

Abstract
By examining the consequences both of changes of [K+]o and of point mutations in the outer pore mouth, our goal was to determine if the mechanism of the block of Kv1.5 ionic currents by external Ni2+ is similar to that for proton block. Ni2+ block is inhibited by increasing [K+]o, by mutating a histidine residue in the pore turret (H463Q) or by mutating a residue near the pore mouth (R487V) that is the homolog of Shaker T449. Aside from a slight rightward shift of the Q-V curve, Ni2+ had no effect on gating currents. We propose that, as with Ho+, Ni2+ binding to H463 facilitates an outer pore inactivation process that is antagonized by Ko+ and that requires R487. However, whereas Ho+ substantially accelerates inactivation of residual currents, Ni2+ is much less potent, indicating incomplete overlap of the profiles of these two metal ions. Analyses with Co2+ and Mn2+, together with previous results, indicate that for the first-row transition metals the rank order for the inhibition of Kv1.5 in 0 mM Ko+ is Zn2+ (KD ∼ 0.07 mM) ≥ Ni2+ (KD ∼ 0.15 mM) > Co2+ (KD ∼ 1.4 mM) > Mn2+ (KD > 10 mM).
INTRODUCTION
Kv1.5 (KCNA5) channels, which are expressed in cardiac myocytes (Fedida et al., 1993; Tamkun et al., 1991) and in smooth muscle cells of airways, the intestine, and the vasculature (Adda et al., 1996; Clement-Chomienne et al., 1999), are members of a major structural class of K+ channels in which the α-subunit consists of six transmembrane (TM) segments with a pore-forming or P-region positioned between transmembrane segment five (S5) and segment six (S6). A characteristic feature of the 6TM-1P subunit is the charge-bearing segment four (S4) domain whose movement upon membrane depolarization (Baker et al., 1998; Larsson et al., 1996) is linked to the opening of the activation gate, which is believed to comprise the cytoplasmic ends of the four S6 regions in the tetrameric channel assembly. Macroscopic currents through Kv1.5 channels resemble delayed rectifier currents. Thus, with a strong sustained depolarization, channel activation is rapid and voltage-dependent whereas inactivation is voltage-independent and occurs on a timescale of seconds.
Kv1.5 channels exhibit only outer pore (P/C-type) inactivation (Fedida et al., 1999) and in this regard are different from Shaker channels, which also show inner pore (N-type) inactivation (Hoshi et al., 1991). The term C-type inactivation was coined to describe the slow inactivation process in Shaker that was uncovered when ball-and-chain or N-type inactivation was removed (ShakerIR) by deletion of the cytoplasmic N-terminal residues 6–46. C-type inactivation is coupled to channel activation and is believed to involve a conformational change in the outer pore mouth that extends to the selectivity filter delimited by the highly conserved GYG sequence. Because C-type inactivated ShakerIR (Starkus et al., 1997) and Kv1.5 (Wang et al., 2000a) channels are able to conduct Na+ ions, the current view is that the conformational change at the outer pore mouth involves an incomplete constriction rather than a complete collapse. An important consequence of C-type inactivation is a leftward shift of the gating charge versus voltage relationship, or Q-V curve, and charge immobilization (Fedida et al. 1996, Olcese et al., 1997).
In ShakerIR channels the residue at position 463 in the S6 segment was the first shown to influence the rate of C-type inactivation (Hoshi et al., 1991). Subsequently, point mutations of the threonine residue (T449) in the outer pore mouth were shown to dramatically accelerate (T449E, T449A, T449K) or slow (T449Y, T449V) C-type inactivation (Lopez-Barneo et al., 1993). In Kv1.5 channels the residue homologous to T449 is R487 and it has been shown that inactivation is substantially slowed in Kv1.5 R487V when Na+ is the charge carrier but not when K+ is the permeant ion (Fedida et al., 1999; Wang et al., 2000a).
The finding that ShakerIR/Kv1.5 channels with the pore mutation W434F/W472F were Na+- but not K+-conductive and showed wild-type gating charge behavior, including gating charge immobilization after channel inactivation (Chen et al., 1997; Olcese et al., 1997), was one of the first indications of the complexity of outer pore inactivation. To account for the properties of the ShakerIR W434F nonconducting mutant it was proposed that there was also a so-called P-type inactivation process that prevented K+ conduction but that was different from C-type inactivation in that it did not affect gating charge movement (Olcese et al., 1997; Yang et al., 1997). Restoration of ionic current in the double mutant Shaker W434F, T449Y supports the hypothesis that enhanced inactivation accounts for the ShakerIR W434F conductance loss (Yang et al., 2002).
An intriguing divergence in the structure-function relationships of Kv1.5 and ShakerIR is seen in the response to extracellular acidification. In Kv1.5 external protons cause, in addition to a rightward shift of the g-V curve that is often referred to as the gating shift, a concentration-dependent decrease of the maximum macroscopic conductance (gmax) as well as an acceleration of the inactivation rate of residual currents (Steidl and Yool, 1999; Kehl et al., 2002). In contrast, in ShakerIR channels increasing [H+]o does not reduce gmax but the gating shift and the speeding of inactivation are observed (Perez-Cornejo, 1999; Starkus et al., 2003). A number of lines of evidence now support the view that protonation of a histidine residue (H463), the equivalent of Shaker F425, in the pore turret (S5-P linker) plays an important role in the proton-induced conductance loss/block in Kv1.5. Thus, in the Kv1.5 H463Q mutant there is a large rightward shift of the concentration dependence of the Ho+ block (Kehl et al., 2002). The finding that the Ho+ block is antagonized by Ko+ and is also reduced in the R487V mutant (Jäger and Grissmer, 2001; Kehl et al., 2002) has suggested that the protonation of H463 facilitates an inactivation process requiring R487. An alternative explanation involving direct pore block by protons has been ruled out on the basis of single channel recordings (Kwan et al., 2003) and the finding that the Na+ current through inactivated Kv1.5 channels is maintained after extracellular acidification (Zhang et al., 2003).
Additional support for a crucial role of H463 in the Ho+-induced decrease of gmax is provided by reports showing that divalent cations known to bind to histidine residues also affect Kv1.5 currents. Harrison et al. (1993) first reported that extracellular Zn2+ blocks Kv1.5 currents and, as with the Ho+ block, this effect of Zn2+ is inhibited either by increasing Ko+ or by mutating H463 and/or R487 (Kehl et al., 2002). Ni2+ is also a histidine ligand and although it too has been reported to block Kv1.5 currents expressed in Chinese hamster ovary (CHO) cells (Perchenet and Clement-Chomienne, 2001), the mechanism of, and the molecular determinants for, the block have not been resolved. To test the hypothesis that the mechanistic basis for the Ni2+ block is essentially the same as that outlined above for Zn2+ and Ho+, we set out in this study to address the following questions. Is the block of Kv1.5 by Ni2+ antagonized by increasing [K+]o? Does Ni2+ speed the inactivation rate of residual Kv1.5 currents? Is the effect of Ni2+ affected either by mutating H463, a putative Ni2+ coordination site, or by mutating R487, a site implicated in the regulation of outer pore inactivation? Are gating currents affected by Ni2+? And finally, is the blocking effect of Ni2+ replicated by other divalent cations such as Co2+, Cd2+, and Mn2+?
MATERIAL AND METHODS
Cell preparation
As described previously (Wang et al., 2000a), wild-type (wt) and mutant human Kv1.5 channels, henceforth referred to simply as Kv1.5 channels, were studied in a human embryonic kidney cell line (HEK293) (Wang et al., 2000b). Cells were dissociated for passage by using trypsin-EDTA and were maintained in minimum essential medium (MEM), 10% fetal bovine serum, penicillin-streptomycin, and 0.5 mg ml−1 gentamicin in an atmosphere of 5% CO2 in air. All tissue culture supplies were obtained from Invitrogen (Burlington, Ontario, Canada).
