

Abstract
Rhodopsin is the only member of the pharmacologically important superfamily of G-protein-coupled receptors with a known structure at atomic resolution. A molecular dynamics model of rhodopsin in a POPC phospholipid bilayer was simulated for 15 ns, revealing a conformation significantly different from the recent crystal structures. The structure of the bilayer compared with a protein-free POPC control indicated hydrophobic matching with the nonpolar interface of the receptor, in agreement with deuterium NMR experiments. A new generalized molecular surface method, based on a three-dimensional Voronoi cell construction for atoms with different radii, was developed to quantify cross-sectional area profiles for the protein, lipid acyl chains and headgroups, and water. Thus, it was possible to investigate the bilayer deformation due to curvature of the individual lipid monolayers. Moreover, the generalized molecular surface derived hydrophobic interface allowed benchmarking of the hydropathy sequence analysis, an important structural genomics tool. Five water molecules diffused into internal hydration sites during the simulation, yielding a total of 12 internal waters. The cytoplasmic loops and the C-terminal tail, containing the G-protein recognition and protein sorting sequences, exhibited a high mobility, in marked contrast to the extracellular and transmembrane domains. The proposed functional coupling of the highly conserved ERY motif to the lipid-water interface via the cytoplasmic loops provides insight into lipid effects on G-protein-coupled receptor activation in terms of a flexible surface model, involving the spontaneous monolayer curvature.
INTRODUCTION
Heterotrimeric guanine nucleotide-binding protein (G-protein)-coupled receptors (GPCRs) constitute a large family of sensory proteins, which respond to a variety of different external stimuli such as hormones, neurotransmitters, neuromodulators, odors, and light (Strader et al., 1995; Ballesteros et al., 2001). They act as the interface between a signal and the cellular response mediated by molecular switches, the family of regulatory G-proteins. Cell membrane receptors, many of them GPCRs, are pharmacologically very important, as they comprise ∼45% of all molecular drug targets of current therapies (Drews, 2000). Rhodopsin is the only GPCR whose structure is known at atomic resolution (Palczewski et al., 2000); it is a member of the largest receptor Family A, with 569 possible candidates identified in the human genome (Lander et al., 2001). The opsins and the protein, peptide, and cationic amine receptor subfamilies, constitute ∼90% of all known GPCRs (Palczewski et al., 2000). They share a conserved structure of seven transmembrane (TM) helices, and the sequences of many other GPCRs are used to build homology models based on rhodopsin (Baldwin et al., 1997; Lomize et al., 1999; Stenkamp et al., 2002). Light-induced photoisomerization of the covalently bound ligand of rhodopsin, 11-cis-retinal, to all-trans-retinal is followed by a series of thermal relaxations known in kinetic detail (Lewis and Kliger, 2000), leading to a tertiary structural rearrangement of the protein (Hubbell et al., 2003). With respect to conformational transitions there is a high similarity among the different GPCRs (Ghanouni et al., 2001). The subsequent steps of the rhodopsin-dependent signal transduction cascade (Ebrey and Koutalos, 2001) involve binding of the heterotrimeric G-protein transducin, resulting in GDP–GTP exchange, and dissociation of the subunits. Activation of a phosphodiesterase by the Gα·GTP subunit is followed by subsequent enzymatic processes, which regulate the cytosolic levels of second messengers. These lead ultimately to closing of cation channels and hyperpolarization of the cellular plasma membrane, giving the visual signal. The structural transitions of the primary photoproduct of rhodopsin culminate in the signaling state, involving the metastable intermediates metarhodopsin I (meta I) and metarhodopsin II (meta II), where the latter is the activated receptor conformation (Hofmann et al., 1995).
The thermodynamic equilibrium between the meta I and meta II conformations of photoactivated rhodopsin is strongly dependent on the lipid composition of the membrane (Beach et al., 1984), and a native-like lipid mixture results in optimal function (Wiedmann et al., 1988). Evidence has increasingly accumulated that the functions of other membrane proteins are similarly modulated by the lipid composition (Jensen and Schutzbach, 1984; Navarro et al., 1984; Epand, 1990; Lee, 1998; Perozo et al., 2002). One possibility entails specific protein and lipid interactions that are related to the domain organization of biomembranes (rafts) (Simons and Ikonen, 2000). Yet in case of rhodopsin the membrane lipids are entirely in the fluid, liquid-crystalline (Lα) state at physiological temperature (Deese et al., 1981; Miljanich et al., 1985; Ellena et al., 1986). An alternative is that nonspecific lipid/protein interactions may influence function due to biophysical properties of the membrane lipids, as described by a flexible surface model (FSM) (Gibson and Brown, 1993; Botelho et al., 2002). It has been argued that the microscopic organization of rod outer segment membranes leads to an active control or regulation of rhodopsin function by the local lipid environment (Beach et al., 1984; Baldwin and Hubbell, 1985; Wiedmann et al., 1988; Mitchell et al., 1992; Brown, 1997). Therefore, the standard fluid-mosaic model for cellular membranes, in which fluid-like phospholipid bilayers constitute a matrix for integral and peripheral membrane proteins, has been extended to account for modulation of membrane protein function due to the composition of the lipid matrix (Botelho et al., 2002). Such lipid regulation of protein function has clear implications for human diseases, as revealed by studies of the influences of dietary intake of essential and polyunsaturated fatty acids (Neuringer, 2000), and the well-known effects of cholesterol in cardiovascular dysfunction and various neurological disorders.
This study centers on a biomembrane model of a protein in a native-like phospholipid bilayer environment under physiological conditions, comprising dark-state rhodopsin and POPC at full hydration. The molecular dynamics (MD) model was based on the crystallographic membrane protein structure (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002), supplemented with experimental solid-state 2H NMR data for the phospholipid component. The microscopic structure of the rhodopsin-containing membrane model is founded on a 15-ns molecular dynamics computer simulation in an all-atom representation (Fig. 1), and is interpreted in terms of the hydrophobic protein/lipid interface. The MD simulations show that once rhodopsin is liberated from crystal-packing constraints and embedded in a native-like phospholipid bilayer environment, it relaxes to an ensemble of conformations different from the crystal structure. Information is obtained for additional internal water molecules and for parts of the protein that are crystallographically unresolved (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002). The definition of a new generalized molecular surface (GMS) approach has allowed us to quantify the contact interfaces between the molecular components of a biomembrane simulation in terms of hydropathy analysis. The average lipid monolayer curvature is inferred to be negative, which is relevant to the native membrane environment of rhodopsin, with a high degree of nonlamellar, reverse hexagonal (HII) phase-promoting phospholipids (Deese et al., 1981). We also suggest that association of the membrane-water interface with the cytoplasmic loop C2 and the adjacent ERY sequence motif of rhodopsin can impose stress/strain on the protein, thereby affecting its function. A remarkable stability of the transmembrane (TM) helix bundle in the bilayer is found in comparison to the extramembranous loops, which pertains to the physiological role of the receptor as a single quantum detector (Baylor, 1996). Finally, the current molecular dynamics model serves as a platform for future structure-function studies of rhodopsin and other homologous GPCRs (Huber et al., 2003). Preliminary results of this study have been communicated (Huber et al., 2002a).
FIGURE 1.

Simulated system demonstrating the location of rhodopsin in the bilayer. A side view of rhodopsin in a POPC bilayer membrane is shown (protein/lipid ratio 1:103); two adjacent unit cells indicate the protein with the extracellular (intradiskal) side up and the cytosolic side down. Transmembrane helices are depicted as rods, and the extramembranous loops as strings. White spheres indicate the lipid acyl carbonyl carbons representing the hydrophobic boundary, and the phosphates representing the headgroups are in orange. Red and blue spheres illustrate the ionic atmosphere of the aqueous region, corresponding to Na+ and Cl−, respectively. The palmitoylation of Cys322 and Cys323 is shown by the white space-filling chains penetrating deep into the lower monolayer. The retinal chromophore is depicted in red. A total of ∼45,000 atoms was used to describe the system, which was simulated for 15 ns using a 1-fs time step and full electrostatic calculations (cf. text). The system is shown at an early state of the simulations, after ∼3 ns of equilibration time, in which the cytoplasmic monolayer is not yet fully adapted to the protein.
COMPUTATIONAL AND EXPERIMENTAL METHODS
Modeling and computational aspects
All MD simulations were performed using the program NAMD2 (Kale et al., 1999). The necessary structure topology files were created with the modeling software CHARMM27 (Brooks et al., 1983). Based on previous studies (Huber et al., 2002b), we combined the CHARMM27b5f parameter set (Feller and MacKerell, 2000) with the F3C water model (Levitt et al., 1997) allowing for a fully flexible all-atom model of the whole system. In addition, the retinal force-field parameters from bacteriorhodopsin (Tajkhorshid et al., 2000) were included. Molecular models were qualitatively characterized by the visualization software VMD1.6 (Humphrey et al., 1996). The trajectories were quantitatively analyzed using the in-house developed program package MOLGEOM6 (Huber et al., 2002b), which represents a collection of tools including procedures to calculate segmental volumes by the Voronoi cell method (Goede et al., 1997), group density profiles, dihedral angle probability distributions, orientational order parameters, and GMS (cf. Appendix).
The MD simulation involved an all-atom model of the phospholipid/rhodopsin system (including the covalently bound retinal, the N-terminal acetylation and glycosylations, the C-terminal palmitoylations, and the disulfide bridge) with excess water and physiological salt concentration. To account for the experimentally determined protonation state of specific amino acids, changes of their ionization were performed during refinement of the protein models, as summarized in Table 1. The models were built on the basis of the recent crystal structures of rhodopsin (Palczewski et al., 2000; Teller et al., 2001), in which unresolved parts were added by distance geometry modeling methods (Blaney et al., 1995; Huber et al., 2002b). Briefly, missing residues of the crystal structure (cf. Table 1) were built in an extended conformation. The atoms represented in the crystal structure were treated in a rigid body approximation, fixing all internal degrees of freedom. The preprocessing algorithm of DGEOM95 (Blaney et al., 1995) generated a distance matrix of upper and lower distance limits from the covalent geometry and chirality of the fragments and their atomic van der Waals radii. Additional constraints were provided, describing the covalent bonds linking the fragments and the crystallographic-defined rhodopsin polypeptide chain. The distance geometry calculations generated a set of 20 protein models scored by pseudoenergy values derived from the distance constraints. The lowest energy conformations were free of constraint violations and were inspected for proper geometry and packing. Subsequently, the models were energy minimized with the CHARMM potential energy function, and inserted in an equilibrated bilayer model of POPC. The initial model of the POPC bilayer for the rhodopsin-containing simulation was generated by duplicating a previous, 72-lipid-containing POPC bilayer model (Huber et al., 2002b). Some of the water molecules and 20 lipids in each leaflet of the bilayer, whose excluded volumes overlapped with the rhodopsin molecule, were removed. Subsequently another lipid was taken out of the extracellular side, giving a total of 103 lipids per rhodopsin. The resulting defects in the bilayer structure were annealed with all the rhodopsin atoms constrained to fluctuate about the initial structure. Multiple time-step integration of the equations of motion allowed for a fully flexible covalent bond treatment with a 1.0-fs time step for the fastest degrees of freedom. The thermodynamic ensemble was a constant particle number, normal pressure, surface tension, and temperature (NPγT) ensemble applying Nosé-Hoover dynamics (Feller and Pastor, 1996). The target pressure was 1.0 bar, and target surface tensions for the simulations are given in the Appendix. As a consequence of the periodic boundary conditions, the systems were oriented multibilayer stacks of the pure lipid or the rhodopsin-containing bilayer at full hydration in the liquid-crystalline state. The temperature for all simulated systems was 27°C (300 K), with one exception noted in Table 2. To account for the long-range nature of the Coulombic interactions, a full electrostatics calculation with particle-mesh Ewald lattice summation (Darden et al., 1993) was applied. The three-dimensional (3-D) fast Fourier transform grid size of 80 × 80 × 96 for the rhodopsin-containing systems replaced the 64 × 64 × 64 grid for the reference POPC bilayer system. All other parameters for the MD integration were identical to previous membrane simulations (Huber et al., 2002b).
TABLE 1.
Rhodopsin models
| RhA | RhB | RhC | |
|---|---|---|---|
| Met1 (N-terminus) | 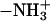 |
−NH(CO)CH3 | −NH(CO)CH3 |
| Asn2 (N-glycosylation) | (Nag)2 | (Nag)2 | (Nag)2 |
| Asn15 (N-glycosylation) | (Nag)2 | (Nag)2Man | (Nag)2Man |
| Asp83 and Glu122 | Deprotonated | Protonated* | Protonated |
| His211 | Protonated | Deprotonated* | Deprotonated |
| Other His† | Protonated | Protonated | Deprotonated |
| Internal water | 0 | 8 | 8 |
| Initial coordinates‡ | 1F88 chains A,C,E | 1HZX chains A,C,E | RhB |
| Missing§ side-chain coordinates | Residues 334–347 | None | None |
| Missing§ residue coordinates | Residues 236–239, 328–333 | Residues 236–240, 331–333 | None |
| Total protein charge | +2 | +2 | −3 |
| Extracellular domain | −3 | −3 | −6 |
| Cytosolic domain | +5 | +5 | +3 |
| Cys322 and Cys323 | Palmitoylated | ||
| Cys110 and Cys187 | Disulfide linkage | ||
| Lys296 | Protonated Schiff base with 11-cis-retinal | ||
| Ala348 | −COO− | ||
| Other Asp and Glu† | Deprotonated | ||
| Other Cys and Lys† | Protonated | ||
| All Arg, Ser, Thr, and Tyr† | Protonated | ||
| Hydrogen coordinates | Generated with PROTONATE | ||
These residues are neutral in the receptor ground state (Beck et al., 1998; Fahmy et al., 1993).
The protonation state of these residues was chosen according to pKa calculations (Wang et al., 2002) for pH 5 (RhA and RhB) and pH 7 (RhC).
The crystal unit cell contains a rhodopsin dimer; the PDB entry numbers are 1F88 (Palczewski et al., 2000) and 1HZX (Teller et al., 2001). The better-resolved monomer is comprised of three chains A, C, and E for the peptide, and two oligosaccharides. The initial coordinates for model RhC were derived from the equilibrated structure of model RhB.
Several residues and side chains were not resolved in the different crystal structures (Palczewski et al., 2000; Teller et al., 2001); their structure was modeled by fitting the missing fragments into the crystal structure by a distance geometry approach (cf. text).
TABLE 2.
