

Abstract
The β-hairpin fold mechanism of a nine-residue peptide, which is modified from the β-hairpin of α-amylase inhibitor tendamistat (residues 15–23), is studied through direct folding simulations in explicit water at native folding conditions. Three 300-nanosecond self-guided molecular dynamics (SGMD) simulations have revealed a series of β-hairpin folding events. During these simulations, the peptide folds repeatedly into a major cluster of β-hairpin structures, which agree well with nuclear magnetic resonance experimental observations. This major cluster is found to have the minimum conformational free energy among all sampled conformations. This peptide also folds into many other β-hairpin structures, which represent some local free energy minimum states. In the unfolded state, the N-terminal residues of the peptide, Tyr-1, Gln-2, and Asn-3, have a confined conformational distribution. This confinement makes β-hairpin the only energetically favored structure to fold. The unfolded state of this peptide is populated with conformations with non-native intrapeptide interactions. This peptide goes through fully hydrated conformations to eliminate non-native interactions before folding into a β-hairpin. The folding of a β-hairpin starts with side-chain interactions, which bring two strands together to form interstrand hydrogen bonds. The unfolding of the β-hairpin is not simply the reverse of the folding process. Comparing unfolding simulations using MD and SGMD methods demonstrate that SGMD simulations can qualitatively reproduce the kinetics of the peptide system.
INTRODUCTION
A β-hairpin is a small protein structure motif in which two β-strands, linked by a turn or a short loop, fold to form hydrogen bonds with each other. Unlike helices, β-hairpins involve interactions between nonlocal amino acids. Experimental studies have shown that β-hairpins possess many characteristics of proteins in their folding behavior, typically, the two-state transition (Munoz et al., 1997; Honda et al., 2000). Therefore, β-hairpin becomes the simplest model for protein folding study.
The mechanism of β-hairpin folding has been the goal of many experimental, computational, and theoretical studies, which have been reviewed by Galzitskaya et al. (2002). However, due to the lack of direct access to the microscopic phenomena, its folding mechanism remains an unanswered question. Many models have been proposed to describe the β-hairpin folding mechanism. Typically, two models, the hydrophobic-core-centric model and the hydrogen-bond-centric model, exist to describe the β-hairpin folding procedure. The hydrogen-bond-centric model assumes that the formation of a folding droplet starting from the β-turn is the determining factor in transition kinetics. The hydrophobic-core-centric model proposes that a core structure formed by side chains from both strands comes first, and then brings the two strands together to form hydrogen bonds.
Molecular simulation of β-hairpin folding at native folding conditions with atomic details can provide direct clues to solve this problem. However, because the timescale of β-hairpin folding is beyond the reach of an all-atom molecular simulation at native folding conditions, people have to perform simulation studies indirectly or with certain simplifications. A typical simplification is using implicit solvation models to replace solvent molecules so that simulation can be sped up significantly. For example, using a solvent-accessible surface area-based solvation model, Wang et al. (1999) simulated the β-hairpin folding of a model peptide, (D–Val)4–Pro–Gly–(Val)4. Schaefer et al. (1998) performed a molecular dynamics simulation of a synthetic β-hairpin forming peptide (BH8) using the analytical continuum solvent potential. Ferrara and Caflisch (2000) simulated the reversible folding of a three-stranded antiparallel β-sheet of a designed 20-residue sequence with an all-atom description and an implicit solvent model. Another strategy is using high temperature to accelerate conformational search. Bonvin and van Gunsteren (2000) performed molecular dynamics simulations for a 19-residue peptide from the α-amylase inhibitor tendamistat in explicit water at high temperatures (360 K and 400 K). In their 30-ns simulations, they observed partial β-hairpin structures briefly. Galzitskaya et al. (2000) studied the β-hairpin folding mechanism through a simulated annealing approach so that some partial β-hairpin conformations could be reached in several nanoseconds.
Due to the difficulty of accessing β-hairpin folding events directly, especially with explicit solvent, many studies address the β-hairpin folding mechanism indirectly. For example, Dinner et al. (1999) performed a multicanonical Monte Carlo simulation on a 16-residue peptide using a Gaussian solvent exclusion model. Pande and Rokhsar (1999) performed unfolding simulations to identify transition states and performed refolding simulations from these transition states to study the folding of a β-hairpin fragment of protein G in explicit water. Bryant et al. (2000) used mechanical forces to unfold a β-hairpin structure in their simulation study. Among indirect simulation approaches, the replica-exchange approach is very attractive for overcoming the multiple-minima problem. Garcia and Sanbonmatsu (2001) and Zhou et al. (2001) applied this method to explore the free energy landscape of a β-hairpin.
It is arguable that these simplifications and indirect simulation approaches may introduce uncertainties in β-hairpin folding studies. Zhou and Berne (2002) demonstrated that the free energy landscape from an implicit solvation model is different from that with explicit water. It has long been recognized that proteins do not search all their conformational space to fold. Therefore, protein folding should not be treated as a nonphysical conformational search problem. High temperature simulations, as well as the replica-exchange method, may not be suitable to address protein folding behavior.
So far, molecular simulation studies with the simplifications or indirect approaches mentioned above have resulted in conflicting observations of the β-hairpin folding mechanism. The hydrogen-bond-centric model was supported by the simulation results of Wang et al. (1999) and Bonvin and van Gunsteren (2000), and the hydrophobic-centric-model was supported by the studies of Dinner et al. (1999), Pande and Rokhsar (1999), Bryant et al. (2000), and Zhou and Linhananta (2002). The results of the replica-exchange simulation by Zhou and co-workers gave a blend of the two models that the hydrophobic core and the β-strand hydrogen bonds form at roughly the same time (Zhou et al., 2001).
Obviously, a direct simulation of β-hairpin folding at native conditions with all-atom details would be highly informative. Recently, we reported a successful simulation of reversible β-hairpin folding of a synthetic peptide in explicit water at native folding conditions through self-guided molecular dynamics (SGMD) simulation (Wu et al., 2002). The SGMD method was developed to enhance systematic motion in molecular systems. The systematic motion, which can be described as an average motion over a certain time period, of a macromolecule is normally very slow as compared to its thermal motion, especially when the molecule is trapped in a local minimum state. By enhancing the slow systematic motion, a protein spends less time in random walk and in local energy minimum states so that the folding timescale is reduced. It has been demonstrated that SGMD simulation is capable of addressing slow events like crystallization (Wu and Wang, 1999; Shinoda and Mikami, 2001), peptide folding (Wu and Wang, 1998, 2000, 2001), and molecular capturing (Varady et al., 2002). This direct access to reversible β-hairpin folding events provides us with an opportunity to examine the β-hairpin folding mechanism. In this work, we present a series of reversible β-hairpin folding events observed in three 300-ns SGMD simulations. Analysis of these reversible folding events provides us structural and energetic insights into the β-hairpin folding mechanism. In addition, to address the concerns about how well a SGMD simulation reproduces the kinetics of an MD simulation, we present two unfolding simulations using the MD and SGMD methods for comparison.
