

Abstract
Understanding the screening by salts of charge-charge interactions in proteins is important for at least two reasons: a), screening by intracellular salt concentration may modulate the stability and interactions of proteins in vivo; and b), the in vitro experimental estimation of the contributions from charge-charge interactions to molecular processes involving proteins is generally carried out on the basis of the salt effect on process energetics, under the assumption that these interactions are screened out by moderate salt concentrations. Here, we explore experimentally the extent to which the screening efficiency depends on the nature of the salt. To this end, we have carried out an energetic characterization of the effect of NaCl (a nondenaturing salt), guanidinium chloride (a denaturing salt), and guanidinium thiocyanate (a stronger denaturant) on the stability of the wild-type form and a T14K variant of Escherichia coli thioredoxin. Our results suggest that the efficiency of different salts to screen charge-charge interactions correlates with their denaturing strength and with the position of the constituent ions in the Hofmeister rankings. This result appears consistent with the plausible relation of the Hofmeister rankings with the extent of solute accumulation/exclusion from protein surfaces.
INTRODUCTION
Electrostatic interactions between charged groups are likely to play essential roles in molecular processes involving proteins, including ligand-binding, protein-protein interactions, and protein folding-unfolding. In fact, recent work supports that charge-charge interactions are the main determinants of the pK values of exposed ionizable groups on protein surfaces (Pace et al., 2002; Sundd et al., 2002; Laurents et al., 2003) and that the surface-charge distribution may be rationally designed for enhanced protein stability and for optimized intermolecular interactions (Grimsley et al., 1999; Ibarra-Molero et al., 1999a; Loladze et al., 1999; Pace, 2000; Perl et al., 2000; Spector et al., 2000; Lee and Tidor, 2001; Nohaile et al., 2001; Perl and Schmid, 2001; Sanchez-Ruiz and Makhatadze, 2001; Ibarra-Molero and Sanchez-Ruiz, 2002; Marshall et al., 2002; Martin et al., 2002; Makhatadze et al., 2003). The experimental estimation of the contribution from charge-charge interactions to processes involving proteins is, therefore, an issue of considerable importance. This estimation is often carried out on the basis of the sodium chloride dependence of the energetic parameters for the process under study, under the assumption that charge-charge interactions are effectively screened out at moderate NaCl concentrations (below 1 M). However, evidence has accumulated over the years indicating that some surface salt bridges can be insensitive to NaCl, even when there is a clear charge-charge, coulombic interaction (Perutz et al., 1985; Yu et al., 1996; Kao et al., 2000; Luisi et al., 2003). Also, the recent work of Dominy et al. (2002) supports that NaCl is likely to screen more efficiently long-range interactions over short-range ones. It appears, therefore, that complete screening of all charge-charge interactions in proteins by moderate concentrations of a given salt (such as NaCl) cannot, in general, be taken for granted.
Here we address a different, but related, issue: the extent to which the charge screening efficiency depends on the nature of the salt. It must be noted that several analyses of experimental data support that denaturants tend to interact preferentially with protein surfaces, whereas stabilizers tend to be preferentially excluded (Arakawa and Timasheff, 1984, 1985; Makhatadze and Privalov, 1992; Bolen and Baskarov, 2001; Courtenay et al., 2001). It appears likely then that denaturing salts (such as guanidinium chloride) accumulate near the surface of proteins and, as a result, that they are very efficient at screening charge-charge interactions in proteins. Indeed, for several proteins, the guanidinium chloride dependence of the denaturation free energy shows abrupt deviations from linearity at low denaturant concentrations (<∼1 M), which have been attributed to the screening of interactions involving charged groups (Santoro and Bolen, 1992; Monera et al., 1994; Ibarra-Molero and Sanchez-Ruiz, 1996; Ibarra-Molero et al., 1999a; Bolen and Yang, 2000; Garcia-Mira and Sanchez-Ruiz, 2001) (in some cases, deviations have been interpreted in terms of ion binding (Greene and Pace, 1974; Santoro and Bolen, 1988; Pace et al., 1990; Hagihara et al., 1993; Mayr and Schmid, 1993; Makhatadze et al., 1998)).
We report here a detailed experimental characterization of the sodium chloride and guanidinium chloride effects on the thermodynamic stability of Escherichia coli thioredoxin and a T14K variant (designed for improved charge-charge interactions on the surface and slightly more stable than the wild-type (WT) form). Our results provide evidence for significant screening by the denaturing salt but not by NaCl and suggest that the efficiency of different salts to screen charge-charge interactions in proteins does correlate with their denaturing strength and, ultimately, with the position of the constituent ions in the Hofmeister series (Baldwin, 1996). We derive additional support for this proposal from experiments on the effect of guanidinium thiocyanate (a stronger denaturant than guanidinium chloride) on the stability of E. coli thioredoxin.
We use in this work a simple electrostatic model (of the Tanford-Kirkwood type) to derive rough theoretical estimates of the contribution from charge-charge interactions to thioredoxin stability and, also, to design the T14K variant. We wish to emphasize, however, that, to estimate the screening efficiency of the different salts, we use an experimental approach based on the premise that screening of charge interactions is reflected in a clearly nonlinear (exponential-like) dependence of denaturation free energy in the ∼0–1 M salt concentration range. Experimental detection of such “exponential-like” dependence is straightforward in the case of nondenaturing salts, such as NaCl. In the case of guanidinium chloride and guanidinium thiocyanate, on the other hand, the screening contribution to denaturation ΔG is superimposed on a large decrease of ΔG with salt concentration associated with the denaturing character of these salts; detection of the screening effect in these cases can be more conveniently carried out on the basis of m values (the derivatives −∂ΔG/∂[salt]) derived from experimental differential scanning calorimetry (DSC) data, as we showed a few years ago (Ibarra-Molero and Sanchez-Ruiz, 1996).
MATERIALS AND METHODS
Electrostatic calculations
Calculations of the energies of charge-charge interactions were carried using our implementation of the Tanford-Kirkwood model with the solvent accessibility correction of Gurd, as we have previously described in detail (Ibarra-Molero et al., 1999a). Calculations on variants of thioredoxin, in which neutral-residue→{Glu or Lys} mutations (see lower panel in Fig. 1) were introduced, were based on structures modeled (starting from the WT-thioredoxin structure) with the SwissViewer v3.7b2 program. No structure optimization was performed, but calculations were carried out for all the sterically allowed rotamers of the new side chains.
