe intensities for the last two assumptions (Fig. 8 B). The online monitors are used to monitor the relative average power and peak power from the laser system and to verify that the excitation remains in the safe region.
For ease of understanding, Eq. 2 will be separated into the three main components of any system: probe, interaction, and observation. In this particular case, the probe is potential two-photon excitation generated in the focal volume, which is represented by the first group of terms. The interaction in the focal volume inside the sample is represented by the middle group of terms ([Dye] × 2PEFcs). Here, the variable [Dye] represents the concentration of the fluorophore in the excited volume and the variable 2PEFcs(λex, λem) represents the two-photon excitation fluorescence cross section, which includes the absorption of two-photon excitation, δ2(λex), and the subsequent radiative efficiency of photon emission, η2(λem), of the fluorophore. The observation is the last term, ϕ(λem), and represents the detected incident photon flux converted into an electrical signal.
The average power at the sample, 〈Psam(λex)〉, and the pulse duration at the sample, tp sam(λex), were independently determined to help provide the excitation levels incident for the safe operation to minimize bleaching (Fig. 8) and enable long-term imaging (Fig. 7).
Equation 2 can be simplified to
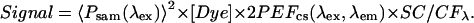 |
(3) |
In our system-scaled equation (following Albota et al., 1998), the constants represent the following terms:
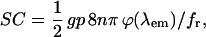 |
(4) |
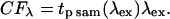 |
(5) |
The accurate determination of the sample average power is summarized below. The values of these parameters are unique for each laser-scanning microscope system.
The average power at the sample, 〈Psam(λex)〉, and the pulse duration at the sample, tp sam(λex), were independently determined to help provide the excitation levels incident for the safe operation to minimize bleaching (Fig. 8) and enable long-term imaging (Fig. 7).
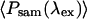 |
Measuring the average power at the sample plane using immersion fluid between the lens and the detector has the following concerns: the measurement may yield local intensities high enough to damage the detector, and silicon detectors are very sensitive to the incident photon angle. For these reasons we used a thermopile detector without immersion fluid to measure the average power at the sample via a 1.2 NA water-immersion objective. An additional scaling factor was required to account for the total internal reflection within the objective lens. The imaging described in this manuscript only required ∼10% of the maximum power, which resulted in fairly low powers at the sample plane. The system was characterized between the monitor diodes and the back aperture plane and sample plane. The scaling factor with the monitors permitted lower average powers (<5 mW) to be measured more accurately.
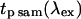 |
The laser bandwidth was measured with a spectrum analyzer (LSA 201, Rees). The laser pulse duration was measured with two-photon absorption autocorrelation using a commercial device (Carpe, APE, Berlin, Germany). A sech2 laser pulse shape was assumed to obtain the pulse durations from the autocorrelation measurements. The autocorrelator included an external signal input for measuring pulse duration differences. The external signal was also derived from two-photon absorption in a GaAsP photodiode. The pulse-duration broadening spectrum of Nikon CFi-60 objective lenses was determined using the before and after pulse duration measurements. The combination of the isolator, cell, and apochromatic objective lens yielded significantly broader pulses at the sample plane, hence the large values presented in this study.