METHODS AND CONDITIONS
Simulation systems
The nine-residue peptide studied here was designed by Blanco et al. (1993), which was modified from the β-hairpin of α-amylase inhibitor tendamistat (residues 15–23). The chemical structure of this peptide is shown in Fig. 1. For the convenience of discussion, we named the amino acids by their three character names and their sequence numbers as Tyr-1, Gln-2, Asn-3, Pro-4, Asp-5, Gly-6, Ser-7, Gln-8, and Ala-9, respectively. We chose this peptide because strong nuclear magnetic resonance (NMR) nuclear Overhauser effect (NOE) evidence indicates that this peptide folds into a β-hairpin structure in an aqueous solution (Blanco et al., 1993). In addition, the small size of this peptide makes a simulation with explicit water not too expensive to perform. The simulation system contains one molecule of the peptide, a single sodium ion, and 725 TIP3P water (Jorgensen et al., 1983) molecules (Fig. 2).
FIGURE 1.

The chemical structure of the synthetic peptide.
FIGURE 2.

The simulation system in a periodic cubic boundary. The peptide is shown as thick sticks, the sodium ion is shown as a sphere, and the water molecules are shown as thin sticks. Carbon, oxygen, nitrogen, hydrogen, and sodium atoms are colored as green, red, blue, white, and yellow, respectively.
Simulation conditions
Three 300-ns simulations, labeled as A, B, and C, with different starting conformations are reported here. Simulation A was started with an extended conformation. The backbone dihedral angles, φ for dihedral angle C–N–Cα–C and ψ for dihedral angle N–Cα–C–N, are 180° except for the φ-dihedral angle of Pro-4, which is determined by the ring structure of the proline side chain. Simulation B was started from a high temperature coil conformation, which was generated by a high temperature simulation of the aqueous system from system A at 600 K for 1000 picoseconds (ps) followed by 100-ps equilibrium at 274 K. The starting conformation of simulation C was generated from a high temperature simulation of the peptide in a vacuum. The peptide alone was simulated at 1000 K for 1000 ps and then solvated with a sodium ion and 725 TIP3P water molecules followed by a 100-step steepest gradient energy minimization. All the three simulations were performed with a cubic periodic boundary condition at constant temperature (274 K) and constant volume (29 × 29 × 29 Å3). The AMBER force field (Cornell et al., 1995) (PARM96) was used to describe the system, and the particle-mesh Ewald method (York et al., 1993) was used for the electrostatic interaction calculation. The self-guided molecular dynamics (SGMD) simulation method (Wu and Wang, 1998) was used with a local sampling time of 0.2 ps and a guiding factor of 0.1. Simulation conformations and energy data were stored every 10 ps for postsimulation analysis.
Conformational analysis
Because the conformational free energy, which determines the conformational distribution of a peptide, cannot be calculated directly from simulation conformations, we use the Generalized-Born (GB) model to estimate the solvation free energy (Jayaram et al., 2000) in our postsimulation analysis. The MM_PBSA module provided with the AMBER6 program (Perlman et al., 1995) was used for this calculation. The conformational free energy is calculated using
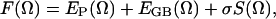 |
where Ω represents a peptide conformation, F, EP, EGB, and S are the conformational free energy, intrapeptide interaction, GB electrostatic interaction, and solvent-accessible surface area, respectively. σ is the surface tension coefficient and takes a value of 0.0072 kcal/mol Å2.
Hydrogen bonds are an important property for secondary structure description. We define a hydrogen bond between an oxygen atom and a polar hydrogen atom when their distance is shorter than 2.4 Å. A β-hairpin structure has hydrogen bonds between its two strands. These hydrogen bonds are called interstrand hydrogen bonds. The peptide studied here has a proline at position 4 (Fig. 1), which is designed to form the turn structure of the β-hairpin. Therefore, we define an interstrand hydrogen bond as the backbone hydrogen bond between residues before Pro-4, i.e., Tyr-1, Gln-2, and Asn-3, and those after Pro-4, i.e., Asp-5, Gly-6, Ser-7, Gln-8, and Ala-9. An interstrand hydrogen bond is denoted by the residue indexes of its bonding atoms as iO-jH, where iO is the residue index of the oxygen atom and jH is the residue index of the hydrogen atom. For example, 3–6 represents a hydrogen bond between the carbonyl oxygen of residue 3 and the amide hydrogen of residue 6, and 6–3 represents the hydrogen bond between the amide hydrogen of residue 3 and the carbonyl oxygen of residue 6.
A β-hairpin structure can be uniquely defined by its interstrand hydrogen bonds. For convenience, we define a hydrogen bond pattern as one or more interstrand hydrogen bonds within a β-hairpin structure. A hydrogen bond pattern is expressed as a group of interstrand hydrogen bonds enclosed by a pair of braces. For example, the hydrogen bond pattern 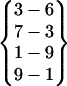 represents a β-hairpin structure with four interstrand hydrogen bonds, between O of Asn-3 and H of Gly-6, between H of Asn-3 and O of Ser-7, between O of Tyr-1 and H of Ala-9, and between H of Tyr-1 and O of Ala-9. A hydrogen bond pattern can have one or more parent patterns, which have only some of its hydrogen bonds. For example,
represents a β-hairpin structure with four interstrand hydrogen bonds, between O of Asn-3 and H of Gly-6, between H of Asn-3 and O of Ser-7, between O of Tyr-1 and H of Ala-9, and between H of Tyr-1 and O of Ala-9. A hydrogen bond pattern can have one or more parent patterns, which have only some of its hydrogen bonds. For example, 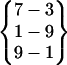 ,
, 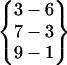 , and
, and 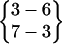 are all the parent pattern of
are all the parent pattern of 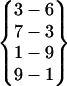 . Because interstrand hydrogen bonds are frequently forming and breaking during a simulation, even at a stable β-hairpin structure, a β-hairpin structure may transit from one hydrogen bond pattern to some of its parent patterns.
. Because interstrand hydrogen bonds are frequently forming and breaking during a simulation, even at a stable β-hairpin structure, a β-hairpin structure may transit from one hydrogen bond pattern to some of its parent patterns.
RESULTS AND DISCUSSIONS
The three 300-ns simulations provide abundant structural and energetic information for the aqueous system. Here, we focus our analysis and presentation on β-hairpin folding events to highlight the understanding gained from these simulations.
The folding of the peptide into a β-hairpin structure is characterized by the formation of interstrand hydrogen bonds. Fig. 3 shows the number of interstrand hydrogen bonds observed during these three simulations. Each peak in Fig. 3 represents a β-hairpin folding-unfolding event. It is clear that there are β-hairpin structures folding and unfolding repeatedly during these simulations.