Point mutations of the wt Kv1.5 α-subunit in the plasmid expression vector pcDNA3 were made using the Quikchange Kit (Stratagene, La Jolla, CA) to convert the histidine (H) residue at position 463 to glutamine (Q) (H463Q) or the arginine (R) at position 487 to valine (R487V). Stable transfections of HEK293 cells were made using 0.8 μg of Kv1.5 H463Q or Kv1.5 R487V cDNA and 2 μL of Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Geneticin (0.5 mg/mL) was added 48 h after transfection. Because Shaker-related channels such as Kv1.5 are homotetramers (MacKinnon, 1991) a given point mutation will exist in each of the four subunits of the channel assembly.
Recording solutions
The standard bathing solution contained, in mM, 140 NaCl, 3.5 KCl, 10 Hepes, 2 CaCl2, 1 MgCl2, and 5 glucose and its pH was adjusted to 7.4 with NaOH. Hepes was replaced by Mes when the pH of the extracellular solution was <6.8 in the experiments directly comparing the proton block and the Ni2+ block. Where the effect of the external concentration of potassium ([K+]o) on the divalent metal cation block was examined, a nominally K+-free solution was made by substituting NaCl for KCl and, for [K+]o >3.5 mM, NaCl was replaced by KCl. The standard patch pipette solution for recording K+ currents contained 130 KCl, 4.75 CaCl2 (pCa2+ = 7.3), 1.38 MgCl2, 10 EGTA, and 10 Hepes and was adjusted to pH 7.4 with KOH. Solutions of divalent metal ions were made by dilution of 0.1–1-M stock solutions of the chloride salt in distilled water. At pH 7.4 the concentration of Ni2+ that can be used was limited to 10 mM or less by virtue of the solubility product for Ni(OH)2 (∼2 × 10−16).
Mouse fibroblasts expressing Kv1.5 channels at a low density were used to record unitary currents from outside-out patches. The inside face of the patch was exposed to standard patch pipette solution and the outside face was exposed to standard bath solution either with or without added Ni2+.
For gating current recordings the bath solution contained, in mM, 140 NMGCl, 1 MgCl2, 10 Hepes, 2 CaCl2, and 10 glucose and the pH was adjusted to 7.4 with HCl. The patch pipette solution contained 140 NMGCl, 1 MgCl2, 10 Hepes, and 10 EGTA and was adjusted to pH 7.2 with HCl. Chemicals were purchased from Sigma Aldrich Chemical (Mississauga, Ontario, Canada).
Signal recording and data analysis
Macroscopic currents were recorded at room temperature (20–22°C) using the patch-clamp technique primarily in the whole-cell configuration. In some of the cell lines expressing mutant Kv1.5 channels at a high level, i.e., the H463Q and some of the R487V mutants, the large amplitude of the whole-cell currents necessitated recording macroscopic currents from outside-out patches. Voltage clamp experiments were done with an EPC-7 patch-clamp amplifier and Pulse+PulseFit software (HEKA Electronik, Lambrecht, Germany). Patch electrodes were made from thin-walled borosilicate glass (World Precision Instruments, Sarasota, FL) and had a resistance of 1.0–2.5 MΩ measured in the bath with standard internal and external solutions. Typically, 80% series resistance compensation was used and an on-line P/N method, for which the holding potential was −100 mV and the scaling factor was 0.25, was used to subtract the leak current as well as any uncompensated capacitive currents. Current signals filtered at 3 kHz (−3 dB, 8-pole Bessel) were digitized (16 bit) at a sampling interval of 100 μs (10 kHz). Voltages have been corrected for the liquid junction potentials.
In an experiment, a section of glass coverslip with cells attached to it was placed in the recording chamber (0.5 ml vol) and was continuously perfused with bathing solution. After recording currents in the control solution the inflow was switched to the test solution and once 5–6 ml had been flushed through the bath the treated responses were recorded. Recovery currents were taken after flushing the bath with 5–6 ml of control solution. If the recovery currents were not within ±15% of the pretreatment amplitudes the data for that cell were discarded. By this criterion most cells showed recovery.
To quantify the effect of Ni2+ and other metal cations on Kv1.5, tail currents were recorded at −40 or −50 mV after depolarizing prepulses of differing magnitude. Peak tail current amplitudes were obtained by fitting a polynomial function and taking the fitted value for the maximum current. After normalization of tail currents either to the maximum current of the control or the treated response, data points were fitted to a single Boltzmann function:
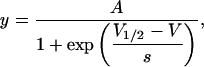 |
(1) |
where, when y is the current normalized with respect to the control response, A is the proportion of the control gmax. When y is the current normalized with respect to the maximal treated current, A is the best fit value for the normalized maximal response and ideally has a value of unity. V1/2 is the half-activation potential or midpoint of the activation curve, V is the voltage during the prepulse, and s is the slope factor, in mV, reflecting the steepness of the voltage dependence of gating.
To quantify gating charge movement during activation, charge-voltage (Qon-V) curves were generated by time integration of on-gating currents as described previously (Chen et al., 1997). Activation gating in Kv1.5 is best fit by the sum of two Boltzmann functions where the larger component, known as Q2, represents ∼80% of the total charge movement (Hesketh and Fedida, 1999). However, for simplicity, Q-V data obtained at pH 7.4 and 5.4 were fitted to Eq. 1 where y is the charge moved, A is the maximal charge (Qmax), and V is the voltage at which the on-gating charge (Qon) is evoked. V1/2 represents the midpoint of the Q-V curve and s reflects the steepness of the voltage dependence of charge movement.
Concentration-response data were fitted to the Hill equation:
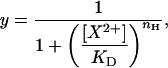 |
(2) |
where y is the proportion of the control gmax, KD is the equilibrium dissociation constant for the test cation (X2+), and nH is the Hill coefficient reflecting the number of test cations binding per channel.
Microscopic currents were low-pass filtered at 3 kHz (8-pole Bessel), sampled at 10 kHz and digitally filtered at 1 kHz for the data analysis using TAC and TACFit (Bruxton, Seattle, WA). Leak and uncompensated capacitive currents were subtracted using a template generated from blank sweeps. Half-amplitude threshold analysis was used to idealize single channel recordings for the generation of dwell-time histograms.
Data are expressed as the mean ± SE except for the values obtained by nonlinear least-squares fitting routines (Igor, Wavemetrics, Lake Oswego, OR) which are expressed as the mean ± SD. The paired-sample t-test was used to compare the inactivation rates of residual currents in Ni2+ and Ho+. A P-value of 0.05 or less was considered significant.
RESULTS
Shown in Fig. 1 A are traces confirming the block of Kv1.5 currents by external Ni2+. From a holding potential of −80 mV and with 0 mM Ko+, currents were evoked by a family of 300 ms depolarizations from −45 to +35 mV with a cycle length of 5 s. Tail currents were recorded at −40 mV. After obtaining the control responses, the perfusate was switched to a test bathing solution containing 0.1 mM Ni2+ and then to one containing 1 mM Ni2+. Complete recovery was obtained after returning to Ni2+-free solution. As noted previously (Perchenet and Clement-Chomienne, 2001), and in contrast to the effects with Zn2+ (Zhang et al., 2001a), with Ni2+ there was neither a significant change of the activation kinetics nor an obvious effect on the decay of residual pulse currents. The effect of Ni2+ on the current behavior during longer depolarizing pulses is examined below (Fig. 4).
FIGURE 1.

