

Abstract
For the purpose of equilibrium and kinetic folding-unfolding studies, the SH3 domain of α-spectrin (spc-SH3) has long been considered a classic two-state folding protein. In this work we have indeed observed that the thermal unfolding curves of spc-SH3 measured at pH 3.0 by differential scanning calorimetry, circular dichroism, and NMR follow apparently the two-state model when each unfolding profile is considered individually. Nevertheless, we have found that protein concentration has a marked effect upon the thermal unfolding profiles. This effect cannot be properly explained in terms of the two-state unfolding model and can only be interpreted in terms of the accumulation of intermediate associated states in equilibrium with the monomeric native and unfolded states. By chemical cross-linking and pulsed-field gradient NMR diffusion experiments we have been able to confirm the existence of associated states formed during spc-SH3 unfolding. A three-state model, in which a dimeric intermediate state is assumed to be significantly populated, provides the simplest interpretation of the whole set of thermal unfolding data and affords a satisfactory explanation for the concentration effects observed. Whereas at low concentrations the population of the associated intermediate state is negligible and the unfolding process consequently takes place in a two-state fashion, at concentrations above ∼0.5 mM the population of the intermediate state becomes significant at temperatures between 45°C and 80°C and reaches up to 50% at the largest concentration investigated. The thermodynamic properties of the intermediate state implied by this analysis fall in between those of the unfolded state and the native ones, indicating a considerably disordered conformation, which appears to be stabilized by oligomerization.
INTRODUCTION
A few dozen proteins have to date been reported as folding in a two-state process (Jackson, 1998), amongst which the folding-unfolding properties of the Src-homology region 3 (SH3) domains have probably been the most extensively studied (Viguera et al., 1994; Grantcharova and Baker, 1997; Grantcharova et al., 1998; Guijarro et al., 1998a; Martinez et al., 1998; Plaxco et al., 1998; Martinez and Serrano, 1999). Kinetic and calorimetric criteria are usually applied to determine whether or not a folding reaction follows a two-state model (Privalov, 1979; Jackson and Fersht, 1991). For the SH3 domain of α-spectrin (spc-SH3) these two criteria have been fulfilled under all the conditions investigated so far (Viguera et al., 1994) and no equilibrium or kinetic intermediates have been detected to date. In contrast to the two-state behavior of the folding-unfolding reactions of spc-SH3, the study of the amide hydrogen-deuterium exchange (HX) measured by NMR under native conditions indicates that this domain, similar to that in other proteins, undergoes a considerable variety of conformational fluctuations, ranging from local motions to large structural disruptions affecting the domain's core and even extending above the transition-state energy barrier (Sadqi et al., 1999, 2002a,b; Englander, 2000; Casares et al., 2003). It has been reported for several proteins that as destabilizing conditions are approached, e.g., by a change in pH, temperature, pressure, or concentration of denaturants, the large conformational heterogeneity of native fluctuations, as probed by HX measurements, is progressively replaced by a few conformational states, including partially folded states, or just the globally unfolded state (Bai et al., 1995; Chamberlain et al., 1996; Fuentes and Wand, 1998; Milne et al., 1999). In a previous paper we reported the temperature dependence of HX in the spc-SH3 domain at pH 3.0 (Sadqi et al., 2002a). As the temperature approached thermal unfolding conditions the HX data indicated the existence of a significant population of states with Gibbs energies clearly lower than that of the globally unfolded state. These types of state might have remained undetectable by the standard analysis of folding-unfolding data as was suggested previously by Englander and colleagues (Mayne and Englander, 2000).
In standard HX experiments the concentration of sample is usually high due to the relatively low sensitivity of NMR. It is of interest therefore to explore the possibility that partially folded intermediates of spc-SH3 might become stabilized by intermolecular association favored by the high concentrations used in the NMR experiments. The self-association of folding intermediates and denatured states of proteins and protein fragments has been reported previously (Filimonov et al., 1993; Pecorari et al., 1996; Uversky et al., 1998; Kuznetsova et al., 1999; Liu et al., 1999; Uversky et al., 1999; Ye and Wang, 2001). The oligomeric states have been often described as an association of folding intermediates with residual super-secondary structure (Filimonov et al., 1993), which in certain cases appears as native-like (Alexandrescu et al., 2000; Ye and Wang, 2001). Even CI2, another classic two-state protein, can form transient aggregates during folding under suitable conditions (Silow et al., 1999). The association of folding intermediates has special relevance in that with some proteins the association process proceeds further to the formation of large aggregates and amyloid fibrils (Dobson, 1999; Rochet and Lansbury, 2000), even in many proteins that are not related to any amyloid-related disease, including the PI3-SH3 domain. Fibril formation appears to depend upon the accumulation of unstable, partially structured, intermediate states stabilized by self-assembly into an oligomerization nucleus (Rochet and Lansbury, 2000). Studies of association of folding intermediates are, therefore, of increasing importance to understand the details of these misfolding processes.
We present here the results of a detailed study into the effects of increasing protein concentration upon the thermal unfolding equilibrium of the spc-SH3 domain using a combination of calorimetry and spectroscopic techniques. We have also tested the presence of oligomeric species using chemical cross-linking and pulse-field gradient NMR diffusion measurements. The results confirm the existence of partially unfolded oligomeric states in equilibrium with the native and unfolded states. Using a simple mathematical model we have estimated the thermodynamic properties of these intermediates, which may help to shed light upon their conformational properties and their possible role in the mechanisms of aggregation of these small domains.
MATERIAL AND METHODS
Protein samples and chemicals
Wild-type chicken α-spectrin SH3 domain was obtained as described elsewhere (Sadqi et al., 1999). Samples were prepared for experimental work by extensive dialysis against a large volume of 20 mM glycine pH 3.0, except for the samples in deuterated buffer, which were prepared by dissolving the lyophilized protein directly in the buffer and subsequently readjusting their pH (direct pH-meter reading). Sample concentration was measured by UV absorption at 280 nm using an extinction coefficient of 16,147 for the native protein (Viguera et al., 1994).
Perdeuterated glycine for NMR samples came from Cambridge Isotope (Andover, MA). Deuterium oxide and other deuterated compounds were from Sigma (St. Louis, MO). Glutaraldehyde for cross-linking experiments was grade I (Sigma). The remaining chemicals used were all of analytical grade.
Differential scanning calorimetry
High-sensitivity differential scanning calorimetry (DSC) was done with a VP-DSC microcalorimeter (Microcal, Northampton, MA). The scan rate was usually 1°C/min, except for experiments made specifically to test any possible effect of scan rate, where it was varied between 0.5°C/min and 1.5°C/min. The experiments were performed at pH 3.0 in 20 mM glycine buffer and the concentration of protein was varied from 0.03 mM to 4.85 mM. After baseline subtraction, the temperature dependence of the molar partial heat capacity (Cp) of spc-SH3 was calculated from the DSC thermograms using Origin (OriginLab, Northampton, MA) as described elsewhere (Privalov and Potekhin, 1986). Preliminary fittings of the Cp curves using the two-state unfolding model were made as described elsewhere (Viguera et al., 1994).