FIGURE 3.

The number of interstrand hydrogen bonds during the three SGMD simulations. Interstrand hydrogen bonds are hydrogen bonds between the backbone atoms of Tyr-1–Gln-2–Asn-3 and backbone atoms of Gly-6–Ser-7–Gln-8–Ala-9. Each peak in the plot represents a β-hairpin folding-unfolding event. Thirteen folding-unfolding events are identified as labeled and are analyzed in this article.
Repeated folding and unfolding is called reversible folding, which is essential for successful simulation studies of protein folding. Without reversible folding events, a simulation could either end with a compact structure, which could be a state entrapped in a local minimum, or with a coil state, from which it is hard to tell whether the peptide does not fold or the simulation is not long enough. In real biologic systems, proteins are believed to exist in equilibrium between the unfolded state and the folded state with continuous folding and unfolding transitions, i.e., proteins undergo continuous reversible folding processes. Therefore, an informative simulation of protein folding should reproduce reversible folding events.
In Fig. 3 we identified 13 reversible folding events with two or more interstrand hydrogen bonds. These folded species are labeled as Fold 1 through Fold 13. Table 1 lists the hydrogen bond patterns and time frames. As can be seen, there were several types of β-hairpin structures folded during these simulations. The repeated occurrence of a β-hairpin with an interstrand hydrogen bond pattern of 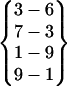 (Fold 2, Fold 6, Fold 11, and Fold 12) in these simulations (see Table 1) indicates that reversible folding of this β-hairpin structure has been achieved.
(Fold 2, Fold 6, Fold 11, and Fold 12) in these simulations (see Table 1) indicates that reversible folding of this β-hairpin structure has been achieved.
TABLE 1.
Time frames, interstrand hydrogen bond patterns, and average energies of the folded species as labeled in Fig. 3
| Start time, ps | End time, ps | EP, kcal/mol | EGB, kcal/mol | Esa, kcal/mol | FGB, kcal/mol | Hydrogen bond pattern | |
|---|---|---|---|---|---|---|---|
| Sim. A | 0 | 300,000 | −185.28 ± 0.21 | −261.03 ± 0.19 | 8.56 ± 0.00 | −437.75 ± 0.05 | {…} |
| Fold 1 | 15,320 | 16,030 | −193.14 ± 9.83 | −254.02 ± 8.35 | 8.30 ± 0.14 | −438.87 ± 4.89 | 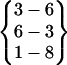 |
| Fold 2 | 20,720 | 27,870 | −246.16 ± 0.85 | −209.56 ± 0.80 | 7.41 ± 0.01 | −448.30 ± 0.22 | 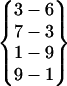 |
| Fold 3 | 105,900 | 107,960 | −262.42 ± 0.90 | −189.89 ± 0.83 | 7.57 ± 0.01 | −444.73 ± 0.33 | 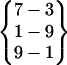 |
| Fold 4 | 148,790 | 149,730 | −243.86 ± 2.00 | −208.85 ± 1.85 | 7.72 ± 0.03 | −445.00 ± 0.66 | 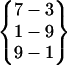 |
| Fold 5 | 165,660 | 168,920 | −283.09 ± 0.81 | −171.33 ± 0.73 | 7.81 ± 0.01 | −446.62 ± 0.32 | 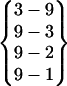 |
| Fold 6 | 184,350 | 191,970 | −269.80 ± 0.69 | −185.36 ± 0.63 | 7.38 ± 0.01 | −447.78 ± 0.21 | 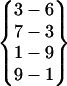 |
| Fold 7 | 255,070 | 255,600 | −246.13 ± 2.05 | −203.46 ± 1.73 | 7.78 ± 0.03 | −441.81 ± 0.88 | 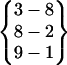 |
| Sim. B | 0 | 300,000 | −181.95 ± 0.17 | −264.23 ± 0.16 | 8.54 ± 0.00 | −437.64 ± 0.04 | {…} |
| Fold 8 | 8,930 | 10,110 | −271.00 ± 1.17 | −181.90 ± 1.07 | 7.59 ± 0.03 | −445.31 ± 0.42 |
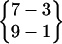 or or 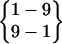
|
| Fold 9 | 44,240 | 49,780 | −208.62 ± 0.99 | −237.61 ± 0.92 | 8.06 ± 0.01 | −438.17 ± 0.24 | 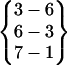 |
| Fold 10 | 180,180 | 180,180 | −219.42 ± 0.00 | −229.91 ± 0.00 | 7.97 ± 0.00 | −441.36 ± 0.00 | 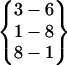 |
| Fold 11 | 211,420 | 215,880 | −266.76 ± 0.62 | −187.85 ± 0.53 | 7.42 ± 0.01 | −447.19 ± 0.20 | 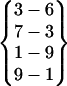 |
| Sim. C | 0 | 300,000 | −177.70 ± 0.15 | −266.78 ± 0.14 | 8.79 ± 0.00 | −435.70 ± 0.04 | {…} |
| Fold 12 | 72,030 | 73,050 | −260.30 ± 2.77 | −193.72 ± 2.00 | 7.53 ± 0.02 | −446.49 ± 1.17 | 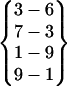 |
| Fold 13 | 74,300 | 78,920 | −250.09 ± 1.66 | −200.97 ± 1.56 | 7.86 ± 0.01 | −443.20 ± 0.29 | 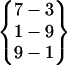 |
In the following sections, we are going to address the β-hairpin folding mechanism by answering some basic questions about the folding of this peptide: What is the folded structure? Why does this peptide fold into a β-hairpin? How does this peptide fold into a β-hairpin? And how does the β-hairpin unfold?
What is the folded structure?
The success of a simulation study of a real system is determined by two factors: 1), an accurate model system and 2), an efficient simulation method. The model system used to represent a real system should be accurate enough to reproduce the behavior of the real system. On the other hand, the simulation method used should be efficient enough to reach the conformational state or timescale to be studied.
Previously, timescale has been the major obstacle in protein folding studies because normal MD simulations with explicit water can only reach a submicrosecond timescale, far less than the timescale for actual protein folding, which normally is above milliseconds. In a SGMD simulation, the system undergoes an enhanced conformational search, which brings slow events like the β-hairpin folding of this peptide to a reachable timescale. Once the folded state is reachable, immediate questions arise: What is the folded structure? And how does it compare with the real system? This comparison would be important to validate the force field and simulation conditions.