FIGURE 1.

(Upper panel) Bar graph of energies due to charge-charge interactions of all ionizable residues in the thioredoxin molecule at pH 7 as calculated using our implementation of the Tanford-Kirkwood model (see Materials and Methods for details). Positive values of Eq-q indicate that the amino acid side chains are involved in predominantly destabilizing charge-charge interactions, whereas negative values of Eq-q correspond to the amino acid side chains that are involved in predominantly stabilizing interactions. (Lower panel) Charge-charge interaction energies calculated for variants of thioredoxin in which Lys or Glu have been substituted for surface neutral polar residues. ΔEq-q is the difference between the total charge-charge interaction energy in the native state calculated for the variant and that corresponding to the WT form. The values actually plotted are −ΔEq-q, that is, the calculated contributions from charge-charge interactions in the native state to the mutation effect on denaturation ΔG. The calculations were performed for all sterically allowed rotamers of the newly introduced side chains; the average values and the corresponding standard deviations are shown.
Accessible surface areas (ASA) were calculated using a modification of the Shrake-Rupley algorithm (Shrake and Rupley, 1973), which randomly places 2000 points in the expanded van der Waals sphere representing each atom. A radius of 1.4 Å for the solvent probe and the Chothia set (Chothia, 1976) for the protein atoms were used. Residue accessibilities were calculated as the ratio between the side-chain ASA in the native structure and that in Gly-X-Gly tripeptide.
Site-directed mutagenesis
Oligonucleotides used for mutagenesis were obtained from Genotek (Ottawa, Canada). Mutation in the codon corresponding to position 14 in the amino acid sequence of thioredoxin was introduced by the QuikChange site-directed mutagenesis method developed by Stratagene (La Jolla, CA). Briefly, the QuikChange method is based on polymerase chain reaction amplification using two complementary oligonucleotide primers containing the desired mutation. The parental nonmutated DNA is finally digested by an endonuclease. Mutation was verified by DNA sequence analysis.
Protein expression and purification
Plasmid pTK100 encoding wild-type thioredoxin (a gift from Dr. Maria Luisa Tasayco) was transformed into E. coli JF521 strain for protein overexpression. Cells were grown, starting from single colonies, at 37°C in Luria broth with 40 μg/mL of kanamycin to select for the plasmid-bearing cells. The final 750-fold dilution of the cell broth was allowed to grow during 12 h after stationary phase was reached. After centrifugation, cell pellets were frozen at −20°C until purification. Protein purification protocol was as follows. Briefly, cells were thawed, resuspended in 1 mM EDTA, 30 mM TRIZMA buffer, pH 8.3, and lyzed using a French press. The cell debris was centrifuged and the supernatant was collected and stirred with streptomycin sulfate (10% w/v) at 4°C overnight to precipitate nucleic acids. The filtered supernatant was then loaded onto a 2 L Sephacryl S-100 high resolution (Amersham Pharmacia Biotech, Upsala, Sweden) gel filtration column equilibrated in 1 mM EDTA, 30 mM TRIZMA buffer, pH 8.3. Thioredoxin fractions were identified by SDS-PAGE, pooled, and applied to a 250 mL Fractogel EMD DEAE (M) (Merck, Darmstadt, Germany) ion exchange column equilibrated in 1 mM EDTA, 30 mM TRIZMA buffer, pH 8.3. The protein was eluted by a linear gradient between 0 and 0.5 M NaCl. The proteins were pure as measured by SDS-PAGE gel densitometry. The molecular weight of pure proteins was confirmed by mass spectrometry. Thioredoxin concentration was determined spectrophotometrically at 280 nm using a published value of the extinction coefficient (Holmgren and Reichard, 1967).
Reagents and experimental conditions
Guanidinium chloride was ultrapure grade from Pierce (Rockford, IL). NaCl was analysis grade from Merck. Deionized water was used throughout. Aqueous stock solutions of WT and T14K thioredoxin were prepared by exhaustive dialysis against 5 mM HEPES, pH 7.0. Stock solutions of 6 M guanidinium chloride in HEPES buffer and 6 M NaCl in HEPES buffer were prepared as described previously (Ibarra-Molero et al., 1999b). Guanidinium chloride concentrations were determined from refraction index measurements (Pace et al., 1989) using an Atago (Tokyo, Japan) R 5000 hand refractometer. Guanidinium thiocyanate was ultrapure grade from Sigma (St. Louis, MO) and its solutions were prepared by weight.
Measurements of pH values for guanidinium salts solutions were carried out after calibration of the glass-electrode cell with aqueous standard buffers. No pH corrections (Garcia-Mira and Sanchez-Ruiz, 2001; Acevedo et al., 2002) were applied and, therefore, the pH value of 7 given for the guanidinium salts solutions is actually a pH-meter reading value or “apparent” pH value (see Garcia-Mira and Sanchez-Ruiz, 2001, for further discussion). We note that, in any case, the pH-dependence of thioredoxin denaturation energetics appears to be rather small, in the neighborhood of pH 7 (see Georgescu et al., 2001).
Circular dichroism
The experiments were carried out using a Jasco (Tokyo, Japan) J-715 spectropolarimeter equipped with a PTC-348WI temperature control unit.
Two sets of thermal unfolding experiments were collected to study guanidinium chloride concentration effects in the far and near ultraviolet (UV) regions, respectively. The change in circular dichroism (CD) signal within the temperature range of 20–85°C was monitored at both 222 and 280 nm, using an equilibration time of 60 s, signal-averaging time of 4 s, a bandwidth of 2 nm, and 1.0 nm step size. Thermal scans were done at 0, 0.5, 1, and 1.6 M guanidinium chloride. In the far-UV CD experiments, the protein concentration was ∼0.2 mg/mL and 1 mm cell path length was used. In the near-UV CD thermal melts, the protein concentration was ∼0.9 mg/mL and 10 mm cell path length was used. All transitions were highly reversible as was shown by the recovery of the CD signal after cooling the protein solution from 85°C to 20°C. Fittings of the two-state model to the CD signal versus temperature profiles were carried out assuming linear pre- and posttransition baselines and that the denaturation enthalpy does not significantly change within the narrow temperature range of the transition (see Ibarra-Molero and Sanchez-Ruiz, 1997, for further details).