Ni2+ block of Kv1.5 currents in 0 mM Ko+. (A) Control currents evoked by a family of 300-ms depolarizations from −45 to +35 mV, here shown in 10-mV increments, from a holding potential of −80 mV. Tail currents were recorded at −40 mV. Perfusion of solution containing 0.1 mM Ni2+ and then 1 mM Ni2+ caused a concentration-dependent inhibition of the current. Recovery traces illustrate the complete reversal of the Ni2+ block. (B and C). Ni2+ decreases gmax and shifts the g-V curve slightly rightward. (B) Peak tail current at −40 mV after a 300-ms depolarization to the voltage indicated on the x axis. Note the absence of any voltage dependence of the inhibition between 0 and 50 mV. (C) The g-V relationship derived by normalizing tail currents with respect to the maximum tail current shows that Ni2+ caused a 10-mV shift of the half-activation voltage. Current tails in 0.5 mM Ni2+ were too small to be unequivocally analyzed.
FIGURE 4.

A comparison of the residual current behavior, done in 3.5 mM Ko+ with a concentration of Ho+ (A) or Ni2+ (B) estimated to block 80–95% of the channels, reveals divergent effects on inactivation. From a holding potential of −80 mV, the voltage protocol consisted of a 5-s step to 50 mV followed by 50-ms steps at variable intervals to 50 mV to monitor recovery from inactivation. Current during the 5-s pulse is shown expanded on the right side of the figure. (A) At pH 5.4 the inactivation of current during the 5-s pulse is well fitted by a single exponential with a time constant of 91 ms. Peak currents, marked by the filled circles, that were evoked by the 50-ms test pulses were fitted to a single exponential with a time constant of 4.3 s. (B) In the same cell after switching to solution containing 2 mM Ni2+ at pH 7.4, inactivation was ≈20 times slower (τinact = 1.69 s) and recovery from inactivation was ≈5 times slower (τrecovery = 24.8 s) than at pH 5.4.
Increasing [K+]o causes a rightward shift of the concentration dependence of the Ni2+ block
To quantify the block by Ni2+, g-V curves were constructed from peak tail currents as described in the Methods section. Fig. 1 B plots the peak tail current amplitude versus the pulse voltage for the same cell in 0 mM Ko+ without Ni2+ and with 0.25 or 0.5 mM Ni2+. In this cell 0.25 mM and 0.5 mM Ni2+ decreased the maximum tail current, and by extension the maximum conductance (gmax), by ∼70% and 90%, respectively. To more clearly illustrate the effect of Ni2+ on the midpoint (V1/2) of the g-V curve, the currents in panel B were normalized with respect to the maximum current for the same treatment group and are presented in panel C. It is evident that Ni2+ caused a rightward shift of the g-V curve and this is assumed to reflect a change of surface charge due to screening and/or binding to the channel. With 0.25 mM Ni2+ the shift of V1/2 determined from the best fit of the g-V data to the Boltzmann function was 10.6 ± 0.9 mV (n = 4). The gating shift with 0.5 mM Ni2+ was not determined because the standard deviation in the fitted values for V1/2 was quite large.
Fig. 2 shows the concentration-response relationship for the block of Kv1.5 by Ni2+ and the influence of [K+]o thereon. Panel A illustrates representative current traces from three different cells in 0 mM (left), 3.5 mM (middle), and 140 mM (right) Ko+. In the absence of Ni2+ (−Ni2+) the current in each of the Ko+ concentrations had a similarly slow rate of decay. The inward tail current recorded at −40 mV in 140 mM Ko+ is due to the shift of EK to ∼0 mV. To produce a similar degree of block in the three different experiments it was necessary to increase the Ni2+ concentration to offset the effect of increasing [K+]o. Note that in each example the Ni2+ block was not associated with an acceleration of pulse current decay. The latter observation, together with the fact that the tail current decay was not slowed, as best seen with the traces in 140 mM Ko+, supports the conclusion that a block of the open channel occurring with intermediate-to-slow kinetics (vis à vis the activation rate) is not involved. For the graph in Fig. 2 B the gmax relative to the control value has been plotted against the concentration of Ni2+ for experiments in which [K+]o was 0 mM (open circles), 3.5 mM (open triangles), or 140 mM (open squares). The solid lines overlaying the three data sets represent the best fit to Eq. 2. With 0 mM Ko+ the KD for the Ni2+ block was 0.15 ± 0.01 mM and nH was 1.3 ± 0.1. Increasing [K+]o to 3.5 mM increased the KD to 0.44 ± 0.02 mM and nH was 1.6 ± 0.2. With 140 mM Ko+ the KD was 3.1 ± 0.3 mM and nH was 0.9 ± 0.1. These results clearly demonstrate that, as with the block by Ho+ and Zn2+, the block of Kv1.5 by Ni2+ is antagonized by increasing [K+]o.
FIGURE 2.

Increasing [K+]o changes the concentration dependence of the block of Kv1.5 by Ni2+. (A) Representative traces obtained from three different cells showing, superimposed, the currents evoked in the Ko+ concentrations indicated either without (−) or with (+) the Ni2+ concentration indicated. The voltage protocol consisted of a 300-ms step from −80 mV to 50 mV followed by a step to −40 mV. Increasing [K+]o necessitates a higher concentration of Ni2+ to produce roughly the same degree of block. The time calibration is the same for the three sets of traces. (B) The concentration-response relationship for Ni2+ in 0, 3.5, and 140 mM Ko+ shows that increasing [K+]o from nominally K+-free to 3.5 mM shifted the KD from 0.15 ± 0.01 mM to 0.44 ± 0.02 mM. Increasing Ko+ to 140 mM shifted the KD for the Ni2+ block to 3.1 ± 0.3 mM. Each point represents the mean ± SE of measurements from three to seven cells.
The time courses of the onset and the offset of the Ni2+ block are similar
Using a fast solution application system, Perchenet and Clement-Chomienne (2001) noted that the offset of the Ni2+ block was rapid but that the onset was comparatively much slower. They found, with test pulses delivered at 15-s intervals and using 1 mM Ni2+ and 5 mM Ko+, that steady-state block was reached only after 5–7 min. Because a similar phenomenon is not seen with Ho+ or Zn2+, we felt it was important to characterize the time dependence of the Ni2+ block and did so by comparing the time course of the current inhibition by Ni2+ with that by Ho+. Graphs summarizing the outcome of this comparison are shown in Fig. 3. For each graph, the peak tail current, measured at −40 mV after a 300-ms pulse to 50 mV applied at 10-s intervals, was plotted against the elapsed time. In Fig. 3 A, Ni2+ and Ho+ were applied for the duration indicated by the horizontal bar at concentrations of 150 μM and 0.16 μM (pH 6.8) (Kehl et al., 2002), respectively, and in 0 mM Ko+ to cause roughly 50% block of the current at the steady state. In four such experiments we consistently found that the time courses for the onset and offset of the block by Ni2+ and Ho+ were similar. Because the failure to uncover any asymmetry in the on- and off-time courses might be attributed to the absence of Ko+, experiments were also done with 5 mM Ko+ which necessitated using higher concentrations of Ni2+ and Ho+ to compensate for the effect of Ko+ on the block. Fig. 3 B shows that the outcome was still the same: after switching from the control to the test perfusate the relaxation to the steady state was complete in less than a minute, a time frame that appears to reflect primarily the dynamics of solution exchange in the bath and is much shorter than the onset noted by Perchenet and Clement-Chomienne (2001).
FIGURE 3.