Thermal unfolding followed by circular dichroism
Circular dichroism (CD) measurements were made in a Jasco J-715 spectropolarimeter (Jasco, Easton, MD) equipped with a temperature-controlled cell holder. The cell path length was 1 mm or 10 mm, depending upon the protein concentration used and the spectral region to be observed. Thermal unfolding was studied by monitoring the CD signal at 222 nm in the far-ultraviolet (UV) wavelength range, and at 270 nm and 294 nm in the near-UV range, during temperature scans at a scan rate of 1°C/min. The actual temperature of the sample was measured by immersing a PT-100 probe inside the cell and registered by the instrument's computer. Experimental conditions were identical to those of the DSC experiments. The concentration of protein was 0.028 mM or 0.14 mM for measurements at 222 nm, and 0.14 mM or 0.69 mM for measurements at both 270 nm and 294 nm. Baseline CD temperature scans were recorded with pure buffer solutions and subtracted from the CD traces obtained with the protein solutions. Preliminary fitting of the CD thermal traces using a two-state unfolding model was made as described elsewhere (van Nuland et al., 1998).
Thermal unfolding followed by nuclear magnetic resonance
Thermal unfolding was monitored using NMR on a Bruker AMX-500 spectrometer (Billerica, MA). Protein solutions were prepared in 10% D2O/90% H2O, 20 mM perdeuterated glycine, pH 3.0 (direct pH-meter readings without isotopic correction). Sample concentration was either 0.68 mM or 5.4 mM. A set of one-dimensional NMR spectra at different temperatures between 20.7°C and 81.3°C was acquired using a sweep width of 6024 Hz and 4096 data points. Spectra were sine-bell apodized, Fourier transformed, and baseline corrected using the MestRe-C program (University of Santiago de Compostela, Spain). For each NMR spectrum the intensity of several well-resolved NMR signals was determined by fitting the peaks to Lorentzian functions. This procedure allowed us to measure accurately the intensities of partially overlapping signals. The intensity of the signals of Ala55-CβH3, Leu33-CδH3, and Trp41-NɛH, corresponding to the native state, was monitored as a function of temperature. Two signals corresponding to Trp41-NɛH and Trp42-NɛH in nonnative states were also observed. The data were initially fitted on the basis of the two-state unfolding model using the same procedure as for the CD thermal unfolding experiments.
Chemical cross-linking
Cross-linking experiments with spc-SH3 in 20 mM glycine, pH 3.0, at four sample concentrations of between ∼0.04 mM and 3.5 mM and temperatures of 20°C and 50°C were conducted to identify the presence of spc-SH3 oligomers in solution. Glutaraldehyde was used as cross-linking reagent according to the procedure described elsewhere (Jaenicke and Rudolph, 1989). An identical procedure was repeated with lysozyme solutions in 20 mM glycine, pH 2.0, at similar sample concentrations. This monomeric protein was used as a control for unspecific cross-linking. Samples were incubated at the desired temperature in Eppendorf tubes in a thermostatic bath for 10 min. Glutaraldehyde was added to a final concentration of 1% w/v and the samples were left to incubate in the bath for 3 min. Cross-linking was fixed by adding freshly prepared NaBH4 solution in 0.1 M NaOH to a final concentration of 50 mM. Samples were left to incubate for a further 20 min. Protein was precipitated by the addition of a 10% sodium deoxycholate solution to a final 0.01% w/v and a 78% trichloracetic acid solution to a final 1.5% w/v, followed by further incubation in ice for 10 min. Protein precipitates were collected by centrifugation; the pellets were washed carefully with water and redissolved for SDS-PAGE analysis.
Pulse-field gradient NMR spectroscopy
Pulse-field gradient NMR (PFG-NMR) diffusion measurements were made with the pulse gradient stimulated echo longitudinal encode-decode (PG-SLED) sequence (Wilkins et al., 1999) on a Bruker Avance DRX750 spectrometer. The protein concentrations were ≈0.5 mM and ≈5 mM. Samples were prepared in 20 mM perdeuterated glycine, pH 3.0, either in 7% D2O/93% H2O or in 100% D2O. Thirty microliters of a 1% dioxan solution was added to the sample. 1H chemical shifts are expressed relatively to dioxan (3.75 ppm). Each diffusion data set consisted of a series of 40 one-dimensional 1H-NMR spectra with 2.5% increments of the maximum gradient strength from 2.5 to 100% collected with a three-axis gradient probe. The gradient pulse length, δ, was optimized to obtain a total decay of 80–90% of the initial signal, a typical value being 4 ms. The echo time, Δ, for diffusion was set to 100 ms and NMR spectra were acquired with 8 K complex points and a sweep width of 10,000 Hz. The water resonance was suppressed by weak presaturation during the 1-s relaxation delay. The data were processed with Felix from Biosym Technologies (San Diego, CA).
The intensity of the signals of the protein (I) in different spectral regions (e.g., amide/aromatic (7.8–6.4 ppm) and aliphatic groups (from 3.5 to −0.5 ppm)) as a function of gradient strength (g) were fitted using Origin (OriginLab) to
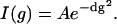 |
(1) |
This equation enables us to obtain the observed decay rate, d, which is proportional to the translational diffusion coefficient, D. The resulting decay rate of the protein, dprot, was then used to fit the change in intensity of the dioxan signal at 3.75 ppm to a double Gaussian function. This procedure was motivated by the need of eliminating the contribution of protein signals to the dioxan signal intensity due to overlap.
The protein hydrodynamic radius (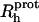 ) was calculated as described elsewhere (Wilkins et al., 1999) using the following equation:
) was calculated as described elsewhere (Wilkins et al., 1999) using the following equation:
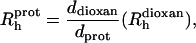 |
(2) |
where ddioxan and 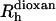 are, respectively, the decay rate and the radius of the dioxan molecule, which was calculated as being 2.12 Å (Wilkins et al., 1999).
are, respectively, the decay rate and the radius of the dioxan molecule, which was calculated as being 2.12 Å (Wilkins et al., 1999).
RESULTS
Thermal unfolding of spc-SH3 monitored by CD and 1H-NMR spectroscopy
The existence of equilibrium folding intermediates of proteins has been revealed on many occasions by discrepancies between the thermal unfolding profiles obtained when using different techniques. We have investigated the thermal unfolding of spc-SH3 at pH 3.0 by monitoring the temperature dependence of the CD signal at 222 nm, in the far-UV region of the spectrum. The experiments were made with sample concentrations of 0.028 mM and 0.14 mM. The spectral changes induced by temperature at 222 nm probe the disruption of the secondary structure of the domain. We also monitored the thermal unfolding of 0.14 mM and 0.69 mM samples at both 294 nm and 270 nm in the near-UV region. The changes in this wavelength region are sensitive above all to the disordering of the tertiary structure. The CD spectra of samples heated up to 95°C and then cooled down to 20°C were indistinguishable from the spectra obtained before heating, which indicates the reversibility of the unfolding under these conditions.