Simulation details
| Segment | Protein model | Nc* | Ne | Nw | N+ | N− | γ/mN m−1 |
|---|---|---|---|---|---|---|---|
| L0 | – | 36 | 36 | 1831 | – | – | – |
| L1 | – | 36 | 36 | 1831 | – | – | 45 |
| L2 | – | 36 | 36 | 1831 | – | – | 32 |
| L3 | – | 36 | 36 | 1831 | – | – | 0 |
| L4† | – | 36 | 36 | 1831 | – | – | −‡ |
| P0§ | – | 72 | 72 | 3662 | – | – | – |
| P1¶ | RhA | 52 | 52 | 7209 | 11 | 13 | 45 |
| P2‖ | RhA | 52 | 52 | 8320 | 20 | 22 | 32–45 |
| P3 | RhA | 52 | 51** | 8320 | 20 | 22 | 32 |
| P4 | RhB†† | 52 | 51 | 8328‡‡ | 20 | 22 | −500–32 |
| P5 | RhC§§ | 52 | 51 | 8328 | 20 | 17 | 0–32 |
| P6¶¶ | RhC | 52 | 51 | 8328 | 20 | 17 | 0 |
The number of molecules in the simulation unit cell: Nc, lipids in the cytoplasmic monolayer; Ne, lipids in the extracellular monolayer; Nw, water; N+, cations; and N−, anions.
The equilibration of segment L3 continued after segment L4 branched off the main trajectory.
The segment L4 was simulated with a constant area per lipid of 62.4 Å2.
The initial membrane was created by doubling and rotating unit cell of L0.
The rhodopsin model RhA was inserted, overlapping lipid and water molecules were removed, and ions and more water were added. The initial 100 ps of segment P1 were performed at a temperature of 37°C (310 K), and all other segments (L1–L4 and P2–P6) were simulated at 27°C (300 K).
More water and ions were added.
One lipid in the extracellular monolayer was extracted from the membrane and subsequently removed from the aqueous space.
The model RhB replaced RhA in the membrane.
The internal water molecules of RhB and RhC are included in Nw.
The model RhC replaced RhB in the membrane.
The production period of the MD simulations is designated as the 5-ns segment P6 of the 15-ns MD simulations.
Rhodopsin recombinants and sample preparation
Rhodopsin-containing membranes with deuterium-labeled lipids were prepared as follows. The lipid studied was 1-perdeuteriopalmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC-d31; Avanti Polar Lipids, Alabaster, AL), in which the sn-1 chain was predeuterated. Rhodopsin was isolated from rod outer segment membranes prepared from bovine retinas (W. L. Lawson, Co., Lincoln, NE), purified, and recombined with POPC-d31 (1:100 molar ratio) by detergent dialysis as described (Botelho et al., 2002). The resulting membrane vesicles were concentrated by ultracentrifugation (100,000 × g for 1 h) and resuspended in a small volume of 67 mM sodium phosphate buffer, pH 7.0, prepared from 2H-depleted H2O (Aldrich, WI), containing 1 mM EDTA. The samples were stored in an argon atmosphere at −80°C and used directly for the NMR experiments. The A280/A500 ratio from ultraviolet-visible spectroscopy was 1.7 ± 0.1 for the recombinant membranes, and 2.4 ± 0.1 for the ROS membranes. The rhodopsin/POPC membranes used for the 2H NMR experiments were characterized in terms of their regenerability, viz. 11-cis-retinal binding to the apoprotein opsin, as well as formation of meta II after flash photolysis (Botelho et al., 2002). Photoactivation of rhodopsin in the recombinants was determined by titration of the 478-nm absorbance from flash-photolysis spectroscopy between pH 5 and pH 8 and 27°C, as described (Gibson and Brown, 1993; Botelho et al., 2002). Approximately equal amounts of the meta I and meta II photolysis products were obtained at pH 7, in agreement with previous work (Mitchell et al., 1992). Moreover, the regeneration of opsin in the POPC membranes with 11-cis-retinal was comparable to the isolated native ROS membranes (95 ± 5%) (Gibson and Brown, 1993). The rhodopsin recombinant membranes were analyzed for a homogenous protein/lipid ratio by isopycnic sucrose density ultracentrifugation, resulting in a single sharp band for the rhodopsin/POPC membranes. The protein/lipid ratio of these bands was determined by optical determination of the rhodopsin concentration and lipid phosphorus analysis, giving a molar ratio of 1:100 ± 10, in agreement with earlier work on rhodopsin/egg PC membranes (Gibson and Brown, 1993). A rhodopsin-free sample of POPC-d31 was prepared as a control following the same dialysis procedure. Alternatively multilamellar lipid vesicles were prepared from ∼30 mg of POPC-d31 in the above buffer (66 wt %), and subjected to multiple freeze-thaw-vortex cycles under argon to ensure complete dispersion of the lipid (Huber et al., 2002b). All samples were checked after the NMR experiments by thin-layer chromatography as described (Brown et al., 2002), and revealed no contamination or lipid degradation.
Solid-state deuterium NMR spectroscopy
Deuterium NMR spectroscopy of the lipid dispersions at 11.7 T employed a Bruker AMX-500 spectrometer using the solid-state quadrupolar echo method with composite pulses, as described (Huber et al., 2002b). Briefly, the 2H NMR spectra of the randomly oriented bilayer membrane dispersions of POPC-d31 in the presence or absence of rhodopsin were numerically deconvoluted (de-Paked). The C–2H segmental order parameters were calculated from the observed quadrupolar splittings, using a static quadrupolar coupling constant of 170 kHz for the aliphatic C–2H bonds. The assignments were made in reversed order from the methyl terminus toward the plateau region. The effect of rhodopsin on the lipid order parameter profiles was investigated by 2H NMR at pH 7 over the temperature range from −43 to 37°C for each sample, and the data determined at 27°C were compared with the MD simulations at the same temperature.
RESULTS
All-atom molecular dynamics model of membrane-bound rhodopsin
In what follows, we present a refined model for dark-adapted rhodopsin in the native-like environment of a bilayer membrane comprising POPC in the liquid-crystalline (Lα) state at full hydration. The model is based on a 15-ns molecular dynamics computer simulation, utilizing an all-atom representation of the system. During the first 5 ns of the MD simulations, several modifications were applied to the initial model to obtain an accurate description of the real system. In particular, care had to be been taken to choose the correct protonation state of titratable amino acids, to solvate internal cavities with water, to adjust the number of lipids in each of the monolayers, and to find an area per lipid that reproduces the experimental observables. The model was equilibrated for a further 5 ns, and the final 5 ns of the 15-ns trajectory, designated segment P6 (cf. Appendix) were used for the analysis of the protein structure, the lipid order parameters, and the generalized molecular surface of the various interfaces of the biomembrane system. Likewise, the protein-free POPC bilayer was simulated for 39 ns, and a5-ns segment, designated L4, with a similar area per lipid was used for comparison of the order parameters. The details of the protein models are described in Table 1, and a complete description of the bilayer systems and equilibration can be found in the Appendix.
Comparison of calculated and experimental order parameter profiles for rhodopsin/POPC membranes
Fig. 2 a shows the results of theoretical MD simulations for the sn-1 acyl chain order parameters of POPC in the rhodopsin-containing membrane model (segment P6), and the protein-free lipid control (segment L4) (cf. Appendix). The average area per lipid for the rhodopsin simulation was 62.6 Å2, with some uncertainty introduced by the vase-shape protein inclusion (vide infra). The lipid control simulation segment was performed at a constant area per lipid of 62.4 Å2. As can be seen in Fig. 2 a, the orientational order of the lipid acyl chain segments C8–C15 is reduced due to rhodopsin incorporation in the MD model system. How do the MD simulations correspond to the experimental effects of rhodopsin on POPC acyl chain order parameters? In this regard, solid-state deuterium nuclear magnetic resonance (2H NMR) allows investigation of the bilayer structure (Petrache et al., 2001) under physiological conditions (hydration, ionic strength, and temperature). The acyl chain order parameters obtained by 2H NMR spectroscopy of deuterated POPC-d31 bilayers in the presence of rhodopsin can be compared with the order parameters calculated from the MD simulations (Huber et al., 2002b). The order parameter profiles are sensitive to the average bilayer structure (Petrache et al., 2001), and can help to guide the choice of simulation parameters, such as the applied surface tension, as well as judge the quality of the final MD model.
FIGURE 2.

Comparison of POPC acyl chain order parameters from MD simulations and solid-state deuterium (2H) NMR experiments. (a) Simulated order parameter profile for sn-1 chain of POPC at 27°C: (red −○−) presence of rhodopsin at zero surface tension (simulation segment P6; cf. Table 2) with an average cross-sectional area per lipid in the acyl chain region of 62.6 Å2; (black −□−) order parameter profile in absence of rhodopsin at a constant area per lipid of 62.4 Å2 (simulation segment L4). (b) Experimental 2H NMR sn-1 chain order parameter profile of POPC-d31 in the presence (red −○−) and absence (black −□−) of rhodopsin at 27°C and pH 7, with a lipid-to-protein ratio of 100:1. Insert: numerically deconvoluted solid-state 2H NMR spectra of POPC-d31 in the absence (black line) and presence of rhodopsin (red line). The order parameters nearest the aqueous interface show little effect of rhodopsin, whereas the carbon segments C8–C15 are more disordered.
The influence of rhodopsin on a bilayer membrane comprising POPC-d31, in the liquid-crystalline (Lα) state at full hydration, was studied experimentally by solid-state 2H NMR spectroscopy (at 27°C and pH 7). As shown in Fig. 2 b, the effect of rhodopsin on the acyl chain order parameter profile of the lipid bilayer resembles the simulated system. The insert shows the numerically deconvoluted (de-Paked) solid-state 2H NMR spectra of POPC-d31, with and without rhodopsin. The order parameter profiles were obtained from the quadrupolar splittings, as described (Huber et al., 2002b). There is a small disordering effect of rhodopsin on the order parameter profiles for chain segments C7–C15, in qualitative agreement with the MD simulations. The similarity of the experimental order parameters close to the headgroups in the presence and absence of rhodopsin, cf. Fig. 2 b, suggests equivalent packing in this region. Evidently rhodopsin induces some disorder, where the largest effect is in vicinity of chain segment C10. For a lipid/rhodopsin ratio of 100:1, approximately half of the lipid acyl chains are in the boundary layer that directly solvates the hydrophobic surface of rhodopsin; therefore the actual effects of the protein surface are greater than indicated by the average order parameters. The overall shape of the order profiles and the progressive reduction along the chains manifest an increase in configurational freedom with depth in the bilayer, due to chain terminations (Petrache et al., 2001). We suggest that a greater lateral freedom of the acyl segments, together with the vase-like shape of the protein inclusion, results in larger order parameter differences in the middle part of the chains. On the other hand, the overall resemblance of the order parameter profiles indicates matching of the hydrophobic thickness of the bilayer to rhodopsin. The change of the membrane thickness with temperature is less in the presence of rhodopsin, and at 27°C there is an apparent match of the bilayer thickness with and without protein (not shown). It is interesting that the POPC bilayer in simulation segment L3, which has a zero surface tension as in rhodopsin simulation segment P6 (cf. Appendix) equilibrates at the same temperature to a significantly smaller area per lipid. This suggests that the bilayer tends to match the hydrophobic thickness of rhodopsin, leading to a difference in area per lipid, which is in turn inversely proportional to the bilayer thickness.
Formulation of a generalized molecular surface based on Voronoi polyhedra
Currently there is no generally accepted standard method to describe the molecular packing of the protein, lipids, and water in biomembrane simulations. Hence, we developed a novel volumetric method based on a generalization of Voronoi polyhedra to differently sized atoms (Richards, 1985) to compute the molecular cross-sectional area and the solvent contact interface, the generalized molecular surface (cf. Appendix). The GMS quantifies the interface of rhodopsin with lipid and water, and allows for further analysis in terms of a hydropathy profile. One can then consider how the shape of rhodopsin in the membrane corresponds to the partial cross-sectional areas of the lipids (cf. Fig. 3), that is to say, how the transmembrane protein is embedded in the lipid bilayer. A related aspect is how rhodopsin is related to the lipid composition of its native membranes. For instance, in the case of a rodlike or cylinder-shaped protein inclusion in a bilayer membrane, the cross-sectional area profile as a function of depth (z) within the membrane would be uniform over the length of the protein, and zero elsewhere. A second case would be a globular or ellipsoidal protein having a parabolic cross-sectional area distribution over the length of the protein, with a maximum at the center of the protein. The third case is that of a vase-shaped protein inclusion, with larger cross-sectional area at the height of the lipid headgroups than in the center of the bilayer. Below we consider how application of the GMS approach can be used to quantify the various interfaces and describe the bilayer deformation due to rhodopsin.
FIGURE 3.

Lateral deformation of the bilayer membrane due to the protein inclusion. (a) The GMS method was applied to determine the partial contributions of the different components of the membrane system to the cross-sectional area as a function of position along the bilayer normal. The center of mass of rhodopsin defines the origin of the coordinate system; positive z values are on the extracellular side and negative z values are on the cytoplasmic side. The grayscale of the transverse planes indicates the different components (from light to dark: anions, cations, water, glycerophosphocholine, acyl ester, acyl chain hydrocarbon, and protein atoms) of the Voronoi regions. Rhodopsin is depicted in terms of its backbone secondary structure elements, and the internal water molecules are shown as van der Waals sphere (CPK) models. The central slice shows only three components; viz. atoms from the protein, acyl chain hydrocarbon, and a single internal water. The slices through the headgroup regions illustrate the heterogenity in this region with all components present. (b) Quantitative results from the GMS analysis. The cross-sectional area as a function of slice position along the z axis in the unit cell is plotted for the four principal components: aqueous solvent (blue; AW(z), all water and salt atoms), hydrophobic part of the phospholipids (pale yellow; AH(z), all aliphatic acyl chain atoms), polar part of the phospholipids (cyan; AP(z), all lipid atoms except the aliphatic acyl chain atoms), and protein (black; AR(z), including posttranslational modifications). Note that the bilayer is shifted toward the extracellular domain of rhodopsin (cyan and yellow curve), where the protein has a vase-like shape, with larger cross-sectional area values close to the lipid/water interfaces than in the bilayer center.
Bilayer deformation: the lipid/protein interface
The determination of the cross-sectional area profiles of the membrane protein and lipid components is the first application of the GMS method (Fig. 3). It is shown below that the cross-sectional area profile of rhodopsin AR(z) is indicative of a vase-shaped protein, with larger cross-sectional area in the headgroup region of the bilayer than in the hydrophobic bilayer interior. A rectangular intersection of the simulation unit cell with a plane perpendicular to the z axis at position zi relative to the center of mass of rhodopsin can be analyzed in terms of the generalized Voronoi regions of the different components of the system (cf. Appendix). In Fig. 3, the direction of the z axis is from the c-side to the e-side of rhodopsin, where the cytosolic (c) domain is at negative z values, and the extracellular (e) domain is at positive z values. The method is illustrated in Fig. 3 a by three representative sections, with a grayscale corresponding to the different components. The (partial) cross-sectional area of each component as a function of the section position z is plotted in Fig. 3 b; here AR(z) (black) designates the protein solute rhodopsin; AP(z) (cyan), the lipid polar headgroups; AH(z) (pale yellow), the lipid hydrophobic acyl chains; and AW(z) (blue), the aqueous solvent.