Fig. 4 shows the representing structures from these folding events. A representing structure is the conformation that has the smallest root mean-square deviation from the average structure calculated over the folded period. β-hairpin structures with the same interstrand hydrogen bond pattern are similar to each other. As can be seen, of the 13 folded structures, Fold 2, Fold 6, Fold 11, and Fold 12 have the same hydrogen bond pattern, 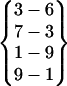 , and are very similar to each other. Therefore, they are categorized into one cluster (cluster I). Fold 3, Fold 4, Fold 8, and Fold 13 also have the same hydrogen bond pattern,
, and are very similar to each other. Therefore, they are categorized into one cluster (cluster I). Fold 3, Fold 4, Fold 8, and Fold 13 also have the same hydrogen bond pattern, 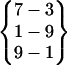 (in Fold 8 these three interstrand hydrogen bonds did not coexist during its folded period), and belong to another cluster (cluster II). The rest of the folded structures, Fold 1, Fold 5, Fold 7, Fold 9, and Fold 10, have their own interstrand hydrogen bond patterns. They do not belong to these two clusters and are different from each other.
(in Fold 8 these three interstrand hydrogen bonds did not coexist during its folded period), and belong to another cluster (cluster II). The rest of the folded structures, Fold 1, Fold 5, Fold 7, Fold 9, and Fold 10, have their own interstrand hydrogen bond patterns. They do not belong to these two clusters and are different from each other.
FIGURE 4.

Representative conformations of each folded structure. Only backbone atoms are shown. Atoms are colored as described in the legend of Fig. 2 except for the structures of cluster I and cluster II. Hydrogen bonds are marked by dotted lines.
Based on the frequency of folding and the duration of the folded periods (Table 1), it is clear that cluster I represents the major folded structure. Cluster I structures have a turn structure involving residues Asn-3, Pro-4, Asp-5, and Gly-6 with a hydrogen bond between the carbonyl oxygen of Asn-3 and the amide hydrogen of Gly-6 {3–6}. This turn is a common type turn and cannot directly link a tight β-hairpin structure because of the conflict between the twist of the turn structure and that of β-strands (Sibanda and Thornton, 1985; Richardson and Richardson, 1989). Gly-6 serves as a bulge to correct this twist conflict. The two β-strands, Tyr-1–Gln-2–Asn-3 and Ser-7–Gln-8–Ala-9, are hydrogen-bonded with each other through interstrand hydrogen bonds between Tyr-1 and Ala-9, 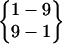 , and between Asn-3 and Ser-7, {7–3}.
, and between Asn-3 and Ser-7, {7–3}.
Cluster II structures are also visited in all three simulations. But their folded periods are much shorter than those of cluster I structures (see Table 1). The hydrogen bond pattern of cluster II is 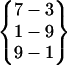 , which is similar to that of cluster I except for the turn hydrogen bond, {3–6}. From Fig. 4 we can see that residues Pro-4, Asp-5, and Gly-6, form a loop structure, linking the two β-strands. When examining the structures, we find that, in addition to the difference in the turn structure, the two clusters are also different in their side-chain packing. Fig. 5 highlights the side-chain difference between these two clusters. As can be seen from Fig. 5, in cluster I, the side chain of Gln-2 is on top of that of Gln-8, whereas in cluster II, it is the other way around. Also, due to the difference between the turn and the loop structure, the side chain of Asp-5 points upward in cluster II but points away in cluster I.
, which is similar to that of cluster I except for the turn hydrogen bond, {3–6}. From Fig. 4 we can see that residues Pro-4, Asp-5, and Gly-6, form a loop structure, linking the two β-strands. When examining the structures, we find that, in addition to the difference in the turn structure, the two clusters are also different in their side-chain packing. Fig. 5 highlights the side-chain difference between these two clusters. As can be seen from Fig. 5, in cluster I, the side chain of Gln-2 is on top of that of Gln-8, whereas in cluster II, it is the other way around. Also, due to the difference between the turn and the loop structure, the side chain of Asp-5 points upward in cluster II but points away in cluster I.
FIGURE 5.

Comparison of the side-chain packing in cluster I and cluster II structures. Only heavy atoms are shown. The side chains of Gln-2, Asp-5, and Gln-8 are colored purple.
Other β-hairpin structures are only observed once, suggesting they are not dominant folded states. Fold 1 is a tight β-hairpin structure with a hydrogen bond pattern of 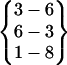 . The turn structure on Asn-3–Pro-4–Asp-5–Gly-6 is not an ideal common type turn. The carbonyl oxygen of Asn-3 and the amide hydrogen of Gly-6 are oriented toward the solvent. Because of the conflict between the turn structure and the twist of β-strands, the β-hairpin cannot extend to form the H1-O8 hydrogen bond, {8–1}. Fold 5 is a β-hairpin structure with a glycine turn-like structure on residue Pro-4–Asp-5–Gly-6–Ser-7. A glycine turn often has a glycine at the third position, as in this case. However, the turn hydrogen bond O4–H7 never forms throughout its folded period. Its hydrogen bond pattern is
. The turn structure on Asn-3–Pro-4–Asp-5–Gly-6 is not an ideal common type turn. The carbonyl oxygen of Asn-3 and the amide hydrogen of Gly-6 are oriented toward the solvent. Because of the conflict between the turn structure and the twist of β-strands, the β-hairpin cannot extend to form the H1-O8 hydrogen bond, {8–1}. Fold 5 is a β-hairpin structure with a glycine turn-like structure on residue Pro-4–Asp-5–Gly-6–Ser-7. A glycine turn often has a glycine at the third position, as in this case. However, the turn hydrogen bond O4–H7 never forms throughout its folded period. Its hydrogen bond pattern is 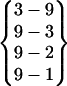 . Obviously, the reason that this β-hairpin structure is relatively stable is because the carboxyl group at the C-terminal forms three hydrogen bonds with the N-terminal strand. Fold 7 does not have a turn structure. Its hydrogen bond pattern is
. Obviously, the reason that this β-hairpin structure is relatively stable is because the carboxyl group at the C-terminal forms three hydrogen bonds with the N-terminal strand. Fold 7 does not have a turn structure. Its hydrogen bond pattern is 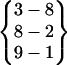 . A loop in helical turn conformation links Asn-3 to Gln-8. This structure formed only very briefly, indicating it is not stable. Fold 9 has a hydrogen bond pattern of
. A loop in helical turn conformation links Asn-3 to Gln-8. This structure formed only very briefly, indicating it is not stable. Fold 9 has a hydrogen bond pattern of 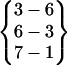 . Like Fold 1, it has a tight β-turn structure. Fold 10 is a tight β-hairpin with a β-turn similar to Fold 1 and Fold 9 but is very unstable.
. Like Fold 1, it has a tight β-turn structure. Fold 10 is a tight β-hairpin with a β-turn similar to Fold 1 and Fold 9 but is very unstable.