Additionally, far-UV CD spectra (from 260 to 210 nm) and near-UV CD spectra (from 325 to 260 nm) of wild-type and T14K thioredoxin were monitored in the absence and in the presence of guanidinium chloride at 20°C and 85°C, using a bandwidth of 1 nm, an average of 4 scans, and 1.0 nm step size.
Differential scanning calorimetry
DSC experiments were carried out with a VP-DSC calorimeter from MicroCal (Northampton, MA) at a scan rate of 1.5 K/min. Protein solutions for the calorimetric experiments were prepared by exhaustive dialysis against the buffer (5 mM HEPES, pH 7.0). The samples were degassed at room temperature before the calorimetric experiments. Calorimetric cells (operating volume ∼0.5 ml) were kept under an excess pressure of 30 psi to prevent degassing during the scan. In all measurements, the buffer from the last dialysis step was used in the reference cell of the calorimeter. Several buffer-buffer baselines were obtained before each run with a protein solution to ascertain proper equilibration of the instrument. In most experiments, a reheating run was carried out to determine the reversibility of the denaturation process. Finally, an additional buffer-buffer baseline was obtained immediately after the protein runs to check that no significant change in instrumental baseline had occurred. When working with aqueous solutions, the level of instrumental baseline reproducibility attained was excellent and similar to that we have recently described (see Fig. 2 in Irun et al., 2001). However, as we have pointed out (Plaza del Pino and Sanchez-Ruiz, 1995; Ibarra-Molero et al., 1999b) baseline reproducibility is significantly poorer in the presence of cosolvents. This prevents us from obtaining absolute heat capacity values in NaCl and guanidinium salt solutions, although it does not compromise the calculation of denaturation enthalpies and denaturation temperatures from the analysis of the transitions.
FIGURE 2.

Near-UV (upper panel) and far-UV (lower panel) CD spectra for WT thioredoxin (solid symbols) and the T14K variant (open symbols) in their native and denatured states at pH 7. The different symbols refer to the guanidinium chloride concentration: 0 M (circles), 0.5 M (hexagons), 1 M (squares), and 1.6 M (triangles). Native-state spectra were obtained at 20°C and denatured-state spectra at 85°C.
A protein concentration dependence for thioredoxin denaturation temperature has been reported in the literature and attributed to protein dimerization (Ladbury et al., 1993). Therefore, we carried out all the DSC experiments at comparatively low protein concentrations: ∼0.5 mg/mL or below in some cases. We found no protein concentration effects on denaturation energetics within the 0.1–0.5 mg/mL range. Fittings of theoretical models to the heat capacity profiles were performed using programs written by us in the MLAB environment (Civilized Software, Silver Spring, MD). The general approach used in the two-state fittings was as described previously (Ibarra-Molero et al., 1999b).
Gibbs energy calculations
Protein stability curves (plots of denaturation ΔG versus temperature) for aqueous solutions (i.e., in the absence of NaCl or guanidinium salts) were calculated from DSC data under two different assumptions: I), assuming that the denaturation heat capacity change is temperature independent and using the constant-ΔCP integrated Gibbs-Helmholtz equation with the ΔCP value obtained by extrapolating to the transition Tm the pre- and posttransition baselines; and II), taking into account the potential temperature dependence of ΔCP and assuming, for the purposes of ΔCP(T) calculation, that the heat capacity of the denatured state is equal to that expected for a fully solvated unfolded state as estimated from the group contributions given by Makhatadze and Privalov (1990) (the native-state and unfolded-state heat capacities we used are those shown in Fig. 5 of Georgescu et al., 2001). Further details about the calculation of protein stability under different assumptions for the temperature dependence of ΔCP can be found elsewhere (Ibarra-Molero and Sanchez-Ruiz, 1996; Ibarra-Molero et al., 1999b). Here, we simply point out that the procedures used (I and II) involve different assumptions and that the comparison of the two stability curves obtained provides an estimate of the uncertainty involved in the ΔG calculation from DSC data in this case (see Results for further details).
FIGURE 5.

(Upper panel) Plots of denaturation temperature at pH 7 versus salt (NaCl, guanidinium chloride, or guanidinium thiocyanate) concentration for WT thioredoxin. The circles are the experimental data and the lines represent the best fits of a straight line (NaCl and guanidinium thiocyanate) and a third-order polynomial (guanidinium chloride) to the experimental data. The Tm versus C (NaCl or guanidinium chloride) profiles for the T14K variant are very close to the ones shown for the WT form and have been omitted for the sake of clarity. (Lower panel) Plots of denaturation enthalpy versus denaturation temperature for WT thioredoxin (solid symbols) and the T14K variant (open symbols). Circles refer to experiments carried out in the presence of GdnCl. The two straight solid lines are drawn to guide the eye; actually, the ΔH values obtained in the presence of GdnCl for WT thioredoxin and the T14K variant appear to be the same within experimental uncertainty (which is ∼±10 kJ/mol). Squares correspond to experiments in the presence of GdnSCN. The dashed line corresponds to the best fit of a second-order polynomial to the experimental data and is drawn to guide the eye. It must be noted that the slopes of these plots must not be assigned to ΔCp values, since each point corresponds to a different cosolvent concentration (see Plaza del Pino and Sanchez-Ruiz, 1995, for details).
The effects of the T14→K mutation and of NaCl on denaturation ΔG were very small and, therefore, we could use the Schellman equation (Schellman, 1987) to calculate them without introducing significant errors. In the terminology of this work, the Schellman equation can be written as,
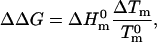 |
(1) |
where ΔΔG is the perturbation Gibbs energy, 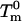 and
and 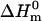 are the denaturation temperature and denaturation enthalpy change (at the denaturation temperature) for the unperturbed protein, and ΔTm is the perturbation effect on denaturation temperature. Note that Eq. 1 provides the ΔΔG value at the temperature
are the denaturation temperature and denaturation enthalpy change (at the denaturation temperature) for the unperturbed protein, and ΔTm is the perturbation effect on denaturation temperature. Note that Eq. 1 provides the ΔΔG value at the temperature 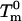 . If the perturbation is the T14→K mutation, the unperturbed protein is the WT form, and ΔTm is the difference Tm(T14K) − Tm(WT). If the perturbation is the presence of NaCl, the unperturbed protein is the WT form in the absence of salt and ΔTm is the difference Tm(WT in the presence of salt) − Tm(WT in the absence of salt); then application of Eq. 1 allows us to obtain ΔΔG = ΔG(WT in the presence of NaCl) − ΔG(WT in the absence of salt); the calculation actually yields ΔG(WT in the presence of NaCl), since at the denaturation temperature of the unperturbed protein ΔG(WT in the absence of salt) = 0. The NaCl concentration dependence of ΔG for WT thioredoxin denaturation was obtained in this way.
