

Abstract
Transcriptional regulation is a fundamental mechanism of living cells, which allows them to determine their actions and properties, by selectively choosing which proteins to express and by dynamically controlling the amounts of those proteins. In this article, we revisit the problem of mathematically modeling transcriptional regulation. First, we adopt a biologically motivated continuous model for gene transcription and mRNA translation, based on first-order rate equations, coupled with a set of nonlinear equations that model cis-regulation. Then, we view the processes of transcription and translation as being discrete, which, together with the need to use computational techniques for large-scale analysis and simulation, motivates us to model transcriptional regulation by means of a nonlinear discrete dynamical system. Classical arguments from chemical kinetics allow us to specify the nonlinearities underlying cis-regulation and to include both activators and repressors as well as the notion of regulatory modules in our formulation. We show that the steady-state behavior of the proposed discrete dynamical system is identical to that of the continuous model. We discuss several aspects of our model, related to homeostatic and epigenetic regulation as well as to Boolean networks, and elaborate on their significance. Simulations of transcriptional regulation of a hypothetical metabolic pathway illustrate several properties of our model, and demonstrate that a nonlinear discrete dynamical system may be effectively used to model transcriptional regulation in a biologically relevant way.
INTRODUCTION
An emerging theme in modern biology is the development of accurate experimental techniques for monitoring cellular behavior (e.g., see Schena et al., 1996; Brown and Botstein, 1999; Turner and Varshavsky, 2000; Zhu et al., 2001; Baldi and Hatfield, 2002). Although current techniques are mostly used to identify molecular markers for certain types of disease (e.g., cancer; see Golub et al., 1999; Bittner et al., 2000; Kobayashi et al., 2003b), it is the monitoring and modeling of cellular behavior that could mostly benefit from them.
An important cellular process under investigation is transcriptional regulation. Understanding the biological mechanisms underlying transcriptional regulation may lead to significant advances in cell biology, drug development, and medicine. It is becoming increasingly clear that, to enrich our knowledge about transcriptional regulation and understand the role it plays in cellular function, we need to construct a sufficiently predictive mathematical model for such a process, derived from basic biological principles. Moreover, experimental and computational techniques should be developed to estimate the underlying structure of the model and its parameters. Model simplicity, via reasonable biological assumptions and approximations, is important, due to limited biological knowledge of the mechanisms underlying transcriptional regulation, and difficulties of current technologies in measuring underlying parameters. If the model is sufficiently predictive, we may use it as a computational tool (even in the absence of exact parameter values) to simulate biological scenarios (e.g., steady-state analysis, mutation effects, knock-out studies, perturbation effects, homeostatic and epigenetic regulation, etc.), and generate hypotheses pertaining to the mechanisms underlying transcriptional regulation and control. This plan seems to be easier, faster, and cheaper to implement in silice (i.e., on a digital computer by simulation) than in vivo or in vitro.
There have been considerable efforts to build models for transcriptional regulation (e.g., see Thomas and D'Ari, 1990; Kauffman, 1993; Smolen et al., 2000; Gibson and Mjolsness, 2001; Hasty et al., 2001a; Savageau, 2001; de Jong, 2002; Shmulevich et al., 2002; for reviews of such models and several references). Most models can be categorized as being “qualitative” or “quantitative.” The former models emphasize structural information sharing among genes and lack detailed quantitative description of transcriptional regulation. The later models focus on a quantitative description of transcriptional regulation and are often more biologically oriented than qualitative models. The Boolean network (Kauffman, 1993) is a good example of a qualitative model, whereas, transcriptional regulation models based on ordinary differential equations (ODEs) (Chen et al., 1999) are typical examples of quantitative models.
Typically, a qualitative model (like a Boolean network) is a “coarse” approximation of transcriptional regulation. It may provide some insights into the underlying mechanisms of transcriptional regulation, but it may also lead to biologically erroneous conclusions (e.g., see Hatzimanikatis and Lee, 1999). However, qualitative models may be used to predict steady-state behavior of transcriptional regulation. This is a useful property, because cells are often observed at steady state.
Cells may often transition to different states, due to environmental perturbations or genetic instability, which may result in differentiation during development, irreversible adjustments, or disease. Therefore, it is important to design transcriptional regulation models that sufficiently predict transient as well as steady-state behavior. It is believed that ODE-based models can accomplish this goal (e.g., see Hammond, 1993; Elowitz and Leibler, 2000; Gardner et al., 2000; Yildirim and Mackey, 2003). Models based on ODEs are considered to be more detailed than qualitative models, but require structural knowledge of the transcriptional machinery and of several biological parameters (e.g., identification of promoters, regulatory regions, transcription factors, mRNA decay rates, etc.). This knowledge is not currently available for most organisms, and it is thought to be the main disadvantage of ODE-based models. However, several current efforts are geared toward determining the structure of the transcriptional machinery and estimating its parameters (e.g., see Hammond, 1993; Endy et al., 1997; Arkin et al., 1998; Tavazoie et al., 1999; Akutsu et al., 2000b; Gardner et al., 2000; Turner and Varshavsky, 2000; Lee et al., 2002; Ronen et al., 2002; Wang et al., 2002). For these reasons, ODE-based models are becoming increasingly attractive as models for transcriptional regulation.
An attractive feature of a Boolean network is that it dynamically relates the state of transcriptional regulation at time t to its state at time t − Δt, for some Δt > 0. The state of transcriptional regulation is summarized by binary-valued variables, which are dynamically related from t − Δt to t by means of Boolean functions. In this formulation, the analysis and simulation of transcriptional regulation employs theoretical and computational tools from discrete dynamical systems theory (e.g., see Sandefur, 1993), specialized to the Boolean case.
On the other hand, ODE-based models represent transcriptional regulation by a (usually large) system of nonlinear ODEs. According to this formulation, the state of the system is summarized by real-valued variables, with regulatory interactions taking the form of differential and nonlinear functional relationships. Due to the size and nonlinear structure of the system, it is not in general possible to develop mathematical techniques for its analysis. In this case, analysis is done by means of numerical techniques and computer simulations. In particular, the system may be solved by a numerical technique, like a Runge-Kutta or a predictor-corrector method (e.g., see Meir et al., 2002). Although these methods lead to general analysis and simulation techniques for transcriptional regulation, they may not be efficient, and direct biological interpretation of the various terms in the resulting equations may not be possible.
As noted in Meir et al. (2002), instead of using general techniques, it may be more preferable to derive a numerical approach to transcriptional regulation by exploiting the specific nature of the problem at hand. In this article, we investigate the possibility of doing so, by replacing an ODE-based model for transcriptional regulation with a nonlinear discrete dynamical system that is “biologically transparent,” in the sense that the resulting equations preserve the biological relevance and structure of the original model. This allows us to construct a biologically relevant quantitative model for transcriptional regulation that, like the Boolean network, enjoys attractive dynamical properties and is amenable to efficient simulation and analysis.
The system proposed in this article is directly obtained from a well-known model of transcriptional regulation based on ODEs. The ODE-based model is derived for a large population of cells by applying simple arguments of chemical kinetics on the processes of transcription and translation. It is required that the cell population is large, because the derivation of the ODE-based model relies on the Boltzmann distribution of statistical mechanics, which specifies how energy is distributed in a large population of identical molecules at statistical equilibrium. Because the ODE-based model is central to our work, we show in the next section how this model is derived from first principles. The purpose of our discussion is to clarify the limitations of modeling transcriptional regulation by means of ODEs, and to establish terminology and notation.
In the third section, we show how to model transcriptional regulation by means of a discrete dynamical system. We view the processes of transcription and translation as being discrete, and replace the actual transcriptional machinery with one for which the speeds of transcription and translation, as well as the delays in cis-regulation, are constant and equal to their mean values. We refer to this as an “average” transcriptional machinery. Therefore, the discrete dynamical system derived in this section models an “average” behavior of transcriptional regulation. The system is obtained by discretizing the ODE-based model discussed in the previous section. The discretization step is taken to be the time δt that it takes the RNA polymerase II to read one nucleotide. Moreover, we assume that, for each t = δt, 2δt, …, both the fraction of DNA templates committed to the transcription of a given gene and the mRNA concentration associated with that gene, remain constant within the time interval [t − δt, t). The resulting dynamical system is referred to as a discrete transcriptional regulatory system. It is specified by means of parameters that characterize transcription, translation, and degradation, by functionals that characterize cis-regulation, and by time delays.
In the fourth section, we discuss the steady-state behavior of the discrete model under consideration. Our discussion is motivated by the fact that the steady-state behavior of a model for transcriptional regulation may be used to characterize the cell's phenotype, and focuses on three results. The first result shows that, at steady state, the mRNA concentration vector of the discrete model “decouples” from the steady-state protein concentration vector, in the sense that one vector can be derived as a solution of a system of (nonlinear in general) equations without knowledge of the other vector. The second result shows that there is a one-to-one correspondence between the steady-state mRNA concentration vector and the steady-state protein concentration vector. This suggests that, at steady state, mRNA expression data may be used to characterize protein activity, provided that a sufficiently good estimate of the steady-state mRNA concentration vector can be inferred from such data (this also requires that the model parameters associated with translation are known). The final result shows that the discrete model has the same steady-state behavior as the associated ODE-based model. This result, together with several computational advantages underlying the discrete model, indicates that it may be more preferable to use the proposed discrete dynamical system as a model for transcriptional regulation, than the original ODE-based model.
In the next section, and by using classical arguments from chemical kinetics, we specify the nonlinearities underlying cis-regulation and include both activators and repressors as well as the notion of regulatory modules in our formulation. The derivation is based on the assumption that regulatory proteins are free to bind at several distinct sites in a promoter's regulatory region, and on the assumption that different proteins do not interact with each other or affect each other's binding affinity. Moreover, the inclusion of repressor proteins in the formulation focuses on a specific repression mechanism by which, when a repressor protein binds on a DNA template, it either blocks the recruitment of the transcription initiation complex on the promoter or prevents the release of RNA polymerase II. Finally, we show how to model cis-regulation organized in a modular fashion. According to this organization, transcriptional activity of a given gene may be controlled by a set of distinct modules, with each module asserting its own transcriptional control, independently of other modules.
In the sixth section, we discuss several properties of the proposed discrete model, related to homeostatic and epigenetic regulation as well as to Boolean networks, and elaborate on their significance. In particular, the structure of the discrete model under consideration predicts a specific response of transcriptional regulation to changes in the cellular environment, and suggests that mRNA and protein degradation, together with the rates of mRNA and protein synthesis, may play an important role in homeostatic regulation. We show that the functional form of cis-regulation is scale-invariant. This property implies that an increase (decrease) in the rates of translation, accompanied by a proportional decrease (increase) in the affinity constants underlying the binding of proteins on a promoter's regulatory region, does not change the steady-state mRNA concentration but proportionally increases (decreases) the steady-state protein concentration. It also implies that an increase (decrease) in the rates of transcription, accompanied by a proportional decrease (increase) in the affinity constants, proportionally increases (decreases) both the steady-state mRNA and protein concentrations. These properties suggest that the rates of transcription and translation, together with the affinity constants, may play an important role in epigenetic regulation. We also discuss the problem of specifying the underlying parameters, we briefly remark on the appropriateness of the Hill function as a model for cis-regulation, and introduce a parameter that provides a trade-off between model accuracy and computational efficiency. Finally, we provide a mathematical argument that indicates a limitation of using a Boolean network as a model for transcriptional regulation.
In the seventh section, we present simulations, based on transcriptional regulation of a hypothetical metabolic pathway, that illustrate several aspects of the proposed discrete model. By varying the parameters of the model, and observing how these changes affect mRNA and protein activity, we demonstrate that the nonlinear discrete dynamical system proposed in this article may effectively be used to model transcriptional regulation in a biologically relevant way.
Finally, in the last section, we summarize our conclusions.
We believe that the main contribution of this work is to show that, by using available biological information pertaining to the processes of transcription, translation, and cis-regulation, we can derive a nonlinear discrete dynamical system that may serve as a promising and testable model for transcriptional regulation. Our theoretical discussions and simulations indicate that the proposed model is capable of sufficiently predicting basic biological function and producing biologically relevant responses. Finally, the discrete dynamical nature of the proposed model makes it very attractive for large-scale computational analysis and simulation studies of transcriptional regulation.
REVIEW OF A CONTINUOUS MODEL
To model transcriptional regulation, we consider a large population 𝒞 of genetically identical cells that express the same set of G (distinct) genes, and denote those genes by 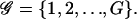 We take the population to be large because the derivation of the continuous model discussed here (as well as the model for cis-regulation discussed later in this article) uses the Boltzmann distribution of statistical mechanics. The Boltzmann distribution specifies how energy is distributed in a large population of identical molecules (DNA templates, mRNAs, and regulatory proteins in our case) at statistical equilibrium. We view transcriptional regulation as a complex system of interacting genes and regulatory proteins (transcription factors), whose state at time t is summarized by the G × 1 vectors r(t) and p(t), given by
We take the population to be large because the derivation of the continuous model discussed here (as well as the model for cis-regulation discussed later in this article) uses the Boltzmann distribution of statistical mechanics. The Boltzmann distribution specifies how energy is distributed in a large population of identical molecules (DNA templates, mRNAs, and regulatory proteins in our case) at statistical equilibrium. We view transcriptional regulation as a complex system of interacting genes and regulatory proteins (transcription factors), whose state at time t is summarized by the G × 1 vectors r(t) and p(t), given by
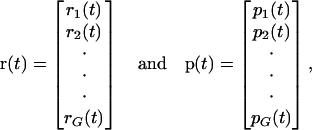 |
where ri(t) and pi(t) are the concentrations in 𝒞, at time t, of the mRNAs and regulatory proteins produced by the ith gene (measured in mol/L or molarity M; the concentrations considered in this article are with respect to the total cellular volume in 𝒞). We consider systems that are “complete,” in the sense that p consists of all proteins that regulate transcription of the mRNAs in r. For ease of presentation, we focus on a transcriptional machinery that is “isolated,” in the sense that it is not subject to external inputs. If necessary, our formulation can be modified to consider those cases as well (see the example depicted in Fig. 5).