We also used one-dimensional 1H-NMR to monitor unfolding by measuring the intensity of several well-resolved signals as a function of temperature, as described in Material and Methods. These experiments were carried out with protein concentrations of 0.68 mM and 5.4 mM. The NMR spectra were registered between 20.7 and 81.3°C. After measuring the spectrum at the highest temperature, the sample was cooled down to 20.7°C and another spectrum was registered. Even at the highest sample concentration of 5.4 mM, the spectra before and after heating were virtually identical and the sample remained clear with no visible signs of aggregation. No evidence of either line broadening or signal displacement due to rapid exchange were observed in the NMR spectra.
The unfolding profiles were fitted initially using the two-state unfolding model (see Table 1). When considered individually all the fittings are excellent (not shown), thus apparently agreeing with the previously reported two-state behavior for the unfolding of spc-SH3, although very strikingly the Tm values obtained for the different experiments often do not concur. These discrepancies, however, rather than being related to the use of different techniques, seem more to be a simple effect of protein concentration. In fact, the Tm values do agree quite well for experiments made at similar concentrations but using different observables (e.g., far-UV and near-UV CD at 0.14 mM, near-UV CD and 1H-NMR at ∼0.68 mM). Therefore the unfolding Tm depends to a great extent upon the sample concentration, ranging from 54°C at the lowest concentration to ∼47°C at the highest. This result is completely inconsistent with a simple two-state unfolding model, which does not allow for any concentration effect, and suggests that under our experimental conditions the thermal unfolding of spc-SH3 is coupled with some change in the degree of association of the protein. The increase in concentration has a destabilizing effect, which would imply that the associated state is a nonnative state of the domain.
TABLE 1.
Thermodynamic parameters resulting from a two-state analysis of the thermal unfolding profiles of spc-SH3 at pH 3.0, followed by CD, NMR, and DSC
| Observable | Protein concentration (mg·mL−1) | Tm (°C) | ΔHm (kJ × mol−1) |
|---|---|---|---|
| CD wavelength (nm) | |||
| 222 | 0.2 | 54.0 ± 0.4 | 176 ± 5 |
| 222 | 1.0 | 53.5 ± 0.2 | 168 ± 4 |
| 294 | 1.0 | 52.8 ± 0.3 | 170 ± 9 |
| 270 | 5.0 | 51.9 ± 0.4 | 172 ± 12 |
| 294 | 5.0 | 51.3 ± 0.5 | 171 ± 13 |
| NMR native signal | |||
| Ala55-CβH3 | 4.9 | 51.3 ± 2.5 | 146 ± 31 |
| Leu33-CδH3 | 4.9 | 52.0 ± 3.5 | 159 ± 46 |
| Trp41-NɛH | 4.9 | 51.2 ± 3.3 | 202 ± 87 |
| Ala55-CβH3 | 38.9 | 47.1 ± 2.2 | 166 ± 34 |
| Leu33-CδH3 | 38.9 | 45.9 ± 3.6 | 160 ± 41 |
| Trp41-NɛH | 38.9 | 47.6 ± 2.0 | 223 ± 66 |
| DSC | |||
| 0.22 | 56.1 ± 0.1 | 182 ± 1 | |
| 1.1 | 55.9 ± 0.1 | 174 ± 1 | |
| 5.5 | 55.2 ± 0.1 | 164 ± 1 | |
| 15.5 | 54.6 ± 0.1 | 149 ± 1 | |
| 35.0 | 50.2 ± 0.3 | 127 ± 1 |
The heat-capacity change on the unfolding of SH3 has been fixed in the fittings of the CD and NMR profiles at a value of 1.7 ± 0.6 kJ × mol−1, as obtained from the DSC experiments. The uncertainties in the parameters correspond to the standard errors of the fittings.
The effect of concentration on the DSC thermograms of spc-SH3
In previous studies into the thermal unfolding of spc-SH3 by DSC, the sample concentration has not been taken to be a variable and has in general been kept below 0.5 mM (Viguera et al., 1994). Thus we have reinvestigated the thermal unfolding of spc-SH3 using DSC in 20 mM glycine at pH 3.0 and at different concentrations between 0.03 mM and 4.9 mM. The partial molar heat capacity (Cp) curves thus obtained are shown in Fig. 1. The curves were always highly reproducible in a second consecutive scan with the same sample and were not affected by a scan rate between 0.5°C/min and 1.5°C/min. The percentage of reversibility in terms of area of the unfolding transitions was >95% in all the DSC experiments, except at a protein concentration of 4.9 mM, where reversibility was ∼75%. The denaturation peak in the Cp curves clearly broadens and its maximum shifts toward lower temperatures concomitantly with a rise in protein concentration.
FIGURE 1.

Partial molar heat capacity, Cp, versus temperature observed by DSC for spc-SH3 in 20 mM glycine, pH 3.0, at several concentrations of protein sample, which are indicated along each curve. Symbols represent the experimental data. Solid lines correspond to the Cp curves predicted by the three-state model described in the text using the parameters of Table 2 for data set A. Dashed lines represent the best prediction of the whole set of Cp curves using the two-state model.
Despite these observations we fitted each Cp curve on the basis of a two-state model. In a similar way as the results from the unfolding experiments followed by CD or NMR, the individual Cp curves fit the model very well, except for the case of the thermogram at the highest concentration (4.9 mM), which shows a small deviation from the model on the high-temperature side of the transition (data not shown). Once more, the Tm values obtained from the fittings decrease concomitantly with a rise in the concentration of the sample, although to a lesser extent than in the CD or the NMR experiments. Another important observation is that the Tm values found by DSC are generally higher than those obtained either by CD or NMR at similar concentrations. This is probably due to the fact that whereas CD (both in the near and far-UV ranges) and NMR are sensitive to the disruption of the native structure on unfolding, DSC monitors the heat capacity of the protein whatever its state, and nonnative states of the protein that may be undistinguishable by CD and NMR could contribute differently to the enthalpy and the heat capacity of the system.
Taken together, the thermal unfolding profiles of spc-SH3 obtained at pH 3.0 using different techniques and different sample concentrations contradict the hypothesis that this small domain is a two-state folding protein under all conditions. The results suggest the presence of a nonnative associated state of the domain in the unfolding equilibrium, favored by high sample concentrations.
A minimalist unfolding model accounting for the effect of concentration upon the unfolding profiles of spc-SH3
The simplest model that might account for the observed effect can be schematized by
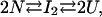 |
where three thermodynamically different states are in equilibrium: the native, N, the unfolded, U, and a dimeric intermediate, I2. We assumed an association degree of two for the sake of simplicity. Two relevant equilibrium constants can thus be defined:
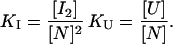 |
The same thermodynamic model, although with different assumptions concerning the nature of the dimeric intermediate state, has been employed elsewhere to describe the thermal unfolding of the chemotactic protein CheY (Filimonov et al., 1993) and of a C-terminal thermolysin fragment (Azuaga et al., 1995). The equations of the model that allow us to predict the thermal unfolding profiles are essentially the same as those described in the above-mentioned papers and are therefore not included here. States I2 and U are characterized, respectively, by Gibbs energies ΔGI and ΔGU, enthalpies ΔHI and ΔHU, and heat capacities, ΔCpI and ΔCpU, all relative to the native state and expressed per mol of monomer. We also defined TU as the temperature at which ΔGU is zero (which would be the equivalent to the Tm for a two-state model) and TI as a reference temperature at which KI has a certain value (Filimonov et al., 1993) taken here for convenience to be 103.