. If the perturbation is the T14→K mutation, the unperturbed protein is the WT form, and ΔTm is the difference Tm(T14K) − Tm(WT). If the perturbation is the presence of NaCl, the unperturbed protein is the WT form in the absence of salt and ΔTm is the difference Tm(WT in the presence of salt) − Tm(WT in the absence of salt); then application of Eq. 1 allows us to obtain ΔΔG = ΔG(WT in the presence of NaCl) − ΔG(WT in the absence of salt); the calculation actually yields ΔG(WT in the presence of NaCl), since at the denaturation temperature of the unperturbed protein ΔG(WT in the absence of salt) = 0. The NaCl concentration dependence of ΔG for WT thioredoxin denaturation was obtained in this way.
Monte Carlo estimates of the errors associated to the reported energetic parameters
All error intervals given in this work have been obtained using the Monte Carlo method. That is, several replicas of each given original data set were randomly generated using suitable distribution functions for the errors associated to the original data; subsequently, the replica data sets were processed in the same manner as the original set and the statistical analysis of the results obtained led to the errors associated to the derived energetic parameters. As an illustrative example, we explain below the Monte Carlo calculation of the errors for the guanidinium chloride m1/2 values.
The original data set for the m value calculation (see Discussion for details) consists of Tm and ΔHm values for different guanidinium chloride concentrations (the experimental C values). The experimental Tm versus C and ΔHm versus C dependencies could be adequately described by first- and third-order polynomials, respectively. We took those polynomials as the starting point for replica generation. That is, we used the polynomials to calculate, for the experimental C values, “error-free” Tm and ΔHm values. To these we added errors randomly generated according to Gaussian distributions of zero mean and standard deviation of 0.36° (for Tm) and 10 kJ/mol (for ΔHm) (these representative standard deviation values were obtained from the analysis of several DSC experiments carried out in the absence of denaturant). In this way, we generated 20 replicas of the original data, which were subjected to the same type of data processing: fitting of third-order polynomial to the Tm versus C dependence, from which we obtained the derivatives dTm/dC, which were used, together with the generated values of Tm and ΔHm, to obtain the m1/2 values. This procedure yielded 20 m1/2 values for each experimental denaturant concentration so that standard deviations could be calculated; these are given as the errors associated to the original m1/2 values.
RESULTS
Theoretical estimates of the charge-charge interactions in the thioredoxin molecule
Fig. 1 (upper panel) shows the energy of charge-charge interactions for individual ionizable residues in the thioredoxin molecule, as calculated by our implementation (Ibarra-Molero et al., 1999a) of the Tanford-Kirkwood model (Tanford and Kirkwood, 1957) (see Materials and Methods for details). The plot shown is similar to those we have previously reported for ubiquitin and other proteins (Ibarra-Molero et al., 1999a; Sanchez-Ruiz and Makhatadze, 2001): a positive value of the interaction energy for a given group means that the group is involved in predominantly destabilizing interactions with groups of alike charge; conversely, negative values for the interaction energy indicate stabilizing interactions with groups of opposite charge. In the case of the ubiquitin molecule, we found (Ibarra-Molero et al., 1999a) several groups with positive interaction energy and, in fact, charge-deletion and charge-reversal mutations on the corresponding positions led to the expected stability enhancements (Loladze et al., 1999). According to the calculations we report here (Fig. 1, upper panel), most ionizable groups in the thioredoxin molecule are involved in clearly stabilizing interactions at pH 7. In fact, it appears that, in this case, the charge distribution is already optimized for stability to a significant extent and the total charge-charge interaction energy (sum of all pairwise interaction energies) for native thioredoxin is estimated to be ∼−22 kJ/mol, that is, significant when compared with typical denaturation Gibbs energy values and stabilizing. Of course, it is conceivable that electrostatic interactions may also occur in the denatured state (Pace et al., 2000; Guzman-Casado et al., 2003) and contribute to the denaturation Gibbs energy change. Nevertheless, it seems at least reasonable to assume in this case that denatured-state charge-charge interactions do not fully cancel the strongly stabilizing native-state interactions. Accordingly, we may expect a significant (and stabilizing) contribution from charge-charge interactions to the thermodynamic stability of thioredoxin.
The design of the T14K variant
Our electrostatic calculations on native thioredoxin (Fig. 1, upper panel) do not suggest any positions in which charge-deletion or charge-reversal mutations could likely lead to very large stability enhancement. We thus turned to consider the creation of additional favorable charge-charge interactions via the introduction of new charged groups (see Sanchez-Ruiz and Makhatadze, 2001, for a general discussion). We carried out electrostatic calculations on modeled variants of thioredoxin (see Materials and Methods for details) in which Glu or Lys residues had been substituted for surface polar residues (taken here as the polar residues with side-chain solvent accessibility >0.5). Calculations were performed for all sterically allowed rotamers of the new side chains, and the results are summarized in the lower panel of Fig. 1 as the difference between the total charge-charge interaction energy calculated for the variant and that corresponding to the WT. Again, all values for this difference are moderate, although the more promising variant appears to be T14K, which shows a calculated charge-charge interaction energy in the native state ∼2.5 kJ/mol lower than that for the WT form. This variant has been obtained and characterized in this work and it is, in fact, slightly more stable than the WT form (see further below). It is to be noted that the newly introduced Lys residue in this variant is fully exposed to the solvent: modeling based on the WT structure shows that all its rotamers are sterically allowed and our ASA calculations indicate an accessibility to the solvent of 0.81 ± 0.04 (average of the values obtained for all rotamers) when taking Gly-X-Gly tripeptides as reference (see Materials and Methods for details). Thus, comparison of the stability of the variant T14K with that of WT thioredoxin provides a suitable model system to test the efficiency of salts to screen a well-exposed charge.