FIGURE 5.

An example of a tRS of a hypothetical metabolic pathway that consists of four genes. In this figure, ⊸ denotes an activator, whereas, ⊣ denotes a repressor.
Given a target gene, we need to mathematically describe how its expression level (i.e., the mRNA concentration produced by this gene) is regulated by the expression levels of other genes. Fig. 1 depicts a block diagram of a model for transcriptional regulation, in which a target gene 3 is directly regulated by two other genes, 1 and 2. By “direct regulation” we mean that changes in the expression levels of genes 1 and 2 may produce a change in the expression level of gene 3 with no mediation from other genes. According to this model, the mRNAs transcribed from genes 1 and 2, with respective concentrations r1(t) and r2(t), at time t, are translated into two regulatory proteins whose concentrations are p1(t) and p2(t). These proteins bind to the control region of gene 3 and regulate the recruitment of general transcription factors and RNA polymerase II (for eukaryotic cells) to the gene's promoter. This step is referred to as cis-regulation. After the general transcription factors and RNA polymerase II have been assembled and positioned on the promoter, the RNA polymerase II initiates transcription of gene 3, whose mRNA concentration at time t is r3(t).
FIGURE 1.

Block diagram of a model for transcriptional regulation. The target gene 3 is directly regulated by two genes 1 and 2. Transcriptional regulation involves three steps: translation, cis-regulation, and transcription.
In the diagram depicted in Fig. 1, we have assumed that mRNAs and proteins do not decay, and that the tasks of translation, cis-regulation, and transcription are completed instantaneously. It is a well-known fact however that mRNAs and proteins are subject to degradation and that the time required to complete transcription and translation is not negligible. Transcription is subject to a time delay for completing RNA chain elongation, whereas, translation is subject to a time delay for completing the elongation phase of protein synthesis. Moreover, and for controlling the assembly of the transcription initiation complex (i.e., the general transcription factors and RNA polymerase II) at the promoter, appreciable time is required for the transport of proteins to the nucleus, for the binding of these proteins to the appropriate DNA regulatory sequences, and for recruiting the general transcription factors at the promoter. These effects can be accounted for, by assuming that translation, cis-regulation, and transcription are subject to time delays τp,i, τc,i, and τr,i, respectively, for i ∈ 𝒢. In general, these delays depend on the particular genes under consideration.
To obtain a model for transcriptional regulation, we need to mathematically describe the three steps of translation, transcription, and cis-regulation. To derive a mathematical model for translation, we adopt the following notation: T, absolute temperature (in degrees Kelvin, K); R, gas constant (1.9872 cal mol−1 K−1); Utr,i, activation energy of translation of the ith mRNA (in cal/mol); Udg,i, activation energy of degradation of the ith regulatory protein (in cal/mol); ri(t | U > Utr,i), concentration, at time t, of ith mRNA molecules in 𝒞 with energy greater than the activation energy Utr,i; pi(t | U > Udg,i), concentration, at time t, of ith regulatory protein molecules in 𝒞 with energy greater than the activation energy Udg,i.
The activation energy depends on the specific aspects of the underlying chemical reaction. By using standard arguments from chemical kinetics (e.g., see Moore and Pearson, 1981; Chapter 5 and Espenson, 1995; Chapter 7), we take the rate of protein synthesis (per second) during translation to be proportional to the concentration of mRNAs with energy >Utr, with proportionality constant αtr. Similarly, we take the rate of protein degradation (per second) to be proportional to the concentration of proteins with energy >Udg, with proportionality constant αdg.
By focusing on the macroscopic behavior of translation during the time interval [t, t + Δt], for some Δt > 0, we can write:
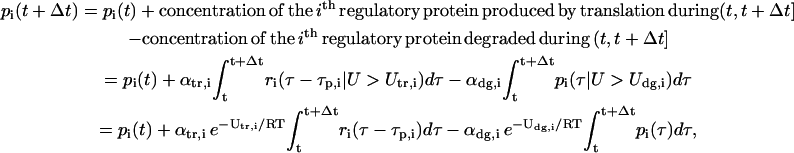 |
(1) |
for  where αtr,i and αdg,i are the proportionality constants (measured in s−1) associated with the two reactions of mRNA translation and protein degradation, respectively. To obtain Eq. 1, we use a fundamental result of statistical mechanics, which states that, in a large population of identical molecules at statistical equilibrium with concentration η, the concentration η(U) of molecules with kinetic energy U is given by the Boltzmann distribution
where αtr,i and αdg,i are the proportionality constants (measured in s−1) associated with the two reactions of mRNA translation and protein degradation, respectively. To obtain Eq. 1, we use a fundamental result of statistical mechanics, which states that, in a large population of identical molecules at statistical equilibrium with concentration η, the concentration η(U) of molecules with kinetic energy U is given by the Boltzmann distribution
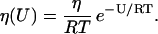 |
This leads to
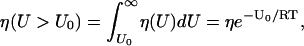 |
(2) |
where η(U > U0) denotes the concentration of molecules in the population with kinetic energy greater than some threshold U0. From Eq. 1, we obtain
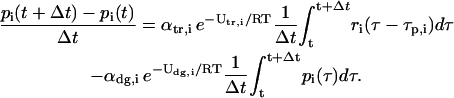 |
(3) |
By taking limits, as 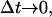 on both sides of Eq. 3, and by setting
on both sides of Eq. 3, and by setting
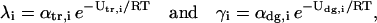 |
(4) |
we obtain the following system of rate equations:
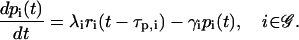 |
(5) |
These first-order ODEs imply that the rate of change in the concentration of the ith regulatory protein at time t is proportional to the expression level ri(t − τp,i) of gene i at time t − τp,i. Moreover, it implies that this protein degrades at a rate 0 < γi < ∞ (in s−1), which is proportional to its concentration. In Eq. 5, 0 < λi < ∞ (in s−1) is the rate of translation; i.e., the proteins synthesized per second from a mol of mRNA.
By following similar arguments, we can show that transcription can be modeled by the following system of rate equations:
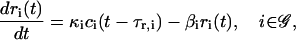 |
(6) |
where 0 ≤ ci(t) ≤ 1 is the fraction, at time t, of DNA templates in 𝒞 that are committed to the transcription of gene i, and 0 < κi < ∞ is the transcription rate of gene i; i.e., the concentration of mRNAs synthesized per second when all DNA templates in 𝒞 are committed to the transcription of gene i (in M s−1). We say that a DNA template is “committed” to the transcription of a gene, if it has successfully recruited the transcription initiation complex and has anchored it at the promoter of that gene. Note that a DNA template that is committed to transcription may not necessarily lead to transcription initiation. For this to happen, the energy of the committed DNA template should be greater than the activation energy of transcription initiation. The first-order ODEs in Eq. 6 imply that the rate of change in mRNA concentration produced from gene i at time t is proportional to the fraction ci(t − τr,i) of DNA templates committed to the transcription of gene i at time t − τr,i. Moreover, they imply that these molecules degrade at a rate 0 < βi < ∞ (in s−1), which is proportional to their concentration.
In general, the cis-regulation of a target gene i may be modeled by the following equations:
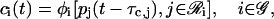 |
(7) |
where φi[·] is a (nonlinear) function, which is specific to the target gene under consideration, and 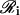 is the set of all genes in 𝒢 that produce proteins, which regulate the transcription of the ith gene. We refer to φi[·] as the cis-regulatory function of gene i. Moreover, we refer to
is the set of all genes in 𝒢 that produce proteins, which regulate the transcription of the ith gene. We refer to φi[·] as the cis-regulatory function of gene i. Moreover, we refer to 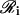 as the regulatory set of gene i and to the genes in
as the regulatory set of gene i and to the genes in 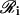 as the regulating genes of gene i. We call the collection
as the regulating genes of gene i. We call the collection 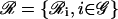 of all regulatory sets a transcriptional regulatory network (tRN). In Eq. 7, we assume that transcription is controlled by the protein products, at times t − τc,j,
of all regulatory sets a transcriptional regulatory network (tRN). In Eq. 7, we assume that transcription is controlled by the protein products, at times t − τc,j, 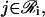 obtained by translating the regulating genes of the target gene i.
obtained by translating the regulating genes of the target gene i.
We note here that several variations of the model governed by Eqs. 5–7 have been proposed in the literature (e.g., see Hargrove and Schmidt, 1989; Mjolsness et al., 1991; Mestl et al., 1995; Endy et al., 1997; Wolf and Eeckman, 1998; Chen et al., 1999; Hatzimanikatis and Lee, 1999; Akutsu et al., 2000b; Cherry and Adler, 2000; von Dassow et al., 2000; Elowitz and Leibler, 2000; Gardner et al. 2000; Hasty et al., 2000; Voit, 2000; Smolen et al., 2000; Gibson and Mjolsness, 2001; Mjolsness, 2001, Vohradský, 2001; Wahde and Hertz, 2001; de Jong, 2002; Yildirim and Mackey, 2003, and the references therein). A limitation of Eqs. 5–7 is that they only apply to a large population cells. Moreover, these equations are derived by employing a macroscopic view of the chemical reactions underlying translation, cis-regulation, and transcription. The resulting ODE-based model oversimplifies the complex structure of a cell's transcriptional activity, by ignoring several factors affecting such activity. For example, Eq. 5 ignores the effects of mRNA transport from the nucleus to the cytoplasm and mRNA localization in the cytoplasm, whereas Eq. 6 does not take into account the mechanisms of RNA processing. Eq. 7 oversimplifies transcriptional control by ignoring, for example, complex interactions, inside the cis-regulatory mechanisms, among regulatory proteins, general transcription factors, RNA polymerase II, chromatin remodeling complexes and DNA, and by ignoring the role that protein-DNA complexes play in transcriptional regulation. Finally, the model does not consider how protein folding affects transcriptional regulation and ignores several biochemical interactions among proteins and interactions between different biological and signaling pathways. Nevertheless, the ODE-based model governed by Eqs. 5–7 provides a “first-order” approximation of transcriptional activity that leads to a mathematically tractable model for transcriptional regulation.
To conclude this section, note that, by solving Eqs. 5 and 6 with respect to pi(t) and ri(t), we obtain
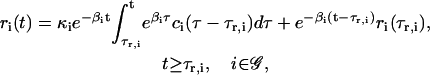 |
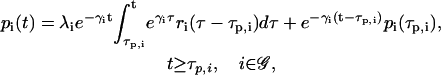 |
which in turn result in
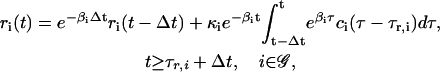 |
(8) |
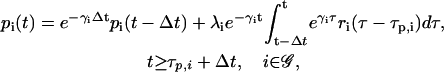 |
(9) |
for some Δt > 0. According to Eq. 8, the concentration ri(t) of the ith mRNA present in the cytoplasm at time t equals the concentration 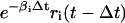 of the mRNA that survives degradation during the time interval [t − Δt, t), plus the concentration
of the mRNA that survives degradation during the time interval [t − Δt, t), plus the concentration 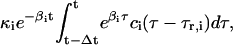 of the new mRNA that is synthesized by transcription and survives degradation during the same interval. According to Eq. 9, the concentration pi(t) of the ith protein present in the cytoplasm at time t equals the concentration
of the new mRNA that is synthesized by transcription and survives degradation during the same interval. According to Eq. 9, the concentration pi(t) of the ith protein present in the cytoplasm at time t equals the concentration 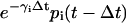 of the protein that survives degradation during the time interval [t − Δt, t), plus the concentration
of the protein that survives degradation during the time interval [t − Δt, t), plus the concentration 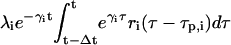 of the new protein that is synthesized by translation and survives degradation during the same interval.
of the new protein that is synthesized by translation and survives degradation during the same interval.
A DISCRETE MODEL
The previous ODE-based model provides a continuous description of transcriptional regulation. However, the processes of transcription and translation may be thought as being discrete. During transcription, the RNA polymerase II moves along the DNA in a stepwise fashion and extends the growing RNA chain by adding one nucleotide at a time (see Alberts et al., 2002, pp. 302–304). Similarly, during translation, a ribosome moves along an mRNA transcript by sequentially processing groups of three nucleotides (codons), and extends the growing polypeptide chain by adding one amino acid at a time (see Alberts et al., 2002, pp. 342–344). These observations, together with the need for solving Eqs. 5–7 using computational techniques, motivates us to derive a discrete model for transcriptional regulation.
The motion of RNA polymerase II along a DNA molecule may not be smooth (see Alberts et al., 2002, p. 313); its speed may depend on time, the particular gene transcribed, and other factors. Moreover, ribosomes may translate with different speeds at individual codons (e.g., see Sørensen and Pedersen, 1991), whereas, for each 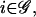 the cis-regulation delay τc,i may fluctuate. To avoid complications, we replace the actual transcriptional machinery with one for which the speeds of transcription and translation, vr and vp, and the cis-regulation delays
the cis-regulation delay τc,i may fluctuate. To avoid complications, we replace the actual transcriptional machinery with one for which the speeds of transcription and translation, vr and vp, and the cis-regulation delays 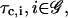 are all constants, taken to be equal to their mean values. We refer to this machinery as an “average” transcriptional machinery. This implies that the discrete dynamical system to be derived in this section will model an “average” transcriptional activity. Because the transcription speed is constant, the transcription delay, τr,i, will be an integer multiple of the time δt that takes the RNA polymerase II to read one nucleotide. For eukaryotic cells, we may take the average transcription speed vr ≅ 20 nucleotides/s (see Alberts et al., 2002, p. 304), in which case δt ≅ 0.05 s, whereas, we may take the average translation speed vp ≅ 2 codons/s (see Alberts et al., 2002, p. 343). For this value of vp, the translation delay, τp,i, is also an integer multiple of δt. Finally, we assume that the “average” transcriptional machinery is also characterized by cis-regulation delays τc,i, i ∈ 𝒢, which are integer multiples of δt as well.
are all constants, taken to be equal to their mean values. We refer to this machinery as an “average” transcriptional machinery. This implies that the discrete dynamical system to be derived in this section will model an “average” transcriptional activity. Because the transcription speed is constant, the transcription delay, τr,i, will be an integer multiple of the time δt that takes the RNA polymerase II to read one nucleotide. For eukaryotic cells, we may take the average transcription speed vr ≅ 20 nucleotides/s (see Alberts et al., 2002, p. 304), in which case δt ≅ 0.05 s, whereas, we may take the average translation speed vp ≅ 2 codons/s (see Alberts et al., 2002, p. 343). For this value of vp, the translation delay, τp,i, is also an integer multiple of δt. Finally, we assume that the “average” transcriptional machinery is also characterized by cis-regulation delays τc,i, i ∈ 𝒢, which are integer multiples of δt as well.
