

Abstract
Cytochrome c oxidase mediates the final step of electron transfer reactions in the respiratory chain, catalyzing the transfer between cytochrome c and the molecular oxygen and concomitantly pumping protons across the inner mitochondrial membrane. We investigate the electron transfer reactions in cytochrome c oxidase, particularly the control of the effective electronic coupling by the nuclear thermal motion. The effective coupling is calculated using the Green's function technique with an extended Huckel level electronic Hamiltonian, combined with all-atom molecular dynamics of the protein in a native (membrane and solvent) environment. The effective coupling between CuA and heme a is found to be dominated by the pathway that starts from HisB204. The coupling between heme a and heme a3 is dominated by a through-space jump between the two heme rings rather than by covalent pathways. In the both steps, the effective electronic coupling is robust to the thermal nuclear vibrations, thereby providing fast and efficient electron transfer.
INTRODUCTION
Cytochrome c oxidase (CCO) is the terminal enzyme in the respiratory chain. The enzyme catalyzes electron transfer (ET) to the molecular oxygen (Babcock and Wikstrom, 1992; Chan and Li, 1990; Berg et al., 2002; Brzezinski et al., 1995; Winkler et al., 1995), a key bioenergetic reaction in aerobic organisms. Fig. 1 shows a schematic view of the key groups involved in ET. A copper dimer (CuA) functions as a primary donor, receiving electrons from cytochrome c molecules. The ET reaction is believed to occur in two subsequent steps: from CuA to heme a, and further to the complex of heme a3 and the molecular oxygen. A proton transfer reaction concomitant to the ET reduces the molecular oxygen to water, releasing the redox energy. This reaction creates and maintains a transmembrane pH gradient, which provides energy for adenosine triphosphate (ATP) synthesis. The produced ATP is then delivered to different locations throughout the cell, providing the necessary energy supply to a broad variety of biochemical reactions (Babcock and Wikstrom, 1992; Chan and Li, 1990; Berg et al., 2002; Brzezinski et al., 1995; Winkler et al., 1995).
FIGURE 1.

Electron transfer pathways in CCO. The relevant pathways are shown as solid black lines (where mediated by covalent bonds) and dashed black lines (where mediated by hydrogen bonds or through-space jumps). The dominant pathways are thicker than the less important ones. The relevant protein residues are shown as thinner tubes colored by atom name, the heme groups are shown as thicker tubes colored by atom name, and the metal atoms are shown as van der Waals spheres colored by atom name.
Because of extreme importance of CCO for the bioenergetic reactions in living cells, the enzyme molecular structure has been extensively studied (e.g., Iwata et al. (1995), Tsukihara et al. (1995, 1996), and Yoshikawa et al. (1998)). Although a number of ET pathways has been suggested, there still is an ongoing discussion over which pathways dominate the ET reaction (Iwata et al., 1995; Ramirez et al., 1995; Williams et al., 1997; Regan et al., 1998; Gamelin et al., 1998; Paula et al., 1999; Farver et al, 2000; Medvedev et al., 2000). In this manuscript, we elucidate the ET pathways in CCO and investigate how thermal motion of the protein atoms affects the interference among the dominant pathway tubes (Regan and Onuchic, 1999). In the conventional theories of biological ET reactions (the Marcus theory (Marcus and Sutin, 1985) and its modifications), nuclear dynamics controls the Franck-Condon factor; however, it was recently shown that nuclear dynamics may also control the effective electronic coupling (Balabin and Onuchic, 2000; Beratan et al, 1987).
METHODS
Following the approach described in Balabin and Onuchic (2000), we calculate the effective electronic couplings between the donors and the acceptors using the Green's function technique with an extended Huckel-like electronic Hamiltonian. To study effects of nuclear motion on the electronic coupling, we perform molecular dynamics (MD) simulations of the protein in a native (membrane and solvent) environment.
Molecular dynamics simulations
A CCO structure from bovine heart in the fully oxidized state (Yoshikawa et al., 1998; Protein Data Bank code 2OCC) was simulated. The protein was embedded into a rectangular patch of phosphatidylethanolamine membrane. (See http://www.ks.uiuc.edu/~ilya/Membranes for details of the membrane structure.) The lipid molecules that overlapped with the proteins were removed. The protein-lipid complex was then solvated in a rectangular volume of preequilibrated TIP3 water molecules. Sodium and chlorine ions were added, corresponding to an ionic strength of 0.025 M/L. The final system measured 116 × 85 × 133 Å in size and included 122,084 atoms.