To apply these equations to an analysis of the thermal unfolding profiles described here we made two assumptions. Firstly, we took it that both CD and NMR are only sensitive to the disappearance of the native state and do not distinguish between nonnative states. This is strictly true for the unfolding profiles followed by monitoring the intensity of a NMR signal corresponding to the native conformation, as it is the case here. It is also an appropriate assumption for the near-UV CD signal because folding intermediates or protein aggregates do not usually exhibit CD in this wavelength range (Kelly and Price, 1997). In contrast, nonnative states contribute to the far-UV CD. Nevertheless, the unfolding profile of spc-SH3 obtained at 222 nm and 0.14 mM matches those of the same concentration obtained at 270 nm or 294 nm very closely. The second of our assumptions, which was also made with the CheY protein (Filimonov et al., 1993), was that the intermediate and unfolded states have the same heat capacity functions, i.e., ΔCpI = ΔCpU. This approximation was motivated by the fact that each of these two heat-capacity changes would be estimated with an extremely large error if we tried to determine them simultaneously from the unfolding curves, which do not contain enough information about them. This approximation implies the assumption that the amounts of surface exposed to the solvent by the intermediate and the unfolded states are similar.
With this model it was possible to predict the unfolding profiles given the following set of parameters: the two reference temperatures, TI and TU; the enthalpy values for the intermediate, ΔHI, and for the unfolded, ΔHU, states given at TI and TU, respectively; the heat-capacity change for both the I2 and U states at a certain temperature (50°C for convenience), ΔCp; and finally the Cp function of the native state, considered as being a linear function of temperature. The Cp function of the unfolded state was calculated as described elsewhere (Makhatadze and Privalov, 1990).
The fitting of the thermal unfolding profiles using the model equations was made using two different approaches: firstly, the global fitting of a set of Cp curves at different concentrations and secondly, simultaneous fitting of the unfolding profiles obtained by different techniques at similar concentrations. Following these two approaches we combined the unfolding profiles in five different data sets (from A to E), as described in Table 2. The fittings of data sets A and C are shown in Figs. 1 and 2, respectively. The resulting parameters of all the fittings are included in Table 2. In the fittings of data set D and E we needed to fix the parameters corresponding to the intermediate state, i.e., ΔHI and TI, which were taken from the fitting of data set A. This was motivated by the highly indeterminate nature of these parameters in the fittings, due to the small population of the intermediate state at these low concentrations, as shown below. In fact, the fitting of data sets D and E using the two-state model also gave satisfactory results (data not shown).
TABLE 2.
Thermodynamic parameters obtained from the simultaneous fitting of different sets of thermal unfolding profiles of SH3 at pH 3.0, using the three-state model described in the text
| Data set* | TI (°C) | TU (°C) | ΔHI (TI) (kJ × mol−1) | ΔHU (TU) (kJ × mol−1) | ΔCp (50°C) (kJ × K−1 × mol−1) |
|---|---|---|---|---|---|
| A | 56.6 ± 0.3 | 55.5 ± 0.2 | 119 ± 1 | 181 ± 2 | 2.2 ± 0.2 |
| B | 57.0 ± 0.2 | 53.6 ± 0.3 | 120 ± 1 | 166 ± 5 | 2.8 ± 0.1 |
| C | 56.6 ± 0.8 | 54.6 ± 0.2 | 104 ± 4 | 180 ± 2 | 2.4 ± 0.1 |
| D | 56.6 (f) | 55.6 ± 0.2 | 119 (f) | 178 ± 1 | 2.2 ± 0.1 |
| E | 56.6 (f) | 55.9 ± 0.1 | 119 (f) | 183 ± 1 | 2.2 ± 0.1 |
For the sake of simplicity, the parameters corresponding to the values of the different observables for the individual states have not been included. The uncertainty of each parameter corresponds to the standard error of the fit. Parameters marked with ‘f’ have been fixed to the corresponding values of data set A.
Data set A consisted of five Cp curves obtained at five different sample concentrations between 0.03 mM and 4.8 mM; data set B consisted of the Cp curve at 4.8 mM plus an unfolding profile obtained by NMR at 5.4 mM; data set C consisted of the Cp curve at 0.76 mM, a near-UV CD curve at 0.69 mM, and an NMR profile at 0.68 mM; data set D consisted of a Cp curve at 0.15 mM, a near-UV CD curve at 0.14 mM, and a far-UV CD curve at 0.14 mM; data set E consisted of a Cp curve at 0.03 mM and a far-UV CD curve at 0.028 mM.
FIGURE 2.

Thermal unfolding profiles of spc-SH3 in 20 mM glycine, pH 3.0, observed by several techniques. (A) Partial molar heat capacity versus temperature obtained by DSC. (B) Far-UV CD at 222 nm versus temperature. (C) Temperature dependence of the 1H-NMR signal intensity corresponding to the Ala55 methyl proton in the native state. Spc-SH3 concentrations were 0.76 mM (A), 0.69 mM (B), and 0.68 mM (C), respectively. Symbols represent the experimental data. Solid lines correspond to the curves predicted by the three-state model described in the text using the parameters of Table 2 for data set C.
As can be seen in Fig. 1, this simple model was capable of predicting correctly the concentration effects observed in the Cp curves. This was not possible using the two-state model, as it is also shown in the same figure. Additionally, the model could explain simultaneously the unfolding profiles observed using different techniques (see Fig. 2), which was impossible using the two-state model at sample concentrations of ∼0.5 mM and higher. The resulting thermodynamic parameters, particularly the two enthalpy values, agree well for the fittings of different data sets. The enthalpy of the intermediate state, which was considered to be a dimer in the model, is closer to that of the unfolded state than to the native one, which suggests that it has a low degree of tertiary structure.
Fig. 3 shows the population of each state of the model as a function of temperature according to the parameters obtained from the fittings. At low concentrations the population of the intermediate state is very small throughout the whole temperature range (≤1% at 0.03 mM and ≤5% at 0.15 mM). This indicates that at these concentrations the two-state model describes quite satisfactorily the unfolding of spc-SH3 under these experimental conditions. At 0.7 mM and above, the population of the intermediate state is quite considerable, reaching a maximum at temperatures of between 50 and 70°C at the highest concentration investigated.
FIGURE 3.

Temperature dependence of the relative populations of the native, N, dimeric intermediate, I2, and unfolded, U, states of spc-SH3 for different protein concentrations according to the model described in the text. The curves have been calculated using the parameters of Table 2 for data set A. Protein concentrations are as follows: 0.03 mM (solid line); 0.15 mM (dashed line); 0.76 mM (dotted line); 2.2 mM (dash-dot line); and 4.9 mM (dash-dot-dot line).