An important aspect is that the area distributions in Fig. 3 allow one to calculate the position of the bilayer relative to the rhodopsin molecule. In Fig. 3 b, integration of the sum of the area distributions of the lipid components, AP(z) and AH(z), gives the volume of the bilayer, which is centered atz = +1.7 Å relative to the center of mass of rhodopsin, taking the different numbers of lipids for each monolayer into account. The following calculations are based on integrations for z values <+1.7 Å for the cytosolic monolayer, and for the extracellular monolayer the integration is for largerz values. The cross-sectional area distributions of the acyl chains AH(z) (pale yellow), Fig. 3 b, are centered at +8.9 Å and −5.7 Å for the extracellular and cytosolic monolayers, respectively. The center of the distribution for the lipid polar headgroups AP(z) (cyan), Fig. 3 b, in the extracellular monolayer is at a z position of +20.4 Å, and the corresponding value for the cytosolic leaflet is at −16.9 Å. The bilayer thickness from the headgroup distributions is 37.3 Å.
What is the average cross-sectional area of rhodopsin for the different regions of the bilayer, viz. corresponding to the polar headgroups and the acyl chains? The weighted average of the cross-sectional area of rhodopsin AR(z), using the partial areas of the lipid polar headgroups AP(z) as weights, gives the cross-sectional protein area in the vicinity of membrane surface. Its value for the extracellular monolayer is 870 Å2, and it is 960 Å2 for the cytoplasmic monolayer. The corresponding area for the acyl chain region of the extracellular monolayer is 905 Å2, whereas it is 858 Å2 for the cytoplasmic monolayer. These weighted average cross-sectional protein area values are subtracted from the unit cell area to obtain the monolayer area values for the different regions of the bilayer. In the headgroup region, the calculations yield a value of 63.3 Å2 for the area per lipid in the extracellular leaflet, and 60.3 Å2 in the cytoplasmic leaflet, indicating that the cytoplasmic monolayer is more condensed. This is perhaps not surprising, because it contains one additional lipid compared to the extracellular monolayer. By contrast, the calculation of the average area per lipid in the hydrophobic lipid acyl chain regions for each monolayer gives almost identical values of 62.3 and 62.6 Å2 for the cytosolic and extracellular leaflets, respectively.
The cross-sectional area profiles for the different components, viz. protein, lipid acyl chains and headgroups, and aqueous solvent, allow one to express the bilayer deformation in terms of the monolayer curvature. We can then gain insight into the relation of the rhodopsin structure to the composition of the native lipid environment (Brown, 1997; Botelho et al., 2002). Briefly, the curvature is related to the different cross-sectional areas in the headgroup and acyl chain regions of a monolayer (Thurmond et al., 1993; Rand and Parsegian, 1997). A concave monolayer, when viewed on the headgroups, corresponds to a negative curvature, and a flat monolayer to zero curvature, i.e., an infinite radius of curvature. How can one define the monolayer curvature for a planar bilayer membrane? This apparent paradox is resolved when we consider the protein interface with water to be in effect positively curved, as in case of a vase-shaped bilayer inclusion, and the lipid component to be negatively curved. Hence a mean curvature of zero is obtained for both components together. The monolayer curvature can be estimated from the ratio of the average area per lipid in the headgroup region and in the acyl chain region, together with the average positions of the polar and hydrophobic parts of the monolayers. The results are expressed as the radius of curvature of the so-called pivotal surface, which in our calculations divides the positions of the centers of the headgroup and acyl chain distributions in a ratio of 1/3 to 2/3, that is, the pivotal surface is closer to the headgroups than the acyl chains. In this way a radius of curvature of −692 Å is obtained for the cytoplasmic monolayer, and +2144 Å for the extracellular monolayer. This is in agreement with the lipid composition of the native rod disk membranes, in which a preferential distribution of nonlamellar phase-promoting lipids is found within the cytoplasmic monolayer, with a tendency to form negatively curved, reverse hexagonal (HII) phases (Deese et al., 1981; Miljanich et al., 1981).
Membrane protein generalized molecular surface compared with hydropathy analysis
Let us next consider the relative exposure of the membrane-embedded rhodopsin molecule to the hydrophobic and polar regions of its environment. The total contact or exposed areas with water or lipid were calculated from the GMS for each residue as described in the Appendix. The GMS area values of the ith residue of rhodopsin at the lipid/protein hydrophobic interface aH(i), the lipid/protein polar interface aP(i), and the protein/water interface aW(i) are shown as a function of sequence position in Fig. 4, a–c, respectively. These interface area functions represent the accessibility of the explicit environment molecules to rhodopsin as obtained from the all-atom MD simulations. Note especially the periodicity of the lipid/protein hydrophobic interface area function aH(i) in the transmembrane helical regions of rhodopsin, Fig. 4 a, which is characteristic for transmembrane helix bundles. The helical domains of the rhodopsin MD structure are indicated by a gray background. The periodicity is also found to extend to the headgroup region of the lipid/protein polar interface aP(i), Fig. 4 b. However it is not as pronounced in the aqueous exposure profile of the protein/water interface aW(i), Fig. 4 c, which is mainly outside of the helical secondary structure in the loop and turn regions.
FIGURE 4.

Hydropathy analysis compared with GMS analysis of the MD model of rhodopsin in the POPC bilayer. (a) Contribution aH(i) to area of lipid/protein hydrophobic interface versus residue position (i). The exposure to the hydrophobic bilayer interior is calculated as the GMS of rhodopsin with the hydrocarbon acyl chain atoms of POPC for each amino acid residue. Posttranslational modifications are not included, for example, the palmitoylations of Cys322 and Cys323 do not appear as peaks. (b) Residue contributions aP(i) to area of lipid/protein polar interface, computed from the GMS of rhodopsin with all lipid atoms excluding the hydrocarbon part of the acyl chains. (c) Contribution aW(i) of individual amino acid residues to area of protein/water interface, calculated as the aqueous solvent-exposed GMS of rhodopsin with internal and external water molecules, as well as with the salt ions that give a very low contribution. (d) Gibbs free energy 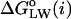 of partitioning 19-residue-long sequence windows into the membrane, obtained from the ratio of the lipid-exposed GMS and the water-exposed GMS of rhodopsin (solid line). The hydropathy index from the GMS was calculated from the protein/water interface, and the sum of the lipid/protein hydrophobic and polar interfaces (cf. text). The results are compared with a hydropathy analysis (White and Wimley, 1999) of the protein amino acid sequence (dotted line). Note the striking similarities of the two plots, in which the null crossover points of both functions are very close to one another, and the locations of the local minima and maxima are similar. (e) The hydrophobic and polar interfaces mapped onto the residue sequence, where the sequence topology scheme of rhodopsin illustrates the protein residues associated with the different GMS interfaces (cf. text). The extracellular e-side and cytoplasmic c-side of the bilayer are depicted, where the light and dark gray regions denote the headgroup and acyl chain regions, respectively. Helices are indicated as cylinders, where the first and last residues are labeled by the residue index. The lipid interface type is denoted by the filling color of the residue symbols, whereas the symbol shape marks the water interfaces. Residues exposed to the bilayer hydrocarbon interior (the lipid/protein hydrophobic interface) are shown in yellow; exposure to the lipid headgroups (the lipid/protein polar interface) is indicated by blue; and any contact with the lipid/protein hydrophobic and polar interfaces, is depicted by green. Protein residues not exposed to water are shown as circles, whereas residues exposed to internal or external water are displayed as squares. The remaining symbols indicate posttranslational modifications, and the charge states of the residues in the MD model are designated by red or blue symbol frames. An important detail is the exposure of the C2 loop residues to both the lipid/protein polar and hydrophobic interfaces, which is related to the model of lipid effects on rhodopsin activation.
of partitioning 19-residue-long sequence windows into the membrane, obtained from the ratio of the lipid-exposed GMS and the water-exposed GMS of rhodopsin (solid line). The hydropathy index from the GMS was calculated from the protein/water interface, and the sum of the lipid/protein hydrophobic and polar interfaces (cf. text). The results are compared with a hydropathy analysis (White and Wimley, 1999) of the protein amino acid sequence (dotted line). Note the striking similarities of the two plots, in which the null crossover points of both functions are very close to one another, and the locations of the local minima and maxima are similar. (e) The hydrophobic and polar interfaces mapped onto the residue sequence, where the sequence topology scheme of rhodopsin illustrates the protein residues associated with the different GMS interfaces (cf. text). The extracellular e-side and cytoplasmic c-side of the bilayer are depicted, where the light and dark gray regions denote the headgroup and acyl chain regions, respectively. Helices are indicated as cylinders, where the first and last residues are labeled by the residue index. The lipid interface type is denoted by the filling color of the residue symbols, whereas the symbol shape marks the water interfaces. Residues exposed to the bilayer hydrocarbon interior (the lipid/protein hydrophobic interface) are shown in yellow; exposure to the lipid headgroups (the lipid/protein polar interface) is indicated by blue; and any contact with the lipid/protein hydrophobic and polar interfaces, is depicted by green. Protein residues not exposed to water are shown as circles, whereas residues exposed to internal or external water are displayed as squares. The remaining symbols indicate posttranslational modifications, and the charge states of the residues in the MD model are designated by red or blue symbol frames. An important detail is the exposure of the C2 loop residues to both the lipid/protein polar and hydrophobic interfaces, which is related to the model of lipid effects on rhodopsin activation.
In what follows, we compare the GMS-based hydropathy analysis of the rhodopsin-containing membrane MD model with the results of a sequence-based hydropathy analysis (Kyte and Doolittle, 1982; Engelman et al., 1986) of the primary structure of rhodopsin, an important method in bioinformatics and structural genomics. In each case a hydropathy index 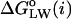 is calculated as a function of sequence position i in the protein, defined as the standard free energy for transferring a potentially membrane-spanning helix of 19 residues, called a window, from the membrane interface to water. Here we use for the rhodopsin sequence-based hydropathy index
is calculated as a function of sequence position i in the protein, defined as the standard free energy for transferring a potentially membrane-spanning helix of 19 residues, called a window, from the membrane interface to water. Here we use for the rhodopsin sequence-based hydropathy index 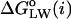 a whole-residue polarity scale,
a whole-residue polarity scale, 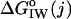 , as experimentally determined from the standard free energy of transfer of a set of model peptides from POPC bilayer membranes to water, accounting for the charge state of protonable residues (White and Wimley, 1999):
, as experimentally determined from the standard free energy of transfer of a set of model peptides from POPC bilayer membranes to water, accounting for the charge state of protonable residues (White and Wimley, 1999):
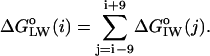 |
(1) |
The calculations were performed for the same ionization of the residues in rhodopsin as in the MD model RhC (Table 1). Note that due to the smoothing, the functional form does not provide a value for the hydropathy index function for the first and last nine residues of a protein. Peaks in the hydropathy plot of greater than a predefined cutoff level, zero for these whole-residue hydropathy analysis methods, are indicative of potential transmembrane helices (White and Wimley, 1999).
As distinct from the sequence-based hydropathy analysis, the GMS can be used to define a hydropathy index 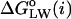 corresponding to the actual interfacial areas of the rhodopsin-containing MD model. The free energy of transfer from membrane to water of a 19-residue sequence window, i.e., the hydropathy index, is related to the probability to find these residues exposed to a membrane environment as compared to the probability for water exposure. Assuming the area of the molecular surface is independent of the type of environment, viz. membrane or water, then the probabilities for membrane and water exposure are proportional to the exposed molecular surface areas. The GMS-derived area exposure functions for the lipid/protein interface, aH(i) and aP(i), and for the protein/water interface, aW(i), are summed independently over the window from residue
corresponding to the actual interfacial areas of the rhodopsin-containing MD model. The free energy of transfer from membrane to water of a 19-residue sequence window, i.e., the hydropathy index, is related to the probability to find these residues exposed to a membrane environment as compared to the probability for water exposure. Assuming the area of the molecular surface is independent of the type of environment, viz. membrane or water, then the probabilities for membrane and water exposure are proportional to the exposed molecular surface areas. The GMS-derived area exposure functions for the lipid/protein interface, aH(i) and aP(i), and for the protein/water interface, aW(i), are summed independently over the window from residue 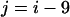 to residue
to residue 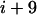 , as in the sequence-based analysis. The ratio of these 19-residue area exposures is interpreted as a mol fraction partition coefficient between the phospholipid and water compartments, which is then related to the standard free energy of transfer from the phospholipid bilayer to water:
, as in the sequence-based analysis. The ratio of these 19-residue area exposures is interpreted as a mol fraction partition coefficient between the phospholipid and water compartments, which is then related to the standard free energy of transfer from the phospholipid bilayer to water:
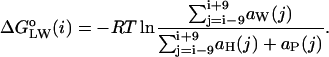 |
(2) |
It should be noted that the identification of the GMS interface with partition coefficients representing free energies of transfer is conceptually different from the relation of molecular surface exposure to the hydrophobic effect. In the latter case, the expulsion of nonpolar compounds from water is considered in terms of the water-to-hydrocarbon transfer free energy, which is related to the interfacial surface tension and the accessible surface area (Dill, 1997; White and Wimley, 1999). On the other hand the GMS-based hydropathy analysis describes partitioning of the molecular components resulting from the MD simulations of rhodopsin. When one compares the partitioning free-energy profile computed in this way (solid line) with the sequence-based hydropathy analysis (dotted line), Fig. 4 d, one can see clear similarities between both. The results clearly reflect the transmembrane helical and loop topology of rhodopsin. A positive value of the hydrophobic moment function corresponds to the transmembrane region, whereas a negative value relates to the aqueous region. Note that the zero crossover points of the hydrophobic moment functions, i.e., the MD-calculated hydropathy profile and the sequence-based hydropathy profile, are very similar, confirming that the actual position of rhodopsin in the biomembrane model is defined by the amino acid sequence. This conclusion is significant to homology-based modeling of GPCRs and bioinformatics in general. The lipid/protein and protein/water interfacial contacts as a function of the rhodopsin sequence are schematically summarized in the form of a two-dimensional topology model in Fig. 4 e. It can be seen that many residues of the transmembrane domain of helices H3 and H7 (indicated in white) are not exposed to the lipid bilayer, resulting in small hydrophobic moments relative to the other transmembrane helices. Moreover, helices H4 and H6 have the shortest transmembrane-spanning sequences corresponding to local thickness variations of the bilayer, a possible origin of rhodopsin dimerization (Fotiadis et al., 2003). Note the association of the cytoplasmic loops C2 and C3 with the lipid/water interface, which is significant for conformational changes due to the membrane environment (vide infra). The organization of the transmembrane helix bundle, which is conserved among the members of the GPCR superfamily (Baldwin et al., 1997), gives rise to a complicated pattern of protein/water and lipid/protein interfaces as described herein.