The simulations were performed using a molecular dynamics program NAMD (Kale et al., 1999) with the all-atom Charmm27 force field (MacKerell et al., 1998), particle mesh Ewald (PME) full electrostatics, periodic boundaries, constant pressure (NpT) ensemble, and a Langevin thermostat. In all simulations, the integrator time step was 1 fs. First, with all protein backbone atoms restrained, the system underwent 1,000 conjugate gradient minimization steps followed by 200 ps of equilibration at 310 K. Then the system was equilibrated for 30 ps with no restraints. The root mean square deviation (RMSD) of the protein Cα atoms from the crystallographic conformation, along with the system volume, energies, temperature, and membrane surface area were monitored and did not show any significant changes after 30 ps. After that, the system was simulated for another 150 ps, and a trajectory frame was saved each 1 ps, providing 150 “snapshots” that described the protein dynamics. Thus only local equilibrium fluctuations are considered in this study. An equilibrium conformation was obtained by aligning and averaging positions of each atom in these “snapshots”. The RMSD between the crystallographic structure and the equilibrium conformation, which describes the overall change in the protein conformation caused by the thermal motion, is ∼1.5 Å. The RMSD between the equilibrium conformation and any of the “snapshots”, which characterizes the average magnitude of each atom's thermal vibrations around its equilibrium position, is ∼0.6 Å. To further investigate conformational changes in protein associated with the thermal motion, RMSDs for each protein residue involved in mediating ET were computed. These RMSDs vary between 1 and 3 Å, depending on the side chain size rather than on the residue location in the protein; therefore, no major conformational changes in the protein are indicated.
Electron transfer reaction rate
Because of the typically large donor to acceptor distance (10–25 Å), most biological ET reactions occur in the weak coupling limit, and their rates are described by the Fermi golden rule (Marcus and Sutin, 1985; Hopfield, 1974; Jortner, 1980; Regan and Onuchic, 1999; Gray and Winkler, 1996; Page et al., 1999; Bertini et al., 1994; Sigel and Sigel., 1991)
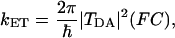 |
(1) |
where TDA is the electronic tunneling matrix element between donor and acceptor, and (FC) is the Franck-Condon density of states that describes the nuclear control of the energy gap. In the weak coupling limit, (FC) is well described by the classical Marcus formula (Marcus and Sutin, 1985):
 |
(2) |
where λ is the reorganization energy, and ΔG0 is the driving force of the reaction. In this article, we focus on qualitative effects of the protein structure and dynamics on the effective tunneling coupling, and we do not aim at obtaining quantitative estimates for the ET reaction rate. As such, we hereafter compute the maximum ET reaction rate, which occurs at the activationless limit (λ + ΔG0 ≈ 0), and the Franck-Condon factor is optimized. Although this assumption may lead to overestimating the reaction rate by up to three orders of magnitude, it does not interfere with the analysis of the effective coupling. Previous studies reported reorganization energies of about 0.3 eV for the reaction step from CuA to heme a (Regan et al., 1998; Randall et al., 2000; Brzezinski, 1996) and about 0.76 eV for the reaction step from heme a to heme a3 (Regan et al., 1998; Brzezinski, 1996; Adelroth et al., 1995). Assuming that the driving force is small, the maximum ET reaction rate is about 2 × 1016 (TDA)2 s−1, whereas the actual rate (computed with nonoptimized Franck-Condon factor) is 2 × 1015 (TDA)2 s−1 for the first step and 2 × 1013 (TDA)2 s−1 for the second step, respectively.
Effective tunneling matrix element calculations
Following the perturbation approach by Marcus (Marcus and Sutin, 1985), the effective tunneling matrix element for a given protein conformation can be calculated as follows:
 |
(3) |
where S and H are the bridge electronic orbital overlap and Hamiltonian matrices, Etun is the donor and acceptor resonant tunneling energy, indexes i and j refer to bridge (protein or cofactor matrix) orbitals, and  is the transformed Green's function of the bridge (Balabin and Onuchic, 1996)
is the transformed Green's function of the bridge (Balabin and Onuchic, 1996)
 |
(4) |
The overlap matrix was computed using a minimum basis set (four valence orbitals per heavy atom and one orbital per hydrogen atom) with the Slater type atomic orbitals. The bridge orbitals included all protein residues that participate in mediating the relevant electron transfer pathways. The relevant protein residues were identified in a way similar to the method described in Siddarth and Marcus (1990). Calculations of TDA between CuA and heme a included residues ArgA438, ArgA439, CysB196, CysB200, GluB198, HisA61, HisB161, HisB204, HisA378, IleB199, MetB207, SerB197, and TyrA440. Calculations of TDA between heme a and heme a3 included residues AlaA375, ArgA438, ArgA439, HisA61, HisA240, HisA290, HisA291, HisA376, HisA378, PheA377, TrpA126, TyrA440, and ValA374. Calculations of TDA between CuA and heme a3 included all of the above residues.