Detection of spc-SH3 oligomers by chemical cross-linking
We employed chemical cross-linking, using glutaraldehyde as reagent, to detect any oligomeric states of spc-SH3. The experiments were made at pH 3.0 in 20 mM glycine buffer at 20°C and 50°C, as described in Material and Methods. The sample concentration was varied over two orders of magnitude. Lysozyme solutions of similar concentrations at pH 2.0 were used as a control for a typically monomeric protein. The results of this experiment are shown in Fig. 4. At 20°C only a very small amount of dimers and traces of higher-order aggregates of spc-SH3 were trapped at the highest concentrations investigated. Some lysozyme dimers are also detected at the highest concentration. At 50°C there was no appreciable cross-linking with spc-SH3 at low concentrations (0.04 mM and 0.15 mM) or with lysozyme at all concentrations, indicating that both proteins are essentially monomeric under these conditions. For spc-SH3 at 0.52 mM, cross-linking detects some population of dimers at 50°C. At the highest concentration investigated (2.6 mM) a considerable population and variety of oligomers was trapped by cross-linking. This does not appear to be an artifact of the high concentration of protein, because lysozyme is not cross-linked at similar concentrations. These results demonstrate the existence of oligomerization at high concentrations of spc-SH3 at 50°C, whereas at 20°C the presence of oligomers is much less evident. At 50°C the trapped oligomers increase in size markedly concomitant with an increase in concentration, going from mainly dimers at 0.52 mM to a whole variety of sizes at a concentration of 2.6 mM.
FIGURE 4.

SDS-PAGE gels of spc-SH3 and lysozyme samples submitted to cross-linking with glutaraldehyde at two different temperatures and several protein concentrations. (A) 20°C; lanes 1–5: lysozyme in 20 mM glycine, pH 2.0; lanes 6–10: spc-SH3 in 20 mM glycine, pH 3.0. Lanes 1 and 6 correspond to samples not cross-linked. Lysozyme concentrations: lane 2, 1.92 mM; lane 3, 0.38 mM; lane 4, 0.07 mM; and lane 5, 0.014 mM. spc-SH3 concentrations: lane 7, 3.4 mM; lane 8, 0.69 mM; lane 9, 0.15 mM; and lane 10, 0.042 mM. (B) 50°C, lanes 1–5: lysozyme in 20 mM glycine, pH 2.0; lanes 6–10: SH3 in 20 mM glycine, pH 3.0. Lanes 1 and 6 correspond to samples not cross-linked. Lysozyme concentrations: lane 2, 0.021 mM; lane 3, 0.063 mM; lane 4, 0.31 mM; and lane 5, 1.8 mM. SH3 concentrations: lane 7, 0.042 mM; lane 8, 0.15 mM; lane 9, 0.53 mM; and lane 10, 2.6 mM. Lane M in both A and B gels corresponds to molecular weight markers.
PFG-NMR diffusion measurements
The apparent hydrodynamic radius (Rh) of spc-SH3 at two temperatures (20°C and 50°C) and at two protein concentrations (0.5 mM and 5 mM) was determined at pH 3.0 in 93% H2O/7% D2O and in 100% D2O, using the PFG-NMR method as described in Materials and Methods. Each set of PG-SLED spectra was first recorded at 20°C and then at 50°C with the same sample. After cooling the sample down to 20°C a third series of spectra were recorded to check for reversibility. No differences were seen between the Rh values obtained at 20°C before and after heating the samples to 50°C.
Fig. 5 A shows two expanded regions of the series of spectra taken at 5 mM concentration in the 93% H2O/7% D2O mixture at 20°C and 50°C. We have used the resonance at −0.28 ppm, corresponding to Ala55-CH3 in the native state, to monitor selectively the diffusion of this state. At 20°C, the indol signal at 9.89 ppm corresponding to Trp41 in the native state is also well resolved. At 50°C there are two additional tryptophan indol signals corresponding to Trp41 and Trp42 in a nonnative state, one of which at 9.86 ppm is well resolved, thus enabling the estimation of the apparent Rh of the nonnative state under these conditions. Fig. 5 B shows the analysis of the diffusion data extracted from the spectra shown in Fig. 5 A at 20°C and 50°C.
FIGURE 5.

(A) Expanded regions of a series of 750 MHz 1D PG-SLED 1H-NMR spectra of 5 mM spc-SH3 in 93%/7% H2O/D2O, 20 mM glycine, pH 3.0. Diffusion at 20°C and 50°C, as indicated in the plot. The correspondences of each signal with either native (N) or denatured (D) states of the spc-SH3 domain are also indicated along the spectra. (B) Analysis of the PFG-NMR diffusion measurements shown in panel A. Solid circles represent the intensity changes of the native signal at −0.29 ppm at both temperatures. Open triangles represent the intensity of the native Trp41 indol signal at 9.89 ppm at 20°C. Open squares represent the intensity of the nonnative tryptophan indol signal at 9.86 ppm at 50°C. The decay of each signal was fitted using Eq. 1 and the fits are shown in solid lines.
At 20°C different native signals decay with approximately the same rate, thus giving a Rh value of 16.5 ± 0.2 Å for native spc-SH3, as calculated using Eq. 2. This value agrees well with a Rh of 15.7 Å estimated for a folded protein of this size using the empirical equation given elsewhere (Wilkins et al., 1999). At 50°C, the decay of the native Ala55-CH3 signal yields an Rh of 12.3 ± 0.4 Å, which is significantly smaller than at 20°C. This value is equal to the value of 12.3 Å estimated for the radius of gyration (Rg) based on its relationship with the hydrodynamic radius, Rg = ρRh, where ρ reflects the shape of the molecule and equals (3/5)1/2 for spherical molecules (Burchard et al., 1980). The drop in Rh for the native state with temperature is not gradual but occurs abruptly just above 45°C according to diffusion measurements performed at several temperatures between 20 and 50°C (data not shown).
The analysis of the decay of the tryptophan indol signal at 9.86 ppm in the series of PG-SLED spectra at 50°C results in a substantially larger Rh of 21.8 ± 0.8 Å. The hydrodynamic radius of the fully unfolded chain of spc-SH3 can be estimated from empirical relationships as being 23.2 Å (Wilkins et al., 1999), which is only slightly larger than our experimental result for the nonnative state of spc-SH3. Using a similar empirical equation given by the same authors, the hydrodynamic radius of a globular folded dimer of spc-SH3 would be 19.2 Å, this time slightly smaller than the experimental Rh.
To determine more accurately the diffusion rate of the fully unfolded state of spc-SH3 we lowered the pH from 3.0 to 1.0 to destabilize the protein and thereby shift the denaturation transition to a lower temperature. Under these conditions the DSC denaturation transitions show that spc-SH3 is almost completely unfolded at 50°C (data not shown), which enables us to analyze the diffusion rate of the fully unfolded state by integrating the aromatic/amide and aliphatic regions of the PG-SLED spectra. This analysis resulted in an Rh of 21.7 ± 0.2 Å for the fully unfolded state, which is very close to the value of 21.8 ± 0.8 Å estimated by the analysis of the single tryptophan indol resonance at pH 3.0 (Fig. 5).