Circular dichroism studies
Far-UV and near-UV CD spectra of the WT form of thioredoxin and the T14K variant under conditions in which both proteins are in the native state (pH 7, 20°C, guanidinium chloride concentrations within the 0–1.6 M range) are shown in Fig. 2. It appears that both the T14K mutation and the guanidinium chloride concentration have little effect on these native-state CD spectra.
Due to technical limitations, we could not carry out CD spectra determinations at temperatures >90°C. The spectra for the denatured states of WT and T14K shown in Fig. 2 were obtained at 85°C and in the presence of 1 M and 1.6 M guanidinium chloride (so that the denaturation temperature is clearly <70°C; see further below). The denatured-state far-UV CD spectra are similar for WT and T14K and suggestive of some kind of residual structure in the denatured state. Actually, there appears to be only a small difference between the native-state and the denatured-state far-UV CD spectra in the 210–260 nm region. On the other hand, the near-UV signal is essentially absent in the denatured state for both proteins (Fig. 2) and the ellipticity at 280 nm provides a sensitive probe to follow thermal denaturation, as is shown in Fig. 3.
FIGURE 3.

Thermal denaturation of WT thioredoxin and the T14K variant at pH 7 as followed by the ellipticity at 280 nm. The numbers alongside the denaturation profiles stand for the guanidinium chloride concentration in the solution. The solid line represents in both cases (WT and T14K) the best fit of the two-state model to the denaturation profile for 1.6 M guanidinium chloride (see Materials and Methods for details) for which both pre- and posttransition baselines are observed. From these fittings, we obtain Tm = 58°C and ΔHm = 223 kJ/mol for the WT form, and Tm = 58°C and ΔHm = 235 kJ/mol for the T14K variant. These noncalorimetric estimates are in good agreement with the calorimetric values shown in Fig. 5.
The effect of guanidinium chloride on the thermal denaturation of WT thioredoxin and the T14K variant as followed by DSC
We have carried out DSC experiments for WT thioredoxin and the T14K variant at pH 7 and in the presence of several guanidinium chloride concentrations within the 0–2 M range. The upper panel in Fig. 4 shows some representative examples of the DSC profiles obtained. Under the conditions studied, the thermal denaturation of both WT thioredoxin and the T14K variant was highly reversible. As we have previously noted (Ibarra-Molero et al., 1999b), reproducibility of the instrumental baseline recorded with guanidinium chloride solutions in the calorimetric cells is poor, a fact that prevents us from calculating protein absolute heat capacities values from the DSC thermograms obtained in the presence of this denaturant. On the other hand, baseline reproducibility is excellent for aqueous buffers (see Materials and Methods) and we found no significant effect of the T14K mutation on the absolute heat capacities of the native and thermally denatured states (results not shown), although the variant is slightly more stable than the WT form as shown by a somewhat higher value of the denaturation temperature (Fig. 4, lower panel). It must be noted that the difference in denaturation temperature between T14K and WT thioredoxin is small, but significant and reproducible: we carried out three sets of DSC experiments with the two protein forms and found the three following values for the denaturation temperature difference (ΔTm = Tm(T14K) − Tm(WT)): 1.0 K, 1.0 K, and 1.1 K.
FIGURE 4.

(Upper panel) Representative examples of the DSC profiles obtained in this work for the thermal denaturation of WT thioredoxin and the T14K variant at pH 7. The concentration of sodium chloride or guanidinium chloride is indicated. The profiles have been shifted in the y axis for display purposes. The circles are the experimental heat capacity data obtained after correcting for the instrumental baseline and normalizing to a mole of protein. The solid thin lines represent the best nonlinear, least-squares fits of the two-state equilibrium model to the experimental data. (Lower panel) Absolute heat capacity versus temperature profiles for the thermal denaturation of WT thioredoxin and the T14K variant at pH 7, in the absence of salt.
Fittings of the DSC transitions for both proteins were carried out on the basis of the two-state model. Fits were always excellent (see Fig. 4 for illustrative examples and Materials and Methods for details on the fitting procedure) and the energetic parameters derived from them were consistent with the thermal denaturation profiles determined on the basis of near-UV CD (see legend to Fig. 3 for details). The values for the denaturation temperature derived from the fittings are plotted against denaturant concentration in the upper panel of Fig. 5: as was to be expected, for both proteins the Tm value decreases sharply with increasing guanidinium chloride concentration. The lower panel of Fig. 5 shows plots of the denaturation enthalpy (values obtained from the fittings of the DSC profiles) versus denaturation temperature (Tm) for both proteins; it is to be noted that each data point in these plots belongs to a different denaturant concentration and, as a result, the slopes of such plots must not be automatically equated to the corresponding heat capacity changes (see Plaza del Pino and Sanchez-Ruiz, 1995).
The effect of guanidinium thiocyanate on the thermal denaturation of WT thioredoxin as followed by DSC
We have carried out DSC experiments for WT thioredoxin at pH 7 and in the presence of several guanidinium thiocyanate concentrations within the 0–0.8 M range. It must be noted that, for guanidinium thiocyanate concentrations >1 M, the denaturation temperature values get close to room temperature, the denaturation enthalpies become small (which implies very broad transitions) and no useful information can be derived from the DSC thermograms—hence, the comparatively narrow range of guanidinium thiocyanate concentration studied.
The statements we have made in the previous section regarding baseline reproducibility, reversibility, and the two-state fittings also apply here. The values for the denaturation temperature and the denaturation enthalpy of WT thioredoxin in the presence of guanidinium thiocyanate are given in Fig. 5.
The effect of sodium chloride on the thermal denaturation of WT thioredoxin and the T14K variant as followed by DSC
We have carried out DSC experiments for WT thioredoxin and the T14K variant at pH 7 and in the presence of several sodium chloride concentrations within the 0–2 M range (see Fig. 4 for representative examples), and fittings of the DSC profiles were carried out on the basis of the two-state model. Only a very small effect of NaCl on the DSC profiles was found. The denaturation temperature values show a rather small (and essentially linear) increase with NaCl concentration (see upper panel of Fig. 5) and the denaturation enthalpy values do not change significantly within the 0–1 M NaCl concentration range, although they appear to decrease slightly with NaCl concentration >1 M (results not shown).