A comparison of the time course of the onset and offset of the inhibition of Kv1.5 by Ni2+ and Ho+. Concentrations of Ni2+ or Ho+ producing ∼50% steady-state inhibition with 0 mM Ko+ (A) and, in a different cell, with 5 mM Ko+ (B) were used. Peak tail currents measured at −40 mV after a 300-ms depolarization to 50 mV applied at a 10-s interval are plotted against the elapsed time. The horizontal bar indicates the duration of each application. The results do not reveal any obvious asymmetry in the onset versus the offset of the block either with or without Ko+. As with Ho+, the time course for the development or reversal of the Ni2+ block was similar and in both cases is presumed to reflect the time course of solution exchange in the bath.
Ni2+ block is associated with a slight acceleration of inactivation of residual currents
In addition to blocking Kv1.5 currents, extracellular acidification accelerates the rate of inactivation of residual currents (Kehl et al., 2002; Steidl and Yool, 1999) and this was the motivation for determining if there was a similar association between block and inactivation with Ni2+. Our approach to addressing this question was to use cells expressing Kv1.5 channels at a very high density so that despite the reduction of gmax by 80–95% the residual currents were virtually unfettered by endogenous HEK currents and could therefore be unambiguously analyzed. The voltage protocol consisted of a 5-s step from −80–50 mV followed by brief depolarizations to 50 mV to track recovery from inactivation (Fedida et al., 1999). An interval of 120 s between the 5-s pulses was used to permit complete recovery from inactivation in the experiments with Ni2+. Initially, these experiments were done with 0 mM Ko+ but the interpretation of the data was confounded by a very slowly rising phase of current with a time constant of 1–1.5 s in 1 mM Ni2+ and 200–300 ms at pH 5.9, which followed a normally activating component of current (not shown). This slow component was not observed with 3.5 mM Ko+, consequently this was the [K+]o used when comparing the effects on inactivation of concentrations of Ho+ and Ni2+ that reduce gmax by 80–95% (Fig. 2 and Kehl et al., 2002). Results representative of those obtained in five experiments with 2 mM Ni2+ and five experiments with pH 5.4 are shown in Fig. 4, A and B, where the effects of Ho+ and Ni2+, respectively, were tested on the same cell. At pH 5.4 the inactivation of the residual current during the 5-s pulse was well fitted by a single exponential with a time constant of 91 ms and the steady-state current was ∼25% of the peak amplitude. At pH 5.4 the mean inactivation time constant (τinact) at 50 mV was 101 ± 3 ms (n = 5 cells). In Fig. 4 A recovery from inactivation, tested by 50 ms depolarizations delivered from 0.5 s up to 96 s after the 5-s depolarization, was fitted to a single exponential with a time constant of 4.3 s. The mean τrecovery at pH 5.4 was 4.2 ± 0.1 s. In contrast, currents recorded after switching from pH 5.4 solution to perfusate containing 2 mM Ni2+ at pH 7.4 (Fig. 4 B) showed much slower inactivation as well as slower recovery from inactivation: τinact = 1.69 s and τrecovery = 24.8 s. In the five cells tested with 2 mM Ni2+ the mean value for τinact and τrecovery was 1.71 ± 0.07 s and 23.5 ± 2.1 s, respectively. Because of their very large amplitude, currents in Ni2+-free medium at pH 7.4 could not be recorded from these cells, however, the best fit to a single exponential of the current decay during 7–10 s depolarizations to 60 mV at pH 7.4 in Kv1.5 is typically of the order of 2–3 s (Kehl et al., 2002) and the τrecovery measured at −80 mV in 5 mM Ko+ and using a similar voltage protocol is 1.1 s (Fedida et al., 1999).
The KD for the Ni2+ block is increased in the H463Q and R487V mutants
We next examined the effect of Ni2+ in Kv1.5 channels in which either a putative Ni2+ binding site in the S5-P linker (turret) was mutated to a glutamine residue (H463Q) or the residue analogous to Shaker T449 was changed from arginine to valine (R487V). To circumvent the potential problem of changes of the Ko+-dependence of the block relief, the analysis of the effect of Ni2+ on currents from these mutated channels was done with 0 mM Ko+. Concentration-response curves for the Ni2+ block of currents from Kv1.5 H463Q (filled squares) and Kv1.5 R487V (filled circles) are shown superimposed in Fig. 5. As with the block by Ho+ and Zn2+ (Kehl et al., 2002), the concentration dependence for the block by Ni2+ was shifted substantially to the right by either mutation. In Kv1.5 R487V the KD was estimated to be 2.8 ± 0.004 mM or roughly 20-fold higher than in wt Kv1.5. With Kv1.5 H463Q the concentration dependence of the block was much more shallow (nH ∼ 0.4) than in wt Kv1.5 and the KD was estimated by extrapolation to be 24 ± 8 mM, which is from 100- to 200-fold higher than in wt Kv1.5.
FIGURE 5.

Ni2+ sensitivity is reduced in Kv1.5 H463Q and Kv1.5 R487V. Experiments with the mutant channels were done with 0 mM Ko+ to preclude a change of the Ko+ binding as the basis for the change of the sensitivity to Ni2+. Because Ni2+ is known to bind to histidine (H) residues, a mutant was constructed in which glutamine (Q) was substituted for H463, a residue in the S5-P linker that forms part of the outer pore vestibule. Kv1.5 H463Q (▪) was from 100- to 200-fold less sensitive to Ni2+ (KD = 24 ± 8 mM; nH = 0.4 ± 0.04) compared to the wt Kv1.5 responses (dashed line taken from Fig. 2) measured in the same recording condition, i.e., 0 mM Ko+. In another mutant construct, the arginine (R) residue near the entrance to the pore mouth that has been implicated, by alignment with Shaker T449, in the outer pore inactivation mechanism, was mutated to valine. The sensitivity of Kv1.5 R487V currents (•) to Ni2+ was ∼20-fold less (KD = 2.8 ± 0.004 mM; nH = 0.7 ± 0.001) than that measured under the same recording conditions in wt Kv1.5. The dotted line, which was taken from Fig. 2, represents the line fitted to the block of wt Kv1.5 in 140 mM Ko+.
Ni2+ decreases channel availability
Macroscopic current amplitude (I) is, in general, the product of the number of channels available (N), the single channel current (i), and the channel open probability (Po)(I = NPoi). To gain a clearer insight into which of these variables was affected by Ni2+ ions, recordings were made from outside-out patches containing a single channel. Fig. 6 A shows representative, consecutive control sweeps evoked by a 300-ms depolarization from −80 mV to 100 mV applied at a frequency of 0.1 Hz. As reported previously (Chen and Fedida, 1998), channel openings occurred in bursts of varying duration and within bursts channel closings were frequent but brief. With seconds-long pulses (not shown) we observed closed states with longer mean dwell times, which are assumed to reflect a multistep inactivation pathway. Double-Gaussian fits to the control all-points amplitude histogram (e.g., Fig. 6 C) indicates an open channel current (i) of 1.7 ± 0.1 pA (n = 8 patches). After switching to medium with 0.5 mM Ni2+, which is the KD for the block in 3.5 mM Ko+ (Fig. 2), there was no significant change of the single-channel current (e.g., Fig. 6 D; 1.6 ± 0.1 pA, n = 6 patches), and the Po in sweeps containing channel activity was also not significantly affected (Po,-Ni = 0.64 ± 0.06 versus Po,+Ni = 0.61 ± 0.06). There were, however, many more blank sweeps in the presence of the Ni2+ (Fig. 6 B). Channel availability (N), defined as the number of sweeps with channel activity divided by the total number of sweeps, decreased significantly from the control value of 0.90 ± 0.06 (n = 6 patches) to 0.43 ± 0.14 (n = 6 patches) in Ni2+.
FIGURE 6.