In contrast to the results of the analysis of individual resonances, the Rh values obtained by fitting the intensity changes of both the amide/aromatic and aliphatic regions of the spectra at pH 3.0 and 50°C depend to a great extent upon the concentration of spc-SH3 (14.4 Å for 0.5 mM and 17.3 Å for 5 mM). This result can be explained by the difference between the two samples in their relative contribution of nonnative (unfolded and/or oligomeric) species to the overall intensity decay. Because the radii of nonnative proteins are longer than that of the native state, a higher population of nonnative species caused by a rise in concentration will increase the Rh values obtained in the PG-SLED experiments.
Assuming only two distinguishable states with substantially different hydrodynamic radii at pH 3.0 and 50°C (native and denatured states), and using the experimentally estimated decay rates for the denatured state (dD) and native state (dN) from the individually resolved NMR signals, as described above, the complete amide/aromatic and aliphatic regions of the PG-SLED spectra can be fitted to the equation:
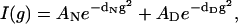 |
(3) |
where AN and AD are the relative contributions to the decay in intensity of the native and denatured states, respectively (see also Material and Methods). At 50°C and 5 mM this analysis results in a native state population of 36% and a denatured state population of 64%, whereas at 50°C and 0.5 mM the populations are 80% and 20%, respectively. These relative populations of native and nonnative states at 50°C agree very well with those found in the analysis of the thermal unfolding profiles shown above (see Fig. 3). This strong dependence of the relative contributions of the states upon protein concentration once more tends to support the participation of the oligomerization equilibrium in the overall unfolding of spc-SH3.
DISCUSSION
The spc-SH3 domain has long been considered an archetypal two-state folding protein (Viguera et al., 1994; Martinez et al., 1998; Martinez and Serrano, 1999). Other SH3 domain variants such as Fyn (Plaxco et al., 1998; Filimonov et al., 1999), Abl (Filimonov et al., 1999), PI3 (Guijarro et al., 1998a), or Src (Grantcharova and Baker, 1997; Grantcharova et al., 1998) have also been shown in other studies to undergo simple two-state folding and unfolding without the presence of intermediates under a variety of conditions.
We have provided here several lines of evidence to show that at pH 3.0 and protein concentrations of ≈0.5 mM and above, the two-state unfolding model cannot properly describe the thermal unfolding of spc-SH3. The thermal unfolding profiles observed by CD, both in the far-UV and near-UV region, and by one-dimensional 1H-NMR do not in general match the results of the DSC thermograms as the protein concentration increases, which suggests the presence of equilibrium intermediates. In addition, the unfolding curves depend very much upon the concentration of the protein, which indicates that protein association is coupled to the unfolding of the domain. The rise in concentration destabilizes the native state, which implies that the state undergoing association is nonnative.
Despite these results, the two-state model accurately describes each one of the individual unfolding profiles, including the Cp curves obtained by DSC, even under conditions where associated states are considerably populated. The deviation from the two-state model does not manifest itself in the shape of the unfolding curves but in the consistency between the thermodynamic parameters obtained at different concentrations and/or using different techniques. This result suggests that the calorimetric criterion for two-state unfolding may not be valid under some conditions, such as those of our experiments in this work, as has been suggested elsewhere (Zhou et al., 1999).
We propose therefore a very simple three-state model for the thermal unfolding of spc-SH3, which assumes the presence of an equilibrium dimeric intermediate state. The model fits very well and simultaneously the unfolding profiles obtained by spectroscopic and calorimetric techniques and explains the apparent discrepancies that exist when the profiles are fitted using the two-state unfolding model. It also satisfactorily accounts for the observed effect of protein concentration on the unfolding curves. The model predicts a significant population of the dimeric intermediate state at concentrations of ∼0.5 mM and higher and at temperatures between ∼45 and 80°C (Fig. 3). For instance, at 50°C and 0.76 mM the population of the intermediate state is ∼10%, whereas at the same temperature and 4.9 mM it reaches 36%. At concentrations lower than ∼0.15 mM, on the other hand, the population of the intermediate state is lower than 5% throughout the whole temperature range and therefore under these conditions the unfolding process of spc-SH3 is adequately described by the two-state model. This agrees with previous studies of the folding-unfolding of spc-SH3 under similar conditions, in which the sample concentration was not considered as a variable and in any case was usually kept quite low (Viguera et al., 1994; Martinez et al., 1998).
The application of the three-state model allows us to characterize the thermodynamic properties of the intermediate state. The enthalpy change from the native state to the dimeric intermediate state, expressed per mol of monomer, is ≈110 kJ × mol−1 at ≈57°C (Table 2), which is little more than half that of the enthalpy change of global unfolding. The entropy change from the native to the intermediate state is also relatively high (T × ΔSI ≈ 100 kJ × mol−1 at 57°C) and there is a large enthalpy-entropy compensation for this equilibrium, similar to what is always found in protein unfolding. All this suggests a partially disordered conformation for the intermediate state.
A major assumption of this model was to consider an association degree of 2 for the intermediate state. In fact, the results of the cross-linking experiments indicate the presence of a significant population of dimers at 50°C and a concentration of 0.53 mM, whereas only monomers are detected below 0.15 mM at the same temperature. At a concentration of 2.6 mM, however, a broad range of oligomeric species were trapped by cross-linking, suggesting that at these concentrations the association equilibrium is more complicated than the simple dimerization allowed for by the model. Nevertheless, due to its irreversible character, we cannot exclude the possibility that chemical cross-linking is actually driving the formation of oligomers and larger aggregates.
In contrast to the native state, the PFG-NMR diffusion measurements did not permit us to distinguish between the hydrodynamic radii of the unfolded state and any oligomeric species present in the unfolding equilibrium at 50°C. This may be due to the existence of fast chemical exchange in the NMR timescale between these states and/or to the similarity in their translational diffusion coefficients. By selectively monitoring nonnative resonances we could estimate an apparent Rh value of 21.8 Å, which agrees well with the empirical correlation for highly unfolded proteins (Wilkins et al., 1999), and with the Rh value of spc-SH3 measured under highly destabilizing conditions. It is tempting to assign this Rh value to the monomeric unfolded state. Nevertheless, an analysis of the decays in intensity of the whole aromatic and aliphatic regions of the spectra using a double Gaussian function (Eq. 3) revealed that there is a marked dependency upon protein concentration in the relative amplitudes of the intensity decays corresponding to the native and nonnative states, and their values were entirely consistent with those deriving from the analysis of the thermal unfolding profiles. This strongly supports the existence of a population of oligomers that increases concomitantly with protein concentration in the unfolding equilibrium of spc-SH3, which is also reflected in the concentration dependence of the hydrodynamic properties of the domain.