The β-hairpin structure of this peptide has been detected by NMR experiments (Blanco et al., 1993). Fig. 6 compares the observed NMR NOEs and the average proton-proton distances averaged over all the folded periods of cluster I structures. For comparison, the average proton-proton distances of cluster II structures are also shown in Fig. 6. Theoretically, an NOE observation indicates a short distance between the corresponding atom pair. Normally, an NOE is observable when the distance between an atom pair is <5 Å, and the shorter the distance is, the stronger the NOE will be. However, many other factors affect the strength of NOEs. When interpreting NOE data, we should keep in mind that the strength of NOEs only provides qualitative information about atom pair distances. As can be seen from Fig. 6, in cluster I structures, the distances of all atom pairs with observable NOEs fall well within or around the observable range (5 Å). On the contrary, the folded structures in cluster II violate several long-range NOEs (Fig. 6, distances in bold numbers). Therefore, the cluster I structure is the folded structure identified in the NMR experiment. This agreement suggests that the force field used in the simulation (AMBER PARM96) can correctly describe the folded structure of this peptide system. Even though Garcia and Sanbonmatsu (2002) claim that PARM96 biases the β-strand conformation, our simulations have shown that this force field can reproduce the high helix content for a helix folding peptide (Wu and Wang, 2001). Nonetheless, in our simulations, only ∼2–7% of conformations are β-hairpins, far lower than the number observed in the NMR experiment (Blanco et al., 1993), which indicates that the force field needs improvement. We have tried the AMBER PARM99 force field for the same system and at the same simulation condition, but no β-hairpin structure was observed in our 200-ns simulation. It should be noted that the fact that cluster II structures do not show certain NOEs does not mean these structures do not exist. This peptide in solution most likely has many possible conformations, as well as the unfolded structures, as suggested by Blanco et al. (1993).
FIGURE 6.

Comparison of the experimental NOEs and the average distances of corresponding atom pairs of cluster I and cluster II (in parentheses). The distances violating the NOEs are shown in bold numbers.
Predicting protein structures has been one of the ultimate goals of molecular simulation. With accurate force field and sufficient computing resources, molecular simulation can produce the folded structure of proteins. To predict folded structures from simulations, we not only need a simulation long enough to reach the folded state, but we also need to identify the folded structures from simulation conformations. These reversible folding simulations shown here are a typical example to predict or identify folded structures from simulations.
Why does it fold to a β-hairpin?
Protein native states are widely viewed to be the global free energy minima, and protein folding is a free energy downhill process. For this peptide aqueous system, is the β-hairpin the global minimum state?
Because the conformational free energies cannot be calculated directly from an MD simulation, we used an implicit solvation model, the Generalized-Born model plus the surface tension, to estimate solvent contribution as described in Methods and Conditions. The averages of the conformational free energy and each of its components were calculated for each folded species, as well as the whole simulations, and the results are also listed in Table 1.
It is clear from Table 1 that the structures of cluster I (Folds 2, 6, 11, and 12) have the lowest conformational free energies. In other words, cluster I represents the free energy minimum state among the conformations searched during these simulations. Even though these three 300-ns SGMD simulations cannot guarantee that all of the important states have been explored, the diversity of starting conformations and the convergence of the minimum free energy structures suggest that cluster I is very likely the global free energy minimum state. All other folded species have lower conformational free energies than the simulation averages, indicating they are local free energy minimum states.
The decrease of the conformational free energy during the β-hairpin folding comes from the combined changes in intrapeptide interaction and solvent contribution. Using Fold 2 as an example, the average free energy is 10.5 ± 0.2 kcal/mol lower than the average free energy of simulation A. Of this free energy difference, −61.0 ± 1.0 kcal/mol is contributed from intrapeptide potential energy, whereas 51.5 ± 1.0 kcal/mol comes from solvent electrostatic interaction, and only −1.1 ± 0.0 kcal/mol is from the surface tension. It is clear that the driving force for the β-hairpin folding is the intrapeptide interaction. The solvent electrostatic interaction strongly opposes the folding, and the surface tension favors the folded state but has a very limited contribution. The same result is observed for all other folded species.
According to the solvent-peptide interaction, we know that the solvation effect favors conformations with polar groups exposed to solvent and disfavors compact structures with many polar groups inaccessible to solvent. If a peptide goes to a compact structure with its polar groups buried inside, it has to gain enough interaction energy to compensate for the loss in solvation energy. Because the solvation energy loss is large, 51.5 ± 1.0 kcal/mol in the case of Fold 2, only very limited compact structures can result in enough intrapeptide interaction to compensate solvation energy loss. From Table 1, we can see that Fold 5 is the one with the lowest intrapeptide energy. However, it has the highest solvation energy, which makes the conformational free energy of Fold 5 higher than those of cluster I structures. The global minimum structure (cluster I structures) is not the one with the strongest intrapeptide interaction. It is the balance of the intrapeptide interaction and solvent interaction that determines the conformational free energy. Therefore, the solvation effect prevents the peptide from folding to structures whose gain in intrapeptide interaction cannot compensate for the loss in solvation interaction. In other words, the solvation effect restricts the conformational space for the peptide to fold and prevents the peptide from folding into misfolded structures.
The simulations also provide a structural insight into why this peptide folds into a β-hairpin. It is well known that the amino acid sequence of a protein determines its folded structure. Analysis of our simulation conformations shows that the conformational distribution of an amino acid in an unfolded state contains certain structural characteristics which may determine the folded structure. Fig. 7 shows the φ−ψ dihedral angle distributions of Gln-2, Asp-3, Ser-7, and Gln-8, obtained from our simulations. Because the folded structures represent only ∼2–7% of the conformations in these three simulations, the distributions describe mainly the unfolded states.
FIGURE 7.

The φ–ψ distributions of Gln-2, Asn-3, Ser-7, and Gln-8 observed in our simulations.
Comparing these φ−ψ distributions, we can see that the two glutamine residues, Gln-2 and Gln-8, are very different from each other. Gln-2 distributes mainly in the vicinity of a β-strand conformation (φ = −120°, ψ = 120°), whereas Gln-8 spreads over every region. This distribution difference is the result of the difference in their neighboring amino acids or the difference in amino acid sequences. Particularly, Asn-3 is completely restricted to the region near a β-strand. In protein structures, statistical data indicates that Asn has a significant population in all regions, especially in the right-handed and left-handed α-helical regions (Richardson and Richardson, 1989), which, in this case, is very different from the distribution of Asn-3. This conformational constraint comes from the solvent environment. It is the water environment that causes this sequence-dependent conformational preference. As a result, the N-terminal residues of this peptide are strongly restrained to a β-strand conformation, which prohibits the formation of a helical structure. In other words, in the unfolded state, the peptide contains a certain number of local structures in its N-terminal residues. These local structures greatly limit the foldable conformational space of the peptide. It is this structural confinement of the unfolded state that makes the β-hairpin a favored structure to fold.
How does the peptide fold to a β-hairpin?