Identification of internal water molecules
An important aspect of the refined rhodopsin structure is the identification of four internal water molecules together with further internal cavities, which are not accessible to the protein surface (Teller et al., 2001). The presence of voids and internal cavities may be significant for distinguishing regions of potential mobility implicated in the functional mechanism of rhodopsin activation. How can one identify such internal water molecules? The detection of internal water molecules constitutes an important application of our GMS method. In this context, a clear definition of the term internal is necessary: an internal volume has to be surrounded by only the protein. By this definition any lipid molecule of the surrounding membrane is considered as external. Moreover, any nonprotein molecule in contact with any previously assigned external molecule can likewise be assigned as external. Here a molecule was designated in contact with another molecule if a nonzero contact area of the GMS was formed by both molecules. The consequent application of this iterative definition for external molecules results in a negative selection of the protein and any internal molecules, where the internal and external space is given by the connectivity of the network. Initially, eight molecules buried in the rhodopsin structure, four of them corresponding to the locations proposed from analysis of the crystal structure (Teller et al., 2001), and four additional water molecules occupying empty cavities in the protein, were placed in the refined crystal structure (Teller et al., 2001) utilizing a combination of the programs SOLVATE (Grubmüller, 1996) and DOWSER (Zhang and Hermans, 1996). These internal water molecules were introduced into the simulation at segment P4 together with model RhB, cf. Appendix. The hydrophilicity of the internal cavities of rhodopsin was then further explored using the GMS approach. One of the initially placed water molecules dissociated from its cavity, and five additional water molecules entered the protein from the bulk solvent. Thus, in the equilibrated rhodopsin MD model, a total of 12 internal water molecules could be identified by this protocol. The distribution of these internal waters within the rhodopsin molecule is visualized by their GMS with the protein in Fig. 5 a. Together with the water-exposed residues found throughout the transmembrane region (Fig. 4), a more or less continuous network of residues in contact with water is evident, even though the water molecules are not arranged in an uninterrupted chain.
FIGURE 5.

The principal interfaces of a membrane protein as a result of the generalized molecular surface analysis. Each of the four views is rotated about the vertical axis by 90°. (a) The GMS interface of rhodopsin with the internal water molecules identified by the simulations, where the protein is indicated by a simplified secondary structure depiction (cf. text). (b) The GMS interface of rhodopsin with the acyl chain hydrocarbon layer (red), the acyl chain ester group atoms (gray), the glycerophosphocholine headgroup atoms (orange), and the external, or bulk, water molecules (yellow). The protein/water interface (yellow) of the extracellular and cytoplasmic domains of rhodopsin comprises 62% of the total area, which is twice as large as the lipid/protein hydrophobic interface (red) with 31% of the area. The remaining 7% corresponds to the lipid/protein polar interface (gray and orange areas). (c) The GMS area values for the lipid/protein hydrophobic interface mapped in red/white shading onto a Connolly-type molecular surface with basis color blue. For the rest of the MD simulation unit cell the generalized Voronoi regions are shown in cross sections. The Voronoi regions are gray-coded for the different components of the environment of rhodopsin (from dark to light: hydrocarbon, ester, and glycerophosphocholine headgroup atoms of the phospholipids, water, anions, and cations). (d) Simplified representation of the lipid/water interfaces calculated as described in the text (gray- and cyan-colored surfaces, the acyl chain carbonyl atoms and the headgroup phosphorus and nitrogen atoms, respectively). Rhodopsin is depicted by a simplified secondary structure representation, and its internal water molecules are shown in a CPK-type van der Waals sphere representation. The GMS method allows one to quantify the protein/water and lipid/protein interfaces at virtually any level of detail.
Interfaces among protein, water, and lipid components revealed by generalized molecular surface approach
The standard fluid mosaic model for biological membranes involves a two-dimensional solution of (oriented) lipids and peripheral and integral membrane proteins. Generally, a transmembrane protein has interfaces both with water and with the lipid bilayer, and the boundary of the lipid/protein and protein/water interfaces coincides with the lipid/water interface of the bilayer near the protein (Gibson and Brown, 1993; Brown, 1997). Above we have characterized quantitatively the relevant interfaces utilizing the GMS method, as illustrated in Fig. 5. More precisely, one can distinguish the internal and external protein/water interfaces, and the lipid/protein polar and hydrophobic interfaces. The foregoing development, Fig. 5 a, provides a description of the internal protein/water interface, which is the interface of rhodopsin with the internal water molecules (indicated in blue). The other interfaces are depicted in Fig. 5 b, including the external protein/water interface (shown in yellow), the lipid/protein polar interface (gray and orange), and the lipid/protein hydrophobic interface (red). The visualization of the GMS reveals homogeneous regions of polar and hydrophobic contacts, but due to the computational complexity it is difficult to display hyperboloid surface elements. One possibility is to map the contact area values of the GMS interface onto a conventional Connolly-type molecular surface, as illustrated in Fig. 5 c for the lipid/protein hydrophobic interface. The GMS area value of a particular protein atom determines the color of that fraction of the Connolly-type molecular surface of the protein that is closest. A blue color corresponds to no exposure to the lipid/protein hydrophobic interface, and the range from white to red represents an increasing degree of exposure. The probe radius of 1.5 Å, typically used for Connolly-type molecular surfaces, is equivalent to the effective size of a water molecule. Some of the crevices sampled by such a probe are too small for an acyl chain. Atoms deep in those crevices might not contribute to the direct contact interface with the lipid acyl chain hydrocarbon atoms, as defined by the GMS method. These give rise to blue-colored interruptions of the white/red-colored hydrophobic interface map. Despite these limitations, there is an important technical advantage of a GMS mapped on a Connolly-type molecular surface, as the molecular graphics program VMD 1.6 (Humphrey et al., 1996) provides an efficient implementation of color-shaded molecular surfaces with interactive visualization.
How is the structure of the surrounding bilayer affected by the above rhodopsin interfaces? In Fig. 5 c planar slices through the system are shown, where the gray maps depict the Voronoi regions of the components utilized for the GMS computation. It can clearly be seen that the hydrocarbon part of the lipid bilayer forms a sharp interface with the aqueous solvent and the polar lipid headgroups; it is essentially free of polar components such as water or lipid headgroups. In what follows, we refer to this as the hydrophobic lipid/water interface, which also marks the relatively sharp transmembranous lipid/protein hydrophobic interface. The latter is illustrated by the GMS mapped on the Connolly-type molecular surface (vide supra). We note that the lipid/water interfaces, as identified by the GMS method, can be compared with other definitions of the bilayer membrane geometry. For instance, the Luzzati thickness of a bilayer is the lipid head-head separation derived from electron density profiles, and the hydrophobic thickness is relevant for hydrophobic matching of proteins. Implicit in these definitions are two different lipid/water interfaces, which are approximated by the average positions of the headgroup atoms or the acyl chain carbonyl atoms, respectively (Huber et al., 2002b). We refer to these interfaces as the PN-type and COO-type lipid/water interfaces. They are calculated for each monolayer from the positions of the phosphorus and nitrogen atoms of the headgroups for the PN-type, and the carbonyl carbons of the acyl chain ester groups for the COO-type.
With the above definition, the MD model can be analyzed in terms of the PN-type and COO-type lipid/water interfaces, using the distance-weighted averages of the corresponding atom positions in each monolayer. Briefly, a surface z(x, y) as a function of xy position approximating N individual atoms, having coordinates (xi, yi, zi) for the ith atom, is given by:
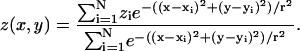 |
(3) |
The nonlocal functional form, a sum of Gaussian-type radial weighting functions with width r, enables one to effectively extrapolate the lipid/water interfaces, as approximated by these surfaces, into the protein interior. Note that the surfaces are calculated under periodic boundary conditions. In Fig. 5 d, the approximation of the lipid/water PN-type interface is shown as the cyan-colored surfaces, and the lipid/water COO-type interface is depicted by the gray-shaded surfaces. These interfaces can be compared with the boundaries of the Voronoi regions from the GMS calculations, as illustrated by the slices through the bilayer in Fig. 5 c. Consequently, the PN-type and COO-type interfaces approximate the all-atom GMS in terms of a simple continuum approximation, and can be utilized for visualization of local membrane deformations.
Structural fluctuations of rhodopsin
In the previous sections, our focus has been on the influences of rhodopsin on the phospholipid bilayer. How is the protein structure affected due to the bilayer environment? Here one should remember that the x-ray structure of rhodopsin corresponds to a nonnative medium, for example its photoactivation leads to disruption of the protein crystals (Okada and Palczewski, 2001). The mobility of the equilibrated structure of rhodopsin in the membrane was studied by computing the rms fluctuations of the Cα backbone structure versus the average equilibrated structure for the last 5 ns of the simulation (segment P6; cf. Appendix). In Fig. 6 a, the rms fluctuation values from the MD model (shown in red) are juxtaposed with those calculated from the crystallographic Debye-Waller B-factors (black), which are related to the variances of the atom positions. The B-factors were averaged for all six available crystallographic rhodopsin structures (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002). The rms fluctuations are measures of mobility that can be compared for the MD-generated structure and for the crystallographic structure. Note that the rms fluctuations calculated from the crystallographic B-factor for a particular atom are only valid in the absence of static disorder, so that a constant offset is zsometimes subtracted (Bernèche and Roux, 2000). However, subtraction of a constant would imply a nonuniform static disorder over the whole molecule, so that such a correction does not seem theoretically well justified. Consequently the data shown in Fig. 6 a are not offset corrected. The general trends of the rms fluctuations from the crystallographic B-factors and the MD-generated ensemble are comparable, as shown in Fig. 6 a. The rms plot from the B-factors has some gaps, as some of the rhodopsin atoms in the crystal structures are either missing, or no B-factor was assigned (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002). It is interesting that the helical domains of rhodopsin exhibit relatively small fluctuations, whereas the extramembranous domains, especially the cytoplasmic side of rhodopsin, show higher mobility. Next we shall analyze the rms deviations of these structures relative to the crystal structure.
FIGURE 6.

Quantitative analysis of the rhodopsin MD structure as compared with the crystal structure, indicating backbone fluctuations and deviations, and secondary structure stability. (a) Polypeptide backbone mobility shown by root mean square (rms) fluctuation of Cα carbons from the equilibrated MD ensemble for the last 5-ns segment (P6) of 15-ns simulation (red −□−), and calculated from the crystallographic B-factors (black line; cf. text). For clarity, the helical domain structure in parts a and b is indicated by a gray background in the plots. (b) Backbone Cα atom rms deviations for simulation segment P6 versus the crystal structure (blue −○−) in comparison with rms fluctuations from MD (red line) and B-factors (gray line). (c) Secondary structure analysis calculated with STRIDE (Frishman and Argos, 1995) for the rhodopsin polypeptide chain versus simulation time. The color code indicates the secondary structures: purple, α-helical (62%); pink, 310-helical (0.11%); yellow, β-sheet (3.8%); white, coil (16%); green, turn (18%); brown, isolated bridge (0.004%); and red,π-helix (0%). Various segments of the trajectory are indicated (cf. Appendix). Note the difference of the secondary structure for model RhA in the region of cytoplasmic loop C3 compared to the later models RhB and RhC. In the second half of the C3 loop, the MD model shows fluctuations between coil and turn structures, and the rms fluctuations have the largest values after the C-terminal tail, which also oscillates between coil and turn conformations. Other regions with large rms deviations from the crystal structure appear relatively stable in terms of their secondary structure. The helical domains remain for the length of the simulations, except for ends and breaks in helices H5 and H7, which persist over the time course of the simulations.
Deviations from the crystal structure
The ensemble of structures from the last 5 ns of the MD membrane model of rhodopsin was compared with the better-resolved monomer in the refined crystal structure (Teller et al., 2001), which is the basis for rhodopsin models RhB and RhC. In Fig. 6 b, the rms deviations of the MD-generated ensemble (segment P6; cf. Appendix) relative to the crystal structure (blue) are compared with the rms fluctuations relative to the average MD structure (red), and with the B-factor derived rms fluctuations (gray). The rms deviation versus the crystal structure shows several significant differences due to the membrane, where the overall rms deviation of the Cα atoms is 1.86 Å. The rms deviation plot has gaps for those atoms that are not resolved in the crystal structure. In several regions, the rms deviation is higher than the rms fluctuations from either the MD model or theB-factor. In particular, the equilibrium positions of the Cα atoms in residues 142–145 in loop C2, residue 241 in loop C3, and residues 323–327 and 335–337 in the C-terminal tail were different to a confidence level larger than 99% (cf. Appendix). On the other hand, no differences with a confidence level >68% were found for all residues in the extracellular loop E1 and in the helices H1 and H2, as well as in the extracellular portions of helices H3 and H7. It is not possible to estimate the significance of structural deviations for residues 228–240, and 331–333 due to the lack of coordinates or B-factor values for these positions. TheC-terminal tail exhibits rms deviations up to 13 Å, the loop C3 deviates by a maximum 9 Å, and the loop C2 and the cytoplasmic region of loop E2 both deviate maximally by 5 Å, whereas the central parts of the TM helices reveal rms deviation values as low as 0.5 Å. Clearly the conformation of the cytoplasmic loops is very important with respect to light-induced conformational changes and G-protein recognition.