The Hamiltonian matrix was computed using a standard extended Huckel parametrization (Yates, 1978): the diagonal matrix elements were the ionization potentials of the respective atom, Hii = αi, and the off-diagonal elements were assigned as Hij = (K/2) (αi + αj) Sij, where K = 1.5 was the standard Huckel constant (Yates, 1978). The overlap and Hamiltonian matrices were converted into the bonding-antibonding orbital basis using the standard transformations (Naray-Szabo et al, 1987). Although calculations of the effective electronic coupling with the above Hamiltonian cannot provide quantitative accuracy, they have proved, in the past experience of us (Balabin and Onuchic, 1996, 2000) and others (Regan and Onuchic, 1999; Medvedev et al, 2000), to provide reasonable estimates and to capture effects of the protein structure and dynamics.
Nuclear dynamics effects on the tunneling matrix element
Thermal motion of the protein atoms influences the overlap and the Hamiltonian matrices, therefore modulating the effective electronic coupling TDA. To investigate how this modulation affects the ET reaction rate, one needs to calculate the average square effective coupling (see article (Balabin and Onuchic, 2000) and the supporting information therein):
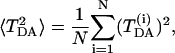 |
(5) |
where  is an effective coupling computed for the i-th protein “snapshot” conformation. The dynamical effects can be quantified by means of the following coherence parameter (Balabin and Onuchic, 2000):
is an effective coupling computed for the i-th protein “snapshot” conformation. The dynamical effects can be quantified by means of the following coherence parameter (Balabin and Onuchic, 2000):  . When the interference among the ET pathways is predominantly constructive, TDA is robust to small changes of the protein conformation, and C is close to 1. In the opposite limit, when there is a pronounced destructive interference among the pathways, TDA is sensitive to details of the protein conformation, and C is close to 0. Therefore, C quantitatively describes the sensitivity of the effective coupling to the thermal motion of the protein atoms, characterizing the regime of the interference among the ET pathways (Balabin and Onuchic, 2000). To separate effects of the overall protein conformation change (the equilibrium conformation at 310 K versus the crystallographic conformation) from those of the thermal vibrations around the equilibrium, we computed the effective couplings for the crystallographic and the equilibrium conformations (Eq. 3) as well as the dynamic coupling (Eq. 5).
. When the interference among the ET pathways is predominantly constructive, TDA is robust to small changes of the protein conformation, and C is close to 1. In the opposite limit, when there is a pronounced destructive interference among the pathways, TDA is sensitive to details of the protein conformation, and C is close to 0. Therefore, C quantitatively describes the sensitivity of the effective coupling to the thermal motion of the protein atoms, characterizing the regime of the interference among the ET pathways (Balabin and Onuchic, 2000). To separate effects of the overall protein conformation change (the equilibrium conformation at 310 K versus the crystallographic conformation) from those of the thermal vibrations around the equilibrium, we computed the effective couplings for the crystallographic and the equilibrium conformations (Eq. 3) as well as the dynamic coupling (Eq. 5).
Ligand-metal bonds
ET from CuA is mediated by the copper ligands, which form metal bonds with the copper atoms. The coupling provided by these bonds (HDi in Eq. 3) is usually substantially weaker than the one provided by typical covalent bonds. In our calculations, these couplings are estimated using the metal bond covalencies (Gamelin et al., 1998; Williams et al., 1997; Solomon et al., 2000; Randall et al., 2000):
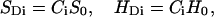 |
(6) |
where S0 and H0 are the average values for the nearest neighbor orbital overlaps and couplings (Balabin and Onuchic, 1998), and Ci are the respective covalencies. Following the recent experimental data and ab initio calculation results (Gamelin et al., 1998; Williams et al., 1997; Solomon et al., 2000; Randall et al., 2000; George et al., 2001), we used the covalency values of 0.23 for the cysteins, 0.04 for the the histidine, and 0.01 for the glutamine.