For each 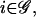 we make the following two assumptions: 1) for each t = δt, 2δt, …, the fraction ci of DNA fragments committed to the transcription of gene i remains constant in the time interval [t − δt, t); and 2) for each t = δt, 2δt, …, the mRNA concentration ri remains constant in the time interval [t − δt, t).
we make the following two assumptions: 1) for each t = δt, 2δt, …, the fraction ci of DNA fragments committed to the transcription of gene i remains constant in the time interval [t − δt, t); and 2) for each t = δt, 2δt, …, the mRNA concentration ri remains constant in the time interval [t − δt, t).
In view of the small value of δt, as compared to the large timescale of transcription (recall that δt ≅ 0.05 s in eukaryotic cells, as compared to the duration of a typical transcription reaction, which ranges from minutes to hours), we may shift all transcription commitments within the interval (t − δt, t), for t = δt, 2δt, …, to time t, with negligible effects on transcription. Therefore, we may approximately assume that no new DNA templates commit to transcription within the time interval (t − δt, t), which explains assumption 1. On the other hand, and due to the fact that the transcription delay τr,i is an integer multiple of δt, new mRNAs are synthesized only at integer multiples of δt. This, together with the previous observation, implies that new mRNAs are synthesized only at times t = δt, 2δt, …. Moreover, experimental evidence suggests that mRNA half-lives are much larger than δt (e.g., see Wang et al., 2002, and compare the value δt ≅ 0.05 s for eukaryotic cells with the mRNA half-lives in yeast, which range from ∼3 min to >90 min). In view of these observations, and assumption 1, we may conclude that assumption 2 is a reasonable assumption as well.
We can now employ the previous two assumptions to show that, by using δt as a basic discretization step and by replacing the actual transcriptional machinery with an “average” one, Eqs. 5–7 can be transformed into a discrete dynamical system that can effectively simulate transcriptional regulation in an iterative fashion. From assumptions 1 and 2, we have that
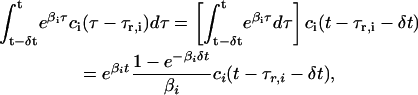 |
and
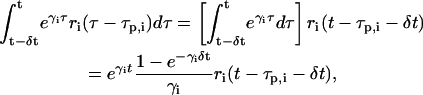 |
for i ∈ 𝒢, which, together with Eqs. 8 and 9, with Δt = δt, result in
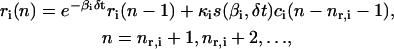 |
(10) |
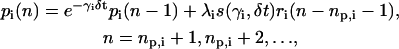 |
(11) |
where one iteration corresponds to the time step δt, nr,i = τr,i/δt, np,i = τp,i/δt, and
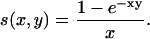 |
(12) |
Moreover, from Eq. 7, we have that
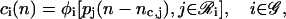 |
(13) |
where nc,j = τc,j/δt.
According to Eq. 10, the concentration ri(n) of the ith mRNA present in the cytoplasm at step n equals the concentration 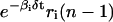 of the mRNA that survives degradation from step n − 1 to step n, plus the concentration κis(βi, δt)ci(n − nr,i − 1) of the new mRNA that is synthesized by transcription and survives degradation between these two steps. According to Eq. 11, the concentration pi(n) of the ith protein present in the cytoplasm at step n equals the concentration
of the mRNA that survives degradation from step n − 1 to step n, plus the concentration κis(βi, δt)ci(n − nr,i − 1) of the new mRNA that is synthesized by transcription and survives degradation between these two steps. According to Eq. 11, the concentration pi(n) of the ith protein present in the cytoplasm at step n equals the concentration 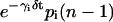 of the protein that survives degradation from step n − 1 to step n plus the concentration λis(γi, δt)ri(n − np,i − 1) of the new protein that is synthesized by translation and survives degradation between these two steps.
of the protein that survives degradation from step n − 1 to step n plus the concentration λis(γi, δt)ri(n − np,i − 1) of the new protein that is synthesized by translation and survives degradation between these two steps.
In the following, and to ease notation, we take the time delays τp,i, τc,i, and τr,i to be independent of i. In this case, Eqs. 10, 11, and 13 can be written in the following compact form:
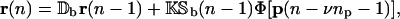 |
(14) |
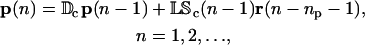 |
(15) |
where ν = (τr + τc)/τp. In Eqs. 14 and 15, Db, Dc, K, and L are G × G diagonal matrices, given by
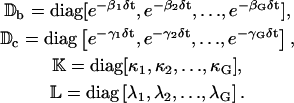 |
Moreover, Sb(n) and Sc(n) are G × G diagonal matrices, given by
 |
where
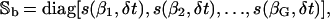 |
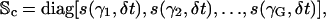 |
and
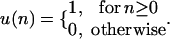 |
Finally, Φ[p(n − νnp − 1)] is an G × 1 vector-valued functional whose ith element is 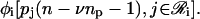 We refer to Φ[·] as the cis-regulatory functional.
We refer to Φ[·] as the cis-regulatory functional.
The iterations suggested by Eqs. 14 and 15 are depicted in Fig. 2, when τp = δt, τc = 3δt, and τr = 2δt. These iterations are initialized with an mRNA concentration vector r(0) and a protein concentration vector p(0). This implies that the fate of gene expression is determined by the initial concentrations of mRNAs and proteins. Matrices Db and Dc model mRNA and protein degradation, respectively, whereas, matrices K and L model the rate of transcription and translation, respectively. For 0 ≤ n ≤ τp/δt = np only degradation is present. Translation of mRNAs to proteins takes place for n ≥ τp/δt + 1 = np + 1, whereas, transcription takes place for n ≥ (τc + τr)/δt + 1 = νnp + 1. Note that the flow graph depicted in Fig. 2 has a modular structure; it consists of individual stages, with the nth stage being the (nonlinear in general) multi-input/multi-output system depicted in Fig. 3 (for n ≥ νnp + 1), where d denotes delay, such that d−mx(n) = x(n − m).
FIGURE 2.

Iterative implementation of transcriptional regulation governed by Eqs. 14 and 15, when τp = δt, τc = 3δt, and τr = 2δt. The implementation is initialized with an mRNA concentration vector r(0) and a protein concentration vector p(0). Matrices Db and Dc model mRNA and protein degradation, respectively, whereas, matrices K and L model the rate of transcription and translation, respectively. For 0 ≤ n ≤ τp/δt = 1 only degradation is present. Translation of mRNAs to proteins takes place for n ≥ τp/δt + 1 = np + 1 = 2, whereas, transcription takes place for n ≥ (τc + τr)/δt + 1 = νnp + 1 = 6.
FIGURE 3.

The nth stage (for n ≥ νnp + 1) of the flow graph depicted in Fig. 2.
The model suggested by Eqs. 10–13 requires knowledge of the cis-regulatory functionals 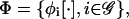 the degradation parameters
the degradation parameters 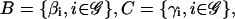 the transcription and translation rates
the transcription and translation rates 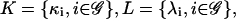 and the delays
and the delays 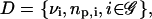 where νi = (τr,i + τc,i)/τp,i. We refer to the collection
where νi = (τr,i + τc,i)/τp,i. We refer to the collection 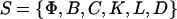 as a (discrete) transcriptional regulatory system (tRS).
as a (discrete) transcriptional regulatory system (tRS).
STEADY-STATE BEHAVIOR
An important issue associated with a tRS is whether or not the iterations suggested by Eqs. 10–13 converge to a steady state and, if they do, to characterize that state. In most cases, and in the absence of external control, for a tRS to be biologically plausible, it is required that the mRNA and protein concentration vectors r(n) and p(n) converge, as 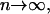 to a steady-state mRNA concentration vector
to a steady-state mRNA concentration vector  and a steady-state protein concentration vector
and a steady-state protein concentration vector 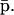 In this case,
In this case,
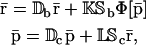 |
(16) |
from which we have that
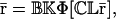 |
(17) |
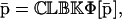 |
(18) |
where
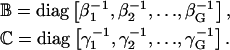 |
This shows that  is a fixed-point attractor of the functional Ψr[·], given by
is a fixed-point attractor of the functional Ψr[·], given by
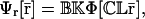 |
(19) |
whereas, 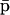 is a fixed-point attractor of the functional Ψp[·], given by
is a fixed-point attractor of the functional Ψp[·], given by
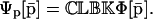 |
(20) |
We refer to Ψr[·] as the “genomic regulatory functional” and to Ψp[·] as the “proteomic regulatory functional” because the first functional can be used to determine the steady-state mRNA concentration vector, whereas, the second functional can be used to determine the steady-state protein vector.
The fixed-point attractors  and
and 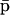 may be used to characterize the cell's phenotype. This is based on the assumption that cells may be differentiated by the concentrations of regulatory proteins synthesized at steady state (or, equivalently, by the concentrations of the corresponding mRNAs), which give each cell type its unique characteristics; e.g., see Kauffman (1993) (Chapter 12) and Alberts et al. (2002) (pp. 375–376). It is believed that the transcriptional regulatory machinery of a given organism is hardwired in its DNA. This implies that regulation of transcription is controlled by the same mechanisms, irrespective of cell type. We may however view cell differentiation as being achieved by transcriptional regulation, which guides the tRS to reach steady-state mRNA and protein concentration values
may be used to characterize the cell's phenotype. This is based on the assumption that cells may be differentiated by the concentrations of regulatory proteins synthesized at steady state (or, equivalently, by the concentrations of the corresponding mRNAs), which give each cell type its unique characteristics; e.g., see Kauffman (1993) (Chapter 12) and Alberts et al. (2002) (pp. 375–376). It is believed that the transcriptional regulatory machinery of a given organism is hardwired in its DNA. This implies that regulation of transcription is controlled by the same mechanisms, irrespective of cell type. We may however view cell differentiation as being achieved by transcriptional regulation, which guides the tRS to reach steady-state mRNA and protein concentration values  and
and 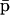 that uniquely characterize the cell type. In this case, the driving force of cell differentiation is said to be “epigenetic” regulation.
that uniquely characterize the cell type. In this case, the driving force of cell differentiation is said to be “epigenetic” regulation.
An implication of Eqs. 17 and 18 is that, at steady state, the mRNA concentration vector  “decouples” from the protein concentration vector
“decouples” from the protein concentration vector 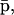 in the sense that
in the sense that  can be obtained as a solution of the system of (nonlinear in general) Eq. 17, without knowledge of
can be obtained as a solution of the system of (nonlinear in general) Eq. 17, without knowledge of 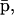 whereas,
whereas,  can be obtained as a solution of the system of (nonlinear in general) Eq. 18, without knowledge of
can be obtained as a solution of the system of (nonlinear in general) Eq. 18, without knowledge of 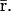 It is however important to keep in mind that, despite this “decoupling,” computation of the steady-state mRNA concentration vector
It is however important to keep in mind that, despite this “decoupling,” computation of the steady-state mRNA concentration vector  (and the steady-state protein concentration vector
(and the steady-state protein concentration vector 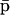 ) requires knowledge of the transcription parameters B, K, Φ and the translation parameters C, L, because Eq. 17 (and Eq. 18) depends on those parameters. Note also that there is a one-to-one correspondence between the fixed-point attractors of the genomic and proteomic regulatory functionals, because (recall Eq. 16)
) requires knowledge of the transcription parameters B, K, Φ and the translation parameters C, L, because Eq. 17 (and Eq. 18) depends on those parameters. Note also that there is a one-to-one correspondence between the fixed-point attractors of the genomic and proteomic regulatory functionals, because (recall Eq. 16)
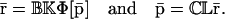 |
(21) |
The second equation above implies that, at steady state, 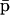 may be determined from
may be determined from 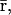 provided that the underlying translation parameters C, L are known. This observation suggests that mRNA expression data, obtained by means of microarray gene expression profiling, may be used to characterize protein activity at steady state, provided that the translation parameters C, L are known, and a sufficiently good estimate of the steady state mRNA concentration vector
provided that the underlying translation parameters C, L are known. This observation suggests that mRNA expression data, obtained by means of microarray gene expression profiling, may be used to characterize protein activity at steady state, provided that the translation parameters C, L are known, and a sufficiently good estimate of the steady state mRNA concentration vector  can be inferred from such data.
can be inferred from such data.
Because Eqs. 10–13 have been obtained by discretizing Eqs. 5–7, it is of interest to investigate how the steady-state behavior of the discrete tRS is related to the steady-state behavior of the continuous tRS. From Eqs. 10–12, we can show that
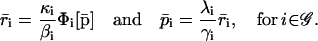 |
(22) |
On the other hand, if 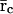 and
and 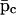 are the steady states of Eqs. 5 and 6, then (by setting the derivatives in Eqs. 5 and 6 equal to zero), we obtain
are the steady states of Eqs. 5 and 6, then (by setting the derivatives in Eqs. 5 and 6 equal to zero), we obtain
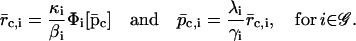 |
(23) |
Eqs. 22 and 23 verify that the discrete tRS has the same steady states as the continuous tRS, and show that the steady-state behavior of the discrete tRS is identical to that of the continuous tRS.