Ni2+ effects at the single channel level. (A) Shown here are 10 representative and consecutive control sweeps in a one-channel, outside-out patch that were evoked by a 300-ms pulse from −80 mV to 100 mV applied at 0.1 Hz and with [K+]i = 140 mM and [K+]o = 3.5 mM. Data were digitally filtered at 1 kHz. (B) From the same patch as in A, 10 consecutive sweeps evoked with the same voltage protocol but with 0.5 mM Ni2+ in the external perfusate. The main effect of Ni2+ is to reduce channel availability. Representative all-point amplitude histograms from a different one-channel patch in control and 0.5 mM Ni2+-containing perfusate are shown in panels C and D, respectively. A double-Gaussian fit to data gave a mean current in each case of 1.6 pA.
Ni2+ causes a rightward shift of the Qon-V curve but does not affect Qmax
A possible explanation for the current block by Ni2+ is that one or more transitions in the gating pathway is prevented. To address that possibility, gating currents were recorded in an HEK293 cell line expressing Kv1.5 W472F channels. The W472F mutation produces channels that are not K+ conductive, but that have normal gating currents. Fig. 7 A shows gating current traces in control solution and in 1 mM Ni2+. On-gating currents were evoked by 12-ms pulses between −60 and 130 mV from a holding potential of −100 mV. In the control traces, charge movement was first evident at ∼−50 mV and the peak amplitude and decay rate increased as the intensity of the depolarization increased. After depolarizations up to −10 mV the off-gating current at −100 mV was rapid (e.g., Fig. 7 B, upper traces) but after stronger depolarizations there was a clear rising phase to the off-gating current and the peak current was substantially smaller and occurred much later (e.g., Fig. 7 B, lower traces) than was the case following steps to −10 mV or less. This pronounced change of off-gating current after stronger depolarizations has been attributed at least in part to a weakly voltage-dependent transition in the return pathway between the open and closed states (Perozo et al., 1993). To construct the charge-voltage (Q-V) curves shown in Fig. 7 C, on-gating currents were integrated and Qon was normalized with respect to the control maximal charge movement (Qmax). Although charge movement is better fitted by a double-Boltzmann function to account for a smaller component of charge movement with depolarizations up to −20 mV (Hesketh and Fedida, 1999), the data of Fig. 7 C were fitted to a single Boltzmann function.
FIGURE 7.

To determine if the conductance loss caused by Ni2+ was due to an inhibition of transitions in the activation pathway, the effect of 1 mM Ni2+ on gating charge movement in Kv1.5 W472F, a nonconducting mutant, was examined. Internal and external permeant ions were replaced by NMG+ to prevent ionic currents through endogenous HEK293 channels. The family of traces in the top of panel A shows control on-gating currents evoked between −60 and 90 mV from a holding potential of −100 mV and off-gating currents at −100 mV; the lower traces of panel A show the gating currents in 1 mM Ni2+. Control and treated traces, taken at the voltages indicated to account for the gating shift, have been superimposed in B to show that the kinetics of the on- and off-gating currents are not substantially affected by Ni2+. (C) The Qon-V curve constructed from six cells by integrating the on-gating currents and normalizing with respect to the control Qmax confirms that, although Ni2+ caused an ∼10-mV rightward shift of the V1/2, the Qmax did not decrease. Fitting to a Boltzmann function gave control and treated V1/2 values of −6.8 ± 1.2 mV and 2.2 ± 0.8 mV, respectively, and values of 7.0 ± 1.4 mV and 9.2 ± 1.2 mV for s.
In six experiments of the type illustrated in Fig. 7, the control V1/2 and s were −6.8 ± 1.2 mV and 7.0 ± 1.4 mV. After switching to 1 mM Ni2+ V1/2was 2.2 ± 0.8 mV and s was 9.2 ± 1.2 mV. The difference in V1/2 between 1 mM Ni2+ and control medium was 9.0 ± 3.0 mV. Aside from this gating shift, the gating current was essentially unaffected by 1 mM Ni2+. In contrast to the situation with 1 mM Zn2+ where Qmax decreased by ∼15% (Zhang et al., 2001b), Qmax was unchanged by 1 mM Ni2+.
Co2+ and Cd 2+, but not Mn2+, block Kv1.5
Other divalent transition metals that can bind to histidine include Cu2+, Fe2+, Co2+, Cd2+, and Mn2+. Because a precipitate formed with Cu2+ and Fe2+, only the effects of Co2+, Cd2+, and Mn2+ could be compared to those of Ni2+. The experimental protocol was the same as that described for Fig. 1 and was confined to tests with a 0-mM Ko+ solution. Fig. 8 A shows a representative example of the effect of Co2+ on currents evoked by the voltage protocol illustrated above the control responses. Switching from the control solution to one containing 0.1 mM Co2+ had no significant effect on the current but 10 mM Co2+ decreased the peak tail current after a +60-mV pulse by >90%. Virtually complete recovery occurred after returning to the control solution. Fitting of g-V curves (not shown) to Eq. 1 revealed that V1/2 shifted by 11.4 ± 0.9 mV with 1 mM Co2+ and by 25.3 ± 1.3 mV with 10 mM Co2+. Neither concentration of Co2+ significantly affected the slope factor of the g-V curve (not shown). A fit of the Hill equation to the concentration-response data for Co2+ (Fig. 8 B) gave an estimate for nH of 1.3 ± 0.1 and a KD (1.4 ± 0.1 mM) that was roughly 10 times larger than that for Ni2+ under the same recording conditions.
FIGURE 8.

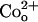 also causes a concentration-dependent block of Kv1.5 currents but is an order of magnitude less potent than Ni2+. Shown in panel A are control and treated current traces evoked in 0 mM Ko+ with the voltage protocol indicated above the control responses. In contrast to the ∼35% block of the current with 0.1 mM Ni2+ (see Fig. 2), 0.1 mM Co2+ had no effect. However, with 10 mM Co2+ the maximum peak tail current amplitude decreased by ∼90%. As with Ni2+, the block by Co2+ was completely reversible. (B) Concentration-response data obtained in 0 mM Ko+, with each point representing the mean ± SE of measurements in three to eight cells, were fitted to Eq. 2, which gave a KD of 1.4 ± 0.1 mM and an nH of 1.3 ± 0.1.
also causes a concentration-dependent block of Kv1.5 currents but is an order of magnitude less potent than Ni2+. Shown in panel A are control and treated current traces evoked in 0 mM Ko+ with the voltage protocol indicated above the control responses. In contrast to the ∼35% block of the current with 0.1 mM Ni2+ (see Fig. 2), 0.1 mM Co2+ had no effect. However, with 10 mM Co2+ the maximum peak tail current amplitude decreased by ∼90%. As with Ni2+, the block by Co2+ was completely reversible. (B) Concentration-response data obtained in 0 mM Ko+, with each point representing the mean ± SE of measurements in three to eight cells, were fitted to Eq. 2, which gave a KD of 1.4 ± 0.1 mM and an nH of 1.3 ± 0.1.
The effects of Cd2+ are not illustrated but closely resembled those of Co2+. The KD was 1.5 ± 0.4 mM and the nH was 1.3 ± 0.3. In 1 mM Cd2+ the V1/2 for the g-V relationship was shifted rightward by 19.5 ± 1.2 mV.
Of the divalent cations we tested for an ability to block Kv1.5, Mn2+ proved to be the least effective. At 10 mM, the highest concentration used, gmax was 73 ± 3% of the control value. The midpoint of the g-V relationship was shifted rightward by 21.5 ± 0.7 mV (n = 3).