Pathways model analysis
The Pathways model (Beratan et al., 1991; Betts et al., 1992; Curry et al., 1995) has provided a simple but efficient way to estimate the effective electronic coupling in proteins. It assumes that the effective coupling between the donor and the acceptor is proportional to a product of all partial decays in the electronic path (Beratan et al., 1991; Betts et al., 1992):
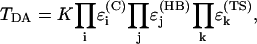 |
(7) |
where 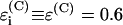 is a decay per covalent bond mediated step,
is a decay per covalent bond mediated step, 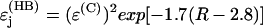 is a decay per hydrogen bond mediated step (R is the distance between the heavy atoms, Å), and
is a decay per hydrogen bond mediated step (R is the distance between the heavy atoms, Å), and 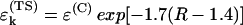 is the decay per through-space jump (R is the jump distance, Å). Consistently with earlier estimates (Regan et al., 1999; Priyadarshy et al., 1998; Curry et al., 1995; Page et al., 1999), we assume that the maximum rate is ∼1013 s−1 at the donor to acceptor separation of 3 Å (two covalent bond steps), leading to the following approximation that relates the pathway structure with the maximum (not adjusted for the Franck-Condon factor) ET reaction rate:
is the decay per through-space jump (R is the jump distance, Å). Consistently with earlier estimates (Regan et al., 1999; Priyadarshy et al., 1998; Curry et al., 1995; Page et al., 1999), we assume that the maximum rate is ∼1013 s−1 at the donor to acceptor separation of 3 Å (two covalent bond steps), leading to the following approximation that relates the pathway structure with the maximum (not adjusted for the Franck-Condon factor) ET reaction rate:
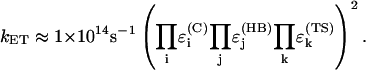 |
(8) |
The Pathways model estimates were additionally verified by computing the extended coupling at the extended Huckel level for isolated individual pathways, one pathway at a time. Only atoms that mediate the pathway were included in the calculations. Effects of protein structural groups on the effective coupling were investigated by computing the effective coupling for the core path alone (no side groups), the core path with its nearest neighbor (NN) atoms (chemically bound to the core path atoms), and the core path with the NN and next to NN (chemically bound to the NN atoms) atoms, as well as with the full pathway structure (Balabin and Onuchic, 1998).
RESULTS AND DISCUSSION
Electron transfer from CuA to heme a
The effective coupling between CuA and heme a and the corresponding maximum ET rate computed at the extended Huckel level are shown on Fig. 2. Because the effective coupling depends on the electron tunneling energy, the calculations were performed for a range of tunneling energies corresponding to the energy gap between σ and π orbitals (from −11 to −9 eV) (Balabin and Onuchic, 2000). It has been shown that the accuracy of extended Huckel level calculations of TDA can be improved by adjusting the tunneling energy based on independent spectral data (Siddarth and Marcus, 1990). In this article, however, we focus on the sensitivity of TDA to the tunneling energy and perform the calculations for a relevant tunneling energy range and not for a single, even though adjusted, tunneling energy.
FIGURE 2.

Electron transfer from CuA to heme a. The tunneling energy range shown corresponds to the main energy gap. The typical donor and acceptor energies are a few eV above the HOMO (∼−9 eV). (Top panel) The coherence parameter is high in the entire tunneling energy range. (Bottom panel) The effective donor to acceptor electronic coupling and the corresponding maximum reaction rate for the crystallographic conformation (dotted line), equilibrium conformation (dashed line), and dynamic (solid line).
The relevant donor and acceptor energies are close to the lowest π orbital level (about −9 eV) (Balabin and Onuchic, 2000). At these energies, the dynamical coupling (see Eq. 5) is close to the one for the equilibrium conformation (Eq. 3), and neither one significantly depends on Etun. The effective coupling for the crystallographic conformation is smaller than either of the above couplings by at least an order of magnitude, and it does not significantly depend on Etun either. The coherence parameter is considerably large at all tunneling energies, indicating that the interference among the dominant pathways is constructive, and the effective coupling is robust to the thermal motion of the protein atoms. The major effect of the protein dynamics is shortening of some through-space jump distances, which increases the effective coupling (Balabin and Onuchic, 2000). The dynamical coupling translates into the maximum ET rate of ∼6 × 104 s−1, which is in a close agreement with the experimentally observed rate of ∼2 × 104 s−1 (Farver et al., 2000; Brzezinski, 1996; Adelroth et al., 1995). Notably, the much smaller maximum ET rate for the crystallographic structure illustrates how calculations based only on the crystallographic conformation can be grossly misleading (Balabin and Onuchic, 2000).
Protein groups between CuA and heme a mediates several possible pathways from CuA to heme a, which start on different copper ligands (Fig. 1). Two pathways that start on HisB204 and CysB196, respectively, have been proposed before (Ramirez et al., 1995; Williams et al., 1997). Using the Pathways analysis, we identified two other potentially relevant pathways that start on CysB200 and GluB198, respectively. (Independently of our work, the CysB200 pathway was suggested in Medvedev et al. (2000)). The effective coupling mediated by each of the above pathways and the corresponding maximum rate computed for the crystallographic and the equilibrium conformations using the Pathways model are presented in Table 1.
TABLE 1.
Pathways model estimates for the electron transfer reaction from CuA to heme a in the crystallographic and equilibrium conformations
| Ligand | No. of steps | kET, s−1 (cryst.) | kET, s−1 (equil.) |
|---|---|---|---|
| CysB196 | 24 + 2 + 0 | 3 × 102 | 2 × 102 |
| GluB198 | 16 + 1 + 1 | 1 × 105 | 5 × 101 |
| CysB200 | 18 + 1 + 1 | 2 × 104 | 7 × 10° |
| HisB204 | 14 + 2 + 0 | 9 × 105 | 5 × 106 |
The second column shows the numbers of covalent steps, hydrogen bond steps, and through-space jumps in each pathway. The steps are counted from CuA to Fe of heme a.