Now we come to the question of how the peptide goes from the unfolded state to a β-hairpin. First, let us examine what the unfolded state is. During the three simulations, the folded state (cluster I) only accounted for ∼2–7% of all conformations. That is, these simulations searched mainly the unfolded state. We calculated the population of conformations as a function of the number of intrapeptide hydrogen bonds, and the result is shown in the lower panel of Fig. 8. As can be seen, the most populated conformations have two or three intrapeptide hydrogen bonds. The fully hydrated conformations, which do not have any intrapeptide hydrogen bonds, account for ∼10% of the simulation conformations. This result indicates that the peptide is likely to have some intrapeptide interactions in the unfolded state. Other than the cluster I structures, the β-hairpin structures, shown in Fig. 4, represent some local minimum structures in the unfolded state.
FIGURE 8.

The populations and average energies of the peptide at a different number of intrapeptide hydrogen bonds during each SGMD simulation.
The upper panel of Fig. 8 shows the average conformational free energies (F), as well as intrapeptide interactions (Ep) and solvent contributions (EGB), as a function of the number of intrapeptide hydrogen bonds (NHB). As the number of intrapeptide hydrogen bonds increases, the average intrapeptide interaction energy decreases, whereas the average solvation energy increases. The combination is that the average conformational free energy decreases slightly as the number of intrapeptide hydrogen bonds increases. In other words, the intrapeptide interaction promotes more intrapeptide hydrogen bonds, whereas the solvation interaction prevents the formation of intrapeptide hydrogen bonds. The combined result is that an increase in intrapeptide hydrogen bonds corresponds to a decrease in the conformational free energy. The fully hydrated coil conformations with zero intrapeptide hydrogen bond represent a high conformational free energy state.
From the unfolded state to the folded state, the peptide is changing from conformations with some intrapeptide interactions to conformations with native interactions. In most cases, the intrapeptide interactions in the unfolded state are not native interactions. Therefore, to fold from the unfolded state, the peptide needs to get rid of these non-native interactions first and gain the native interactions later.
To examine the interaction change during β-hairpin folding, we calculated the number of backbone hydrogen bonds, side-chain hydrogen bonds, intrapeptide hydrogen bonds, peptide-water hydrogen bonds, and total peptide hydrogen bonds during each folding period. Fig. 9 shows these hydrogen bond numbers from 10,000 ps to 30,000 ps in simulation A, which covers Fold 1 and Fold 2 periods. As discussed above, Fold 1 represents a local minimum structure with many non-native interactions, and Fold 2 represents the global minimum state.
FIGURE 9.

The numbers of hydrogen bonds from 10,000 ps to 30,000 ps in simulation A. The shadowed areas mark the folded periods of Fold 1 and Fold 2. HBMM, HBSP, HBPP, HBPW, and HBP are numbers of hydrogen bonds between backbone atoms, between side chains and any peptide atoms, between any two peptide atoms, between the peptide and water, and between the peptide and any atoms, respectively.
As can be seen from Fig. 9, from Fold 1 (∼15,320–16,030 ps) to Fold 2 (∼20,700–27,870 ps), all intrapeptide hydrogen bonds of Fold 1 broke at ∼18,000 ps and were replaced by peptide-water hydrogen bonds. From 18,000 ps to 19,600 ps, the peptide was in a fully hydrated state with few intrapeptide hydrogen bonds. Starting from 19,650 ps, intrapeptide interactions increased until the peptide reached the folded structure. For all other reversible folding events, we also observed the same phenomenon that fully hydrated conformations were visited before each folding event. We concluded that the solvent-peptide interaction plays an important role to prepare the peptide to fold. In the case of this small peptide, a fully hydrated conformation is the result of such preparation. Of course, not all fully hydrated conformations will lead to a folding process.
It would be interesting to examine the interactions that induce the folding process. As can be seen from Fig. 9, the local minimum state, Fold 1, was reached at 15,320 ps when the interstrand hydrogen bond number jumped to 3; and the global minimum state, Fold 2, was reached at 20,700 ps when the interstrand hydrogen bond number jumped to 4. The numbers of sidechain-peptide hydrogen bonds became significant as early as 11,000 ps for Fold 1 and 20,000 ps for Fold 2. These interactions involved one or several side chains of Gln-2, Asn-3, Ser-7, and Gln-8. In every folding event, the peptide was found to form sustained side-chain hydrogen bonds before β-hairpin folding, indicating that side-chain interactions played an important role in the peptide folding.
From Fig. 9 we can see that the number of intrapeptide hydrogen bonds changes opposite to the number of peptide-water hydrogen bonds so that the number of total peptide hydrogen bonds remains nearly constant during each folding event. This is the result of the balance between the intrapeptide interaction and the peptide-solvent interaction. Each successful folding should compensate for the loss in the peptide-solvent interaction through the gain in the intrapeptide interaction. The side-chain interactions (including sidechain-sidechain and sidechain-backbone interactions) are an immediate way to replace peptide-water interaction in the early folding stage. Backbone-backbone interaction can only form at the last stage of the folding when the peptide is almost a compact structure. From a fully solvated structure to a folded structure, the peptide must give up many peptide-water interactions by replacing them with the sidechain-peptide interactions, some of which will be replaced by backbone-backbone hydrogen bonds before reaching the final folded structure.
Fig. 10 shows conformational changes of the peptide from the unfolded state to the folded state, Fold 2, in simulation A. The conformation at 17,000 ps has some non-native interactions, e.g., between the side chain of Tyr-1 and the C-terminal. These non-native interactions disappear when the peptide becomes fully hydrated (18,400 ps). At 19,700 ps, side-chain interactions involving Gln-2, Asn-3, and Gln-8 bring the two strands together. At 20,700 ps the two strands form interstrand hydrogen bonds, and the peptide folds into a β-hairpin structure. These conformational changes present us with a clear picture of how the peptide folds into a β-hairpin.
FIGURE 10.

Typical conformational changes during a β-hairpin folding. The conformation at 17,000 ps represents an unfolded structure with some intrapeptide interactions. At 184,000 ps the peptide becomes fully hydrated. Side-chain interactions bring the two strands together at 19,700 ps. At 20,700 ps the peptide reaches a β-hairpin structure. Only heavy atoms are shown. Backbone atoms are shown as thick sticks. Atoms are colored as described in the legend of Fig. 2.
The 13 β-hairpin folding events observed in our three SGMD simulations provide us with a direct way to examine current β-hairpin folding models. The hydrogen-bond-centric model emphasizes the role of hydrogen bonds in β-hairpin folding. This model assumes that a turn structure forms first, which brings two β-strands together to form a β-hairpin. Analysis of the 13 β-hairpin folding events shows that the formation of the interstrand hydrogen bonds does not follow a certain route. It can start from the turn structure, from the tails, or from the middle of the strands. Six of the 13 β-hairpins folded in our simulations, Folds 3, 4, 5, 7, 8, and 13, do not have a turn structure, which clearly demonstrates that a turn structure is not necessary for the β-hairpin folding. Therefore, our observation does not support the hydrogen-bond-centric model.