It is noteworthy that the MD simulation of rhodopsin in a native-like membrane environment yields a new equilibrium conformation versus the crystal structure. This MD model can be considered as a refined structure for rhodopsin in a native-like bilayer environment. The packing in the crystal unit cell results in a large contact area among neighboring proteins. Moreover a number of detergent molecules (β-nonylglucoside), are present, together with the crystallization adjuvant heptane-1,2,3-triol, and several heavy atoms such as zinc and mercury (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002). The large rms deviations involving the loop regions might reflect the removal of these packing constraints and the heavy atoms found in the crystal together with newly formed interactions with the phospholipid bilayer. One might argue that the observed conformational changes versus the crystal structure are an artifact due to model building. Especially the conformation of the missing residues 236–240 in loop C2 and 330–334 in the C-terminal tail had to be surmised by a distance geometry approach. A particular problem was that the available crystal structures (Palczewski et al., 2000; Teller et al., 2001) came to conflicting conclusions about the direction of the turn from loop C3 into helix H6 around residue 241, hence the resulting models of residues 236–240 were on opposite sides of helix H6. It was possible to close these gaps in the polypeptide chain without sterical overlap of the modeled structure for all models. However, the MD simulations of the protein models in the bilayer environment resulted in a significant structural rearrangement of the modeled fragments and the adjacent residues on a timescale of several nanoseconds. Because the computational cost of the simulations prohibited exhaustive testing of the equilibration properties of alternative starting configurations, we could not investigate the effect of different starting structures. The MD simulations starting with model RhA based on the first structure (Palczewski et al., 2000) were not long enough to obtain an equilibrium conformation for this region. The subsequent models RhB and RhC were based on the second structure (Teller et al., 2001), and the MD simulations yielded an equilibrium position of this region approximately half-way between both crystal structures. The fluctuations of this turn are quite large, which might be relevant for the rotation of helix H6 upon receptor activation (Hubbell et al., 2003). On the other hand, a detailed inspection of the resulting model gave a plausible explanation for the structural changes: several residues increased dramatically their interaction with the phospholipid/water interface. For example, the initial portion of the C-terminal tail bends toward the bilayer, leading to a deeper insertion in the bilayer of the palmitoyl chain on residue Cys323 and to a salt-bridge formation of residue Lys325 with a lipid phosphate group. Several residues away, the side chain of Lys339 also forms another salt bridge to an adjacent phospholipid, but the residue remains close to the position in the crystal structure. The peptide loop between these two anchor points exhibits a new conformational equilibrium with small rms fluctuations but large rms deviations relative to the crystal structure (cf. Fig. 6 b). As the cysteine palmitoylations can be viewed as an anchor for the amphipathic helix H8, the presence of the bilayer also leads to appreciable deviations of this helix. The C-terminal tail beyond residue Lys339 is more or less unstructured and very flexible, in agreement with its function as a protein trafficking signal (Sung et al., 1994). The strong interaction of Lys339 with a phospholipid is remarkable given that recent solution-state 15N NMR spectroscopy data for rhodopsin show high mobility of this residue in dodecylmaltoside micelles, which lack phosphate headgroups (Klein-Seetharaman et al., 2002).
Similar rearrangements in the cytoplasmic loops C2 and C3 and in the adjacent helices result in optimized hydrophobic and electrostatic interactions with the adjacent phospholipids. In particular, Lys141, Phe146, His152, Tyr223, Phe228, Lys231, and Arg252 exhibit movement toward the lipid/water interface, whereas Glu150 and Glu232 shift away from the bilayer toward the aqueous space. This demonstrates that the conformations of the cytoplasmic loops C2 and C3 in the crystal structure are not optimized for the presence of a bilayer. It is particularly interesting that Arg252 exchanges its counterion Glu232 in the crystal structure in favor of the negative partial charge of the phosphate group in the zwitterionic lipid POPC. By contrast, the anionic amino acyl residues exhibit no pronounced interaction with the corresponding positive partial charge of the rather hydrophobic choline group. In summary, we suggest that the observed structural changes in the functionally important cytoplasmic domain of rhodopsin are related to the transfer from the crystal packing into the phospholipid bilayer environment, and that the residue hydrophobicity (White and Wimley, 1999) provides the driving free energy.
Stability of secondary structure elements
As described above, the crystal structure and the MD-equilibrated model exhibit different conformations as a function of the sequence of rhodopsin. In addition, the MD simulations allow one to characterize the trajectories of the secondary structure fluctuations. The dependence of the secondary structure for each residue as a function of time is shown in Fig. 6 c. The relation of the simulation time to the different rhodopsin models (RhA, RhB, and RhC; cf. Table 1) and the different segments of the simulations (P1–P6; cf. Appendix) are indicated in the figure. In Fig. 6 c, the stability or time invariance of the α-helical, the β-strand or extended, and several of the turn and coil structures can be inferred from the noninterrupted band patterns. However, there are several regions with fluctuations between two or more secondary structures. For example, the ends of some α-helices, especially H3, H4, and H7, are dynamic and depart partially from a helical backbone conformation. Furthermore, helices H5 and H7 exhibit a break around His211 and Lys296, respectively, which is possibly related to the nearby proline residues Pro215 and Pro303. This can be seen from the interrupted α-helical patterns at sequence positions 211 and 296. The two antiparallel β-strands in the N-terminal tail are slightly more mobile than the second pair of β-strands in the E2 loop. The structure of the loop C3 and the C-terminal tail is very important, and it is interesting to follow the equilibration of the models in the regions of the crystallographically unresolved residues. The initial models were generated by fitting the missing peptide fragments into the open gaps in the rhodopsin crystal structure by distance geometry, and were very similar to results calculated with the homology modeling program MODELER (Baker and Sali, 2001) (not shown). It is clear that for these regions (residues 236–239 and 328–333) a new conformational equilibrium emerged after several nanoseconds of simulation. Such large structural rearrangements do not typically result from the standard homology modeling approach for membrane proteins (Stenkamp et al., 2002). Rather, only MD simulations allow for such structural relaxations within a meaningful explicit representation of the molecular membrane environment. This observation is important with regard to GPCR structures based on homology models derived from the rhodopsin structure (Huber et al., 2003).
Equilibrium structure of rhodopsin
The rather substantial differences in the mobility of the structural domains of rhodopsin led to a further analysis of its structure. The ensemble of rhodopsin conformations from the last 5 ns of the MD model (segment P6) was aligned together with the different crystal structures that are currently available to identify the structural changes. A representative set of six conformations from the MD model and the six protein molecules from the three crystal structures, having two monomers per unit cell, are shown in Fig. 7. The spectral color of the MD-generated structures indicates the position in the sequence, starting from blue for the N-terminus to red for the C-terminus, with the crystal structures shown in gray for comparison. The 11-cis-retinal ligand can be identified in the side view, Fig. 7 a, and extracellular view, Fig. 7 b. Visual inspection of the rhodopsin structural fluctuations compared to the crystal structures reveals a high stability of the transmembrane helical fold. By contrast, significant mobility and deviations from the crystal structures are evident in the extramembranous loop portions, with larger deviations and fluctuations in the cytoplasmic regions, Fig. 7 c, versus the extracellular regions, Fig. 7 b. Appreciable differences are found in cytoplasmic loops C2 and C3, in the C-terminal tail, in the cytoplasmic ends of the transmembrane helices H4, H5, H6, and H7, and in the cytoplasmic helix H8. The light arrows indicate the cytoplasmic loops C2 and C3, as well as the C-terminal tail, which exhibit the largest deviations relative to the crystal structures.
FIGURE 7.

Conformational snapshots from the MD model compared with the crystal structures. A set of five representative conformations from the MD simulations, sampled every 1 ns during the last segment (P6) of the 15-ns simulation, is compared with the set of six rhodopsin monomers from the three published crystal structures (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002). (a) Side view with the extracellular side up and the cytosolic side down; (b) view from the extracellular side; and (c) view from the cytoplasmic side. All structures were aligned relative to the equilibrated orientation of rhodopsin in the MD membrane model to minimize the overall rms deviation. The crystal structures are shown in gray, and the MD-derived ensemble is shown color-coded by the sequence index (blue, N-terminal; green, central; and red, C-terminal). A remarkable stability of the protein around the covalently bound ligand 11-cis-retinal (shown in a bond representation in gray for the crystal structure, and in red for the MD model) is evinced by the small deviations of the crystal and membrane structures, and by the small fluctuation amplitudes of the different MD conformers. The extracellular domain is likewise very similar to the crystal structure, except at loop E2. The light arrows mark the loop C2 and helix H8, which have the most significant deviations from the crystal structure. These regions are relevant for the G-protein recognition by the activated receptor. The cytoplasmic loop C2 (dark green ) has a large deviation from the crystal structure and moves toward the lipid/water interface. This loop is part of the lipid/protein polar and hydrophobic interfaces (cf. Fig. 4). The changes induced by the membrane environment relative to the crystal structure give insight into the possible mechanisms of the lipid influences on receptor activation.
DISCUSSION
The availability of crystal structures of rhodopsin (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002) provides a unique opportunity to understand structure/activity relationships in visual pigments and related G-protein-coupled receptors (Ballesteros et al., 2001; Okada and Palczewski, 2001; Sakmar et al., 2002). In this work, several interrelated problems have been addressed, which center about the 3-D structure of rhodopsin in the membrane-bound state. These include the relation of the structure from x-ray crystallography to the membrane-equilibrated conformation of rhodopsin; the characterization of the different phospholipid bilayer membrane and water interfaces and their connection to hydropathy analysis and bioinformatics; and the influences of the tightly regulated ROS membrane lipid composition on the function of rhodopsin (Botelho et al., 2002). In the MD simulations the interfaces of the protein, membrane, and water components of the biomembrane are characterized by a new GMS method. It is interesting that the cross-sectional area of rhodopsin in the interfacial region is appreciably larger compared to the bilayer interior, especially for the cytoplasmic side of rhodopsin. The corresponding deformation of the lipid bilayer surrounding the protein as described by a flexible surface model could be the origin for the selection of nonlamellar phase-promoting lipids in the native rod disk membranes. It has been suggested that phosphoethanolamine headgroups together with polyunsaturated DHA chains, in the cytoplasmic leaflet of the bilayer (Miljanich et al., 1981), give a curvature elastic stress (frustration) that results in nonspecific membrane deformation connected to rhodopsin function (Wiedmann et al., 1988; Gibson and Brown, 1993; Brown, 1997; Botelho et al., 2002).
Bilayer deformation and lipid-rhodopsin interactions
As a counterpart to the MD simulations, the membrane deformation due to lipid/rhodopsin interactions has been experimentally investigated by solid-state 2H NMR spectroscopy (Brown, 1996). The effect of rhodopsin is a small but selective perturbation of the acyl chain order parameter profile of the POPC bilayer, corresponding to segments C9–C15. It is interesting to note that surface-bound peptides such as LL37 induce similar order parameter changes (Henzler Wildman et al., 2003). This increased disorder of the acyl chain segments can be interpreted as a lateral expansion of the hydrophobic interior of the membrane close to the center of the bilayer. The theoretical order parameter profiles calculated from the MD models of POPC bilayers with and without rhodopsin (Huber et al., 2002b) are in qualitative agreement with these experimental findings. The volume decomposition of the membrane model by the GMS method reveals a vase-shaped intramembranous rhodopsin contour, where the cross-sectional area of rhodopsin is larger close to the lipid/water interface than in the bilayer interior. This result is in agreement with the lateral expansion of the lipids in the bilayer interior, as inferred from the solid-state 2H NMR results. The importance becomes clearer when we consider the native lipid environment of rhodopsin in the rod outer segment disk membrane. Here the bilayer has a high degree of bulky polyunsaturated DHA chains, which together with the small phosphoethanolamine headgroups adapt optimally to the shape of rhodopsin.
The local bilayer deformation around the protein is illustrated by the relation of the lipid/water interface to the rhodopsin structure. In this case, the results show some local deformation due to the loop structures of the membrane-embedded photoreceptor. The interfacial surfaces of the individual lipid monolayers are constructed from the position of the acyl chain carbonyl atoms of both chains of the phospholipids. This definition follows from the insight obtained from pure bilayer simulations, where the boundary between the aqueous and hydrocarbon compartments coincides with the density distribution maximum of the acyl chain carbonyl groups (Huber et al., 2002b). Analysis of these carbonyl interfaces reveals that the bilayer is ∼3 Å thicker close to helix H2 compared to the regions near helices H6 and H7 and between helices H4 and H5, a possible motif related to rhodopsin dimer formation (Fotiadis et al., 2003). The amphipathic helix H8 is located approximately at the level of the phosphocholine headgroup surface, and the pair of palmitoylated cysteines at the end of H8 is anchored within the membrane. The carbonyl group of the palmitoyl chain at Cys322 reaches almost to the carbonyl surface of the lipid acyl chains, whereas the palmitoyl chain at Cys323 extends one segment out of the hydrocarbon part of the membrane. In general, the ends of the TM helices of rhodopsin coincide with the phosphocholine headgroup surface, and thus the loop structures are almost exclusively outside the membrane. This is a consequence of the polar ends of an α-helix, where the amide and carbonyl groups of the peptide backbone are not usually joined by interresidue hydrogen bonds, as in the helix center. An exception is the cytoplasmic side of H6, which extends considerably into the cytoplasmic space. The TM helix arrangement can be idealized as a helix bundle with a collective twist. This arrangement should have an interfacial area larger at the helix ends than in the center (Cantor, 1999), comparable to our finding of a vase-shaped hydrophobic part of rhodopsin.
Benchmarking of hydropathy analysis
What are the protein sequence motifs that are associated with the different interfaces described by the GMS analysis? The relation of the protein sequence to the various surfaces is of particular importance, because chemical reactivity is dependent on the residue type and exposure. Consequently, the sequence dependence of the surface accessibility of a membrane protein such as rhodopsin is very important for interpreting structural changes related to protein function (Ballesteros et al., 2001; Hubbell et al., 2003). Moreover, quantitative analysis of the interfaces allows the calculation of a hydropathy index that can be compared with the conventional sequence-based hydropathy approach. The GMS analysis of the biomembrane model can thus be considered as benchmarking of the well-studied rhodopsin system as a paradigm for structural genomics and bioinformatics applications (vide supra). The availability of MD models to study lipid/protein interactions allows one to revisit the underlying principles of hydropathy analysis in atomic detail. An important finding is that a sequence-based hydropathy profile predicts very well the location of the lipid/water interface relative to the protein, as found in the equilibrated rhodopsin-containing membrane MD model. Moreover, detailed knowledge of the residues exposed to the lipid bilayer, especially in the loop regions, is valuable for the interpretation of lipid effects on rhodopsin activation.
Internal water molecules and polar pathway through rhodopsin
Several internal water molecules have been identified in the different crystal structures of rhodopsin (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002). In the MD model of rhodopsin eight water molecules were placed in internal cavities, where the equilibration resulted in dissociation of a single water molecule. During the preparation of this manuscript, a new rhodopsin structure (PDB entry number 1L9H, 2.6-Å resolution) was published (Okada et al., 2002), and the assignment of the seven stable water molecules found by the algorithmic approach (Huber et al., 2002a) as applied to the previous structure (PDB entry number 1HZX, 2.8-Å resolution) (Teller et al., 2001), was verified. However, five additional water molecules diffused from the bulk water into internal sites, so that 12 internal waters were present in the final equilibrated structure. It is noteworthy that two shorter MD simulations of rhodopsin, centered on isomerization of retinal, resulted in 10 and 19 internal water molecules, respectively (Röhrig et al., 2002; Saam et al., 2002). Alternative methods for water placement based on the thermodynamic stability of various locations as determined by free energy perturbation methods have been developed (Roux et al., 1996), but these are beyond the present scope of this work. The water molecules found herein are in contact with polar residues, and suggest the possibility of a polar pathway through the protein. Importantly, we found a string of three internal water molecules connecting the two pairs of β-sheets in the extracellular domain, which might have functional implications by mediating water exchange with the retinal binding site.