The values relevant for the current study are given below along with additional system values:
Wavelength of illumination (λex) = 810 nM
Average power at the sample 〈Psam(λex)〉 = 2.3 mW
Pulse duration at the sample (tp sam) = 393 fs
Laser bandwidth = 9 nM
Laser pulse repetition frequency (fr) = 80 MHz
Pulse shape factor (gp) = 0.576 (Hopt and Neher, 2001)
Radius of illumination in back focal plane (r) = 4 mM
Effective lens back-aperture radius = 3 mM
Object lens transmission = 75%
Object lens numerical aperture = 1.2
Sample refractive index (n) = 1.333
Fluorophore concentration = 30 μM
Pixel dwell time = 2.6 μs/pixel
Pixel size = 0.2 μm
Cell exposure time/scan (tc) = 0.9 ms
References
- Albota, M., C. Xu, and W. W. Webb. 1998. Two-photon excitation cross-sections of bimolecular probes from 690 to 960 nM. Appl. Opt. 37:7352–7356. [DOI] [PubMed] [Google Scholar]
- Baylor, S. M., and S. Hollingworth. 2000. Measurement and interpretation of cytoplasmic [Ca2+] signals from calcium-indicator dyes. News Physiol. Sci. 15:19–26. [PubMed] [Google Scholar]
- Bliton, C., and D. E. Clapham. 1993. Optical modifications enabling simultaneous confocal imaging with dyes excited by ultra-violet and visible wavelength light. J. Microsc. 169:15–26. [Google Scholar]
- Bliton, A. C., and J. D. Leichleiter. 1995. Optical considerations at ultraviolet wavelengths in confocal microscopy. In The Handbook of Biological Confocal Microscopy, 2nd ed. J. Pawley, editor. Klewer-Plenum Press, New York. 431–443.
- Dempster, J., J. Graham, J. Sinclair, G. L. Smith, and M. V. Thomas. 2002. Demonstration of integrator unit and data acquisition software for laser-scanning microscopy. J. Physiol. 543P:66P. [Google Scholar]
- Dixon, A., G. Hogg, and N. Louahab. 2001. Design optimization of a commercial multi-photon laser scanning microscope system. Proc. SPIE. 4262:62–71. [Google Scholar]
- Grynkiewicz, G., M. Poenie, and R. Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Haugland, R. P. 2002. Handbook of Fluorescent Probes and Research Products. Molecular Probes, Eugene, OR.
- Helmchen, F., D. W. Tank, and W. Denk. 2002. Enhanced two-photon excitation through optical fiber by single-mode propagation in a large core. Appl. Opt. 41:2930–2934. [DOI] [PubMed] [Google Scholar]
- Hockberger, P. E. 2002. A history of ultraviolet photobiology for humans, animals and microorganisms. Photochem. Photobiol. 76:561–579. [DOI] [PubMed] [Google Scholar]
- Hockberger, P. E., T. A. Skimina, V. E. Centonze, C. Lavin, S. Chu, S. Dadras, J. K. Reddy, and J. G. White. 1999. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proc. Natl. Acad. Sci. USA. 96:6255–6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopt, A., and E. Neher. 2001. Highly nonlinear photodamage in two-photon fluorescence microscopy. Biophys. J. 80:2029–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S., A. A. Heikal, and W. W. Webb. 2002. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J. 82:2811–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keelan, J., N. J. Allen, D. Antcliffe, S. Pal, and M. R. Duchen. 2001. Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J. Neurosci. Res. 66:873–884. [DOI] [PubMed] [Google Scholar]
- Koester, H. J., D. Baur, R. Uhl, and S. W. Hell. 1999. Ca2+ fluorescence imaging with pico- and femtosecond two-photon excitation: signal and photodamage. Biophys. J. 77:2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi, M., S. Hollingworth, A. B. Harkins, and S. M. Baylor. 1991. Myoplasmic calcium transients in intact frog skeletal muscle fibers monitored with the fluorescent indicator Furaptra. J. Gen. Physiol. 97:271–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba, K., and S. Nakayama. 1998. Two-photon laser-scanning microscopy: tests of objective lenses and Ca2+ probes. Neurosci. Res. 32:281–294. [DOI] [PubMed] [Google Scholar]