In a recent paper (Sadqi et al., 2002a), using amide hydrogen-deuterium exchange under the same experimental conditions as those of this work, we reported that near the unfolding temperature there were nonnative conformational states of spc-SH3 with lower Gibbs energy than that of the unfolded state. The results of this work confirm this report because the concentration of sample in the HX experiments was ≈4.5 mM and therefore the population of intermediate state above ≈35°C would be significant (see Fig. 3). Interestingly, using heteronuclear NMR, Serrano and colleagues (Kortemme et al., 2000) have observed that under mildly acidic conditions the spc-SH3 domain has a marked propensity to adopt conformations that resemble the transition state of folding. It is thus likely that in the highly concentrated NMR samples the structural propensity of the spc-SH3 polypeptide chain is stabilized by oligomerization.
Another example of association of an SH3 domain is Eps8-SH3, which has been shown to form stable intertwined, domain-swapped dimers (Kishan et al., 2001). Each SH3 motif in the dimer is formed by the interaction between sequences 1–35 and 40–64 from each polypeptide chain, with an overall fold very similar to the monomer. According to its thermodynamic properties the associated intermediate state of spc-SH3 would be much more unstructured than those of Eps8, although they might share a similar overall topology.
A more dramatic example of association has been found in the PI3-SH3 domain. This homologous domain forms amyloid fibrils under acid conditions, in which the domain is an ensemble of partially folded states (Guijarro et al., 1998b). In contrast, the spc-SH3 domain does not form amyloid fibrils even under conditions where partially folded oligomers are highly populated, as we show here. In a recent study using chimeras of spc-SH3 and PI3-SH3 domains that exchange their n-src loops, Serrano and colleagues have justified this discrepancy in behavior in terms of differences in the amino acid sequence of the domain cores, which may shift the subtle balance of solubility of intermediates toward the formation of either ordered fibrils or amorphous aggregates (Ventura et al., 2002). Additionally, a theoretical study using molecular dynamics simulations of the aggregation of src-SH3 has proposed that amyloidogenesis of SH3 domains may occur through highly unstructured conformations with a solvent-exposed hydrophobic core (Ding et al., 2002). Thus, it is increasingly evident that the characterization of associating folding intermediates and a further understanding of factors that may shift the equilibrium between different classes of intermediates, i.e., productive or unproductive for large aggregates, is crucial to increase our understanding of the mechanisms of amyloidogenesis.
In conclusion, we provide here a thermodynamic characterization of partially folded associated states of the spc-SH3 domain, previously considered an archetypical two-state folding protein. These intermediates can exist at mildly acid pH and temperatures between 45 and 80°C in partially disordered conformations stabilized by high protein concentrations. The existence of these intermediates was not discernible from single thermal unfolding experiments, which appear to reflect two-state transitions, but needed the combined use of several techniques and an analysis of the effect of protein concentration to detect them.
Acknowledgments
We thank Dr. A. Haidour of the Scientific Instrument Centre of the University of Granada for his technical assistance in part of the NMR experiments. We also thank Dr. J. Trout for revising our English text.
This work has been financed by grants HPRN-CT-2002-00241 from the European Union and BIO2000-1459 from the Spanish Ministry of Science and Technology. The 750-MHz NMR spectra were recorded at the SON NMR Large Scale Facility in Utrecht, which is funded by the Access to Research Infrastructures program of the European Union (HPRI-CT-1999-00005).
S. Casares and M. Sadqi contributed equally to this work.
Mourad Sadqi's present address is Dept. of Chemistry and Biochemistry, University of Maryland, College Park, MD 20742 USA.
References
- Alexandrescu, A. T., F. P. Lamour, and V. A. Jaravine. 2000. NMR evidence for progressive stabilization of native-like structure upon aggregation of acid-denatured LysN. J. Mol. Biol. 295:239–255. [DOI] [PubMed] [Google Scholar]
- Azuaga, A. I., F. Conejero-Lara, G. Rivas, V. De Filippis, A. Fontana, and P. L. Mateo. 1995. The thermodynamics of association and unfolding of the 205–316 C-terminal fragment of thermolysin. Biochim. Biophys. Acta. 1252:95–102. [DOI] [PubMed] [Google Scholar]
- Bai, Y., T. R. Sosnick, L. Mayne, and S. W. Englander. 1995. Protein folding intermediates: native-state hydrogen exchange. Science. 269:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchard, W., M. Schmidt, and W. H. Stockmayer. 1980. Information on polydispersity and branching from combined quasi-elastic and integrated scattering. Macromolecules. 13:1265–1272. [Google Scholar]
- Casares, S., M. Sadqi, O. Lopez-Mayorga, J. C. Martinez, and F. Conejero-Lara. 2003. Structural cooperativity in the SH3 domain studied by site-directed mutagenesis and amide hydrogen exchange. FEBS Lett. 539:125–130. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A. K., T. M. Handel, and S. Marqusee. 1996. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Biol. 3:782–787. [DOI] [PubMed] [Google Scholar]
- Ding, F., N. V. Dokholyan, S. V. Buldyrev, H. E. Stanley, and E. I. Shakhnovich. 2002. Molecular dynamics simulation of the SH3 domain aggregation suggests a generic amyloidogenesis mechanism. J. Mol. Biol. 324:851–857. [DOI] [PubMed] [Google Scholar]
- Dobson, C. M. 1999. Protein misfolding, evolution and disease. Trends Biochem. Sci. 24:329–332. [DOI] [PubMed] [Google Scholar]
- Englander, S. W. 2000. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 29:213–238. [DOI] [PubMed] [Google Scholar]
- Filimonov, V. V., A. I. Azuaga, A. R. Viguera, L. Serrano, and P. L. Mateo. 1999. A thermodynamic analysis of a family of small globular proteins: SH3 domains. Biophys. Chem. 77:195–208. [DOI] [PubMed] [Google Scholar]
- Filimonov, V. V., J. Prieto, J. C. Martinez, M. Bruix, P. L. Mateo, and L. Serrano. 1993. Thermodynamic analysis of the chemotactic protein from Escherichia coli, CheY. Biochemistry. 32:12906–12921. [DOI] [PubMed] [Google Scholar]
- Fuentes, E. J., and A. J. Wand. 1998. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry. 37:9877–9883. [DOI] [PubMed] [Google Scholar]
- Grantcharova, V. P., and D. Baker. 1997. Folding dynamics of the src SH3 domain. Biochemistry. 36:15685–15692. [DOI] [PubMed] [Google Scholar]
- Grantcharova, V. P., D. S. Riddle, J. V. Santiago, and D. Baker. 1998. Important role of hydrogen bonds in the structurally polarized transition state for folding of the src SH3 domain. Nat. Struct. Biol. 5:714–720. [DOI] [PubMed] [Google Scholar]
- Guijarro, J. I., C. J. Morton, K. W. Plaxco, I. D. Campbell, and C. M. Dobson. 1998a. Folding kinetics of the SH3 domain of PI3 kinase by real-time NMR combined with optical spectroscopy. J. Mol. Biol. 276:657–667. [DOI] [PubMed] [Google Scholar]
- Guijarro, J. I., M. Sunde, J. A. Jones, I. D. Campbell, and C. M. Dobson. 1998b. Amyloid fibril formation by an SH3 domain. Proc. Natl. Acad. Sci. USA. 95:4224–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, S. E. 1998. How do small single-domain proteins fold? Fold. Des. 3:R81–R91. [DOI] [PubMed] [Google Scholar]
- Jackson, S. E., and A. R. Fersht. 1991. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry. 30:10428–10435. [DOI] [PubMed] [Google Scholar]
- Jaenicke, R., and R. Rudolph. 1989. Folding proteins. In: Protein Structure: A Practical Approach. T. E. Creighton, editor. IRL Press (Oxford University Press), Oxford, UK. 191–223.