Besides fixed-point attractors, the tRS may be subject to limit-cycle attractors, which lead to oscillatory behavior. A tRS with limit-cycle attractors may be useful for modeling periodic cellular behavior, such as cell cycle control or circadian rhythms; see Kauffman (1993) (Chapter 12), and Elowitz and Leibler (2000); Smolen et al. (2000); Goldbeter et al. (2001); Hasty et al. (2001a,b); Tyson et al. (2001). In this article, we do not consider limit-cycle attractors (however, see Fig. 11 d).
FIGURE 11.

Evolutions of mRNA concentrations of the tRS depicted in Fig. 5, when τr = 2000 s, τp = 200 s, τc = 2400 s, and: (a) κ1 = 0.01 pM s−1 and λ = 0.05 s−1, (b) κ1 = 0.001 pM s−1 and λ = 0.05 s−1, (c) κ1 = 0.01 pM s−1 and λ = 0.2 s−1, (d) κ1 = 0.001 pM s−1 and λ = 0.2 s−1.
A MODEL FOR cis-REGULATION
The cis-regulatory functions φi[·] in Eq. 7 are at the core of a tRS, because these functions specify how proteins regulate transcription. In this section, we derive a form for these functions by using simple arguments from chemical kinetics (see also Hill, 1985; Wang et al., 1999). Keep in mind however that the resulting model oversimplifies cis-regulation, because cis-regulation is controlled by rather complicated biochemical interactions (e.g., see Holstege et al., 1998).
To model cis-regulation, we consider again a large population 𝒞 of cells, and assume at the moment that the promoter of a given target gene is controlled by two regulatory proteins P1 and P2, with concentrations p1 and p2, respectively. Moreover, we assume that protein P1 is free to bind at anyone of S1 distinct sites of the promoter's regulatory region, whereas, protein P2 is free to bind at anyone of S2 distinct sites, with the binding sites of P1 being different than that of P2. Let D[s1, s2] be a DNA template with s1 out of the S1 sites being occupied by P1 and s2 out of the S2 sites being occupied by P2. The binding of proteins P1 and P2 at the promoter's regulatory region can be described by means of the following reversible reactions:
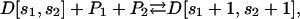 |
(24) |
for  If we assume that P1 and P2 do not interact with each other or affect each other's binding activity, then Eq. 24 can be sequentially written as
If we assume that P1 and P2 do not interact with each other or affect each other's binding activity, then Eq. 24 can be sequentially written as
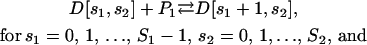 |
(25) |
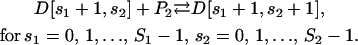 |
(26) |
In the following, d[s1, s2] denotes the concentration of DNA templates in 𝒞 with s1 out of the S1 sites being occupied by P1 and s2 out of the S2 sites being occupied by P2. Moreover, Ubd,i denotes the activation energy (in cal/mol) for a regulatory protein Pi to bind on a DNA template, whereas, Uds,i denotes the activation energy (in cal/mol) for Pi to dissociate itself from the template.
At equilibrium, the concentration of free regulatory proteins P1 that bind (per second) on DNA templates D[s1, s2] to produce DNA templates D[s1 + 1, s2] by means of the forward reaction in Eq. 25 must equal the concentration of regulatory proteins P1 freed (per second) by the backward reaction. By using molecular collision theory (e.g., see Moore and Pearson, 1981, Chapter 4), it can be shown that the first concentration is proportional to the concentration of those proteins P1 with kinetic energy >Ubd,1 times the concentration of sites available for P1 to bind to, with proportionality constant αbd,1 (measured in M−1s−1). Because each DNA template D[s1, s2] has S1 − s1 sites available for P1 to bind to, the concentration of available binding sites for P1 is (S1 − s1)d[s1, s2]. In this case, the concentration of free regulatory proteins P1 that bind (per second) on DNA templates D[s1, s2] to produce DNA templates D[s1 + 1, s2] by means of the forward reaction in Eq. 25, is given by
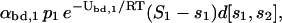 |
where we have used Eq. 2.
On the other hand, the concentration of regulatory proteins P1 freed (per second) by the backward reaction in Eq. 25 is proportional to the concentration of bound P1 molecules on the DNA template D[s1 + 1, s2] with kinetic energy >Uds,1, with proportionality constant αds,1 (measured in s−1). Because each DNA template D[s1 + 1, s2] contains s1 + 1 bound P1 molecules, this concentration is given by
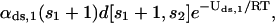 |
where we have used again Eq. 2. Therefore, at equilibrium, we have that
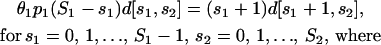 |
(27) |
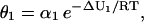 |
(28) |
with α1 = αbd,1/αds,1 and ΔU1 = Ubd,1 − Uds,1 being the binding free energy. The parameter θ1 (measured in M−1) is characteristic to the binding sites and is referred to as “affinity constant.” At equilibrium, and when Ubd,1 = Uds,1, the values of p1 (S1 − s1) d[s1, s2] and (s1 + 1) d[s1 + 1, s2] must be equal; therefore, α1 = 1 M−1. In addition, because Ubd,1 ≤ Uds,1, we have that 1 ≤ θ1 ≤ ∞.
A similar argument applies to Eq. 26 and leads to
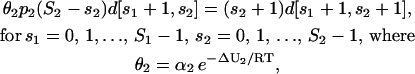 |
(29) |
with ΔU2 = Ubd,2 − Uds,2 and α2 = 1 M−1.
From Eqs. 27 and 29, it can be shown that
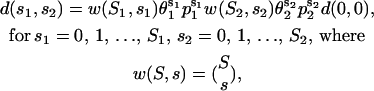 |
(30) |
are the so-called Binomial coefficients. If we ignore additional processes underlying cis-regulation (see Wang et al., 1999 for such processes) and assume that, for transcription to be initiated, it is necessary (but not sufficient) that a DNA template is bound by at least one P1 protein or one P2 protein, then the fraction c of DNA templates in 𝒞 committed to the transcription of the target gene will be given by
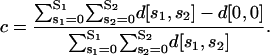 |
(31) |
The previous assumption agrees with the fact that transcription in eukaryotic cells can be initiated only in the presence of activator proteins (e.g., see Alberts et al., 2002, pp. 312–313). Eq. 31, together with Eq. 30, leads to
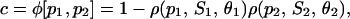 |
where
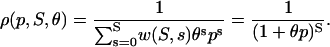 |
(32) |
Our discussion so far has been based on the assumption that regulatory proteins activate transcription; i.e., binding of regulatory proteins on a DNA template recruits the transcription initiation complex and initiates transcription. However, cis-regulation may also be controlled by regulatory proteins that repress transcription. Although eukaryotic genes employ several mechanisms for repressing transcription, we only consider here the mechanism by which a repressor protein binds on the DNA template, and either blocks the recruitment of the transcription initiation complex to the promoter, or prevents the release of the RNA polymerase II (see Alberts et al., 2002, pp. 405–406). This implies that, once a repressor protein binds on a DNA template, transcription cannot be initiated by that template, and leads to a simple model for the repression of transcription. Keep in mind however that, if necessary, it may be possible to derive models for other repression mechanisms as well.
If we assume that the previous protein P2 is a repressor, then the repression mechanism under consideration implies that transcription may be initiated only if a DNA template is free of protein P2 and there is at least one activator protein P1 bound to it. Then, the fraction c of DNA templates committed to the transcription of the target gene will be given by
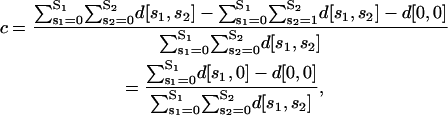 |
which leads to
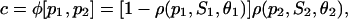 |
as opposed to Eq. 31. In general, if the promoter of a given target gene is controlled by activators P1, P2,…, Pk and repressors Pk+1, Pk+2,…, PJ, it can be shown that
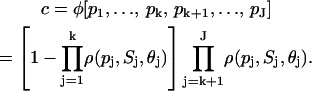 |
(33) |
It is believed that the organization of cis-regulation is modular; see Davidson (2001) (Chapter 1), Alberts et al. (2002) (pp. 408–413), Arnone and Davidson (1997), and Bolouri and Davidson (2002). This means that the regulatory region of a target gene may be partitioned into several entities (modules), with each entity being associated with different sets of regulatory proteins, which may assert a different type of control on transcription. This modular structure allows a gene to express itself under different conditions and different contexts.
We now derive a model for modular cis-regulation. For the purpose of our discussion below, we define a module as being that section of the regulatory region of a target gene, together with the associated regulatory proteins, which, at a given time, controls the promoter of that gene. For simplicity, we assume at the moment that the promoter of a given target gene is controlled by either one of two distinct modules, mod 1 and mod 2. We assume that a regulatory protein P1 may bind at anyone of S1 distinct sites of mod 1, whereas, a regulatory protein P2 may bind at anyone of S2 distinct sites of mod 2. For i = 1, 2, let di[si] be the concentration of all DNA templates in 𝒞, with si out of the Si sites of mod i being occupied by protein Pi. If we assume that transcription is initiated by mod i, and if ci is the fraction of the DNA templates committed to the transcription of the target gene due to the binding of Pi on mod i, then
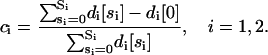 |
(34) |
If we now assume that, at a given time, transcription in the cell population 𝒞 may be initiated by either one of the two modules being occupied by at least one regulatory protein, then the fraction c of the DNA templates committed to the transcription of the target gene will be given by
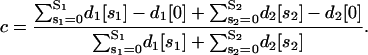 |
(35) |
From Eqs. 34 and 35, we obtain
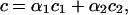 |
where
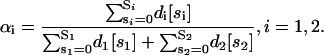 |
Therefore, we can model modular cis-regulation as a weighted summation of separately asserted cis-regulations by each module. Note that α1 + α2 = 1.
The previous discussion can be generalized to include several modules and regulatory proteins. If we assume that the promoter of a target gene i is controlled by Mi modules, we can model transcriptional regulation by means of the following equation (recall Eq. 7):
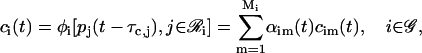 |
(36) |
where
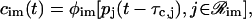 |
(37) |
for m = 1, 2, …, Mi, i ∈ 𝒢, and αim(t), m = 1, 2, …, Mi, are nonnegative weights such that
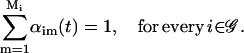 |
In Eq. 36, 0 ≤ αim(t) ≤ 1 quantifies the contribution of the mth module to the transcriptional regulation of the ith gene at time t, whereas, in Eq. 37, ℛim is the set of all genes in 𝒢 that produce the regulatory proteins associated with this module. Note that 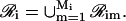
According to Eq. 33, a general form for the cis-regulatory function φim[·] is given by
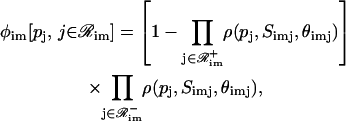 |
(38) |
for some 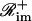 and
and 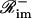 such that ℛim+∩ℛim−=
such that ℛim+∩ℛim−=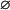 and
and 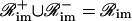 . In Eq. 38, Simj and θimj are the number of binding sites and the affinity constants, respectively, associated with the jth protein that controls the mth module of the ith promoter. Note that, when
. In Eq. 38, Simj and θimj are the number of binding sites and the affinity constants, respectively, associated with the jth protein that controls the mth module of the ith promoter. Note that, when 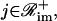 the jth protein acts as an activator, whereas, when
the jth protein acts as an activator, whereas, when 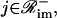 the jth protein acts as a repressor. It is assumed here that, if ℛim+=, then
the jth protein acts as a repressor. It is assumed here that, if ℛim+=, then 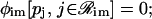 i.e., transcription is not initiated if the associated regulatory region does not contain binding sites for activator proteins, in accordance with the fact that transcription in eukaryotic cells cannot be initiated in the absence of activator proteins.
i.e., transcription is not initiated if the associated regulatory region does not contain binding sites for activator proteins, in accordance with the fact that transcription in eukaryotic cells cannot be initiated in the absence of activator proteins.
The choice in Eq. 38 is a simplified format for the cis-regulatory function of eukaryotic genes and agrees with the belief that activators and repressors work synergistically so that their joint effect is multiplicative; e.g., see Alberts et al. (2002) (p. 405); Herschlag and Johnson (1993); Savageau (2001). The basic ingredient of this model is the function ρ(p, S, θ), p ≥ 0, given by Eq. 32. For given values of S and θ, the functions ρ(p, S, θ) and 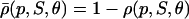 model the fraction of DNA templates in 𝒞 committed to the transcription of a target gene, whose transcription is respectively controlled by a repressor or an activator protein that binds at S distinct sites of the control region with affinity constant θ. Fig. 4 depicts ρ(p, S, θ) and
model the fraction of DNA templates in 𝒞 committed to the transcription of a target gene, whose transcription is respectively controlled by a repressor or an activator protein that binds at S distinct sites of the control region with affinity constant θ. Fig. 4 depicts ρ(p, S, θ) and 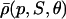 as a function of log10p, for several values of S and for θ = 108 M−1. As the protein concentration p increases, ρ monotonically decreases, whereas,
as a function of log10p, for several values of S and for θ = 108 M−1. As the protein concentration p increases, ρ monotonically decreases, whereas, 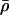 monotonically increases. This implies that the rate of transcription monotonically decreases as a function of repressor concentration, whereas, it monotonically increases as a function of activator concentration. For a given protein concentration, ρ decreases as a function of S (or remains constant at saturating points), whereas,
monotonically increases. This implies that the rate of transcription monotonically decreases as a function of repressor concentration, whereas, it monotonically increases as a function of activator concentration. For a given protein concentration, ρ decreases as a function of S (or remains constant at saturating points), whereas, 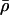 increases as a function of S (or remains constant at saturating points). This implies that the number of regulatory binding sites influence transcription, with more sites resulting in lower transcription rates for the case of repression, and higher transcription rates for the case of activation. Note also that the higher the number of regulatory binding sites, the lower the protein concentration required to produce appreciable repression or activation. Finally, if we interchange p with θ, Fig. 4 suggests that stronger binding affinity produces lower transcription rates for a repressor and higher transcription rates for an activator. These observations have been shown to be consistent with experimental biological evidence (e.g., see Wang et al., 1999 and the references therein), and support the use of Eq. 38 as a plausible choice for cis-regulation. Finally, note that
increases as a function of S (or remains constant at saturating points). This implies that the number of regulatory binding sites influence transcription, with more sites resulting in lower transcription rates for the case of repression, and higher transcription rates for the case of activation. Note also that the higher the number of regulatory binding sites, the lower the protein concentration required to produce appreciable repression or activation. Finally, if we interchange p with θ, Fig. 4 suggests that stronger binding affinity produces lower transcription rates for a repressor and higher transcription rates for an activator. These observations have been shown to be consistent with experimental biological evidence (e.g., see Wang et al., 1999 and the references therein), and support the use of Eq. 38 as a plausible choice for cis-regulation. Finally, note that 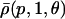 is the well-known Michaelis-Menten function of enzyme kinetics.
is the well-known Michaelis-Menten function of enzyme kinetics.