To further investigate the interference regime among these pathways, extended Huckel level calculations of the effective coupling were performed for each isolated pathway. In the crystallographic conformation, the pathways that start on GluB198 and HisB204 provide effective couplings with same phase and similar magnitude, thereby together dominating the ET reaction. In the equilibrium conformation, the length of the hydrogen bond between HisB204 and ArgA438 shortens by ∼0.5 Å, increasing the maximum ET rate via the pathway that starts on HisB204 by almost an order of magnitude. Conversely, the distance of the through-space jump between IleB199 and ArgA438 increases in the equilibrium conformation by >2 Å, substantially decreasing the contribution of the pathway that starts on GluB198 to the effective coupling. The contributions of the other two pathways remain small.
Electron transfer from heme a to heme a3
Fig. 3 shows the effective coupling between heme a and heme a3 calculated at the extended Huckel level. At all energies, the dynamic effective coupling is close to that for the equilibrium conformation, neither one significantly depending on Etun. The effective coupling for the crystallographic conformation approaches the above couplings at the most relevant tunneling energies close to −9 eV. Thus, the pathway interference regime is constructive, and the effective coupling is robust to the thermal motions and the tunneling energy.
FIGURE 3.

Electron transfer from heme a to heme a3. The tunneling energy range shown corresponds to the main energy gap. The typical donor and acceptor energies are a few eV above the HOMO (∼−9 eV). (Top panel) The coherence parameter is high in the entire tunneling energy range. (Bottom panel) The effective donor to acceptor electronic coupling and the corresponding maximum reaction rate for the crystallographic conformation (dotted line), equilibrium conformation (dashed line), and dynamic (solid line). Thermal motion has little effect on the effective coupling and the reaction rate. Both panels indicate that the electron transfer is dominated by a few pathway tubes that interfere constructively.
The computed maximum ET reaction rate is ∼3 × 108 s−1, substantially exceeding the experimentally observed rate of ∼3 × 105 s−1 (Brzezinski, 1996; Adelroth et al., 1995; Verkhovsky et al., 1992). This difference may be attributed to a nonoptimized Franck-Condon factor (Eq. 2) (Regan et al., 1998). Recently, Verkhovsky and others (Verkhovsky et al, 2001) suggested that ET from heme a to heme a3 includes two kinetically different components: the ultrafast (∼108 s−1) transfer, which is energetically optimized, and the regular transfer, which is controlled by either heme-protein relaxation or CO dissociation from CuB. Although this conclusion remains controversial (Namslauer et al., 2002), our calculations indirectly support the possibility of the ultrafast ET component.
Protein groups surrounding heme a and heme a3 mediate a variety of different pathways (Fig. 1). Two pathways that have been suggested before include the fully covalent pathway, which is mediated by HisA378, PheA377, and HisA376 (Regan et al., 1998), and the direct pathway that includes a through-space jump between the δ propionates of the two hemes (Medvedev et al., 2000). Using the Pathways analysis, we identified two other relevant pathways: one mediated by ArgA438, which includes two hydrogen bonds, and another mediated by HisA378, AlaA375, and HisA376, which includes one hydrogen bond. The numbers of steps and the corresponding maximum rates for these pathways are shown in Table 2. In the crystallographic structure, the covalent pathway and the direct pathway interfere constructively, dominating the effective coupling. Thermal motion shortens the distance of the through-space jump (CMD-HMD[123] groups in either heme) to ∼2 Å (Fig. 4) (and possibly less, on longer timescales), increasing the corresponding ET reaction rate by an order of magnitude and making the direct pathway dominant (Fig. 3). Recently, it was proposed (Zheng et al., 2003) that the ET from heme a to heme a3 is facilitated by water molecules inside the protein. Although inner water is certainly a possibility in CCO, its influence on the ET between the two hemes appears unlikely. As shown in Fig. 5, the protein groups surrounding the dominant direct pathway are strongly hydrophobic; in addition, distances between the two hemes do not leave enough space for water molecules.
TABLE 2.
Pathways model estimates for the electron transfer reaction from heme a to heme a3 in the crystallographic and equilibrium conformations.
| Pathway | Steps | kET, s−1 (cryst.) | kET, s−1 (equil.) |
|---|---|---|---|
| Covalent | 16 + 0 + 0 | 8 × 106 | 8 × 106 |
| Direct | 10 + 0 + 1 | 7 × 106 | 7 × 107 |
| Via AlaA375 | 15 + 1 + 0 | 1 × 106 | 3 × 105 |
| Via ArgA438 | 16 + 2 + 0 | 2 × 105 | 2 × 106 |
The second column shows the numbers of covalent steps, hydrogen bond steps, and through-space jumps in each pathway. The steps are counted from Fe of heme a to Fe of heme a3.