- Kelly, S. M., and N. C. Price. 1997. The application of circular dichroism to studies of protein folding and unfolding. Biochim. Biophys. Acta. 1338:161–185. [DOI] [PubMed] [Google Scholar]
- Kishan, K. V., M. E. Newcomer, T. H. Rhodes, and S. D. Guilliot. 2001. Effect of pH and salt bridges on structural assembly: molecular structures of the monomer and intertwined dimer of the Eps8 SH3 domain. Protein Sci. 10:1046–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortemme, T., M. J. Kelly, L. E. Kay, J. Forman-Kay, and L. Serrano. 2000. Similarities between the spectrin SH3 domain denatured state and its folding transition state. J. Mol. Biol. 297:1217–1229. [DOI] [PubMed] [Google Scholar]
- Kuznetsova, I. M., A. G. Biktashev, S. Y. Khaitlina, K. S. Vassilenko, K. K. Turoverov, and V. N. Uversky. 1999. Effect of self-association on the structural organization of partially folded proteins: inactivated actin. Biophys. J. 77:2788–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T., P. A. Pemberton, and A. D. Robertson. 1999. Three-state unfolding and self-association of maspin, a tumor-suppressing serpin. J. Biol. Chem. 274:29628–29632. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G. I., and P. L. Privalov. 1990. Heat capacity of proteins. I. Partial molar heat capacity of individual amino acid residues in aqueous solution: hydration effect. J. Mol. Biol. 213:375–384. [DOI] [PubMed] [Google Scholar]
- Martinez, J. C., M. T. Pisabarro, and L. Serrano. 1998. Obligatory steps in protein folding and the conformational diversity of the transition state. Nat. Struct. Biol. 5:721–729. [DOI] [PubMed] [Google Scholar]
- Martinez, J. C., and L. Serrano. 1999. The folding transition state between SH3 domains is conformationally restricted and evolutionarily conserved. Nat. Struct. Biol. 6:1010–1016. [DOI] [PubMed] [Google Scholar]
- Mayne, L., and S. W. Englander. 2000. Two-state vs. multistate protein unfolding studied by optical melting and hydrogen exchange. Protein Sci. 9:1873–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, J. S., Y. Xu, L. C. Mayne, and S. W. Englander. 1999. Experimental study of the protein folding landscape: unfolding reactions in cytochrome c. J. Mol. Biol. 290:811–822. [DOI] [PubMed] [Google Scholar]
- Pecorari, F., P. Minard, M. Desmadril, and J. M. Yon. 1996. Occurrence of transient multimeric species during the refolding of a monomeric protein. J. Biol. Chem. 271:5270–5276. [DOI] [PubMed] [Google Scholar]
- Plaxco, K. W., J. I. Guijarro, C. J. Morton, M. Pitkeathly, I. D. Campbell, and C. M. Dobson. 1998. The folding kinetics and thermodynamics of the Fyn-SH3 domain. Biochemistry. 37:2529–2537. [DOI] [PubMed] [Google Scholar]
- Privalov, P. L. 1979. Stability of proteins: small globular proteins. Adv. Protein Chem. 33:167–241. [DOI] [PubMed] [Google Scholar]
- Privalov, P. L., and S. A. Potekhin. 1986. Scanning microcalorimetry in studying temperature-induced changes in proteins. Methods Enzymol. 131:4–51. [DOI] [PubMed] [Google Scholar]
- Rochet, J. C., and P. T. Lansbury, Jr. 2000. Amyloid fibrillogenesis: themes and variations. Curr. Opin. Struct. Biol. 10:60–68. [DOI] [PubMed] [Google Scholar]
- Sadqi, M., S. Casares, M. A. Abril, O. Lopez-Mayorga, F. Conejero-Lara, and E. Freire. 1999. The native state conformational ensemble of the SH3 domain from alpha-spectrin. Biochemistry. 38:8899–8906. [DOI] [PubMed] [Google Scholar]
- Sadqi, M., S. Casares, O. Lopez-Mayorga, and F. Conejero-Lara. 2002a. The temperature dependence of the hydrogen exchange in the SH3 domain of alpha-spectrin. FEBS Lett. 527:86–90. [DOI] [PubMed] [Google Scholar]
- Sadqi, M., S. Casares, O. Lopez-Mayorga, J. C. Martinez, and F. Conejero-Lara. 2002b. pH dependence of the hydrogen exchange in the SH3 domain of alpha-spectrin. FEBS Lett. 514:295–299. [DOI] [PubMed] [Google Scholar]
- Silow, M., Y. J. Tan, A. R. Fersht, and M. Oliveberg. 1999. Formation of short-lived protein aggregates directly from the coil in two-state folding. Biochemistry. 38:13006–13012. [DOI] [PubMed] [Google Scholar]
- Uversky, V. N., A. S. Karnoup, R. Khurana, D. J. Segel, S. Doniach, and A. L. Fink. 1999. Association of partially folded intermediates of staphylococcal nuclease induces structure and stability. Protein Sci. 8:161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky, V. N., D. J. Segel, S. Doniach, and A. L. Fink. 1998. Association-induced folding of globular proteins. Proc. Natl. Acad. Sci. USA. 95:5480–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nuland, N. A., F. Chiti, N. Taddei, G. Raugei, G. Ramponi, and C. M. Dobson. 1998. Slow folding of muscle acylphosphatase in the absence of intermediates. J. Mol. Biol. 283:883–891. [DOI] [PubMed] [Google Scholar]
- Ventura, S., E. Lacroix, and L. Serrano. 2002. Insights into the origin of the tendency of the PI3–SH3 domain to form amyloid fibrils. J. Mol. Biol. 322:1147–1158. [DOI] [PubMed] [Google Scholar]
- Viguera, A. R., J. C. Martinez, V. V. Filimonov, P. L. Mateo, and L. Serrano. 1994. Thermodynamic and kinetic analysis of the SH3 domain of spectrin shows a two-state folding transition. Biochemistry. 33:2142–2150. [DOI] [PubMed] [Google Scholar]
- Wilkins, D. K., S. B. Grimshaw, V. Receveur, C. M. Dobson, J. A. Jones, and L. J. Smith. 1999. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 38:16424–16431. [DOI] [PubMed] [Google Scholar]
- Ye, K., and J. Wang. 2001. Self-association reaction of denatured staphylococcal nuclease fragments characterized by heteronuclear NMR. J. Mol. Biol. 307:309–322. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., C. K. Hall, and M. Karplus. 1999. The calorimetric criterion for a two-state process revisited. Protein Sci. 8:1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]