- Loughrey, C. M., K. E. MacEachern, P. Neary, and G. L. Smith. 2002. The relationship between intracellular [Ca2+] and Ca2+ wave characteristics in permeabilised cardiomyocytes from the rabbit. J. Physiol. 543:859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maravall, M., Z. F. Mainen, B. L. Sabatini, and K. Svoboda. 2000. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys. J. 78:2655–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh, M. A., S. M. Cobbe, and G. L. Smith. 2000. Heterogeneous changes in action potential and intracellular Ca2+ in left ventricular myocyte sub-types from rabbits with heart failure. Cardiovasc. Res. 45:397–409. [DOI] [PubMed] [Google Scholar]
- Miller, D. J., and G. L. Smith. 1984. EGTA purity and the buffering of calcium ions in physiological solutions. Am. J. Physiol. 246:C160–C166. [DOI] [PubMed] [Google Scholar]
- Minta, A., J. P. Kao, and R. Y. Tsien. 1989. Fluorescent indicators for cytosolic calcium based on rhodamine and fluorescein chromophores. J. Biol. Chem. 264:8171–8178. [PubMed] [Google Scholar]
- Ogden, D., K. Khodakhah, T. Carter, M. Thomas, and T. Capiod. 1995. Analogue computation of transient changes of intracellular free Ca2+ concentration with the low affinity Ca2+ indicator Furaptra during whole-cell patch-clamp recording. Pflugers Arch. 429:587–591. [DOI] [PubMed] [Google Scholar]
- Patterson, G. H., and D. W. Piston. 2000. Photobleaching in two-photon excitation microscopy. Biophys. J. 78:2159–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranka, J. K., and A. L. Gaeta. 1997. Autocorrelation measurement of 6-fs pulses based on the two-photon-induced photocurrent in GaAsp photodiode. Optics Lett. 22:1344–1346. [DOI] [PubMed] [Google Scholar]
- Reynolds, D. 2003. A study of organelle calcium dynamics in cardiac muscle. PhD thesis, University of Glasgow, UK.
- Ricken, S., J. Leipziger, R. Greger, and R. Nitschke. 1998. Simultaneous measurements of cytosolic and mitochondrial Ca2+ transients in HT29 cells. J. Biol. Chem. 273:34961–34969. [DOI] [PubMed] [Google Scholar]
- Smith, G. L., and D. J. Miller. 1985. Potentiometric measurements of stoichiometric and apparent affinity constants of EGTA for protons and divalent ions including calcium. Biochim. Biophys. Acta. 839:287–299. [DOI] [PubMed] [Google Scholar]
- Squirrell, J. M., D. L. Wokosin, J. G. White, and B. D. Bavister. 1999. Long-term two-photon fluorescence imaging of mammalian embryos without compromising viability. Nat. Biotechnol. 17:763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann, G. E., F. M. LaFerla, and I. Parker. 2003. Ca2+ signaling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J. Neurosci. 23:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, D. A., and F. S. Fay. 1990. Intracellular calibration of the fluorescent calcium indicator Fura-2. Cell Calcium. 11:75–83. [DOI] [PubMed] [Google Scholar]
- Wokosin, D. L., V. E. Centonze, S. Crittenden, and J. White. 1996. Three-photon excitation fluorescence imaging of biological specimens using an all-solid-state laser. Bioimaging. 4:208–214. [Google Scholar]
- Xu, C., and W. W. Webb. 1996. Measurement of two-photon excitation cross-sections of molecular fluorophores with data from 690 to 1050 nM. J. Opt. Soc. Am. 13:481–491. [Google Scholar]
- Xu, C., W. W. Webb, and J. Guild. 1995. Determination of absolute two-photon excitation cross sections by in situ second order autocorrelation. Opt. Lett. 20:2372–2374. [DOI] [PubMed] [Google Scholar]
- Xu, C., R. M. Williams, W. Zipfel, and W. W. Webb. 1996a. Multiphoton excitation cross-sections of molecular fluorophores. Bioimaging. 4:198–207. [Google Scholar]
- Xu, C., W. Zipfel, J. B. Shear, R. M. Williams, and W. W. Webb. 1996b. Multiphoton fluorescence excitation: new spectral windows for biological nonlinear microscopy. Proc. Natl. Acad. Sci. USA. 93:10763–10768. [DOI] [PMC free article] [PubMed] [Google Scholar]