FIGURE 4.

Time dependence of the through-space jump distance for the dominant pathway from heme a to a3. The distance is measured between the two nearest hydrogen atoms on either heme. Because of the protein thermal vibrations, the distance decreases to 2.0 Å (and possibly smaller on longer timescales) in comparison with the 3.0 Å distance for the crystallographic structure. This decrease makes the direct pathway dominant, providing a robust electron transfer regime. In addition to the hydrophobic protein environment (Fig. 5), little available space is likely to prevent penetration of water molecules in the pathway environment.
FIGURE 5.

Protein environment of the heme rings a and a3. For clarity, heme group tails are not shown. The dominant electron transfer pathway from heme a to a3 (dotted line) is mediated by CMD-HMD[123] groups, which are located in a strongly hydrophobic environment. Polar and charged residues are located close to negatively charged CGD-O[12] D groups, which are not involved in the dominant pathways.
Supplementing the Pathways analysis, extended Huckel calculations of the effective coupling for each of the pathways were performed. The results are in a close agreement with the calculations for the full protein structure as well as the Pathways estimates.
Direct or two-step reaction?
A possibility of the direct ET reaction from CuA to a3 has been brought on by Tsukihara and others (Tsukihara et al., 1995). The reaction could be mediated by four pathways, which start on the copper ligands CysB196, GluB198, CysB200, and HisB204. The effective coupling and the corresponding maximum rate via any of these pathways were estimated at the Pathways level. The ligand-metal bonds were treated as covalent bonds, and the reorganization energy was set to 0.8 eV. For both the crystallographic and equilibrium conformations, the maximum rate for the direct reaction turned out to be >2 orders of magnitude smaller than the rate of the two-step reaction. Even accounting for possible inner water molecules, which could facilitate the effective coupling for the direct ET reaction, the latter does not appear to be relevant.
CONCLUSIONS
The above analysis shows that the ET reactions in CCO occur via distinct ET pathways. The protein and cofactor structure that mediates the pathways is optimized for providing fast, efficient, and robust ET reaction. Thermal motion of the protein and cofactor atoms are a key factor in further optimizing the ET reactions, which is critical for the protein physiological function. Consistently with available experimental data, the ET reaction occurs in two sequential steps rather than directly. ET from CuA to heme a is found to be dominated by the pathway starting at HisB204, and ET from heme a to heme a3 is dominated by a through-space jump between the CMD-HMD[123] groups. The extended Huckel level calculations are in close agreement with both experimental data and the Pathways level estimates. Our calculations support the possibility of the ultrafast ET component between the two hemes recently reported by Verkhovsky and others (Verkhovsky et al., 2001).
Acknowledgments
I.A.B. acknowledges Klaus Schulten, and M.L.T. acknowledges Toshiko Ichiye for providing support at the final phase of the work. I.A.B. thanks Justin Gullingsrud for helpful discussions on MD simulation techniques. We thank Pittsburgh Supercomputer Center for providing supercomputer time for the MD simulations.
This work was supported by the National Science Foundation (grant MCB-0084797 with additional support from PHY-0216576, 0225630, and 0107594). Any opinions, findings, and conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Ming-Liang Tan's present address is School of Molecular Biosciences, Washington State University, Pullman, WA 99164-4660.
Ilya Balabin's present address is P. M. Gross Chemical Laboratory, Duke University, Box 90349, Durham, NC 27708.
References
- Adelroth, P., P. Brzezinski, and B. G. Malmstrom. 1995. Internal electron transfer in cytochrome c oxidase from rhodobacter sphaeroides. Biochemistry. 34:2844–2849. [DOI] [PubMed] [Google Scholar]
- Babcock, G. T., and M. Wikstrom. 1992. Oxygen activation and the conservation of energy in cell respiration. Nature. 356:301–309. [DOI] [PubMed] [Google Scholar]
- Balabin, I. A., and J. N. Onuchic. 1996. Connection between simple models and quantum chemical models for electron-transfer tunneling matrix element calculations: a Dyson's equations-based approach. J. Phys. Chem. 100:11573–11580. [Google Scholar]
- Balabin, I. A., and J. N. Onuchic. 1998. A new framework for electron-transfer calculations: beyond the Pathways-like models. J. Phys. Chem. 102:7497–7505. [Google Scholar]
- Balabin, I. A., and J. N. Onuchic. 2000. Dynamically controlled protein tunneling paths in photosynthetic reaction centers. Science. 290:114–117. [DOI] [PubMed] [Google Scholar]
- Beratan, D. N., J. N. Onuchic, and J. J. Hopfield. 1987. Electron tunneling through covalent and noncovalent pathways in proteins. J. Chem. Phys. 86:4488–4498. [Google Scholar]
- Beratan, D. N., J. N. Betts, and J. N. Onuchic. 1991. Protein electron transfer rates set by the bridging secondary and tertiary structure. Science. 252:1285–1288. [DOI] [PubMed] [Google Scholar]
- Berg, J. M., J. L. Tymoczko, and L. Stryer. 2002. Biochemistry. W. H. Freeman, New York, NY.