FIGURE 4.

The functions ρ(p, S, θ) and 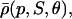 plotted in terms of log10 p, for S = 1, 2, 4, 8, 16, 32 and θ = 108 M−1.
plotted in terms of log10 p, for S = 1, 2, 4, 8, 16, 32 and θ = 108 M−1.
Although we believe that the general trends we have described in this section are true, they may oversimplify cis-regulation. An explanatory model for cis-regulation may need to be placed in a combinatorial setting. For example, two proteins with weak individual DNA bindings at contiguous sites, for example, may have a strong inter-protein binding tendency, and thus remain bound on the DNA as a complex, achieving locational accuracy by essentially achieving more binding interactions with the DNA as a complex than as singletons. This combinatorial structure substantially complicates mathematical modeling of cis-regulation. We have therefore chosen in this article not to consider the role that protein-DNA complexes play in transcriptional regulation.
REMARKS
Homeostatic regulation
The form of the genomic regulatory functional Ψr[·] in Eq. 19 suggests a specific response of transcriptional regulation to changes in the cellular environment. To retain the same level of steady-state mRNA concentration, the tRS may compensate for changes in the underlying parameters by keeping Ψr fixed. In this case, the tRS may compensate for changes in the rates of transcription by appropriate changes in mRNA degradation (and vice versa), so that the product BK remains constant. Moreover, it may compensate for changes in the rates of translation by appropriate changes in protein degradation (and vice versa), so that the product CL remains constant. On the other hand, to retain the same level of steady-state protein concentration, the tRS may compensate for changes in the underlying parameters by keeping Ψp fixed. The form of the proteomic regulatory functional Ψp[·] in Eq. 20 suggests that, for the tRS to retain the same level of steady-state protein concentration, it may adjust the rates of transcription, the rates of translation, or mRNA and protein degradations so that the product CLBK remains constant. Equations 19 and 20 also suggest that changes in the rates of transcription and translation (or in mRNA and protein degradation) that leave the product LK (or the product CB) invariant, have no effect on the steady-state protein concentration (because such changes do not affect Ψp), but may affect the steady-state mRNA concentration (because such changes may affect Ψr). These remarks predict that mRNA and protein degradation, together with the rates of mRNA and protein synthesis, may play an important role in a transcriptional regulation that maintains the levels of mRNA and protein concentrations at or near fixed values (known as homeostatic regulation; see also Hargrove and Schmidt, 1989; Carrier and Keasling, 1997; Grunberg-Manago, 1999; Wang et al., 2002).
Epigenetic regulation
The function φim[·] in Eq. 38 results in a cis-regulatory functional Φ[·] that is scale-invariant, in the sense that its value does not change if pj is multiplied by a constant a and the associated affinity constant θimj is divided by a. This is a direct consequence of the fact that (see Eq. 32) ρ(ap, S, θ/a) = ρ(p, S, θ), for a constant a.
This scaling property has some specific implications on regulation. It can be seen from Eqs. 17 and 18 that, if a tRS with parameters {κi}, {λi}, and {θimj} converges to 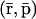 at steady state, then the same tRS with parameters {κi}, {aiλi}, and {θimj/ai} may converge to
at steady state, then the same tRS with parameters {κi}, {aiλi}, and {θimj/ai} may converge to 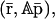 where A = diag[a1, a2, …, aG]. This implies that an increase (decrease) in the rates of translation, accompanied by a proportional decrease (increase) in the affinity constants, does not change the steady-state mRNA concentration but proportionally increases (decreases) the steady-state protein concentration. It can also be seen that the tRS with parameters {aiκi}, {λi}, and {θimj/ai} may converge to
where A = diag[a1, a2, …, aG]. This implies that an increase (decrease) in the rates of translation, accompanied by a proportional decrease (increase) in the affinity constants, does not change the steady-state mRNA concentration but proportionally increases (decreases) the steady-state protein concentration. It can also be seen that the tRS with parameters {aiκi}, {λi}, and {θimj/ai} may converge to 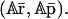 This implies that an increase (decrease) in the rates of transcription, accompanied by a proportional decrease (increase) in the affinity constants, proportionally increases (decreases) both the steady-state mRNA and protein concentrations. These remarks predict that the rates of transcription and translation, together with the affinity constants, may play an important role in a type of transcriptional regulation that changes the state of the tRS from one level of steady-state mRNA and protein concentrations to another (known as epigenetic regulation; see also Hargrove and Schmidt, 1989).
This implies that an increase (decrease) in the rates of transcription, accompanied by a proportional decrease (increase) in the affinity constants, proportionally increases (decreases) both the steady-state mRNA and protein concentrations. These remarks predict that the rates of transcription and translation, together with the affinity constants, may play an important role in a type of transcriptional regulation that changes the state of the tRS from one level of steady-state mRNA and protein concentrations to another (known as epigenetic regulation; see also Hargrove and Schmidt, 1989).
Parameters
To employ a tRS as an accurate predictor of transcriptional regulation, we need to specify the model parameters. Some parameters may be determined directly from available a priori biological knowledge. For example, if the sizes of the genes composing a tRS and the sizes of the associated mRNA products are known, then the discretization step δt and the time delays τr,i and τp,i may be estimated. As we mentioned before, during transcription, the RNA polymerase II may be thought of moving stepwise along the DNA, so that the growing RNA chain is extended one nucleotide at a time. Let vr be the average transcription speed of the RNA polymerase, measured in transcribed nucleotides per second (recall that, for eukaryotic cells, vr ≅ 20 nucleotides/s), and let the size of the regulatory gene i of a given genome be Gi nucleotides. Then,
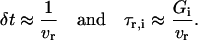 |
On the other hand, during translation, ribosomes may be thought of adding amino acids to a polypeptide chain in a stepwise fashion, with an average speed vp, measured in translated codons per second (recall that, for eukaryotic cells, vp ≅ 2 codons/s). If the size of the mRNA sequence required to encode the ith protein is Ri nucleotides, then
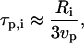 |
because each amino acid is produced from a codon that contains three nucleotides.
Most parameters of a tRS, like the degradation parameters β, γ, the rates of transcription and translation κ, λ, and, most importantly, the cis-regulatory functional Φ[·], are not known a priori. These parameters need to be estimated by means of carefully designed in vivo or in vitro experiments and computational analysis of genomic data (e.g., see Hammond, 1993; Iyer and Struhl, 1996; Arkin et al., 1997, 1998; Endy et al., 1997; Liang et al., 1998; Lorsch and Herschlag, 1999; Tavazoie et al., 1999; Akutsu et al., 2000a,b; Gardner et al., 2000; Turner and Varshavsky, 2000; Voit 2000; Wahde and Hertz, 2001; Caselle et al., 2002; Lee et al., 2002; Ronen et al., 2002; Wang et al. 2002; Yeung et al. 2002; Yildirim and Mackey, 2003, for emerging estimation techniques). This problem is key to one of the most exciting areas of modern biology, which is attracting collaborative efforts between biologists, statisticians, electrical engineers, and computer scientists, and promises to revolutionize biological research (e.g., see Hartwell et al., 1999; VanBogelen et al., 1999; D'haeseleer et al., 2000; Smolen et al., 2000; Endy and Brent, 2001; Hasty et al., 2001b; Somogyi et al., 2001; de Jong, 2002; Michelson, 2002, and the references therein). In the absence of quantitative knowledge for model parameters, the tRS governed by Eqs. 10–13 can be used as a qualitative tool that may provide valuable insights on the behavior and properties of transcriptional regulation (e.g., by means of steady-state analysis, perturbation analysis, computational knock-out studies, and hypothesis testing, as well as by studying the effects that parameters have on the system's dynamic behavior and steady state). Some insights, obtained by means of the tRS proposed in this article, are discussed in the next section.
Implementation
The discrete dynamical system governed by Eqs. 10–13 has been obtained by setting Δt = δt in Eqs. 8 and 9, and by considering assumptions 1 and 2. In view of the fact that the timescales of transcription, translation, and mRNA and protein degradation may be large, assumptions 1 and 2 may be still satisfied if we replace δt with a larger time step Δt = σδt, for some σ > 1, so that all time delays are integer multiples of Δt. In this case, the discrete model will be still governed by Eqs. 10–13, but with δt being replaced by Δt = σδt. The iterations required to simulate transcriptional regulation within a given time interval when Δt = σδt will be less than the iterations required when Δt = δt, reduced by a factor of σ. Parameter σ controls the “resolution” of the discrete model under consideration, and provides a trade-off between simulation accuracy and computational complexity. Clearly, smaller values of σ produce better accuracy but poor computational efficiency, whereas, larger values of σ may reduce accuracy but improve computational efficiency. Note that the steady-state behavior of the model does not depend on σ. Therefore, if the discrete dynamical system converges, it will converge to the same steady-state mRNA and protein concentration vectors, regardless of the particular value of σ.
The Hill function
Instead of ρ(p, S, θ) and 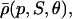 repression and activation are frequently modeled by means of functions
repression and activation are frequently modeled by means of functions
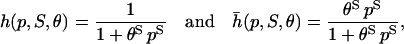 |
(39) |
respectively, where 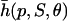 is known as the Hill function (e.g., see Cherry and Adler, 2000; Elowitz and Leibler, 2000; Gardner et al., 2000; von Dassow et al., 2000; Ronen et al., 2002; Yildirim and Mackey, 2003). Note that h(p, 1, θ) = ρ(p, 1, θ) and
is known as the Hill function (e.g., see Cherry and Adler, 2000; Elowitz and Leibler, 2000; Gardner et al., 2000; von Dassow et al., 2000; Ronen et al., 2002; Yildirim and Mackey, 2003). Note that h(p, 1, θ) = ρ(p, 1, θ) and 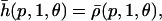 but these functions are different for S > 1. It can be shown (e.g., see Hill, 1985, pp. 64–66) that the functions in Eq. 39 are only appropriate in the limiting case when the only possible binding configurations at a gene's control region are either all sites to be empty or all to be occupied (a condition known as extreme cooperativity). In view of the fact that cis-regulation may not be subject to extreme cooperativity, the use of Eq. 39 for modeling cis-regulation may be limited.
but these functions are different for S > 1. It can be shown (e.g., see Hill, 1985, pp. 64–66) that the functions in Eq. 39 are only appropriate in the limiting case when the only possible binding configurations at a gene's control region are either all sites to be empty or all to be occupied (a condition known as extreme cooperativity). In view of the fact that cis-regulation may not be subject to extreme cooperativity, the use of Eq. 39 for modeling cis-regulation may be limited.
A note of caution
We want to point out here that there have been efforts to build tRSs by means of a functional Ψ[·] that relates the mRNA concentration vector r(n) at step n to the mRNA concentration vector r(n − 1) at step n − 1, by means of the following iterative equation (e.g., see Kauffman, 1993; Weaver et al., 1999; Wahde and Hertz, 2001; Baldi and Hatfield, 2002; de Jong, 2002; Shmulevich et al., 2002; Liebermeister, 2002):
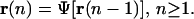 |
(40) |
If we take Ψ[·] to be the genomic regulatory functional Ψr[·] in Eq. 19, then the tRS governed by Eq. 40 will enjoy the same mRNA steady-state behavior (in terms of fixed-point attractors) as the tRS governed by Eqs. 14 and 15. In general however the transient behavior of these two systems will be different. As a matter of fact, the iterations suggested by Eqs. 14 and 15 imply that the value of the mRNA concentration vector r(n) cannot be inferred from knowing only its value at step n − 1. This can be seen by considering the case when np = ν = 0 (no time delays). This implies that
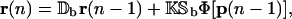 |
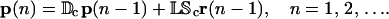 |
From these equations, we have that
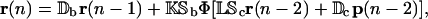 |
which shows that r(n) depends on r(n − 1) and r(n − 2), as well as on p(n − 2). Therefore, transient transcriptional behavior is governed by a tRS whose state at step n requires mRNA concentration at steps n − 1 and n − 2 as well as protein concentration at step n − 2 (see also the related discussion in Hargrove and Schmidt, 1989; Chen et al., 1999; Hatzimanikatis and Lee, 1999). This is different from what Eq. 40 suggests. We therefore conclude that the regulatory system governed by Eq. 40 may only be appropriate for studying steady-state behavior (in terms of fixed-point attractors) and should not be used to study transient transcriptional behavior.
Boolean networks
Boolean networks are attractive models for transcriptional regulation for two main reasons: they are much simpler than the model discussed in this article, and seem to be compatible with the limited nature of gene expression data obtained by current microarray technologies. A Boolean network is based on the premise that the status of a target gene can be represented by a binary variable that takes value 1 if the target gene is active (ON) or 0 if the target gene is inactive (OFF), and that this provides enough information for the status of transcriptional regulation. A Boolean network model relates a binary mRNA concentration vector rb(n) at step n to a binary mRNA concentration vector rb(n − 1) at step n − 1, by means of the following iteration
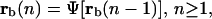 |
for some functional Ψ[·]. In view of our previous discussion, this type of Boolean network may be useful for modeling transcriptional regulation at steady state.