Co2+ and Zn2+ mimic the effect of Ni2+ on Kv1.5 inactivation
Fig. 9 A illustrates representative results of the effect of 10 mM Co2+ on inactivation and recovery from inactivation using a voltage protocol identical to that described for Fig. 4. Again, a slowly rising phase of current seen in 10 mM Co2+, Ko+-free medium (not shown) necessitated recording with 3.5 mM Ko+. In 10 mM Co2+ both the onset of and recovery from inactivation was comparable to that seen with 2 mM Ni2+ (Fig. 4). In the four cells studied with 10 mM Co2+, τinact and τrecovery were 1.3 ± 0.1 s and 24.6 ± 1.7 s, respectively. As noted above, Zn2+ also causes a concentration and Ko+-dependent inhibition of Kv1.5 currents and for that reason its effects on inactivation were also examined (Fig. 9 B). Using a Zn2+ concentration of 2 mM, which is estimated to reduce gmax by 80–90% in 3.5 Ko+, τinact was 1.64 ± 0.3 s and τrecovery was 27.7 ± 2.1 s (n = 5 cells). Thus, a feature that is shared by Ni2+, Co2+, and Zn2+ is an ability to substantially slow recovery from inactivation and to modestly accelerate inactivation. In this regard at least these divalent cations are clearly distinct from extracellular protons that, by comparison, accelerate inactivation to a far greater extent (τinact ≈ 100 ms at pH 5.4) and slow recovery from inactivation much less (τrecovery ≈ 4 s at pH 5.4).
FIGURE 9.

Co2+ and Zn2+ mimic the effect of Ni2+ on macroscopic inactivation. (A) Using a voltage protocol identical to that described for Fig. 4, the τinact with 10 m M Co2+ in 3.5 mM Ko+ was well fitted by a single exponential with a time constant of 1.6 s. The fit of an exponential function to the peak currents evoked by 50-ms test pulses after the 5-s pulse to 50 mV gave a τrecovery of 21.8 s. (B) With 2 mM Zn2+ in 3.5 mM Ko+, τinact was 1.86 s and τrecovery was 29.3 s. Because Zn2+ slowed the activation rate the duration of test pulses used to monitor recovery was increased to 200 ms. These data indicate a clear difference in the effect on Kv1.5 inactivation of divalent cations versus external protons (Fig. 4 A).
DISCUSSION
As reported previously (Perchenet and Clement-Chomienne, 2001), external Ni2+ ions were shown to reversibly block human Kv1.5 currents (Fig. 1). We have also shown here that Ni2+ block is affected by [K+]o (Fig. 2). Thus, with 0 mM Ko+ the KD for the Ni2+ block is ∼150 μM whereas with 3.5 mM Ko+ the KD increases to 400 μM. The latter value is consistent with the KD of 570 μM obtained with 5 mM Ko+ in CHO cells (Perchenet and Clement-Chomienne, 2001). Increasing Ko+ to 140 mM increased the KD to ∼3 mM. The nH of 1.2–1.6 derived from concentration-response data in 0–5 mM Ko+ (see also Perchenet and Clement-Chomienne, 2001)) suggests that the block requires the binding of at least two Ni2+ ions. In the study with Kv1.5 expressed in CHO cells the Ni2+ block was shown, regardless of the pulse frequency, to develop slowly over a 2–5 min period (Perchenet and Clement-Chomienne, 2001) despite the use of a fast drug application system. These data were interpreted to reflect a large disparity in the association and dissociation rate constants for Ni2+ binding to the closed state of the channel. Although we agree that the Ni2+ block can occur from the closed state, we found no evidence for a slow development of that block (Fig. 3).
One possible interpretation of the inhibition of the Ni2+ block by Ko+ is that it reflects an interaction in the channel pore either by competition for the same binding site or by an electrostatic effect between separate Ni2+ and K+ binding sites. However, as noted with the block by Zn2+ and Ho+ (Kehl et al., 2002), the block by Ni2+ shows no voltage dependence over a range of voltages where the open probability is maximal (Perchenet and Clement-Chomienne, 2001). This observation supports the conclusion that the Ni2+ binding site is at least not in a region of the pore that is within the electric field and, by extension, that Ni2+ is not blocking by occlusion of the pore. The fact that Kv1.5 currents are blocked by Ho+, Zn2+, and Cd2+, whereas Shaker channels are not, also suggests a binding site external to the pore (e.g., in the turret) because in Kv1.5 and Shaker there is complete homology from the N-terminal end of the pore helix to the GYG pore signature sequence.
As is the case with the block of Kv1.5 by Ho+ and Zn2+, the sensitivity of Kv1.5 channels to Ni2+ is greatly affected (Fig. 5) either by mutating H463 in the pore turret or by mutating R487, a residue in the outer pore mouth that has been shown in Shaker channels to play a pivotal role in P/C-type inactivation. These results with the 463Q and 487V mutant channels, as well as the sensitivity of the Ni2+ block to Ko+ and the outcome of other substitutions at position 463 (see below), are consistent with a model in which the binding of Ni2+ to one or more H463 residues in the pore turret facilitates an inactivation process that involves the outer pore mouth. Although this model is the same as that proposed for the Ho+ and Zn2+ block of Kv1.5, there is not complete overlap of the effects of these three metal cations. For example, the inactivation rate of the residual currents is markedly different with the divalent cations (Ni2+, Co2+, Zn2+) compared to Ho+ (Figs. 4 and 9). Thus, for example, using concentrations that produce a similar degree of block in 3.5 mM Ko+, the residual currents inactivated roughly 20 times faster with Ho+ (pH 5.4) than with Ni2+ (Fig. 4). Additionally, the shift of the midpoint of the g-V curve and the Qon-V curve by Ni2+ was also much less than with either Ho+ or Zn2+. Finally, the dramatic slowing of the activation rate observed with Zn2+ (Zhang et al., 2001a) is not seen with either Ni2+ or Ho+. It seems unlikely, though we cannot disprove, that these differences are due solely to the nature of ligand coordination by the histidine residues in the turret. Particularly in the case of Ho+, the involvement of additional binding sites seems likely. This is suggested by the fact that although ShakerIR channels are largely resistant to the conductance collapse in low pH, acidification does accelerate current inactivation (Perez-Cornejo, 1999; Starkus et al., 2003). Furthermore, we and others have shown that manipulations that reduce the block of Kv1.5 by metal cations do not affect the gating shift (Kehl et al., 2002; Trapani and Korn, 2003).
From the data in Fig. 5 it is also apparent that neither of the outer pore mutations completely prevents current inhibition by Ni2+. Currents through the Kv1.5 H463Q construct decreased by ∼30% in 5 mM Ni2+ and, as with the Zn2+ block of this mutant channel (Kehl et al., 2002), the nH fitted to the concentration dependence of this block was quite small (∼0.5) suggesting the involvement of a binding site and mechanism of action that is different. In the case of the R487V mutant, the KD and the nH for the Ni2+ block with 0 mM Ko+ are similar to that estimated for wt Kv1.5 in 140 mM Ko+. Paradoxically, neither of these manipulations, increasing [K+]o or mutating R487, substantially affects the inactivation rate of macroscopic currents carried by K+ during sustained depolarizations (Fedida et al., 1999). Although the latter observations might be construed as evidence against an involvement of outer pore inactivation in the Ni2+ block, that is to say neither manipulation can be shown directly to affect the current decay rate, an alternative explanation is that these manipulations inhibit an outer pore inactivation process occurring from a closed state but are much less effective against inactivation from the open state. In this connection, a Ko+-sensitive (KD ∼ 0.8–10 mM) inactivation process occurring from a closed state has been suggested to account for the decline of the macroscopic conductance seen in fast-inactivating ShakerIR T449 mutants when the [K+]o is decreased (Lopez-Barneo et al., 1993) and there is evidence in ShakerIR supporting, not exclusively, “multiple, independent pathways of which C-type is only one” (Yang et al., 1997).