DISCUSSION
Denaturant m values from DSC experiments
The slope of the plot of folding ΔG versus denaturant concentration, known as the m value (Greene and Pace, 1974; Myers et al., 1995), has found widespread application in protein folding studies. Although m values are routinely determined from chemical denaturation experiments, we showed a few years ago (Ibarra-Molero and Sanchez-Ruiz, 1996) that they can also be obtained from DSC experiments performed at different denaturant concentrations using a procedure that is straightforward and, to a large extent, model-independent. Since these “calorimetrically determined” m values play a key role in the discussion of the results reported in this work, we briefly summarize in this section some essential features about their calculation and interpretation.
We take all thermodynamic changes for an equilibrium denaturation process as functions of both temperature (T) and denaturant concentration (C); thus, the denaturation change in Gibbs energy is expressed as
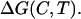 |
(2) |
The values of T and C for which ΔG = 0 (and, consequently, the equilibrium constant for the process is unity) define an equilibrium line in C versus T or T versus C plots. Note that this equilibrium line can be viewed in two entirely equivalent ways: 1), as the effect of denaturant concentration on the denaturation temperature (effect of C on Tm); and 2), as the effect of temperature on the mid-point denaturant concentration (effect of T on C1/2).
The partial derivative of ΔG with respect to C gives, by definition, the denaturant m value,
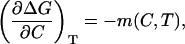 |
(3) |
which, strictly, is a function of C and T, as indicated in the right-hand side of Eq. 3. If, for a given temperature, the m value does not depend significantly on denaturant concentration, then the plot of ΔG versus C for that temperature will be linear down to C = 0 and the linear extrapolation method will be valid. Note, however, that we do not make here any assumptions regarding the denaturant concentration dependence of the m values (actually, we aim at determining such dependence from the experimental DSC data).
We will refer to the m values corresponding to the equilibrium line (i.e., to C and T conditions for which ΔG = 0) as m1/2 values. These m1/2 values can be calculated from experimental DSC profiles using the following equation (for a derivation, see Ibarra-Molero and Sanchez-Ruiz, 1996),
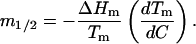 |
(4) |
Equation 4 is rigorous (for two-state equilibrium denaturation) and, we note again, is not based on the linear extrapolation approximation. The calculation of m1/2 from Eq. 4 only requires values of the denaturation enthalpy change at the Tm (equilibrium-line ΔHm values) and the effect of denaturant concentration on denaturation temperature (so that the derivative dTm/dC can be computed). In particular, the value of the denaturation heat capacity is not required.
Equation 4 can be used to calculate m1/2 values for different denaturant concentrations, provided that DSC experiments at those denaturant concentrations have been performed (of course, the m1/2 values belong to the equilibrium line and thus they correspond to different denaturant concentrations and to different temperatures; we expect, however, the effect of denaturant concentration to dominate the change of m1/2 along the equilibrium line: see further below in this Discussion for an illustration). For hen egg-white lysozyme at pH 4.5 (see Fig. 6 in Ibarra-Molero and Sanchez-Ruiz, 1996) we found that the m1/2 values were constant within the experimental scatter for guanidinium chloride concentrations >∼1 M; <1 M, however, the m1/2 values increased sharply as the denaturant concentration approached zero. These results indicate that the guanidinium chloride dependence of ΔG for lysozyme denaturation at pH 4.5 is actually linear over an extended denaturant concentration range, but also that a strong deviation from linearity occurs at low guanidinium chloride concentrations, deviation which is likely due to the screening of charge-charge interactions (for a more detailed discussion, see Ibarra-Molero and Sanchez-Ruiz, 1996, and Ibarra-Molero et al., 1999a). As we discuss below, the same general kind of behavior is found for thioredoxin.
FIGURE 6.

(Upper panel) Plot of m values versus denaturing salt concentration for WT thioredoxin (solid symbols) and the T14K variant (open symbols). Circles and triangles refer to the values calculated in this work from the analysis of DSC experiments carried out at different guanidinium chloride and guanidinium thiocyanate concentrations, respectively. The solid lines represent the best fits of Eq. 5 to the experimental data. The solid square is the m value for WT thioredoxin reported in the literature (Kelley et al., 1987; Santoro and Bolen, 1992) and derived from guanidinium chloride denaturation experiments at 25°C. (Lower panel) Temperature-dependence of the denaturation Gibbs energy changes for WT thioredoxin at pH 7 and zero guanidinium chloride concentration. The lines labeled DSC are the stability curves calculated from calorimetric data using two different assumptions for the temperature-dependence of the denaturation heat capacity change (see Materials and Methods for details): solid line, stability curve calculated using a temperature-independent ΔCP value; dashed line, stability curve calculated taking into account the temperature-dependence of ΔCP and using as the heat capacity of the denatured state the values calculated as sum of group contributions. The circles represent the ΔG values obtained from the m1/2 and C1/2 values (upper panel) using two different procedures: open circles, values calculated assuming that m is constant for each temperature (“linear extrapolation”) and using Eq. 6; solid circles, values calculated assuming that m changes with denaturant concentration as shown in the upper panel (“nonlinear extrapolation”) and using Eq. 8.
Guanidinium chloride m1/2 values for WT thioredoxin and the T14K variant
Fig. 6 (upper panel) shows the m1/2 values calculated for WT thioredoxin and the T14K variant by using Eq. 4 and the ΔHm and Tm data of Fig. 5 (the calculation of the derivative dTm/dC was based on a polynomial fitting to the Tm versus C dependence: see legend to Fig. 5 for details). Clearly, for both proteins, m1/2 changes along the equilibrium line, in particular for low denaturant concentrations (<∼1 M). It is to be noted (see Fig. 6) that the m1/2 values at high C1/2 agree with the value reported in the literature from chemical denaturation studies (Kelley et al., 1987; Santoro and Bolen, 1992). The m1/2 values for both proteins appear to be the same within the experimental scatter and can be adequately described (see Fig. 6, upper panel) by the following empirical equation:
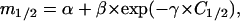 |
(5) |
with α = 13.6 ± 1.2 kJ·mol−1·M−1, β = 13.7 ± 3.6 kJ·mol−1·M−1, and γ = 2.3 ± 0.6 M−1.