To derive a Boolean network model that qualitatively reproduces the steady-state behavior of the discrete dynamical system discussed in this article (and, therefore, the behavior of the corresponding ODE-based model), we need to find a threshold operator H[·] and a functional Ψ[·] so that a binary mRNA concentration vector 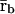 is a fixed-point attractor of Ψ[·] if and only if
is a fixed-point attractor of Ψ[·] if and only if 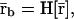 for a fixed-point attractor of the genomic regulatory functional Ψr[·]. We assume that the threshold operator H[·] is such that H[r] = rb, where, for i ∈ 𝒢, rb,i = ∞, if ri is larger than a given threshold value hi (in which case, gene i is thought to be ON), and rb,i = 0, if ri is smaller than the threshold value hi (in which case, gene i is thought to be OFF), and take Ψ[·] = HΨr[·]. Clearly, H[·] should be chosen such that:
for a fixed-point attractor of the genomic regulatory functional Ψr[·]. We assume that the threshold operator H[·] is such that H[r] = rb, where, for i ∈ 𝒢, rb,i = ∞, if ri is larger than a given threshold value hi (in which case, gene i is thought to be ON), and rb,i = 0, if ri is smaller than the threshold value hi (in which case, gene i is thought to be OFF), and take Ψ[·] = HΨr[·]. Clearly, H[·] should be chosen such that:
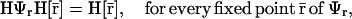 |
(41) |
in which case, every fixed-point attractor  of Ψr[·] will lead to a fixed-point attractor
of Ψr[·] will lead to a fixed-point attractor 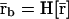 of Ψ[·]. However, given the genomic regulatory functional Ψr[·], we may not be able to find the threshold values
of Ψ[·]. However, given the genomic regulatory functional Ψr[·], we may not be able to find the threshold values 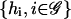 so that Eq. 41 is satisfied. Moreover, it may not be true that every fixed-point attractor of Ψ[·] will correspond to a fixed-point attractor of Ψr[·]. Finally, limit-cycle attractors of the Boolean network may not correspond to limit-cycle attractors of the discrete tRS discussed in this article, and vice versa (see also the discussion in Glass and Kauffman, 1973; Bagley and Glass, 1996). These problems should be seriously considered when a Boolean network is used in place of a discrete tRS (for what might happen if we carelessly do so, see Hatzimanikatis and Lee, 1999).
so that Eq. 41 is satisfied. Moreover, it may not be true that every fixed-point attractor of Ψ[·] will correspond to a fixed-point attractor of Ψr[·]. Finally, limit-cycle attractors of the Boolean network may not correspond to limit-cycle attractors of the discrete tRS discussed in this article, and vice versa (see also the discussion in Glass and Kauffman, 1973; Bagley and Glass, 1996). These problems should be seriously considered when a Boolean network is used in place of a discrete tRS (for what might happen if we carelessly do so, see Hatzimanikatis and Lee, 1999).
AN EXAMPLE
We now present an example of a tRS that consists of four genes, and use this example to illustrate several properties of the proposed model. A graphical representation of the system is depicted in Fig. 5, where ⊸ denotes an activator and ⊣ denotes a repressor. We assume that the tRS regulates a hypothetical pathway, which metabolizes an input substrate to an output product. This is done by means of enzymes whose transcriptional control is regulated by the protein produced from gene 3. Moreover, we assume that the effect of higher input substrate concentration is to increase the transcription rate κ1, whereas, the effect of lower substrate concentration is to reduce κ1. Unless otherwise specified, the parameters associated with this example are taken to be gene independent. These parameters are summarized in Table 1.
TABLE 1.
Parameter values used in simulations
| Figure
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Value | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| Initial mRNA concentration | ri(0) = 1.25 pM, i = 1, 2, 3, 4 | • | • | • | • | • | • | • |
| Initial protein concentration | pi(0) = 2.08 pM, i = 1, 2, 3, 4 | • | • | • | • | • | • | • |
| Affinity constant | θ = 108 M−1 | • | • | • | (b) | • | • | |
| θ = 5 × 107 M−1 | • | |||||||
| 106 M−1 ≤ θ ≤ 1010 M−1 | (a) | |||||||
| Number of binding sites | S = 1 | • | ||||||
| S = 2 | • | |||||||
| S = 4 | • | • | • | • | • | • | ||
| S = 6 | • | |||||||
| S = 8 | • | • | ||||||
| mRNA half-life | ρ = 1200 s | • | • | • | • | • | • | • |
| Protein half-life | π = 3600 s | • | • | • | • | • | • | • |
| Transcription rate: gene 1 | κ1 = 0.02 pM s−1 | (b) | ||||||
| κ1 = 0.01 pM s−1 | (a) | (a) | • | • | (a,c) | • | ||
| κ1 = 0.001 pM s−1 | (b) | • | (b,d) | |||||
| 0.001 pM s−1 ≤ κ1 ≤ 1 pM s−1 | • | |||||||
| Transcription rate: genes 2–4 | κ2 = κ3 = κ4 = 0.10 pM s−1 | (b) | ||||||
| κ2 = κ3 = κ4 = 0.05 pM s−1 | • | (a) | • | • | • | • | • | |
| Translation rate | λ = 0.05 s−1 | • | (b) | • | • | • | (a,b) | |
| λ = 0.10 s−1 | (a) | |||||||
| λ = 0.20 s−1 | (c,d) | • | ||||||
| Time delays | τr = τc = τp = 0 s | • | • | • | • | • | ||
| τr = 2000 s, τc = 200 s, τp = 2400 s | • | • | ||||||
We assume that each cis-regulator is controlled by one module with four binding sites, and set S = 4, θ = 108 M−1, κ2 = κ3 = κ4 = 0.05 pM s−1, and λ = 0.05 s−1. The value of the affinity constant θ corresponds to a binding free energy of ΔU = −11.35 kcal/mol at temperature T = 310.15 K (or 37°C). The values of the transcription rates κ2, κ3, and κ4 correspond to a transcriptional machinery that, on the average, produces one mRNA molecule every 8 s. This value turns out to be typical for yeast cells (Iyer and Struhl, 1996). We also assume that, on the average, the volume of each cell in 𝒞 equals to 4 pL (Alberts et al., 2002; Table 2–4). The translation rate λ is taken to be 10-fold larger than the rate of 0.3/min for translation initiation observed in vitro using a semipurified rabbit reticulocyte system (Lorsch and Herschlag, 1999).
The degradation parameters β and γ are specified by means of the mRNA and protein half-life parameters ρ and π, respectively, which satisfy
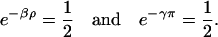 |
In this case,
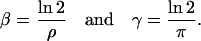 |
We set ρ = 1200 s (20 min) and π = 3600 s (1 h). For clarity of presentation, we set all time delays equal to zero, in which case np = ν = 0. Nonzero time delays complicate the evolutions of mRNA and protein concentrations and make simple descriptions of system behavior rather difficult. Simulation results that include “realistic” nonzero time delays are provided at the end of this section (see Figs. 11 and 12). Due to zero delays, mRNA and protein concentrations reach steady-state values faster than it is biologically expected. The simulations are initialized with mRNA and protein concentrations given by ri(0) = 1.25 pM and pi(0) = 2.08 pM, for i = 1, 2, 3, 4. To obtain these values, we assume that, initially, each gene contributes, on the average, three mRNA and five protein molecules to a cell in 𝒞.
FIGURE 12.

Evolutions of mRNA concentrations of the tRS depicted in Fig. 5, when τr = 2000 s, τp = 200 s, τc = 2400 s, κ1 = 0.01 pM s−1, λ = 0.2 s−1, and for four values of σ.
Fig. 6 a depicts typical evolutions of mRNA and protein concentrations. We assume that the transcription rate of gene 1 is given by κ1 = 0.01 pM s−1. The protein produced by gene 1 enhances transcription of gene 2, which produces enough protein to repress the transcription of gene 4. This results in protein 4 eventually reaching a state of low concentration, which releases the repression of gene 3. Gene 3 is now free to autoregulate, as well as to actively regulate gene 2. The mRNA and protein concentrations of these genes eventually reach appreciable steady-state values. The overall tRS converges to a “high” steady-state 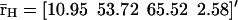 and
and 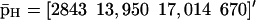 (in pM). Computer simulations indicate that this state is stable, in the sense that, eventually, the tRS drives any perturbation to steady-state concentrations back to their nominal values.
(in pM). Computer simulations indicate that this state is stable, in the sense that, eventually, the tRS drives any perturbation to steady-state concentrations back to their nominal values.
FIGURE 6.

Evolutions of mRNA and protein concentrations of the tRS depicted in Fig. 5. The transcription rate for gene 1 is: (a) κ1 = 0.01 pM s−1, and (b) κ1 = 0.001 pM s−1.
On the other hand, a 10-fold reduction in the transcription rate of gene 1 results in the evolution depicted in Fig. 6 b. In this case, gene 1 cannot sustain its own transcription by autoregulation and eventually reaches a state of zero mRNA and protein concentrations. In turn, the concentration of protein 3 cannot sufficiently increase the expression level of gene 2, to produce enough proteins to repress gene 4, which also reaches a state of zero mRNA and protein concentrations. Because gene 3 is being repressed by gene 4, it gradually produces low mRNA and protein concentrations. The overall tRS converges to a “low” steady-state 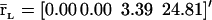 and
and 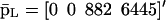 (in pM). Computer simulations indicate that this state is stable as well.
(in pM). Computer simulations indicate that this state is stable as well.
Fig. 7 a depicts the evolutions of mRNA and protein concentrations when λ = 0.10 s−1 and θ = 5 × 107 M−1. This amounts to a twofold increase in the rate of translation and a twofold decrease in the affinity constant. In this case, the steady state mRNA and protein concentration vectors are given by 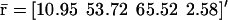 and
and 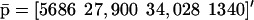 (in pM), respectively. As we have discussed earlier, this change does not affect the steady-state mRNA concentration values, but produces steady-state protein concentration values that are two times larger than the ones depicted in Fig. 6 a. On the other hand, Fig. 7 b depicts the evolutions of mRNA and protein concentrations when κ1 = 0.02 pM s−1, κ2 = κ3 = κ4 = 0.10 pM s−1, and θ = 5 × 107 M−1. This amounts to a twofold increase in the rate of transcription and a twofold decrease in the affinity constant. In this case, the steady-state mRNA and protein concentration vectors are given by
(in pM), respectively. As we have discussed earlier, this change does not affect the steady-state mRNA concentration values, but produces steady-state protein concentration values that are two times larger than the ones depicted in Fig. 6 a. On the other hand, Fig. 7 b depicts the evolutions of mRNA and protein concentrations when κ1 = 0.02 pM s−1, κ2 = κ3 = κ4 = 0.10 pM s−1, and θ = 5 × 107 M−1. This amounts to a twofold increase in the rate of transcription and a twofold decrease in the affinity constant. In this case, the steady-state mRNA and protein concentration vectors are given by 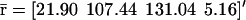 and
and 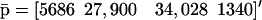 (in pM), respectively. This change produces steady-state mRNA and protein concentration values that are two times larger than the ones depicted in Fig. 6 a.
(in pM), respectively. This change produces steady-state mRNA and protein concentration values that are two times larger than the ones depicted in Fig. 6 a.
FIGURE 7.

Evolutions of mRNA and protein concentrations of the tRS depicted in Fig. 5 for the case of: (a) a twofold increase in the rate of translation and a twofold decrease in the affinity constant, and (b) a twofold increase in the rate of transcription and a twofold decrease in the affinity constant.
The results depicted in Fig. 7 (and, more generally, our discussion about the scaling properties of a tRS) indicate that quantitative steady-state mRNA data alone, like data obtained by DNA chip technologies, may not be sufficient for predicting steady-state protein concentrations (see also Hatzimanikatis and Lee, 1999). It is suggested by Figs. 6 a and 7 a that the same steady-state mRNA concentrations may be associated with substantially different steady-state protein concentrations. Moreover, it is suggested by Fig. 7 that different steady-state mRNA concentrations may be associated with the same steady-state protein concentrations. These observations agree with biological evidence (e.g., see Gygi et al., 1999), and suggest that additional information is needed to predict steady-state protein concentrations from steady-state mRNA concentrations. As a matter of fact, Eq. 21 suggests a precise solution to this problem: to obtain 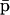 from
from  we need to know the value of the product CL (see Hargrove and Schmidt, 1989, for a similar observation).
we need to know the value of the product CL (see Hargrove and Schmidt, 1989, for a similar observation).
Due to the fact that the protein concentration pi(n) at step n is calculated from the protein concentration  at step n − 1 by adding the amount λis(γi, δt)ri(ℏ − np,i − 1), which is a linear function of the mRNA concentration ri(n − np,i − 1) at step n − np,i − 1 (recall Eq. 11), the protein evolutions depicted in Figs. 6 and 7 are very similar to the corresponding mRNA evolutions. For this reason, and in the rest of the article, we only depict mRNA evolutions.
at step n − 1 by adding the amount λis(γi, δt)ri(ℏ − np,i − 1), which is a linear function of the mRNA concentration ri(n − np,i − 1) at step n − np,i − 1 (recall Eq. 11), the protein evolutions depicted in Figs. 6 and 7 are very similar to the corresponding mRNA evolutions. For this reason, and in the rest of the article, we only depict mRNA evolutions.