As for the hydrophobic-core-centric model, it does not apply to this peptide because it does not have strong enough hydrophobic residues to form a hydrophobic core. The only residue with an aromatic ring is Tyr-1, which points its side chain away from other residues in the folded structure (cluster I). However, if we extend the hydrophobic interaction to the general side-chain interaction, this model provides a reasonable description of the β-hairpin folding processes observed in our simulations. When there are several hydrophobic side chains, forming a hydrophobic core is an effective way to lower the conformational free energy without the loss in peptide-water interaction. Therefore, to be general, side-chain interactions, including both hydrophilic and hydrophobic interactions, bring β-strands together to form a β-hairpin. This conclusion is consistent with Zhou and Linhananta's simulation study (2002).
How does the β-hairpin unfold?
An interesting question in protein folding study is whether the unfolding process is a reversal of the folding process. We performed a SGMD simulation starting from a folded structure taken from Fold 2. Fig. 11 a shows the distances of the hydrogen bonding atom pairs during this simulation. As can be seen, this peptide remains a β-hairpin structure for 1.8 ns before unfolding. After 1.8 ns, all interstrand hydrogen bonds are broken, as evidenced by the increases in these distances. After the break of the interstrand hydrogen bonds, the peptide remains in a relatively compact but mobile structure with the help of the side-chain interactions. At 3.6 ns, the peptide becomes a random coil structure. After an extensive conformational search, this unfolded peptide refolds into a β-hairpin structure at ∼15 ns, which again unfolds at 16.1 ns.
FIGURE 11.

Distances of the backbone hydrogen bonding atoms during simulations starting from the folded state. The dashed lines mark the standard distance (2 Å) to form a hydrogen bond. (a) SGMD simulation; (b) MD simulation.
Similar to the folding processes observed in previous folding simulations, the unfolding process goes through an intermediate state with a significant number of intrapeptide side-chain interactions. The side chains of Gln-2, Asn-3, and Gln-8 are often involved in these interactions. However, the fully hydrated state observed at the beginning of a folding process does not appear right after the unfolding. Therefore, the unfolding process is not simply a reversal of the folding process. Our explanation of this difference is that the intermediate structure through which a folding process goes is unstable and quickly changes to either the folded structure when the folding is successful or an unfolded structure when the folding fails. Even though many collapses are observed in our long SGMD simulations, only a handful of them reach the folded structure. The intermediate state reached from the unfolded state is relatively stable; therefore, the peptide can stay there without going through a fully hydrated state. If the intermediate state reached from the folded state is not stable, the peptide is likely to refold back to its folded state.
To compare our SGMD simulations with conventional MD simulations, we performed MD simulations at exactly the same condition. In an MD simulation starting from the extended conformation, we failed to see any folding event up to 1 ms (1000,000 ps) probably because a 1-ms MD simulation is not long enough to reach the β-hairpin folding event. Fortunately, in an MD simulation starting from the folded conformation, we successfully observed the unfolding of this β-hairpin structure. Fig. 11 b shows the distances of the hydrogen bonding atom pairs during this simulation. As can be seen, the β-hairpin structure unfolded after 31.6 ns (31,600 ps) as evidenced by the increases in these distances. After the break of the interstrand hydrogen bonds, the peptide remained in a relatively compact structure with the help of side-chain interactions. At ∼35 ns, this peptide completely unfolded.
Like the SGMD unfolding simulation shown in Fig. 11 a, this simulation also reached a refolding event at ∼60 ns. The unfolded peptide went through a fully hydrated structure at 58.5 ns, as evidenced by the large distances, before folding into a β-hairpin structure at 61.4 ns when all distances are within the hydrogen bonding range. This β-hairpin structure is not very stable, as the hydrogen bonds, O3-H6 and O7-H3, break frequently. After 66.6 ns, the peptide unfolded to an intermediate structure involving many intrapeptide side-chain interactions. This intermediate unfolded completely at 75.2 ns.
This MD unfolding simulation verified many of our observations in above SGMD simulations. For example, the MD simulation showed that the folding and unfolding of the peptide go through an intermediate state with intrapeptide side-chain interactions. Also, the refolding event in the MD simulation showed that a fully hydrated state occurs at the beginning of the folding process. Comparing Fig. 11, a and b, we can see that the SGMD simulation produces a faster unfolding, refolding, and re-unfolding procedure than the MD simulation. These similarities indicate that, qualitatively, a SGMD simulation can more quickly reproduce the kinetics of an MD simulation.
CONCLUSIONS
The β-hairpin folding of a nine-residue peptide was directly simulated in explicit water at native folding conditions in three 300-ns SGMD simulations. Through structural and energetic analysis of the folding events, we answered some basic questions about the folding of this peptide in water.
This peptide folded into a series of β-hairpin structures in our simulations. The major cluster observed in these simulations agrees well with the NMR experimental observation. Energy calculations demonstrate that this major cluster very likely represents the global free energy minimum state, and other β-hairpin structures are local conformational free energy minimums.
Intrapeptide interactions drive the peptide to fold, and the solvation effect, which resists folding, is believed to prevent the peptide from folding into misfolded structures. The balance of the intrapeptide interaction and the solvation effect makes the folded structure the global minimum. In the unfolded state, the N-terminal residues of the peptide are confined to certain local structures, which make the β-hairpin a favored structure to fold.
In the unfolded state, the peptide often has a certain number of intrapeptide interactions. The peptide goes through a fully solvated state to get rid of these non-native interactions before folding. From a fully solvated structure, the peptide folds into a β-hairpin by replacing some peptide-solvent interactions with intrapeptide interactions. Side-chain interactions bring β-strands together to form a β-hairpin. The interstrand hydrogen bonds form at the last stage of the folding process.
The unfolding of the β-hairpin also goes through an intermediate state with many side-chain interactions. However, the unfolding process is not simply a reversal of the folding process. Comparison simulations with MD and SGMD methods demonstrate that SGMD can qualitatively reproduce the kinetics of the peptide system more quickly.
We should be cautious about the conclusions drawn from this simulation study. First, the peptide studied here is small in comparison to typical β-hairpin motifs in proteins. Further studies with larger peptides would be very helpful to generalize our understanding of the β-hairpin folding. Second, the force field used to model the system has a bias effect (Beachy et al., 1997; Garcia and Sanbonmatsu, 2002), which may distort the description of the peptide system. Third, 300-ns SGMD simulations are not long enough to provide an equilibrium description of the folded and unfolded states. Therefore, further studies are needed to enhance our understanding of the β-hairpin folding mechanism.
Acknowledgments
All simulations reported in this work were performed on the National Institutes of Health Biowulf computer cluster.