It is important to note again that C1/2 changes with temperature (see Fig. 5, upper panel) and, therefore, there could be some doubt as to whether Eq. 5 actually reflects the guanidinium chloride dependence of the m1/2 values or, rather, an effect of temperature on them. This later interpretation, however, is disfavored by the following illustrative calculations (see Appendix 1 for a more formal and rigorous analysis):
Let us assume for the sake of the argument that Eq. 5 reflects exclusively a temperature effect and, consequently, that the m values are denaturant-concentration independent for any given temperature. Then, the linear extrapolation method will be valid and denaturation Gibbs energy at zero denaturant concentration can be calculated (Ibarra-Molero and Sanchez-Ruiz, 1996) as m1/2 × C1/2. That is:
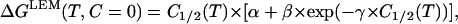 |
(6) |
where ΔGLEM(T, C = 0) is the linear-extrapolation estimate of ΔG at the temperature T and zero denaturant concentration, and C1/2(T) is the C1/2 value at the temperature T.
If, on the other hand, we assume that Eq. 5 represents the actual denaturant-concentration effect, then the temperature effect is not significant, m1/2 and C1/2 in Eq. 5 can be taken simply as m and C, and integration yields the denaturant-concentration dependence of ΔG at any temperature:
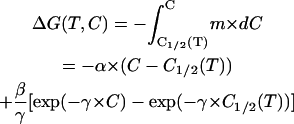 |
(7) |
and substituting C = 0 in this equation we obtain ΔG at zero denaturant concentration:
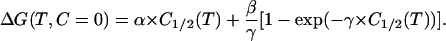 |
(8) |
Both Eqs. 6 and 8 provide denaturation ΔG values in the absence of denaturant; they, however, are based upon different assumptions. The validity of these assumptions may be assessed by comparing the ΔG values calculated on the basis of Eqs. 6 and 8 with those obtained from the DSC profile in the absence of denaturant by using standard thermodynamic procedures (see Materials and Methods for details). Such a comparison is shown in the lower panel of Fig. 6 and supports clearly the validity of Eq. 8 and its underlying assumptions.
The guanidinium chloride concentration dependence of the denaturation Gibbs energy for thioredoxin
From the above calculations, we conclude that, to an acceptable degree of approximation, the dependence of m1/2 with C1/2 shown in the upper panel of Fig. 6 (and described by Eq. 5) reflects the actual denaturant-concentration dependence of the m values. Accordingly, we are justified in using Eq. 7 to calculate the denaturant-concentration dependence of ΔG at constant temperature. The results of such calculation are given in Fig. 7 (upper panel) for the denaturation temperature of WT thioredoxin in the absence of denaturant (note that, for that temperature, C1/2 = 0). As was to be expected from the m1/2 data (Fig. 4, upper panel), the dependence of ΔG with C is linear over an extended denaturant-concentration range, but that there is a clear deviation from linearity below C ∼ 1 M, a behavior similar to that we have previously found for other protein systems (Ibarra-Molero and Sanchez-Ruiz, 1996; Ibarra-Molero et al., 1999a).
FIGURE 7.

(Upper panel) Plots of denaturation Gibbs energy at 88.8°C versus salt (NaCl or guanidinium chloride) concentration. For NaCl, the solid symbols represent the experimental data and the line is the best fit of a straight line to them. For guanidinium chloride, we show (solid line) the ΔG versus C profile calculated using Eq. 7 and based on the m1/2 versus C data of Fig. 6 (upper panel). We also show for guanidinium chloride (dashed line) the dependency given by the linear extrapolation of the high guanidinium chloride concentration data. (Middle panel) Plot of the right-hand side of Eq. 12 versus guanidinium chloride concentration for WT thioredoxin denaturation. Solid symbols refer to guanidinium chloride concentration of 1 M or higher; the solid line represents the best fit of a straight line to those data. The three values corresponding to denaturant concentration <1 M are shown with open circles to highlight deviation from linearity. (Lower panel) Effect of NaCl (solid circles) and guanidinium chloride (open circles) on the effect of the T14→K mutation on thioredoxin stability (ΔΔG = ΔG(T14K) − ΔG(WT)). The error associated with the ΔΔG value in the absence of salt (solid square) has been derived from three sets of DSC experiments with the two protein forms; we believe this error to be roughly representative of those corresponding to the ΔΔG values in the presence of salts.
The deviations from linearity at low guanidinium chloride concentrations can also be clearly detected at the level of the Tm values. Thus, from Eq. 4 the effect of guanidinium chloride concentration on the denaturation temperature is given by,
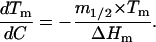 |
(9) |
As shown in the lower panel of Fig. 5, the change of the denaturation enthalpy along the equilibrium line can be adequately described as a linear dependence with the denaturation temperature:
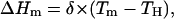 |
(10) |
with δ = 7.0 kJ·K−1·mol−1 and TH = 300 K. TH can be interpreted as the temperature at which ΔHm becomes zero for a given denaturant concentration (CH). On the other hand, we do not interpret δ as a denaturation heat capacity change, since both temperature and guanidinium chloride concentration change along the equilibrium line, and cosolvents (such as denaturants) may affect the denaturation enthalpy value (for a clear example of this, see Plaza del Pino and Sanchez-Ruiz, 1995). Substitution of Eq. 10 into Eq. 9, variable separation and integration (from {TH,CH} to any point in the equilibrium line: {Tm,C}) yields:
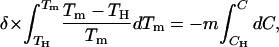 |
(11) |
where we have already introduced the linear approximation; that is, we have assumed that the denaturant m value is constant and, as such, it appears outside the integral in the right-hand side and it is denoted simply as m (rather than as m1/2). The integrals in Eq. 11 are straightforward, and the result of the integration is:
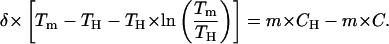 |
(12) |
For the sake of convenience, we will refer to the left-hand-side term in Eq. 12 as F(Tm). Since we know the values of δ and TH, we can calculate F(Tm) for the several experimental Tm values and construct the plot of F(Tm) versus C. According to Eq. 12, if the linear ΔG versus C dependence holds, this plot must be linear with a slope equal to minus the m value. The plot of F(Tm) versus C for WT thioredoxin is shown in the middle panel of Fig. 7. The data points corresponding to the three highest guanidinium chloride concentrations describe a straight line with a slope of −14.6 kJ·K−1·mol−1, in excellent agreement with the high-C m value we have obtained in this work (upper panel of Fig. 6) and with the m values derived from chemical denaturation experiments reported in the literature (Kelley et al., 1987; Santoro and Bolen, 1992). Note that the linear extrapolation value of F(Tm) is 35.3, consistent with a denaturation temperature of 84°C, whereas the experimental Tm value in the absence of denaturant is 88.8°C. That is, the linear extrapolation underestimates the Tm value.