Fig. 8 depicts steady-state (after 48 h) mRNA concentrations for genes 2, 3, and 4, as a function of the transcription rate κ1 of gene 1, and for S = 1, 2, 4, 6, 8. As expected, for small values of κ1 (i.e., for low input substrate concentrations), the tRS reaches a “low” steady state (i.e., low mRNA and protein concentrations for gene 3), whereas, for large values of κ1, the tRS reaches a “high” steady state (i.e., high mRNA and protein concentrations for gene 3). The transition from the “low” to the “high” steady state is sharper for larger values of S (i.e., when more binding sites are available in the regulatory region). Moreover, larger values of S result in smaller “low” steady-state mRNA and protein concentrations for gene 3 and larger “high” steady-state concentrations. When S = 1 (i.e., when the Michaelis-Menten function is used to model cis-regulation), the transition between the “low” and “high” steady-state concentrations is slow. Moreover, the difference between the “low” and “high” mRNA and protein concentrations for gene 3 is very small. This indicates that the Michaelis-Menten function may not be appropriate for modeling cis-regulation, when the input substrate is expected to trigger gene expression in an all-or-none fashion (see also Cherry and Adler, 2000, for a similar remark).
FIGURE 8.

Steady-state mRNA concentrations of the tRS depicted in Fig. 5, as a function of the transcription rate κ1 of gene 1, for S = 1, 2, 4, 6, 8.
For larger values of S, the transition from the “low” to the “high” steady state is sharper, whereas, the difference between these two states is larger. Moreover, the value of κ1 at which this transition occurs decreases as S increases. This is a consequence of the fact that lower protein concentration is required to produce appreciable repression or activation when more binding sites are available in the regulatory region. It is clear from Fig. 8 that, when S = 8, small variations in the value of κ1 around the critical value κ1,c ≅ 10−2.5 pM s−1 (i.e., small variations in input substrate concentrations around a critical value) may produce sharp and fast changes in mRNA and protein concentrations for gene 3 (i.e., may produce sharp changes in the transcriptional control of the metabolic enzymes). In this case, the tRS under consideration may abruptly switch from the “low” to the “high” steady state (and vice versa). Note however that the tRS is robust for values of κ1 that are not in the transition region, in the sense that changes in κ1 produce no changes in steady-state values. Insensitivity of steady-state behavior on certain parameter values is an essential biological property of a tRS, which is related to its robustness (e.g., see the discussion in Kitano, 2002).
Clearly, for large values of S, the tRS under consideration acts as a switch, controlled by the particular value of κ1: for κ1 < κ1,c, the tRS reaches the “low”steady state, whereas, for κ1 > κ1,c, it reaches the “high” steady state. This is illustrated in Fig. 9, which also indicates that the tRS under consideration is robust, in the sense that the system can effectively cope with environmental changes. This is another essential biological property (e.g., see the discussion in Kitano, 2002) illustrated by our example. Temporary changes in external conditions may cause temporary changes in mRNA or protein concentrations. As soon as the external influences disappear, effective transcriptional regulation causes mRNA and protein concentrations to return back to their nominal steady-state values (see also Fig. 10 b).
FIGURE 9.

Illustration of the switching behavior of the tRS depicted in Fig. 5, for S = 8. The tRS switches between the “high” and the “low” steady states as a function of the transcription rate κ1 of gene 1.
FIGURE 10.

(a) Steady-state mRNA concentrations of the tRS depicted in Fig. 5, as a function of the affinity constant. (b) At the normal temperature of 37°C, the tRS approaches the “high” steady state. However, a heat induction at 40°C during a 24-h period results in the tRS to switch to a “lower” steady state. When the temperature reverses back to normal, the tRS stably switches back to the “high” steady state.
Fig. 10 a depicts the steady state (after 48 h) mRNA concentrations as a function of the affinity constant θ. This result indicates that, at large values of the affinity constant, gene 3 promotes activation (or repression) of enzymatic activity in the metabolic pathway under consideration, whereas, at small values of the affinity constant, gene 3 inhibits activation (or repression) of such activity. From Eq. 28, and for α1 = 1, ΔU1 ≤ 0, it is clear that the affinity constant monotonically increases as the temperature decreases. It is therefore expected that, at low temperatures, gene 3 will promote activation (or repression) of enzymatic activity in the metabolic pathway, whereas, at high temperatures, gene 3 will inhibit such activity. However, this may not be true, because parameters β, γ, κ, and λ depend on temperature as well (e.g., recall Eq. 4). Fig. 10 b illustrates how changes in temperature may affect transcriptional regulation. At the normal temperature of 37°C, the tRS under consideration reaches the “high” steady state, whereas, at a temperature of 40°C, the tRS switches to a “higher” steady state. To obtain this result, we consider a 15% decrease in the value of the affinity constant θ, and assume a 20% increase in the values of κ and λ, and no change in the values of β and γ (we assume zero activation energies for mRNA and protein degradation). Fig. 10 b also indicates that the metabolic pathway under consideration is robust to heat induction: the underlying tRS can effectively cope with a temporary increase in temperature, by reversing the mRNA and protein concentrations back to their nominal steady-state values after the temperature returns back to its previous value.
In a real situation, nonzero time delays should be specified. It has been recently shown by Kobayashi et al. (2003a) that, under certain conditions (which are satisfied by the model presented in this article), the steady-state behavior of a tRS with only positive feedback loops (i.e., loops that contain activators and possibly an even number of repressors) does not depend on time delays. However, this is not true when the tRS contains negative feedback loops, in which case time delays may directly affect steady-state behavior. It turns out that the feedback loops of the tRS depicted in Fig. 5 are all positive except one. The loop
 |
involves an activator and a repressor and, therefore, it is a negative feedback loop. Hence, the steady-state behavior of the tRS depicted in Fig. 5 may depend on the time delays.
Fig. 11 depicts typical evolutions of mRNA concentrations of the tRS under consideration, when the time delays are taken to be nonzero and gene independent. We assume that the size of an average gene is ∼40,000 nucleotides, which implies that τr = 2000 s (we take vr = 20 nucleotides/s). We also assume that the same average gene is transcribed to an mRNA of ∼1200 nucleotides long, which is then translated to a protein composed of 400 amino acids. This implies that τp = 200 s (we take vp = 2 codons/s). Finally, we assume that the cis-regulation delay τc is, on the average, ∼20% larger than the transcription delay τr, in which case τc = 2400 s. By comparing the first row of Fig. 11 with Fig. 6, it is clear that it takes longer for the tRS to reach steady state in the case of nonzero time delays. As a matter of fact, for the evolution depicted in Fig. 11 a, it takes ∼72 h to reach steady state, as compared to ∼48 h for the evolution depicted in Fig. 6 a, whereas, for the evolution depicted in Fig. 11 b, it takes more than six days to reach steady state, as compared to ∼24 h for the evolution depicted in Fig. 6 b. Moreover, the evolutions are more complicated in the case of nonzero delays, although the same steady state is reached in both cases, because time delays do not affect the steady-state behavior of the tRS under consideration, as it is clear from Eqs. 17 and 18.
The situation changes if the rate of translation is increased to a value λ = 0.2 s−1. The second row of Fig. 11 depicts typical evolutions of mRNA concentrations for this case. When κ1 = 0.01 pM s−1, the tRS converges to a steady-state mRNA and protein concentration vector after ∼24 h. However, a 10-fold reduction in the value of κ1 results in a tRS that converges to a stable limit-cycle attractor. This is illustrated in Fig. 11 d. Although genes 1 and 2 shut off, genes 3 and 4 initiate a self-sustained oscillation whose cycle is completed in ∼16 h at steady state. This corroborates the fact that a tRS with at least one negative feedback loop may effectively be used to model gene-expression “clocks” (e.g., see Smolen et al., 2000).
We conclude this section with a brief discussion on how the time step Δt, used in the implementation of the discrete dynamical system, affects simulation accuracy and computational efficiency. As we have discussed before, Δt = σδt, where δt = 0.05 s and σ > 1 is a resolution parameter that provides a trade-off between simulation accuracy and computational efficiency. Larger values of σ lead to a more efficient implementation of the tRS at the expense of simulation accuracy. However, due to the slow timescales of transcription, translation, and degradation, as compared to the value of δt, large values of σ can be afforded, without compromising simulation accuracy. This is illustrated in Fig. 12, which depicts four evolutions of mRNA concentrations, obtained by using the same parameters as the ones used in Fig. 11 c, when σ = 1, 300, 6000, 72,000. These values correspond to Δt = 0.05 s, 15 s, 5 min, and 1 h, as well as to 1,728,000, 5760, 288, and 24 iterations, respectively. Note that, as σ increases, simulation accuracy decreases. The results obtained with σ = 6000 or 72,000 are clearly not acceptable, although they may provide a coarse approximation of mRNA evolution. Moreover, these results converge to the desired steady state, because the steady-state behavior of the tRS under consideration does not depend on σ, as it is clear from Eqs. 17 and 18. However, the evolutions obtained with σ = 300 are very close to the ones obtained with σ = 1.
If we assume that the relative errors, of approximating, at each time point, the evolutions of mRNA concentrations obtained when σ = 1 with the evolutions obtained when σ > 1, are statistically independent and identically distributed random variables, then we can estimate their mean value and standard deviation by standard empirical formulas. Fig. 13 depicts such estimates for the example depicted in Fig. 12, as a function of 1 ≤ σ ≤ 400. These results indicate that the number of iterations required for simulating the discrete dynamical model under consideration can be dramatically reduced with only a small compromise in simulation accuracy. For example, and according to Fig. 13, at any time point, the relative error of approximating the mRNA concentration depicted in Fig. 12 when σ = 1 with the one obtained when σ = 300, will on the average be equal to 5 × 10−4, with a standard deviation of ∼10−3. However, with σ = 300, the iterations required for simulating transcriptional regulation are 300 times fewer than the iterations required when σ = 1.
FIGURE 13.

The mean value and the standard deviation of the relative error in approximating, at each time point, the evolutions of mRNA concentrations obtained when σ = 1, with the evolutions obtained when 1 ≤ σ ≤ 400, as a function of σ.
CONCLUSION
In this article, we have considered the problem of modeling transcriptional regulation in a large population of cells. We have adopted a standard model for transcriptional regulation, based on ordinary differential equations that model transcription and translation, coupled with nonlinear equations that model cis-regulation. Simple arguments from chemical kinetics have led us to derive a model for cis-regulation that encompasses both activators and repressors, as well as the notion of regulatory modules. The need to use computational techniques for the analysis and simulation of transcriptional regulation has motivated us to derive a discrete model. Derivation of such a model is possible under certain assumptions and leads to a nonlinear discrete dynamical system, which is easy to implement and can be used to simulate transcriptional regulation in an iterative fashion. Moreover, the steady-state behavior of the proposed discrete dynamical system is identical to that of the continuous model.
We have discussed several mathematical properties of our model and have elaborated on their biological significance. Model implementation requires knowledge of several parameters, which are directly related to the biochemical mechanisms of transcription, translation, and cis-regulation. We have briefly discussed the problem of determining such parameters. We have adopted a hypothetical metabolic pathway, which we use to illustrate several properties of our model and show that a nonlinear dynamical system may effectively be used to quantitatively model transcriptional regulation in a biologically relevant way.
We have derived the proposed model for the case of transcriptional regulation in a large population of cells. However, it is also desirable to describe transcriptional regulation in a single cell, to take into account uncertainties about parameter values, and characterize modeling errors introduced by exemplifying transcriptional regulation. This entails development of a model by means of probabilistic techniques that effectively deals with uncertainty, for which the model presented in this article may serve as an “average” model. Construction of such a model for transcriptional regulation is currently under investigation.
Acknowledgments
The authors express their gratitude to Dr. Michael L. Bittner. His knowledge and expertise have been valuable for formulating our ideas and for completing this article. The authors would also like to thank Lei Xu for pointing out reference Wang et al. (1999).
References
- Akutsu, T., S. Miyano, and S. Kuhara. 2000a. Algorithms for identifying Boolean networks and related biological networks based on matrix multiplication and fingerprint function. J. Comput. Biol. 7:331–343. [DOI] [PubMed] [Google Scholar]
- Akutsu, T., S. Miyano, and S. Kuhara. 2000b. Inferring qualitative relations in genetic networks and metabolic pathways. Bioinformatics. 16:727–734. [DOI] [PubMed] [Google Scholar]
- Alberts, B., A. Johnson, J. Lewis, M. Raff, K. Roberts, and P. Walter. 2002. Molecular Biology of the Cell, 4th Ed. Garland Science, New York.
- Arkin, A., P. Shen, and J. Ross. 1997. A test case of correlation metric construction of a reaction pathway from measurments. Science. 277:1275–1279. [Google Scholar]
- Arkin, A., J. Ross, and H. H. McAdams. 1998. Stochastic kinetic analysis of developmental pathway bifurcation in phage λ-infected Escherichia coli cells. Genetics. 149:1633–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone, M. I., and E. H. Davidson. 1997. The hardwiring of development: organization and function of genomic regulatory systems. Development. 124:1851–1864. [DOI] [PubMed] [Google Scholar]
- Bagley, R. J., and L. Glass. 1996. Counting and classifying attractors in high dimensional dynamical systems. J. Theor. Biol. 183:269–284. [DOI] [PubMed] [Google Scholar]
- Baldi, P., and G. W. Hatfield. 2002. DNA Microarrays and Gene Expression: From Experiments to Data Analysis and Modeling. Cambridge University Press, Cambridge, UK.