Domain flexibility of rhodopsin and functional dynamics
Clearly in membrane systems there is the possibility of a motional hierarchy, encompassing segmental and molecular motions (picosecond to nanosecond range), together with collective motions spanning a broad range of timescales (Brown et al., 2002). Comparison of the MD-generated membrane model to the crystal structure of rhodopsin indicates selective alterations of the protein conformation due to the membrane environment. However, the structure is virtually identical to the crystal structure in the regions close to the chromophore 11-cis-retinal, and near the internal water molecules identified by the GMS method. The average root mean square deviation from the crystal structure during the last 5 ns of the 15-ns MD simulations is 1.86 Å for all Cα atoms of the protein backbone. The local deviations from the crystal structure and fluctuation amplitudes reveal a correspondence of the flexible domains of the protein with the functional domains. Significant domain motions can be identified in loops C2 and C3, the amphipathic helix H8, and the C-terminal tail in the cytoplasmic region. The domain motion of the cytoplasmic loop C2 toward the lipid/water interface in the membrane model relative to the crystal structure occurs in a region that is tightly coupled to the membrane interface, as evident from the GMS analysis. The current MD simulations support the idea that rhodopsin domains of high flexibility in the nanosecond range are functionally important with regard to the mechanism of receptor activation. A possible explanation for the effects of the lipid composition on rhodopsin activation is discussed below.
Implications for mechanism of rhodopsin activation
How does this work contribute to understanding the basis of rhodopsin activation? Extensive experiments (Ghanouni et al., 2001; Koenig et al., 2002; Hubbell et al., 2003) together with modeling approaches have led to various paradigms of photoactivated rhodopsin and its complex with transducin (Arimoto et al., 2001). However, it remains unclear how at the molecular level the activated receptor couples to the G-protein, and the role of membrane lipid interactions requires further study. The principles of GPCR activation can be considered within the concept of functional microdomains in the protein (Sakmar et al., 2002), and the ensemble theory of a continuous distribution of microstates (Kenakin, 2002) in receptor activation. The conformational switch of the receptor involves its ability to catalyze the GDP–GTP nucleotide exchange of the G-protein transducin, and other changes, for example, its susceptibility to be phosphorylated. Pharmacologically, rhodopsin is characterized by the constitutive activity of the ligand-free receptor, and by the inverse, partial, and full agonism due to the bound ligand. The ligand of rhodopsin, 11-cis-retinal, acts as an inverse agonist in the dark state, which after photoisomerization to the all-trans conformation functions as an agonist (Sakmar et al., 2002). Several mutations have been described that change the pharmacological profile of wild-type rhodopsin; for example, 11-cis-retinal can also act as a partial agonist in certain mutant rhodopsins (Sakmar et al., 2002).
However, a number of thermodynamic variables, such as the proton activity, temperature, and composition of the lipid environment, can also modulate the activation of rhodopsin, that is, the fraction of the meta II conformation in the equilibrium of the later photointermediates (Brown, 1997; Botelho et al., 2002). For instance, changing the lipid composition to nonlamellar phase-promoting lipids, having small phosphoethanolamine headgroups or bulky DHA chains, strongly promotes formation of the active meta II state (Wiedmann et al., 1988; Botelho et al., 2002). Likewise increasing the local proton activity, due to acidic buffer conditions or anionic phosphoserine headgroups, stimulates meta II formation (Wang et al., 2002). The pH regulation of the active receptor conformation has been suggested to be related to the ERY microdomain in helix H3 of rhodopsin (Arnis et al., 1994), and to the homologous DR(W/Y) motif of other GPCRs (Ghanouni et al., 2001). In rhodopsin, the ERY motif is comprised of Glu134-Arg135-Tyr136. The crystal structure reveals that Arg135 is in contact with Glu134 of helix H3 and Glu247 in helix H6 in the dark-state conformation of rhodopsin (Palczewski et al., 2000), and moreover the H-bonding environment of Glu134 is thought to be altered in the active receptor conformation (Sakmar et al., 2002). Based on the MD model, we suggest that the lipid effects and the pH regulation of meta II formation share a common mechanism. The MD simulations in the bilayer membrane show that the cytoplasmic loop C2, which follows the ERY motif in helix H3, is shifted toward the lipid headgroups relative to the crystal structure. This can alter the aqueous exposure of Glu134 and thus its accessibility to protonation, changing its electrostatic interaction with Arg135 (internal salt bridge) (Honig and Hubbell, 1984; Sakmar et al., 2002). Additionally, water molecules might affect the local dielectric constant, and thus change the energetic stability of the Glu134-Arg135 interaction. In turn, modulation of the ERY stability could alter the protonation state of the retinal Schiff base with Lys296 via the location of its counterion Glu113, which like the ERY motif is part of helix H3. Yet another structural element that can modulate the receptor activation is the interaction of Glu122 in helix H3 with His211 in helix H5 (Weitz and Nathans, 1993), again underscoring the importance of helix H3 in coupling of the functional microdomains of rhodopsin (Sakmar et al., 2002).
What are the specific findings tending to support such a model for rhodopsin activation? In this regard, the cytoplasmic loop C2 exhibits significant mobility, as inferred from the crystallographic B-factors and the root mean square fluctuations from the equilibrated MD simulation. However, the rms deviations of the MD-derived equilibrium ensemble versus the crystal structure can be as much as 6–7 Å in the case of the C2 loop. This observation points to rather different microstates for the loop C2, an effect that extends toward the ERY motif in helix H3, although smaller in magnitude. Hence it appears likely that the conformation of loop C2 is affected by the membrane phospholipids. Furthermore, the GMS analysis reveals that residues 140–146 interact with the acyl chain ester and headgroups of the cytoplasmic phospholipid monolayer. The extent of this exposure to the lipid/water interface might be affected by nonlamellar phase-promoting lipids having a negative spontaneous curvature. In the planar state, these lipids can produce a smaller lateral pressure in the headgroup region, which is directed toward the protein, due to the smaller headgroup size relative to the acyl chains. This reduction in headgroup lateral pressure can favor extension of the loop C2 of rhodopsin into the lipid/water interface. As a result, the membrane composition may affect the ERY microdomain via the conformation of the cytoplasmic loop C2 as a possible mechanism for lipid effects on GPCR activation (Botelho et al., 2002).
In addition, similar influences of the lipid environment on the cytoplasmic loop C3 and the amphipathic helix H8, which are both involved in G-protein activation (Sakmar et al., 2002), can be inferred from the MD simulations. Loop C3 and helix H8 both exhibit movement after light activation (Hubbell et al., 2003), and helix H8 could act as a membrane-dependent conformational switch (Krishna et al., 2002). The interaction of the cysteine palmitoylations and the proximal residues of the C-terminal tail with the bilayer leads to appreciable deviations of helix H8 from the crystal structure, as shown in Fig. 7, a and c. A high mobility of the distal parts of the C-terminal tail is evident from the large rms fluctuations in the MD ensemble. This region of the C-terminal tail contains the rhodopsin kinase-dependent phosphorylation sites, which are necessary for efficient inactivation of G-protein-dependent signaling due to visual arrestin binding (Sakmar et al., 2002). Other GPCRs exhibit arrestin-dependent endocytotic membrane trafficking, important for spatial and temporal regulation of receptor signaling (Ceresa and Schmid, 2000). Moreover, theC-terminal tail is recognized by the protein-sorting machinery of the cell, and mutations lead to autosomal-dominant retinitis pigmentosa (Sung et al., 1994). Thus, the observed high mobility of the C-terminal tail might be physiologically essential for interaction with different protein components related to sorting and signaling of rhodopsin.
By contrast, the extracellular domain of rhodopsin, including the retinal binding site, exhibits few significant deviations from the crystal structure and only small amplitude fluctuations are observed. This might be related to the very low dark noise of rod cells due to the small thermal isomerization rate of the inverse agonist 11-cis-retinal, with consequences for its function as a single photon receptor (Baylor, 1996). Interestingly, all 12 internal water molecules identified by our modeling approach are located in regions with very small deviations from the crystal structure. Even though only seven waters are confirmed in the latest refined rhodopsin crystal structure (Okada et al., 2002), the five additional internal water molecules are structurally nonperturbing. As a result they may also be present in the crystals, but remain undetected due to the limited resolution of 2.6 Å. In the similar case of bacteriorhodopsin, refinement of the crystal structure to a resolution of 1.55 Å has revealed several additional water molecules that are unresolved at lower resolution (Luecke et al., 1999). The internal water molecules in bacteriorhodopsin have a functional role in the proton transport cycle (Kandori, 2000). In this sense, the internal waters of rhodopsin might be similarly important in the photoactivation mechanism (Okada et al., 2002), as it has been suggested to be a blocked proton pump (Hofmann et al., 1995).
CONCLUSIONS
The combination of MD simulations with 2H NMR spectroscopy provides a framework for structure-function studies of membrane-embedded rhodopsin. In the first applications of a new GMS method, we have been able to determine the hydrophilic and hydrophobic interfaces of rhodopsin with its environment. We have also quantified the membrane deformation and the cross-sectional area of the protein, and have identified water molecules that diffuse into internal hydration sites. The structure of rhodopsin imposes a small positive curvature on the extracellular side of the membrane, whereas on its cytosolic side a negative curvature of the adjacent lipid monolayer is found. These curvatures may be related to the preferential association of rhodopsin with reverse hexagonal (HII) phase-promoting lipids, which have a negative spontaneous monolayer curvature. In turn, there may be implications for the selection of polyunsaturated lipids in the native environment of rhodopsin, and possibly other GPCRs, in biological signaling mechanisms. A number of internal water molecules are evident, suggesting a polar pathway running through the protein. Photoisomerization of retinal may switch the connectivity of the polar channel within rhodopsin. The selective mobility of the cytoplasmic domain is probably important for recognizing other signal transduction components, and for interactions with the sorting machinery of the cell. Finally, we suggest a possible microscopic mechanism for lipid effects on rhodopsin activation, based on site-specific conformational changes of rhodopsin and their relation to the lipid/protein interfaces. The highly conserved ERY motif, which is important for receptor activation, may be functionally coupled to the lipid/water interface via the cytoplasmic C2 loop conformation, underlying the lipid compositional effects on rhodopsin activation.
Acknowledgments
We thank Professor Rosalie K. Crouch for kindly providing 11-cis-retinal, Professor Krzysztof Palczewski for supplying the refined rhodopsin structure coordinates before publication, Professor Thomas P. Sakmar for enlightening discussions, Drs. Gary V. Martinez and Andrey V. Struts for assistance with the NMR spectrometer, and Dr. Horia I. Petrache for many helpful discussions during the course of this work.
Supercomputing time was generously provided through development and production grants (MCB020015P, MCB020017N, and MCB020042P) by the Pittsburgh Supercomputing Center (PSC) and the National Center for Supercomputing Applications (NCSA). A.V.B. is the recipient of a scholarship from CAPES, Brasilia, DF, Brazil. This work was supported by the U. S. National Institutes of Health.
APPENDIX
Equilibration of the membrane models
Here we further describe some of the important technical details of the MD simulations. The model of membrane-embedded rhodopsin employed a large unit cell of a POPC bilayer with twice the number of lipids as in control POPC simulations (Huber et al., 2002b). The initial configuration of a hydrated POPC bilayer membrane, designated as system L0 (Table 2), was taken from previous MD simulations (Huber et al., 2002b). The first rhodopsin model (RhA) was immersed in the membrane bilayer patch (P0), and some of the water molecules and 20 lipids in each leaflet of the bilayer, whose excluded volumes overlapped with the rhodopsin molecule, were removed. The resulting defects in the bilayer structure around rhodopsin were annealed with the rhodopsin atoms constrained to fluctuate about the initial structure. Clearly the equilibrium membrane area depends on the applied surface tension γ, which for a stress-free membrane should be zero. However, introduction of a small positive surface tension is needed to compensate for finite size effects in MD membrane models and thus reproduce experimental quantities (Feller et al., 1997; Huber et al., 2002b). An alternative method is to simulate the membrane at constant area, but this approach requires a priori knowledge of the latter.
In this work we have used an iterative protocol to determine the membrane area. A simulation was performed with an initial guess for the applied surface tension, and after a time of equilibration the lipid order parameter profile was calculated, and compared with experimental 2H NMR data. Depending on the result, the surface tension for the subsequent segment of the membrane was changed to alter the membrane area, and thus the order parameters. However, limitations in the available computer time restricted the ability to converge in such an iterative process. The applied surface tension as a function of simulation time is shown for the rhodopsin-containing membrane in Fig. 8 a. Several changes of the molecular composition of the membrane models were introduced during the initial 5 ns of the simulations, described for completeness in Table 2. The average area per lipid for the rhodopsin-containing bilayer membranes in response to the applied surface tension was used to monitor equilibration of the bilayer structure for the different segments (P1–P6) of the simulations, cf. Fig. 8 b. Note that the last 13.6 ns were performed in the presence of rhodopsin models (RhB and RhC) based on the refined crystal structure (Teller et al., 2001). The last 5 ns are designated as segment P6, which was used for the structural analysis.
FIGURE 8.

Equilibration of the rhodopsin-containing membrane during the molecular dynamics simulations for the different models at variable surface tension. The relation of rhodopsin models (RhA, RhB, and RhC; cf. Table 1) and membrane systems (P1–P6 and L1–L4; cf. Table 2) to the simulation time is identified at the top. (a) Simulation protocol for the rhodopsin-containing MD bilayer model. The applied surface tension γ as a function of simulation time for the different segments is related to the changes in the area per lipid. The analysis of the rhodopsin structure was performed for the last segment P6 at zero surface tension. (b) Equilibration of the area per lipid for the rhodopsin-containing membrane. Note that the short pulse of negative applied surface tension γ = −500 mN m−1 in a resulted in a 5-Å2 reduction of the average area per lipid. A similar area reduction was obtained for a weaker pulse of γ = −32 mN m−1 for 2 ns. The rationale behind the biased simulation with negative surface tension (within segment P4) was to accelerate the rate of area condensation. (c) The applied surface tension γ for segments L1–L3 of the POPC/water simulations. Segment L4 of the POPC/water simulation is omitted from the plot, because it was performed with a constant area-isothermal-isobaric algorithm (Feller et al., 1997) at a normal pressure of 1.0 bar. The area was chosen to correspond to the average area from the equilibrated segment P6 of the rhodopsin simulation. (d) Equilibration of the area per lipid for the POPC/water system. The constant surface area simulation in segment L4 is indicated as a dotted line parallel to segment L3 from 19 ns to 24 ns. Note that for zero surface tension (segments P6 and L3) the area per lipid equilibrates to significantly different values, depending on the presence of rhodopsin. Therefore the constant area simulation (segment L4) was performed to compare the lipid order parameter profiles with the rhodopsin-containing simulation (segment P6) at a similar area per lipid.