- Bertini, I., H. B. Gray, S. Lippard, and J. S. Valentine. 1994. Bioinorganic Chemistry. University Science Books, Mill Valley, CA .
- Betts, J. N., D. N. Beratan, and J. N. Onuchic. 1992. Mapping electron tunneling pathways: an algorithm that finds the minimum length maximum coupling pathway between electron donors and acceptors in proteins. J. Am. Chem. Soc. 114:4043–4046. [Google Scholar]
- Brzezinski, P., M. Sundahl, P. Adelroth, M. T. Wilson,, B. el-Agez, P. Wittung, and B. G. Malmstrom. 1995. Triplet-state quenchng in complexes between Zn-cytochrome-C and cytochrome-oxidase or its Cu-A domain. Biophys. Chem. 54:191–197. [DOI] [PubMed] [Google Scholar]
- Brzezinski, P. 1996. Internal electron-transfer reactions in cytochrome c oxidase. Biochemistry. 35:5611–5615. [DOI] [PubMed] [Google Scholar]
- Chan, S. I., and P. M. Li. 1990. Cytochrome c oxidase: understanding nature's design of a proton pump. Biochemistry. 29:1–12. [DOI] [PubMed] [Google Scholar]
- Curry, W. B., M. D. Grabe, I. V. Kurnikov, S. S. Skourtis, D. N. Beratan, J. J. Regan, A. J. A. Aquino, P. Beroza, and J. N. Onuchic. 1995. Pathways, pathway tubes, pathway docking, and propagators in electron - transfer proteins. J. Bioenerg. Biomembr. 27:285–293. [DOI] [PubMed] [Google Scholar]
- Farver, O., O. Einarsdottir, and I. Pecht. 2000. Electron transfer rates and equilibrium within cytochrome c oxidase. Eur. J. Biochem. 267:950–954. [DOI] [PubMed] [Google Scholar]
- Gamelin, D. R., D. W. Randall, M. T. Hay, R. P. Houser, T. C. Mulder, G. W. Canters, S. de Vries, W. B. Tolman, Y. Lu, and E. I. Solomon. 1998. Spectroscopy of mixed-valence Cu-A-type centers: ligand-field control of ground-state properties related to electron transfer. J. Am. Chem. Soc. 120:5246–5263. [Google Scholar]
- George, S. D., M. Metz, R. K. Szilagyi, H. X. Wang, S. P. Cramer, Y. Lu, W. B. Tolman, B. Hedman, K. O. Hodgson, and E. I. Solomon. 2001. A quantitative description of the ground-state wave function of Cu-A by x-ray absorption spectroscopy: comparison to plastocyanin and relevance to electron transfer. J. Am. Chem. Soc. 123:5757–5767. [DOI] [PubMed] [Google Scholar]
- Gray, H. B., and J. R. Winkler. 1996. Electron transfer in proteins. Annu. Rev. Biochem. 65:537–561. [DOI] [PubMed] [Google Scholar]
- Hopfield, J. J. 1974. Electron transfer between biological molecules by thermally activated tunneling. Proc. Natl. Acad. Sci. USA. 71:3640–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata, S., C. Ostermeier, B. Ludwig, and H. Michel. 1995. Structure at 2.8-angstrom resolution of cytochrome c oxidase from paracoccus denitrificans. Nature. 376:660–669. [DOI] [PubMed] [Google Scholar]
- Jortner, J. 1980. Dynamics of electron transfer in bacterial photosynthesis. Biochim. Biophys. Acta. 594:193–230. [DOI] [PubMed] [Google Scholar]
- Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- MacKerell, A. D., Jr., D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Marcus, R. A., and N. Sutin. 1985. Electron transfers in chemistry and biology. Biochim. Biophys. Acta. 811:265–322. [Google Scholar]
- Medvedev, D. M., I. Daizadeh, and A. A. Stuchebrukhov. 2000. Electron transfer tunneling pathways in bovine heart cytochrome c oxidase. J. Am. Chem. Soc. 122:6571–6582. [Google Scholar]
- Namslauer, A., M. Branden, and P. Brzezinski. 2002. The rate of internal heme-heme electron transfer in cytochrome c oxidase. Biochemistry. 41:10369–10374. [DOI] [PubMed] [Google Scholar]
- Naray-Szabo, G., P. R. Surjan, and J. Angyan. 1987. Applied Quantum Chemistry. Dordrecht, Boston, MA.