References
- Beachy, M. D., D. Chasman, R. B. Murphy, T. A. Halgren, and R. A. Friesner. 1997. Accurate ab initio quantum chemical determination of the relative energetics of peptide conformations and assessment of empirical force fields. J. Am. Chem. Soc. 119:5908–5920. [Google Scholar]
- Blanco, F. J., M. A. Jiménez, J. Herranz, M. Rico, J. Santoro, and J. L. Nieto. 1993. NMR Evidence of a short linear peptide that folds into a β-hairpin in aqueous solution. J. Am. Chem. Soc. 115:5887–5888. [Google Scholar]
- Bonvin, A. M., and W. F. van Gunsteren. 2000. Beta-hairpin stability and folding: molecular dynamics studies of the first beta-hairpin of tendamistat. J. Mol. Biol. 296:255–268. [DOI] [PubMed] [Google Scholar]
- Bryant, Z., V. S. Pande, and D. S. Rokhsar. 2000. Mechanical unfolding of a beta-hairpin using molecular dynamics. Biophys. J. 78:584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force field for the simulation of proteins and nucleic acids. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Dinner, A. R., T. Lazaridis, and M. Karplus. 1999. Understanding beta-hairpin formation. Proc. Natl. Acad. Sci. USA. 96:9068–9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara, P., and A. Caflisch. 2000. Folding simulations of a three-stranded antiparallel beta-sheet peptide. Proc. Natl. Acad. Sci. USA. 97:10780–10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galzitskaya, O. V., J. Higo, and A. V. Finkelstein. 2002. Alpha-helix and beta-hairpin folding from experiment, analytical theory and molecular dynamics simulations. Curr. Protein Pept. Sci. 3:191–200. [DOI] [PubMed] [Google Scholar]
- Galzitskaya, O. V., J. Higo, M. Kuroda, and H. Nakamura. 2000. β-hairpin folds by molecular dynamics simulations. Chem. Phys. Lett. 326:421–429. [Google Scholar]
- Garcia, A. E., and K. Y. Sanbonmatsu. 2001. Exploring the energy landscape of a beta-hairpin in explicit solvent. Proteins. 42:345–354. [DOI] [PubMed] [Google Scholar]
- Garcia, A. E., and K. Y. Sanbonmatsu. 2002. Alpha-helical stabilization by side chain shielding of backbone hydrogen bonds. Proc. Natl. Acad. Sci. USA. 99:2782–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda, S., N. Kobayashi, and E. Munekata. 2000. Thermodynamics of a beta-hairpin structure: evidence for cooperative formation of folding nucleus. J. Mol. Biol. 295:269–278. [DOI] [PubMed] [Google Scholar]
- Jayaram, B., D. Sprous, and D. L. Beveridge. 2000. Solvation free energy of biomacromolecules: parameters for a modified Generalized Born model consistent with the AMBER force field. J. Phys. Chem. B. 102:9571–9576. [Google Scholar]
- Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Munoz, V., P. A. Thompson, J. Hofrichter, and W. A. Eaton. 1997. Folding dynamics and mechanism of beta-hairpin formation. Nature. 390:196–199. [DOI] [PubMed] [Google Scholar]
- Pande, V. S., and D. S. Rokhsar. 1999. Molecular dynamics simulations of unfolding and refolding of a beta- hairpin fragment of protein G. Proc. Natl. Acad. Sci. USA. 96:9062–9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman, D. A., D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham III, S. Debolt, D. Ferguson, G. L. Seibel, and P. A. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties. Comp. Phys. Commun. 91:1–41. [Google Scholar]
- Richardson, J. S., and D. C. Richardson. 1989. Principles and patterns of protein conformation. In Prediction of Protein Structure and the Principles of Protein Conformation. Gerald D. Fasman, editor. Plenum Press, New York and London. 1–98.
- Schaefer, M., C. Bartels, and M. Karplus. 1998. Solution conformations and thermodynamics of structured peptides: molecular dynamics simulation with an implicit solvation model. J. Mol. Biol. 284:835–848. [DOI] [PubMed] [Google Scholar]
- Shinoda, W., and M. Mikami. 2001. Self-guided molecular dynamics in the isothermal-isobaric ensemble. Chem. Phys. Lett. 335:265–272. [Google Scholar]
- Sibanda, B., and J. Thornton. 1985. β−hairpin families in globular proteins. Nature. 316:170–174. [DOI] [PubMed] [Google Scholar]
- Varady, J., X. Wu, and S. Wang. 2002. Competitive and reversible binding of a guest molecule to its host in aqueous solution through molecular dynamics simulation: benzyl alcohol/a-cyclodextrin system. J. Phys. Chem. B. 106:4863–4872. [Google Scholar]
- Wang, H., J. Varady, L. Ng, and S. S. Sung. 1999. Molecular dynamics simulations of beta-hairpin folding. Proteins. 37:325–333. [DOI] [PubMed] [Google Scholar]
- Wu, X., S. Wang, and B. R. Brooks. 2002. Direct observation of the folding and unfolding of a beta-hairpin in explicit water through computer simulation. J. Am. Chem. Soc. 124:5282–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, X.-W., and S. Wang. 1998. Self-guided molecular dynamics simulation for efficient conformational search. J. Phys. Chem. B. 102:7238–7250. [Google Scholar]
- Wu, X.-W., and S. Wang. 1999. Enhancing systematic motion in molecular dynamics simulation. J. Chem. Phys. 110:9401–9410. [Google Scholar]
- Wu, X.-W., and S. Wang. 2000. Folding study of a linear pentamer peptide adopting a reverse turn conformation in aqueous solution through molecular dynamics simulation. J. Phys. Chem. B. 104:8023–8034. [Google Scholar]
- Wu, X., and S. Wang. 2001. Helix folding of an alanine-based peptide in explicit water. J. Phys. Chem. B. 105:2227–2235. [Google Scholar]
- York, D. M., T. A. Darden, and L. G. Pedersen. 1993. The effect of long-range electrostatic interactions in simulations of macromolecular crystals: a comparison of the Ewald and truncated list methods. J. Chem. Phys. 99:8345–8348. [Google Scholar]
- Zhou, R., and B. J. Berne. 2002. Can a continuum solvent model reproduce the free energy landscape of a β-hairpin folding in water? Proc. Natl. Acad. Sci. USA. 99:12777–12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, R., B. J. Berne, and R. Germain. 2001. The free energy landscape for beta hairpin folding in explicit water. Proc. Natl. Acad. Sci. USA. 98:14931–14936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., and A. Linhananta. 2002. Role of hydrophilic and hydrophobic contacts in folding of the second beta-hairpin fragment of protein G: molecular dynamics simulation studies of an all-atom model. Proteins. 47:154–162. [DOI] [PubMed] [Google Scholar]