The rhodopsin-containing membrane simulations (vide supra) were also compared with control simulations of the protein-free phospholipid membranes. The initial POPC bilayer membrane model (Huber et al., 2002b), segment L0, was equilibrated by applying a sequence of different surface tension values, as illustrated in Fig. 8 c. These simulations (L1, L2, and L3) corresponded to a single trajectory based on the starting structure (L0), showing the dependence of the area per lipid on the applied surface tension. Note the timescales necessary for equilibration after reducing the surface tension to zero in segment L3; an area condensation of about 9 Å2 occurred during about 17 ns, followed by 5 ns of fluctuations about the equilibrium value of 56 Å2, cf. Fig. 8 d. However, in this condensed state the order parameters were significantly higher than the experimental 2H NMR values, indicating that zero surface tension is inappropriate for the protein-free bilayer. Because the rhodopsin-containing POPC membrane equilibrated at an area per lipid of about 62.6 Å2, with zero applied surface tension (segment P6), after 19 ns of the trajectory in segment L3 we initiated a 5-ns constant area simulation (L4) of the POPC/water system having 62.4 Å2 per lipid. The L4 segment was used for the order parameter analysis, and for comparison with the rhodopsin-containing system based on the experimental data from solid-state 2H NMR spectroscopy.
Generalized molecular surface approach
An important question in membrane biophysics and structural proteomics is how the various amino acids of membrane proteins, such as the GPCR rhodopsin, contribute to the hydrophobic and polar interfaces. These interfaces determine the solvation energy of the folded membrane protein, and predictions of the topology from sequence data alone are possible with hydropathy analysis methods (White and Wimley, 1999). Interactions of proteins with the molecular environment are intimately related to the molecular surface of the protein, because chemical reactivity, even in the case of noncovalent interactions, is related to the accessibility of the reactants. In this regard, Richards- and Connolly-type surfaces are routinely utilized (Richards, 1985; Connolly, 1993) to interpret crystallographic structures in terms of solvent accessibility and excluded volumes. Schematically, a probe sphere having the size of a water molecule is rolled over the van der Waals surface of a protein. The center of this sphere traces the solvent-accessible or Richards-type surface. The solvent-excluded surface is defined by the surface enveloping the volume that is inaccessible to the volumes of these probe spheres. This surface is called a Connolly-type molecular surface, which can be viewed as the dividing surface between an implicit solvent continuum and the protein.
However, an analysis of solute/solvent separating surfaces with explicit solvent molecules is impossible with the above definitions of a molecular surface, which do not account for explicitly represented solvating molecules. Consequently, we developed a new method to quantify the solute/solvent contact surfaces. The method defines the molecular surface of a solute molecule, in analogy to the Connolly-type molecular surface: any point on the surface has equal orthogonal distances to the closest van der Waals surfaces of: i), the solute, and ii), the solvent. The new method to generate this generalized molecular surface, as implemented in the in-house developed software package MOLGEOM (Huber et al., 2002b), is based on a generalization of Voronoi polyhedra to atoms of different sizes with a modified distance criterion (Goede et al., 1997), resulting in hyperboloid faces of the Voronoi cells. Topologically, these cells are polyhedra, but the edges and faces are curved. The distance to each atom is now defined as the orthogonal distance to the van der Waals sphere of the particular atom. Such generalized Voronoi polyhedra with the new distance criterion are given by the union of all possible points in space having a smaller value of a generalized distance to the central atom than to any other atom. Compared to other approaches dealing with the problem of different atomic radii (Gerstein et al., 1995), the packing of these Voronoi cells does not suffer from so-called vertex errors, that is misallocations of space leading to wrong volumes (Goede et al., 1997). The Connolly-type molecular surface and the Voronoi cells both probe the van der Waals spheres of the molecules, making the extension to the GMS method straightforward.
Mathematically, the definition of the geometry utilized in the GMS method is the following. Any point x in space is related to a particular van der Waals (vdW) sphere i of radius ri and center pi by the general distance di(x) = |x − pi| − ri. This distance di(x) can be interpreted as the radius of another sphere with center x touching the surface of the vdW sphere i. The generalized Voronoi cell Vi of the vdW sphere i is defined by Vi = {x|di(x) ≤ dj(x) ∧ i ≠ j}. The cell Vi is the set of all points x closer to the vdW sphere i than to any other vdW sphere j, as measured by the distance criterion. The generalized Voronoi facet Sij of two vdW spheres i and j is given by Sij = Vi ∩ Vj, the intersection of the Voronoi cells Vi and Vj. It is the set of points equidistant to the surfaces of the vdW spheres i and j (equidistance criterion), where no other vdW sphere k is closer than i and j. The GMS is constructed from the surface of the body formed from the union of all the generalized Voronoi polyhedra belonging to a particular molecule. If the Voronoi polyhedra of two atoms within the same molecule share a common face, the union body no longer contains this face. All unique faces in this union of atomic Voronoi cells form the GMS of that molecule.
The stepwise construction of the separating surfaces (generalized Voronoi facets) and of the generalized Voronoi cells is illustrated in Fig. 9. In the schematic construction in Fig. 9, a–c, the volume of the system comprising four atoms (labeled A–D) with different van der Waals radii is parsed into four generalized Voronoi polyhedra (VA–VD). The separating surfaces between these polyhedra are the generalized Voronoi facets (SAB, SAC, SAD, SBC, and SCD). In Fig. 9 a, the red line corresponds to the generalized Voronoi facet SAB separating the space related to vdW spheres A or B. It can be seen that each of the thin red circles is simultaneously in contact with the vdW spheres A and B, implying that its radius touches both spherical surfaces. Hence their centers correspond to SAB, illustrating the equidistance criterion that divides the cells VA and VB. Fig. 9 b continues the construction from Fig. 9 a, in which the additional vdW sphere C reduces the facet SAB to the solid red line. Furthermore, two new facets SBC (solid green line) and SAC (solid orange line) are created. The thin green circles are simultaneously in contact with the vdW spheres B and C, whereas the thin orange circles touch A and C. The dotted green line is part of the cell VA and the dotted orange line is part of the cell VB, whereas the dotted red line is now part of the cell VC. Note also the central thin black circle in contact with the three vdW spheres A, B, and C, corresponding in 3-D to a common edge of the facets SAB, SAC, and SBC formed pairwise by the cells VA, VB, and VC. In Fig. 9 c the additional vdW sphere D generates in the same manner the facets SAD (light green) and SCD (turquoise). All the points on the separating surface of B and D (blue dotted line) are completely enclosed within the other cells, VA and VC, and hence facet SBD is nonexistent. Note that facet SAC is now limited to the region not within VB or VD. Lastly, Fig. 9 d shows the generalized Voronoi cells VA, VB, VC, and VD that are defined by the set of generalized Voronoi facets. By induction, the result is the generalized Voronoi cells and facets that are the contact surface elements between any number of van der Waals spheres of different sizes.
FIGURE 9.

Definition of the geometry utilized in the generalized molecular surface method. The stepwise construction of the separating surfaces (generalized Voronoi facets) between four van der Waals (vdW) spheres A, B, C, and D in 3-D space is illustrated in a–d. The set of contact elements of a molecule with its explicit (solvent) environment comprises the GMS as described in the text.
How are these generalized Voronoi elements (facets and polyhedra) related to the GMS? Let us next consider that atoms A and B are part of a molecule M, and that C and D are solvent atoms. The union of the generalized Voronoi polyhedra VA and VB is the solvent-excluded volume of molecule M in the framework of the GMS method. In this example, the facets SAD, SAC, and SBC are part of the GMS of the molecule M with the solvent. The area contribution of atom A to the GMS is the sum of the area of facets SAD and SAC, whereas atom B only contributes to the GMS by the area of facet SBC. This example illustrates how the GMS method is related to the generalized Voronoi elements. Furthermore, the GMS can be topologically subdivided to account for complex molecules, such as proteins, comprising a collection of different residues. The GMS contribution of a given residue, within a larger molecule, is that part of the GMS of the larger molecule formed by the generalized Voronoi polyhedra of any atom of this residue. In like manner, supramolecular structures formed by a collection of individual molecules, for example a complex solvent, such as lipid/water mixtures, can be used for the topological subdivision of a GMS. The GMS contribution of a particular solvent molecule in a complex solvent mixture is defined by those parts of the GMS closer to any atom of this particular solvent molecule than any other solvent molecule. The definition of residue, molecule, and supramolecular structure can be very general, because the implementation of the algorithm is based on a context-free group definition. In the hydropathy analysis, for example, one group comprises all water molecules, whereas a second group includes all the acyl chain hydrocarbon atoms of the lipid bilayer. Our current implementation generates the GMS by a finite area element method, with an adaptive mesh refinement by subdivision. Another important detail of the actual realization of the GMS is the distance criterion, which is defined within the framework of the nearest image convention under periodic boundary conditions for orthorhombic unit cells. The flexible design considerations allow application of the new GMS method to many contemporary biomolecular MD simulations.
Estimation of confidence levels for structural deviations
The criterion that the equilibrium average structure from the MD simulation differs from the crystal structure can be estimated from the root mean square values:
 |
(4) |
Here, CL is the confidence limit, rmsd is the rms deviation of the simulated ensemble versus the crystal structure, rmsfMD is the rms fluctuation of the simulated ensemble versus the ensemble average, and rmsfXR is the rms fluctuation as calculated from the crystallographic B-factor. The above formula was used as a simple test of statistically significant differences in the analysis of the data presented in Fig. 6 b; more elaborate statistical methods are beyond the scope of this work.
Abbreviations used: GMS, generalized molecular surface; GPCR, G-protein-coupled receptor; CPK, Corey-Pauling-Koltun; c-side, cytosolic (cytoplasmic, extradiskal) side; CT, carboxyl-terminal; DHA, docosahexaenoic acid; e-side, extracellular (luminal, intradiskal) side; FSM, flexible surface model; egg PC, egg yolk phosphatidylcholine; MD, molecular dynamics; meta I, metarhodopsin I; meta II, metarhodopsin II; NMR, nuclear magnetic resonance; NPγT, constant particle number, normal pressure, surface tension, and temperature; NT, amino-terminal; PDB, Brookhaven Protein Data Bank; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPC-d31, 1-perdeuteriopalmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; PUFA, polyunsaturated fatty acid; vdW, van der Waals; TM, transmembrane; rms, root mean square; ROS, rod outer segment.
References
- Arimoto, R., O. G. Kisselev, G. M. Makara, and G. R. Marshall. 2001. Rhodopsin-transducin interface: studies with conformationally constrained peptides. Biophys. J. 81:3285–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnis, S., K. Fahmy, K. P. Hofmann, and T. P. Sakmar. 1994. A conserved carboxylic-acid group mediates light-dependent proton uptake and signaling by rhodopsin. J. Biol. Chem. 269:23879–23881. [PubMed] [Google Scholar]
- Baker, D., and A. Sali. 2001. Protein structure prediction and structural genomics. Science. 294:93–96. [DOI] [PubMed] [Google Scholar]
- Baldwin, J. M., G. F. X. Schertler, and V. M. Unger. 1997. An alpha-carbon template for the transmembrane helices in the rhodopsin family of G-protein-coupled receptors. J. Mol. Biol. 272:144–164. [DOI] [PubMed] [Google Scholar]
- Baldwin, P. A., and W. L. Hubbell. 1985. Effects of lipid environment on the light-induced conformational changes of rhodopsin. 2. Roles of lipid chain length, unsaturation, and phase state. Biochemistry. 24:2633–2639. [DOI] [PubMed] [Google Scholar]
- Ballesteros, J. A., L. Shi, and J. A. Javitch. 2001. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol. Pharmacol. 60:1–19. [PubMed] [Google Scholar]
- Baylor, D. 1996. How photons start vision. Proc. Natl. Acad. Sci. USA. 93:560–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach, J. M., R. D. Pates, J. F. Ellena, and M. F. Brown. 1984. Flash photolysis studies of rhodopsin function in recombinant membranes. Biophys. J. 45:292a. (Abstr.) [Google Scholar]
- Beck, M., T. P. Sakmar, and F. Siebert. 1998. Spectroscopic evidence for interaction between transmembrane helices 3 and 5 in rhodopsin. Biochemistry. 37:7630–7639. [DOI] [PubMed] [Google Scholar]
- Bernèche, S., and B. Roux. 2000. Molecular dynamics of the KcsA K+ channel in a bilayer membrane. Biophys. J. 78:2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaney, J. M., G. M. Crippen, A. Dearing, J. S. Dixon, and D. C. Spellmeyer. 1995. DGEOM(95): Distance Geometry Program: Quantum Chemical Program Exchange.
- Botelho, A. V., N. J. Gibson, R. L. Thurmond, Y. Wang, and M. F. Brown. 2002. Conformational energetics of rhodopsin modulated by nonlamellar-forming lipids. Biochemistry. 41:6354–6368. [DOI] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Brown, M. F. 1996. Membrane structure and dynamics studied with NMR spectroscopy. In Biological Membranes. K. M. Merz Jr. and B. Roux, editors. Birkhäuser, Boston, MA. 175–252.