- Page, C. C., C. C. Moser, X. Chen, and P. L. Dutton. 1999. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 402:47–52. [DOI] [PubMed] [Google Scholar]
- Paula, S., A. Sucheta, I. Szundi, and O. Einarsdottir. 1999. Proton and electron transfer during the reduction of molecular oxygen by fully reduced cytochrome c oxidase: a flow-flash investigation using optical multichannel detection. Biochemistry. 38:3025–3033. [DOI] [PubMed] [Google Scholar]
- Priyadarshy, S., S. M. Risser, and D. N. Beratan. 1998. DNA-mediated electron transfer. JBIC. 3:196–200. [Google Scholar]
- Priyadarshy, S., S. S. Skourtis, S. M. Risser, and D. N. Beratan. 1996. Bridge-mediated electronic interactions: differences between Hamiltonian and Green function partitioning in a non-orthogonal basis. J. Chem. Phys. 104:9473–9481. [Google Scholar]
- Ramirez, B. E., B. G. Malmstrom, J. R. Winkler, and H. B. Gray. 1995. The currents of life: the terminal electron-transfer complex of respiration. Proc. Natl. Acad. Sci. USA. 92:11949–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall, D. W., D. R. Gamelin, L. B. LaCroix, and E. I. Solomon. 2000. Electronic structure contributions to electron transfer in blue Cu and Cu-A. JBIC. 5:16–29. [DOI] [PubMed] [Google Scholar]
- Regan, J. J., B. E. Ramirez, J. R. Winkler, H. B. Gray, and B. G. Malmstrom. 1998. Pathways for electron tunneling in cytochrome c oxidase. J. Bioenerg. Biomembr. 30:35–39. [DOI] [PubMed] [Google Scholar]
- Regan, J. J., and J. N. Onuchic. 1999. Electron-transfer tubes. Adv. Chem. Phys. 107:497–553. [Google Scholar]
- Siddarth, P., and R. A. Marcus. 1990. Electron-transfer reactions in proteins: a calculation of electronic coupling. J. Phys. Chem. 94:8430–8434. [Google Scholar]
- Sigel, H., and A. Sigel. 1991. Electron Transfer Reactions in Metalloproteins. H. Sigel and A. Sigel, editors. Marcel Dekker, New York, NY.
- Solomon, E. I., D. W. Randall, and T. Glaser. 2000. Electronic structures of active sites in electron transfer metalloproteins: contributions to reactivity. Coord. Chem. Rev. 200:595–632. [Google Scholar]
- Tsukihara, T., H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, and S. Yoshikawa. 1995. Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 angstrom. Science. 269:1069–1074. [DOI] [PubMed] [Google Scholar]
- Tsukihara, T., H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono, and S. Yoshikawa. 1996. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 angstrom. Science. 272:1136–1144. [DOI] [PubMed] [Google Scholar]
- Verkhovsky, M. I., J. E. Morgan, and M. Wikstrom. 1992. Intramolecular electron transfer in cytochrome c oxidase: a cascade of equilibria. Biochemistry. 31:11860–11863. [DOI] [PubMed] [Google Scholar]
- Verkhovsky, M. I., A. Jasaitis, and M. Wikstrom. 2001. Ultrafast haem-haem electron transfer in cytochrome c oxidase. Biochim. Biophys. Acta. 1506:143–146. [DOI] [PubMed] [Google Scholar]
- Williams, K. R., D. R. Gamelin, L. B. LaCroix, R. P. Houser, W. B. Tolman, T. C. Mulder, S. de Vries, B. Hedman, K. O. Hodgson, and E. I. Solomon. 1997. Influence of copper-sulfur covalency and copper-copper bonding on valence delocalization and electron transfer in the Cu-A site of cytochrome c oxidase. J. Am. Chem. Soc. 119:613–614. [Google Scholar]
- Winkler, J. R., B. G. Malmstrom, and H. B. Gray. 1995. Rapid electron injection into multisite metalloproteins: intramolecular electron-transfer in cytochrome-oxidase. Biophys. Chem. 54:199–209. [DOI] [PubMed] [Google Scholar]
- Yates, K. 1978. Huckel Molecular Orbital Theory. Academic Press, New York, NY.
- Yoshikawa, S., K. Shinzawa-Itoh, R. Nakashima, R. Yaono, E. Yamashita, N. Inoue, M. Yao, M. J. Fei, C. P. Libeu, T. Mizushima, H. Yamaguchi, T. Tomizaki, and T. Tsukihara. 1998. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 280:1723–1729. [DOI] [PubMed] [Google Scholar]
- Zheng, X. H., D. M. Medvedev, J. Swanson, and A. A. Stuchebrukhov. 2003. Computer simulation of water in cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 1557:99–107. [DOI] [PubMed] [Google Scholar]