As with some of the T449 mutations in Shaker, there are mutations of Kv1.5 H463 that can dramatically affect outer pore inactivation. For example, mutants in which glycine (G) (Kehl et al., 2002) or arginine (R) (Eduljee et al., 2003) is substituted for H463 display rapidly inactivating currents (τinact = 35–75 ms) and, again as in the Shaker T449X mutants, these rapidly inactivating mutants show a collapse of the macroscopic conductance in 0 mM Ko+. Furthermore, in the H463G mutant the conductance collapse in 0 mM Ko+ is prevented by the R487V mutation (Trapani and Korn, 2003). The outcome of these H463G and H463R mutations is significant because it shows directly that the physicochemical properties of the residue at this position can dramatically affect the time course of open- (and closed-?) state inactivation and thus offers additional support for the proposition that noncovalent chemical modification of H463 by the binding of Ni2+, in addition to other metal cations, can affect inactivation.
Another significant property of the H463G mutant is that Ko+ affects the gmax with a KD of ∼1 mM (Eduljee et al., 2003). This low millimolar KD is comparable not only to that estimated for the fast-inactivating ShakerIR mutants (Lopez-Barneo et al., 1993) but to that obtained for the relief by Ko+ of the Ho+ and Zn2+ block (Kehl et al., 2002; Zhang et al., 2001a). A detailed study of the Ko+-dependence of the Ni2+ block was not undertaken here. However, using the KD of the Ni2+ block in zero and 3.5 mM Ko+, and assuming, for simplicity, a competitive interaction, the KD for the relief of the block by Ko+ is calculated to be ∼1.5 mM. A consistent pattern that emerges from these studies, whether it is the spontaneously occurring conductance collapse in ShakerIR and Kv1.5 mutant channels or the metal ion-induced block/conductance collapse in wt Kv1.5, is that inhibition of the conductance loss occurs with low millimolar Ko+ concentrations and that this inhibition occurs in the absence of a change of the inactivation rate measured during depolarizing pulses. This implies that there is an outer pore inactivation process, perhaps that occurring from the closed state, that is much more sensitive to Ko+ and, we suggest, given that its inactivation rate is not distinguishable from wt Kv1.5 channels, that in Kv1.5 the R487V mutation selectively affects this same inactivation process. With the fast-inactivating ShakerIR mutants, Lopez-Barneo et al. (1993) remarked that the tendency for the conductance to collapse (inactivate from the closed state?) in 0 mM Ko+ is associated with fast current inactivation. This correlation also applies to Kv1.5 H463G where the inactivation rate is some 20-fold faster than in wt Kv1.5 channels but it is much less evident with the Ni2+, Co2+, and Zn2+ block where the inactivation rate of residual currents is only approximately twofold faster than in controls (Figs. 4 and 9).
Particularly in view of the low concentrations of Ko+ needed to relieve the metal cation block, a question that inevitably arises is whether the external K+ binding site can also be populated by outward K+ flux through the open channel. Though it has not been studied for Ni2+ block, our recent finding (Zhang et al., 2003) of virtually identical KDs for the block by Ho+ of outward K+ or Na+ currents argues against a contribution of outward K+ currents in the block relief. One explanation for this apparent absence of an effect of K+ efflux through the open pore is that K+ ions at the outer pore mouth rapidly equilibrate with the external solution. Alternatively, if Ni2+-bound channels are inactivating from a closed state, or if the open time is very brief (Zhang et al., 2003), there would be no opportunity for block relief by outward K+ currents.
A comparison of currents from one channel outside-out patches (Fig. 6) before and after the application of 0.5 mM Ni2+ showed: 1), that open channel current (i) at 100 mV did not change; 2), that the open probability (Po) during 300-ms sweeps containing channel activity was not changed; and 3), channel availability (N) decreased from a value of ≈0.9 in the control to ≈0.4 during treatment. Although a detailed analysis and comparison of open- and closed-time behaviors have not yet been done, these preliminary data are consistent with a model in which Ni2+ binding facilitates a reversible transition from an available to an unavailable (closed-state inactivated?) state.
Gating current analyses (Fig. 7) showed that, as with Ho+ (Kehl et al., 2002), Ni2+ did not affect Qmax. This finding rules out the possibility that the prevention of one or more of the transitions in the activation pathway accounts for the Ni2+-induced decrease of gmax. Ni2+ treatment also caused an ∼10 mV shift of the Qon-V curve but this was much less than the 50–60 mV shift seen with Ho+ or Zn2+ (Kehl et al., 2002; Zhang et al., 2001b). As noted above, it is not clear if this disparity in the gating shift reflects differences in ligand coordination with H463 residues or if the larger shift with Ho+ and Zn2+ reflects interactions with additional binding sites.
Transition metal ions that have now been shown to block Kv1.5 currents are Zn2+, Cd2+, Ni2+, and Co2+ (Fig. 8). For the first-row transition metals the rank order for the inhibition of Kv1.5 in 0 mM Ko+ is Zn2+ (KD ∼ 0.07 mM) ≥ Ni2+ (KD ∼ 0.15 mM) > Co2+ (KD ∼ 1.4 mM) > Mn2+ (KD > 10 mM) and, as such, is in accord with the Irving-Williams order (Glusker, 1991). Zn2+, Ni2+, and Co2+, which are intermediate Lewis acids, are known to bind to the thiolate side group of cysteine and the imidazole nitrogen of the histidine. Zn2+ is also able to bind to carboxylate and carbonyl oxygen atoms. Cd2+, a second-row transition metal, is a soft Lewis acid and typically has a higher affinity for a soft base such as the thiolate ion. Preliminary work with the H463C mutant shows a sensitivity to block by Cd2+ that is greater than for wt Kv1.5.
In Cav2.3 (α1E) channels, external Ni2+ causes, in addition to a rightward shift of the g-V curve, a reduction of the slope conductance with an estimated KI of 300 μM (Zamponi et al., 1996). The blocking reaction appears to be bimolecular and is also affected by the type of permeant ion (e.g., Ca2+ versus Ba2+). It was suggested that the Ni2+ block of CaV2.3 reflected changes of permeation due to direct occlusion of the pore in addition to a possible change of the permeant ion concentration at the pore mouth. In voltage-gated K+ channels, divalent cations have proved to be useful probes of gating and permeation. However, whereas Zn2+ and Cd2+ have been studied in some detail (e.g., Gilly and Armstrong, 1982; Spires and Begenisich, 1994), Ni2+ has been used somewhat sparingly. In HERG K+ channels, external Ni2+, as well as Cd2+, Co2+, and Mn2+, increased the maximum current amplitude, an effect that was imputed to an alteration of inactivation gating (Paquette et al., 1998). Interestingly, in HERG channels mutations at a number of sites in the S5-P linker can dramatically alter inactivation (Liu et al., 2002), a finding that underscores the findings with Kv1.5 that, either by substitution through point mutation, or by chemical modification through ligand binding, residues in this region can profoundly influence the rate and extent of one or more inactivation processes occurring at the outer pore mouth.
Acknowledgments
This work was supported by a grant to S.J.K. from the Natural Sciences and Engineering Research Council (NSERC) of Canada and grants from the Heart and Stroke Foundation of British Columbia and Yukon (HSFBCY), and by the Canadian Institutes of Health Research (CIHR) to D.F. A PhD trainee award from the Michael Smith Foundation for Health Research supported D.C.H.K., and C.E. was in receipt of an NSERC Scholarship.