- Bittner, M., P. Meltzer, Y. Chen, Y. Jiang, E. Seftor, M. Hendrix, M. Radmacher, R. Simon, Z. Yakhini, A. Ben-Dor, N. Sampas, E. Dougherty, E. Wang, F. Marincola, C. Gooden, J. Lueders, A. Glatfelter, P. Pollock, J. Carpten, E. Gillanders, D. Leja, K. Dietrich, C. Beaudry, M. Berens, D. Alberts, V. Sondak, N. Hayward, and J. Trent. 2000. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 406:536–540. [DOI] [PubMed] [Google Scholar]
- Bolouri, H., and E. H. Davidson. 2002. Modeling DNA sequence-based cis-regulatory gene networks. Dev. Biol. 246:2–13. [DOI] [PubMed] [Google Scholar]
- Brown, P. O., and D. Botstein. 1999. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 21:33–37. [DOI] [PubMed] [Google Scholar]
- Carrier, T. A., and J. D. Keasling. 1997. Controlling messenger RNA stability in bacteria: strategies for engineering gene expression. Biotechnol. Prog. 13:699–708. [DOI] [PubMed] [Google Scholar]
- Caselle, M., F. D. Cunto, and P. Provero. 2002. Correlating overrepresented upstream motifs to gene expression: a computational approach to regulatory element discovery in eukaryotes. BMC Bioinformatics. 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T., H. L. He, and G. M. Church. 1999. Modeling gene expression with differential equations. In Proceedings of the Pacific Symposium on Biocomputing. R. B. Altman, K. Lauderdale, A. K. Dunker, L. Hunter, and T. E. Klein, editors. World Scientific, Singapore. 4:29–40. [PubMed]
- Cherry, J. L., and F. R. Adler. 2000. How to make a biological switch. J. Theor. Biol. 203:117–133. [DOI] [PubMed] [Google Scholar]
- Davidson, E. H. 2001. Genomic Regulatory Systems: Development and Evolution. Academic Press, San Diego, CA.
- de Jong, H. 2002. Modeling and simulation of genetic regulatory systems: a literature review. J. Comput. Biol. 9:67–103. [DOI] [PubMed] [Google Scholar]
- D'haeseleer, P., S. Liang, and R. Somogyi. 2000. Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics. 16:707–726. [DOI] [PubMed] [Google Scholar]
- Elowitz, M. B., and S. Leibler. 2000. A synthetic oscillatory network of transcriptional regulators. Nature. 403:335–338. [DOI] [PubMed] [Google Scholar]
- Endy, D., D. Kong, and J. Yin. 1997. Intracellular kinetics of a growing virus: a genetically structured simulation for bacteriophage T7. Biotechnol. Bioeng. 55:375–389. [DOI] [PubMed] [Google Scholar]
- Endy, D., and R. Brent. 2001. Modelling cellular behaviour. Nature. 409:391–395. [DOI] [PubMed] [Google Scholar]
- Espenson, J. H. 1995. Chemical Kinetics and Reaction Mechanisms, 2nd Ed. McGraw-Hill, New York.
- Gardner, T. S., C. R. Cantor, and J. J. Collins. 2000. Construction of a genetic toggle switch in Escherichia coli. Nature. 403:339–342. [DOI] [PubMed] [Google Scholar]
- Gibson, M. A., and E. Mjolsness. 2001. Modeling the activity of single genes. In Computational Modeling of Genetic and Biochemical Networks. J. M. Bower and H. Bolouri, editors. MIT Press, Cambridge, MA. 3–48.
- Glass, L., and S. A. Kauffman. 1973. The logical analysis of continuous, non-linear biochemical control networks. J. Theor. Biol. 39:103–129. [DOI] [PubMed] [Google Scholar]
- Goldbeter, A., D. Gonze, G. Houart, J.-C. Leloup, J. Halloy, and G. Dupont. 2001. From simple to complex oscillatory behavior in metabolic and genetic control networks. Chaos. 11:247–260. [DOI] [PubMed] [Google Scholar]
- Golub, T. R., D. K. Slonim, P. Tamayo, C. Huard, M. Gaasenbeek, J. P. Mesirov, H. Coller, M. L. Loh, J. R. Downing, M. A. Caligiuri, C. D. Bloomfield, and E. S. Lander. 1999. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 286:531–537. [DOI] [PubMed] [Google Scholar]
- Grunberg-Manago, M. 1999. Messenger RNA stability and its role in control of gene expression in bacteria and phages. Annu. Rev. Genet. 33:193–227. [DOI] [PubMed] [Google Scholar]
- Gygi, S. P., Y. Rochon, B. R. Franza, and R. Aebersold. 1999. Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19:1720–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, B. J. 1993. Quantitative study of the control of HIV-1 gene expression. J. Theor. Biol. 163:199–221. [DOI] [PubMed] [Google Scholar]
- Hargrove, J. L., and F. H. Schmidt. 1989. The role of mRNA and protein stability in gene expression. FASEB J. 3:2360–2370. [DOI] [PubMed] [Google Scholar]
- Hartwell, L. H., J. J. Hopfield, S. Leibler, and A. W. Murray. 1999. From molecular to modular cell biology. Nature. 402:C47–C52. [DOI] [PubMed] [Google Scholar]
- Hasty, J., J. Pradines, M. Dolnik, and J. J. Collins. 2000. Noise-based switches and amplifiers for gene expression. Proc. Natl. Acad. Sci. USA. 97:2075–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty, J., F. Isaacs, M. Dolnik, D. McMillen, and J. J. Collins. 2001a. Designer gene networks: towards fundamental cellular control. Chaos. 11:207–220. [DOI] [PubMed] [Google Scholar]
- Hasty, J., D. McMillen, F. Isaacs, and J. J. Collins. 2001b. Computational studies of gene regulatory networks: in numero molecular biology. Nat. Rev. Genet. 2:268–279. [DOI] [PubMed] [Google Scholar]
- Hatzimanikatis, V., and K. H. Lee. 1999. Dynamical analysis of gene networks requires both mRNA and protein expression information. Metab. Eng. 1:275–281. [DOI] [PubMed] [Google Scholar]
- Herschlag, D., and F. B. Johnson. 1993. Synergism in transcriptional activation: a kinetic view. Genes Dev. 7:173–179. [DOI] [PubMed] [Google Scholar]
- Hill, T. L. 1985. Cooperativity Theory in Biochemistry: Steady-State and Equilibrium Systems. Springer-Verlag, New York.
- Holstege, F. C. P., E. G. Jennings, J. J. Wyrick, T.-I. Lee, C. J. Hengartner, M. R. Green, T. R. Golub, E. S. Lander, and R. A. Young. 1998. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 95:717–728. [DOI] [PubMed] [Google Scholar]
- Iyer, V., and K. Struhl. 1996. Absolute mRNA levels and transcriptional initiation rates in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 93:5208–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman, S. A. 1993. The Origins of Order: Self Organization and Selection in Evolution. Oxford University Press, New York.
- Kitano, H. 2002. Systems biology: a brief overview. Science. 295:1662–1664. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., L. Chen, and K. Aihara. 2003a. Modeling genetic switches with positive feedback loops. J. Theor. Biol. 221:379–399. [DOI] [PubMed] [Google Scholar]
- Kobayashi, T., M. Yamaguchi, S. Kim, J. Morikawa, S. Ogawa, S. Ueno, E. Suh, E. Dougherty, I. Shmulevich, H. Shiku, and W. Zhang. 2003b. Microarray reveals differences in both tumors and vascular specific gene expression in de Novo CD5+ and CD5− diffuse large B-cell lymphomas. Cancer Res. 63:60–66. [PubMed] [Google Scholar]
- Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J.-B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford, and R. A. Young. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 298:799–804. [DOI] [PubMed] [Google Scholar]
- Liang, S., S. Fuhrman, and R. Somogyi. 1998. REVEAL, a general reverse engineering algorithm for inference of genetic network architectures. In Proceedings of the Pacific Symposium on Biocomputing. R. B. Altman, A. K. Dunker, L. Hunter, and T. E. Klein, editors. World Scientific, Singapore. 3:18–29. [PubMed]
- Liebermeister, W. 2002. Linear modes of gene expression determined by independent component analysis. Bioinformatics. 18:51–60. [DOI] [PubMed] [Google Scholar]
- Lorsch, J. R., and D. Herschlag. 1999. Kinetic dissection of fundamental processes of eukaryotic translation initiation in vitro. EMBO J. 18:6705–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir, E., E. M. Munro, G. M. Odell, and G. von Dassow. 2002. Ingeneue: a versatile tool for reconstituting genetic networks, with examples from the segment polarity network. J. Exp. Zool. 294:216–251. [DOI] [PubMed] [Google Scholar]
- Mestl, T., E. Plahte, and S. W. Omholt. 1995. A mathematical framework for describing and analysing gene regulatory networks. J. Theor. Biol. 176:291–300. [DOI] [PubMed] [Google Scholar]
- Michelson, A. M. 2002. Deciphering genetic regulatory codes: a challenge for functional genomics. Proc. Natl. Acad. Sci. USA. 99:546–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjolsness, E., D. H. Sharp, and J. Reinitz. 1991. A connectionist model of development. J. Theor. Biol. 152:429–453. [DOI] [PubMed] [Google Scholar]
- Mjolsness, E. 2001. Trainable gene regulation networks with applications to Drosophila pattern formation. In Computational Modeling of Genetic and Biochemical Networks. J. M. Bower and H. Bolouri, editors. MIT Press, Cambridge, MA. 101–117.
- Moore, J. W., and R. G. Pearson. 1981. Kinetics and Mechanism, 3rd Ed. John Wiley, New York.
- Ronen, M., R. Rosenberg, B. I. Shraiman, and U. Alon. 2002. Assigning numbers to the arrows: parameterizing a gene regulation network by using accurate expression kinetics. Proc. Natl. Acad. Sci. USA. 99:10555–10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandefur, J. T. 1993. Discrete Dynamical Modeling. Oxford University Press, New York.
- Savageau, M. A. 2001. Design principles for elementary gene circuits: elements, methods, and examples. Chaos. 11:142–159. [DOI] [PubMed] [Google Scholar]
- Schena, M., D. Shalon, R. Heller, A. Chai, P. O. Brown, and R. W. Davis. 1996. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc. Natl. Acad. Sci. USA. 93:10614–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulevich, I., E. R. Dougherty, and W. Zhang. 2002. From Boolean to probabilistic Boolean networks as models of genetic regulatory networks. Proceedings of the IEEE. 90:1778–1792. [Google Scholar]
- Smolen, P., D. A. Baxter, and J. H. Byrne. 2000. Modeling transcriptional control in gene networks: methods, recent results, and future directions. Bull. Math. Biol. 62:247–292. [DOI] [PubMed] [Google Scholar]
- Somogyi, R., S. Fuhrman, and X. Wen. 2001. Genetic network inference in computational models and applications to large-scale gene expression data. In Computational Modeling of Genetic and Biochemical Networks. J. M. Bower and H. Bolouri, editors. MIT Press, Cambridge, MA. 119–157.
- Sørensen, M. A., and S. Pedersen. 1991. Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons, GAA and GAG are translated with a threefold difference in rate. J. Mol. Biol. 222:265–280. [DOI] [PubMed] [Google Scholar]
- Tavazoie, S., J. D. Hughes, M. J. Campbell, R. J. Cho, and G. M. Church. 1999. Systematic determination of genetic network architecture. Nat. Genet. 22:281–285. [DOI] [PubMed] [Google Scholar]
- Thomas, R., and R. D'Ari. 1990. Biological Feedback. CRC Press, Boca Raton, FL.
- Turner, G. C., and A. Varshavsky. 2000. Detecting and measuring cotranslational protein degradation in vivo. Science. 289:2117–2120. [DOI] [PubMed] [Google Scholar]
- Tyson, J. J., M. T. Borisuk, K. Chen, and B. Novak. 2001. Analysis of complex dynamics in cell cycle regulation. In Computational Modeling of Genetic and Biochemical Networks. J. M. Bower and H. Bolouri, editors. MIT Press, Cambridge, MA. 287–305.
- VanBogelen, R. A., K. D. Greis, R. M. Blumenthal, T. H. Tani, and R. G. Matthews. 1999. Mapping regulatory networks in microbial cells. Trends Microbiol. 7:320–328. [DOI] [PubMed] [Google Scholar]
- Vohradský, J. 2001. Neural network model of gene expression. FASEB J. 15:846–854. [DOI] [PubMed] [Google Scholar]
- Voit, E. O. 2000. Computational Analysis of Biochemical Systems. A Practical Guide for Biochemists and Molecular Biologists. Cambridge University Press, New York.
- von Dassow, G., E. Meir, E. M. Munro, and G. M. Odell. 2000. The segment polarity network is a robust developmental module. Nature. 406:188–192. [DOI] [PubMed] [Google Scholar]
- Wahde, M., and J. Hertz. 2001. Modeling genetic regulatory dynamics in neural development. J. Comput. Biol. 8:429–442. [DOI] [PubMed] [Google Scholar]
- Wang, J., K. Ellwood, A. Lehman, M. F. Carey, and Z.-S. She. 1999. A mathematical model for synergistic eukaryotic gene activation. J. Mol. Biol. 286:315–325. [DOI] [PubMed] [Google Scholar]
- Wang, Y., C. L. Liu, J. D. Storey, R. J. Tibshirani, D. Herschlag, and P. O. Brown. 2002. Precision and functional specificity in mRNA decay. Proc. Natl. Acad. Sci. USA. 99:5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver, D. C., C. T. Workman, and G. D. Stormo. 1999. Modeling regulatory networks with weight matrices. In Proceedings of the Pacific Symposium on Biocomputing. R. B. Altman, K. Lauderdale, A. K. Dunker, L. Hunter, T. E. Klein, editors. World Scientific, Singapore. 4:112–123. [DOI] [PubMed]
- Wolf, D. M., and F. H. Eeckman. 1998. On the relationship between genomic regulatory element organization and gene regulatory dynamics. J. Theor. Biol. 195:167–186. [DOI] [PubMed] [Google Scholar]
- Yeung, M. K. S., J. Tegnér, and J. J. Collins. 2002. Reverse engineering gene networks using singular value decomposition and robust regression. Proc. Natl. Acad. Sci. USA. 99:6163–6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim, N., and M. C. Mackey. 2003. Feedback regulation in the lactose operon: a mathematical modeling study and comparison with experimental data. Biophys. J. 84:2841–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H., M. Bilgin, R. Bangham, D. Hall, A. Casamayor, P. Bertone, N. Lan, R. Jansen, S. Bidlingmaier, T. Houfek, T. Mitchell, P. Miller, R. A. Dean, M. Gerstein, and M. Snyder. 2001. Global analysis of protein activities using proteome chips. Science. 293:2101–2105. [DOI] [PubMed] [Google Scholar]