- Brown, M. F. 1997. Influence of nonlamellar-forming lipids on rhodopsin. Curr. Top. Membr. 44:285–356. [Google Scholar]
- Brown, M. F., R. L. Thurmond, S. W. Dodd, D. Otten, and K. Beyer. 2002. Elastic deformation of membrane bilayers probed by deuterium NMR relaxation. J. Am. Chem. Soc. 124:8471–8484. [DOI] [PubMed] [Google Scholar]
- Cantor, R. S. 1999. The influence of membrane lateral pressures on simple geometric models of protein conformational equilibria. Chem. Phys. Lipids. 101:45–56. [DOI] [PubMed] [Google Scholar]
- Ceresa, B. P., and S. L. Schmid. 2000. Regulation of signal transduction by endocytosis. Curr. Opin. Cell Biol. 12:204–210. [DOI] [PubMed] [Google Scholar]
- Connolly, M. L. 1993. The molecular-surface package. J. Mol. Graph. 11:139–141. [DOI] [PubMed] [Google Scholar]
- Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- Deese, A. J., E. A. Dratz, and M. F. Brown. 1981. Retinal rod outer segment lipids form bilayers in the presence and absence of rhodopsin: a 31P NMR study. FEBS Lett. 124:93–99. [DOI] [PubMed] [Google Scholar]
- Dill, K. A. 1997. Additivity principles in biochemistry. J. Biol. Chem. 272:701–704. [DOI] [PubMed] [Google Scholar]
- Drews, J. 2000. Drug discovery: a historical perspective. Science. 287:1960–1964. [DOI] [PubMed] [Google Scholar]
- Ebrey, T., and Y. Koutalos. 2001. Vertebrate photoreceptors. Prog. Retin. Eye Res. 20:49–94. [DOI] [PubMed] [Google Scholar]
- Ellena, J. F., R. D. Pates, and M. F. Brown. 1986. 31P NMR spectra of rod outer segment and sarcoplasmic reticulum membranes show no evidence of immobilized components due to lipid-protein interactions. Biochemistry. 25:3742–3748. [DOI] [PubMed] [Google Scholar]
- Engelman, D. M., T. A. Steitz, and A. Goldman. 1986. Identifying nonpolar transbilayer helices in amino-acid sequences of membrane proteins. Annu. Rev. Biophys. Biophys. Chem. 15:321–353. [DOI] [PubMed] [Google Scholar]
- Epand, R. M. 1990. Relationship of phospholipid hexagonal phases to biological phenomena. Biochem. Cell Biol. 68:17–23. [DOI] [PubMed] [Google Scholar]
- Fahmy, K., F. Jäger, M. Beck, T. A. Zvyaga, T. P. Sakmar, and F. Siebert. 1993. Protonation states of membrane-embedded carboxylic acid groups in rhodopsin and metarhodopsin-II: a Fourier-transform infrared spectroscopy study of site-directed mutants. Proc. Natl. Acad. Sci. USA. 90:10206–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, S. E., and A. D. MacKerell. 2000. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B. 104:7510–7515. [Google Scholar]
- Feller, S. E., and R. W. Pastor. 1996. On simulating lipid bilayers with an applied surface tension: periodic boundary conditions and undulations. Biophys. J. 71:1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, S. E., R. M. Venable, and R. W. Pastor. 1997. Computer simulation of a DPPC phospholipid bilayer: structural changes as a function of molecular surface area. Langmuir. 13:6555–6561. [Google Scholar]
- Fotiadis, D., Y. Liang, S. Filipek, D. A. Saperstein, A. Engel, and K. Palczewski. 2003. Atomic-force microscopy: rhodopsin dimers in native disc membranes. Nature. 421:127–128. [DOI] [PubMed] [Google Scholar]
- Frishman, D., and P. Argos. 1995. Knowledge-based protein secondary structure assignment. Proteins. 23:566–579. [DOI] [PubMed] [Google Scholar]
- Gerstein, M., J. Tsai, and M. Levitt. 1995. The volume of atoms on the protein surface—calculated from simulation, using Voronoi polyhedra. J. Mol. Biol. 249:955–966. [DOI] [PubMed] [Google Scholar]
- Ghanouni, P., J. J. Steenhuis, D. L. Farrens, and B. K. Kobilka. 2001. Agonist-induced conformational changes in the G-protein-coupling domain of the β2 adrenergic receptor. Proc. Natl. Acad. Sci. USA. 98:5997–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson, N. J., and M. F. Brown. 1993. Lipid headgroup and acyl chain composition modulate the MI-MII equilibrium of rhodopsin in recombinant membranes. Biochemistry. 32:2438–2454. [DOI] [PubMed] [Google Scholar]
- Goede, A., R. Preissner, and C. Frommel. 1997. Voronoi cell: new method for allocation of space among atoms: elimination of avoidable errors in calculation of atomic volume and density. J. Comput. Chem. 18:1113–1123. [Google Scholar]
- Grubmüller, H. 1996. Solvate: A Program to Create Atomic Solvent Models. Version 1.0. Theoretical Biophysics Group, Institut für Medizinische Optik, Ludwig-Maximilians-Universität München, Munich, Germany.
- Henzler Wildman, K. A., G. V. Martinez, M. F. Brown, and A. Ramamoorthy. 2003. Disruption of the hydrophobic core of the lipid bilayer by the human antimicrobial peptide LL-37. Biophys. J. 84:278a. (Abstr.) [Google Scholar]
- Hofmann, K. P., S. Jäger, and O. P. Ernst. 1995. Structure and function of activated rhodopsin. Isr. J. Chem. 35:339–355. [Google Scholar]
- Honig, B. H., and W. L. Hubbell. 1984. Stability of “salt bridges” in membrane proteins. Proc. Natl. Acad. Sci. USA. 81:5412–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell, W. L., C. Altenbach, C. M. Hubbell, and H. G. Khorana. 2003. Rhodopsin structure, dynamics, and activation: a perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein. Chem. 63:243–290. [DOI] [PubMed] [Google Scholar]
- Huber, T., A. V. Botelho, and M. F. Brown. 2002a. Hydrophobic interface of the GPCR prototype rhodopsin. Biophys. J. 82:225a. (Abstr.) [Google Scholar]
- Huber, T., B. S. W. Chang, and T. P. Sakmar. 2003. Structure and dynamics of archosaur rhodopsin and other ancestral visual pigments. Biophys. J. 84:271a. (Abstr.) [Google Scholar]
- Huber, T., K. Rajamoorthi, V. F. Kurze, K. Beyer, and M. F. Brown. 2002b. Structure of docosahexaenoic acid-containing phospholipid bilayers as studied by 2H NMR and molecular dynamics simulations. J. Am. Chem. Soc. 124:298–309. [DOI] [PubMed] [Google Scholar]
- Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- Jensen, J. W., and J. S. Schutzbach. 1984. Activation of mannosyltransferase II by nonbilayer phospholipids. Biochemistry. 23:1115–1119. [Google Scholar]
- Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- Kandori, H. 2000. Role of internal water molecules in bacteriorhodopsin. Biochim. Biophys. Acta. 1460:177–191. [DOI] [PubMed] [Google Scholar]
- Kenakin, T. 2002. Drug efficacy at G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 42:349–379. [DOI] [PubMed] [Google Scholar]
- Klein-Seetharaman, J., P. J. Reeves, M. C. Loewen, E. V. Getmanova, J. Chung, H. Schwalbe, P. E. Wright, and H. G. Khorana. 2002. Solution NMR spectroscopy of [α-15N]lysine-labeled rhodopsin: the single peak observed in both conventional and TROSY-type HSQC spectra is ascribed to Lys-339 in the carboxyl-terminal peptide sequence. Proc. Natl. Acad. Sci. USA. 99:3452–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig, B. W., G. Kontaxis, D. C. Mitchell, J. M. Louis, B. J. Litman, and A. Bax. 2002. Structure and orientation of a G protein fragment in the receptor bound state from residual dipolar couplings. J. Mol. Biol. 322:441–461. [DOI] [PubMed] [Google Scholar]
- Krishna, A. G., S. T. Menon, T. J. Terry, and T. P. Sakmar. 2002. Evidence that helix 8 of rhodopsin acts as a membrane-dependent conformational switch. Biochemistry. 41:8298–8309. [DOI] [PubMed] [Google Scholar]
- Kyte, J., and R. F. Doolittle. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132. [DOI] [PubMed] [Google Scholar]
- Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland et al. 2001. Initial sequencing and analysis of the human genome. Nature. 409:860–921. [DOI] [PubMed] [Google Scholar]
- Lee, A. G. 1998. How lipids interact with an intrinsic membrane protein: the case of the calcium pump. Biochim. Biophys. Acta. 1376:381–390. [DOI] [PubMed] [Google Scholar]
- Levitt, M., M. Hirshberg, R. Sharon, K. E. Laidig, et al. 1997. Calibration and testing of a water model for simulation of the molecular dynamics of proteins and nucleic acids in solution. J. Phys. Chem. B. 101:5051–5061. [Google Scholar]
- Lewis, J. W., and D. S. Kliger. 2000. Absorption spectroscopy in studies of visual pigments: spectral and kinetic characterization of intermediates. Methods. Enzymol. 315:164–178. [DOI] [PubMed] [Google Scholar]
- Lomize, A. L., I. D. Pogozheva, and H. I. Mosberg. 1999. Structural organization of G-protein-coupled receptors. J. Comput. Aided Mol. Des. 13:325–353. [DOI] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291:899–911. [DOI] [PubMed] [Google Scholar]
- Miljanich, G. P., M. F. Brown, S. Mabrey-Gaud, E. A. Dratz, and J. M. Sturtevant. 1985. Thermotropic behavior of retinal rod membranes and dispersions of extracted phospholipids. J. Membr. Biol. 85:79–86. [DOI] [PubMed] [Google Scholar]
- Miljanich, G. P., P. P. Nemes, D. L. White, and E. A. Dratz. 1981. The asymmetric transmembrane distribution of phosphatidylethanolamine, phosphatidylserine, and fatty acids of the bovine retinal rod outer segment disk membrane. J. Membr. Biol. 60:249–255. [DOI] [PubMed] [Google Scholar]
- Mitchell, D. C., J. Kibelbek, and B. J. Litman. 1992. Effect of phosphorylation on receptor conformation: the metarhodopsin I-metarhodopsin II equilibrium in multiply phosphorylated rhodopsin. Biochemistry. 31:8107–8111. [DOI] [PubMed] [Google Scholar]
- Navarro, J., M. Toivio-Kinnucan, and E. Racker. 1984. Effect of lipid composition on the calcium/adenosine 5′-triphosphate coupling ratio of the Ca2+-ATPase of sarcoplasmic reticulum. Biochemistry. 23:130–135. [DOI] [PubMed] [Google Scholar]
- Neuringer, M. 2000. Infant vision and retinal function in studies of dietary long-chain polyunsaturated fatty acids: methods, results, and implications. Am. J. Clin. Nutr. 71:256S–267S. [DOI] [PubMed] [Google Scholar]
- Okada, T., Y. Fujiyoshi, M. Silow, J. Navarro, E. M. Landau, and Y. Shichida. 2002. Functional role of internal water molecules in rhodopsin revealed by x-ray crystallography. Proc. Natl. Acad. Sci. USA. 99:5982–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada, T., and K. Palczewski. 2001. Crystal structure of rhodopsin: implications for vision and beyond. Curr. Opin. Struct. Biol. 11:420–426. [DOI] [PubMed] [Google Scholar]
- Palczewski, K., T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. Le Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto, and M. Miyano. 2000. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 289:739–745. [DOI] [PubMed] [Google Scholar]
- Perozo, E., A. Kloda, D. M. Cortes, and B. Martinac. 2002. Physical principles underlying the transduction of bilayer deformation forces during mechanosensitive channel gating. Nat. Struct. Biol. 9:696–703. [DOI] [PubMed] [Google Scholar]
- Petrache, H. I., A. Salmon, and M. F. Brown. 2001. Structural properties of docosahexaenoyl phospholipid bilayers investigated by solid-state 2H NMR spectroscopy. J. Am. Chem. Soc. 123:12611–12622. [DOI] [PubMed] [Google Scholar]
- Rand, R. P., and V. A. Parsegian. 1997. Hydration, curvature, and bending elasticity of phospholipid monolayers. Curr. Top. Membr. 44:167–189. [Google Scholar]
- Richards, F. M. 1985. Calculation of molecular volumes and areas for structures of known geometry. Methods Enzymol. 115:440–464. [DOI] [PubMed] [Google Scholar]
- Röhrig, U. F., L. Guidoni, and U. Rothlisberger. 2002. Early steps of the intramolecular signal transduction in rhodopsin explored by molecular dynamics simulations. Biochemistry. 41:10799–10809. [DOI] [PubMed] [Google Scholar]
- Roux, B., M. Nina, R. Pomes, and J. C. Smith. 1996. Thermodynamic stability of water molecules in the bacteriorhodopsin proton channel: a molecular dynamics free energy perturbation study. Biophys. J. 71:670–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saam, J., E. Tajkhorshid, S. Hayashi, and K. Schulten. 2002. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophys. J. 83:3097–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmar, T. P., S. T. Menon, E. P. Marin, and E. S. Awad. 2002. Rhodopsin: insights from recent structural studies. Annu. Rev. Biophys. Biomol. Struct. 31:443–484. [DOI] [PubMed] [Google Scholar]
- Simons, K., and E. Ikonen. 2000. How cells handle cholesterol. Science. 290:1721–1726. [DOI] [PubMed] [Google Scholar]
- Stenkamp, R. E., S. Filipek, C. Driessen, D. C. Teller, and K. Palczewski. 2002. Crystal structure of rhodopsin: a template for cone visual pigments and other G protein-coupled receptors. Biochim. Biophys. Acta. 1565:168–182. [DOI] [PubMed] [Google Scholar]
- Strader, C. D., T. M. Fong, M. P. Graziano, and M. R. Tota. 1995. The family of G-protein-coupled receptors. FASEB J. 9:745–754. [PubMed] [Google Scholar]
- Sung, C. H., C. Makino, D. Baylor, and J. Nathans. 1994. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J. Neurosci. 14:5818–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajkhorshid, E., J. Baudry, K. Schulten, and S. Suhai. 2000. Molecular dynamics study of the nature and origin of retinal's twisted structure in bacteriorhodopsin. Biophys. J. 78:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teller, D. C., T. Okada, C. A. Behnke, K. Palczewski, and R. E. Stenkamp. 2001. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry. 40:7761–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurmond, R. L., G. Lindblom, and M. F. Brown. 1993. Curvature, order, and dynamics of lipid hexagonal phases studied by deuterium NMR spectroscopy. Biochemistry. 32:5394–5410. [DOI] [PubMed] [Google Scholar]
- Wang, Y., A. V. Botelho, G. V. Martinez, and M. F. Brown. 2002. Electrostatic properties of membrane lipids coupled to metarhodopsin II formation in visual transduction. J. Am. Chem. Soc. 124:7690–7701. [DOI] [PubMed] [Google Scholar]
- Weitz, C. J., and J. Nathans. 1993. Rhodopsin activation: effects on the metarhodopsin I-metarhodopsin II equilibrium of neutralization or introduction of charged amino acids within putative transmembrane segments. Biochemistry. 32:14176–14182. [DOI] [PubMed] [Google Scholar]
- White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]
- Wiedmann, T. S., R. D. Pates, J. M. Beach, A. Salmon, and M. F. Brown. 1988. Lipid protein interactions mediate the photochemical function of rhodopsin. Biochemistry. 27:6469–6474. [DOI] [PubMed] [Google Scholar]
- Zhang, L., and J. Hermans. 1996. Hydrophilicity of cavities in proteins. Proteins. 24:433–438. [DOI] [PubMed] [Google Scholar]