References
- Adda, S., B. K. Fleischmann, B. D. Freedman, M. Yu, D. W. Hay, and M. I. Kotlikoff. 1996. Expression and function of voltage-dependent potassium channel genes in human airway smooth muscle. J. Biol. Chem. 271:13239–13243. [DOI] [PubMed] [Google Scholar]
- Baker, O. S., H. P. Larsson, L. M. Mannuzzu, and E. Y. Isacoff. 1998. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in Shaker K+ channel gating. Neuron. 20:1283–1294. [DOI] [PubMed] [Google Scholar]
- Chen, F. S. P., D. Steele, and D. Fedida. 1997. Allosteric effects of permeating cations on gating currents during K+ channel deactivation. J. Gen. Physiol. 110:87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F. S. P., and D. Fedida. 1998. On the mechanism by which 4-aminopyridine occludes quinidine block of the cardiac K+ channel, hKv1.5. J. Gen. Physiol. 111:539–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement-Chomienne, O., K. Ishii, M. P. Walsh, and W. C. Cole. 1999. Identification, cloning and expression of rabbit vascular smooth muscle Kv1.5 and comparison with native delayed rectifier K+ current. J. Physiol. 515:653–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eduljee, C., B. Glanville, D. Fedida, and S. J. Kehl. 2003. Mutation of a turret residue H463 in hKv1.5 accelerates slow inactivation and causes a collapse of current in the absence of external K+. Biophys. J. 84:221a. [Google Scholar]
- Fedida, D., R. Bouchard, and F. S. P. Chen. 1996. Slow gating charge immobilization in the human potassium chanel Kv1.5 and its prevention by 4-aminopyridine. J. Physiol. 494:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedida, D., N. D. Maruoka, and S. Lin. 1999. Modulation of slow inactivation in human cardiac Kv1.5 channels by extra- and intracellular permeant cations. J. Physiol. 515:315–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedida, D., B. Wible, Z. Wang, B. Fermini, F. Faust, S. Nattel, and A. M. Brown. 1993. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circ. Res. 73:210–216. [DOI] [PubMed] [Google Scholar]
- Gilly, W. F., and C. M. Armstrong. 1982. Divalent cations and the activation kinetics of potassium channels in squid giant axons. J. Gen. Physiol. 79:965–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glusker, J. P. 1991. Structural aspects of metal liganding to functional groups in proteins. Adv. Protein Chem. 42:1–76. [DOI] [PubMed] [Google Scholar]
- Harrison, N. L., H. K. Radke, M. M. Tamkun, and D. M. Lovinger. 1993. Modulation of gating of cloned rat and human K+ channels by micromolar Zn2+. Mol. Pharmacol. 43:482–486. [PubMed] [Google Scholar]
- Hesketh, J. C., and D. Fedida. 1999. Sequential gating in the human heart K+ channel incorporates Q1 and Q2 charge components. Am. J. Physiol. 277:H1956–H1966. [DOI] [PubMed] [Google Scholar]
- Hoshi, T., W. N. Zagotta, and R. W. Aldrich. 1991. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 7:547–556. [DOI] [PubMed] [Google Scholar]
- Jäger, H., and S. Grissmer. 2001. Regulation of a mammalian Shaker-related potassium channel, hKv1.5, by extracellular potassium and pH. FEBS Lett. 488:45–50. [DOI] [PubMed] [Google Scholar]
- Kehl, S. J., C. Eduljee, D. C. H. Kwan, S. Zhang, and D. Fedida. 2002. Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn2+. J. Physiol. 541:9–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan, D. C. H., D. Fedida, and S. J. Kehl. 2003. Single channel analysis of the inhibition of hKv1.5 current by extracellular protons. Biophys. J. 84:74a. (Abstr.) [Google Scholar]
- Larsson, H. P., O. S. Baker, D. S. Dhillon, and E. Y. Isacoff. 1996. Transmembrane movement of the Shaker K+ channel S4. Neuron. 16:387–397. [DOI] [PubMed] [Google Scholar]
- Liu, J., M. Zhang, M. Jiang, and G. N. Tseng. 2002. Structural and functional role of the extracellular S5-P linker in the HERG potassium channel. J. Gen. Physiol. 120:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo, J., T. Hoshi, S. H. Heinemann, and R. W. Aldrich. 1993. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1:61–71. [PubMed] [Google Scholar]
- MacKinnon, R. 1991. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 350:232–235. [DOI] [PubMed] [Google Scholar]
- Olcese, R., R. Latorre, L. Toro, F. Bezanilla, and E. Stefani. 1997. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 110:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette, T., J. R. Clay, A. Ogbaghebriel, and A. Shrier. 1998. Effects of divalent cations on the E-4031-sensitive repolarization current, I(Kr), in rabbit ventricular myocytes. Biophys. J. 74:1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perchenet, L., and O. Clement-Chomienne. 2001. External nickel blocks human Kv1.5 channels stably expressed in CHO cells. J. Membr. Biol. 183:51–60. [DOI] [PubMed] [Google Scholar]
- Perez-Cornejo, P. 1999. H+ ion modulation of C-type inactivation of Shaker K+ channels. Pflugers Arch. 437:865–870. [DOI] [PubMed] [Google Scholar]
- Perozo, E., R. MacKinnon, F. Bezanilla, and E. Stefani. 1993. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 11:353–358. [DOI] [PubMed] [Google Scholar]
- Spires, S., and T. Begenisich. 1994. Modulation of potassium channel gating by external divalent cations. J. Gen. Physiol. 104:675–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus, J. G., L. Kuschel, M. D. Rayner, and S. H. Heinemann. 1997. Ion conduction through C-type inactivated Shaker channels. J. Gen. Physiol. 110:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus, J. G., Z. Varga, R. Schonherr, and S. H. Heinemann. 2003. Mechanisms of the inhibition of Shaker potassium channels by protons. Pflugers Arch. 447:44–54. [DOI] [PubMed] [Google Scholar]
- Steidl, J. V., and A. J. Yool. 1999. Differential sensitivity of voltage-gated potassium channels Kv1.5 and Kv1.2 to acidic pH and molecular identification of pH sensor. Mol. Pharmacol. 55:812–820. [PubMed] [Google Scholar]
- Tamkun, M. M., K. M. Knoth, J. A. Walbridge, H. Kroemer, D. M. Roden, and D. M. Glover. 1991. Molecular cloning and characterization of two voltage-gated K+ channel cDNAs from human ventricle. FASEB J. 5:331–337. [DOI] [PubMed] [Google Scholar]
- Trapani, J. G., and S. J. Korn. 2003. Effect of external pH on activation of the Kv1.5 potassium channel. Biophys. J. 84:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., J. C. Hesketh, and D. Fedida. 2000a. A high-Na+ conduction state during recovery from inactivation in the K+ channel Kv1.5. Biophys. J. 79:2416–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., X. Zhang, and D. Fedida. 2000b. Regulation of transient Na+ conductance by intra- and extracellular K+ in the human delayed rectifier K+ channel Kv1.5. J. Physiol. 523:575–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., Y. Yan, and F. J. Sigworth. 2002. The Shaker mutation T449V rescues ionic currents of W434F K+ channels. Biophys. J. 82:234a. (Abstr.) [Google Scholar]
- Yang, Y., Y. Yan, and F. J. Sigworth. 1997. How does the W434F mutation block current in Shaker potassium channels? J. Gen. Physiol. 109:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi, G. W., E. Bourinet, and T. P. Snutch. 1996. Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. J. Membr. Biol. 151:77–90. [DOI] [PubMed] [Google Scholar]
- Zhang, S., D. C. H. Kwan, D. Fedida, and S. J. Kehl. 2001a. External K+ relieves the block but not the gating shift caused by Zn2+ in human Kv1.5 potassium channels. J. Physiol. 532:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S., S. J. Kehl, and D. Fedida. 2001b. Modulation of Kv1.5 potassium channel gating by extracellular zinc. Biophys. J. 81:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S., H. T. Kurata, S. J. Kehl, and D. Fedida. 2003. Rapid induction of P/C-type inactivation is the mechanism for acid-induced K+ current inhibition. J. Gen. Physiol. 121:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]