There can be little doubt from all the above (upper panel in Fig. 6 and upper and middle panels in Fig. 7) that the dependence of denaturation ΔG with guanidinium chloride concentration shows a clear deviation from linearity <∼1 M denaturant, and this deviation is such that linear extrapolation from high denaturant concentration underestimates the stability of thioredoxin in the absence of denaturant (see upper and middle panels in Fig. 7). It is intriguing then that Santoro and Bolen in their 1992 work reported the opposite for thioredoxin under similar solvent conditions (pH 7): a low-C deviation from linearity in the ΔG versus C dependence, but in such a way that linear extrapolation overestimated the stability of the protein at zero denaturant concentration. The reason for this discrepancy is not clear to us; perhaps, it is related to the fact that Santoro and Bolen (1992) had to rely on a rather long constant ΔCP Gibbs-Helmholtz extrapolation to obtain the ΔG values at 25°C and low-C, as well as the fact that they had to use an estimate of ΔCP based on the temperature-dependence of ΔH values obtained by changing solvent composition. We emphasize again that the m1/2 calculation given here (based on our work in Ibarra-Molero and Sanchez-Ruiz, 1996) does not rely on Gibbs-Helmholtz extrapolations and does not require the use of a ΔCP value.
The deviation from the linear ΔG versus C relation that we find in this work cannot be explained in terms of specific binding of the denaturant ions to the native protein, since this would lead to increased values for denaturation ΔG in the absence of denaturant upon linear extrapolation from high denaturant concentration (see Mayr and Schmid, 1993, and Appendix 2), which is the opposite effect of what we observe in the upper panel of Fig. 7. On the other hand, it would seem that a decreased linear-extrapolation ΔG value (as shown in Fig. 7) could in principle be explained by the denaturant-binding model:
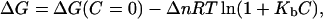 |
(13) |
where Δn is the difference in the number of binding sites between the denatured and the native states, and Kb is the binding constant. However, the denaturant-binding model is not consistent with our experimental m values for thioredoxin denaturation (upper panel in Fig. 6). Thus, using the definition of m (Eq. 3) and Eq. 13, it is straightforward to arrive at the denaturant-concentration dependence of the m value predicted by the binding model:
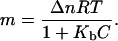 |
(14) |
Note that this equation predicts that m will approach zero upon increasing denaturant concentration, in disagreement with the behavior of the experimental m values, which show clear evidence of approaching an m value of ∼15 kJ·mol−1·M−1 at high denaturant concentration. In fact, Eq. 14 is unable to yield an acceptable fit to the experimental m versus C dependence (results not shown).
Of course, the above analyses do no exhaust all possible models of denaturant action. However, the failure of the above models to account for our experimental data does suggest that the deviation from the linear ΔG versus C dependence at low guanidinium chloride concentration is most likely associated to the screening of charge-charge interactions, a suggestion further supported by the two following facts: 1), for several proteins, sharp changes in calorimetrically determined m values at low-C are observed for guanidinium chloride-induced denaturation, but not for urea-induced denaturation (Ibarra-Molero et al., 2004); and 2), for ubiquitin, the deviations from the linear ΔG versus C dependence at low guanidinium chloride concentration were found to change sign with pH (Ibarra-Molero et al., 1999a) in the way expected for a charge-charge contribution to protein stability.
It must be noted, nevertheless, that the size of the ΔG deviation here is ∼−7 kJ/mol, significantly smaller than the Tanford-Kirkwood estimate of the total energy of charge-charge interactions in the native structure of WT thioredoxin (∼−22 kJ/mol; see also Fig. 1). Several reasons may be adduced to account for this discrepancy: i), The Tanford-Kirkwood model is indeed a very simple one and, perhaps, we should only expect qualitative or semiquantitative predictions from it. ii), Electrostatic interactions in denatured states may be significant (Pace et al., 2000; Guzman-Casado et al., 2003); thus, even if we accepted as exact the Tanford-Kirkwood value for the energy of charge-charge interactions in native thioredoxin, this value would only provide an upper limit (in absolute value) to the contribution of charge-charge interactions to the denaturation Gibbs energy. iii), Guanidinium chloride may not be able to screen out all charge-charge interactions. Actually, this possibility is supported by the analysis of the guanidinium thiocyanate effects that we describe further below in this Discussion.
The sodium chloride concentration dependence of the denaturation Gibbs energy for thioredoxin
Sodium chloride concentrations within the 0–2 M range have a very small effect on the denaturation enthalpy and the denaturation temperature for WT thioredoxin and the T14K variant (see Fig. 5), and calculation of the denaturation ΔG versus sodium chloride concentration for a temperature equal to the denaturation temperature in the absence of salt is straightforward (see Materials and Methods). Such a profile for WT thioredoxin is shown in Fig. 7. There is almost no effect of NaCl on ΔG and, in particular, there is little evidence of a sharp change below ∼1 M salt that could be associated to screening of charge-charge interactions. It appears clear then that the experimental NaCl dependence of thioredoxin stability at pH 7 does not provide evidence for significant screening of charge-charge interactions (see, however, Concluding Remarks).
The effect of the T14→K mutation on thioredoxin stability
The effect of NaCl and guanidinium chloride on the ΔΔG value for the T14→K mutation (ΔΔG = ΔG(T14K) − ΔG(WT)) can be calculated from the experimental Tm and denaturation enthalpy values using a straightforward procedure described in Materials and Methods. The results of such calculation are shown in the lower panel of Fig. 7. It appears that both salts are able to screen out most of the stabilization afforded by the T14→K mutation and, in addition, that they are roughly equally effective in this regard, possibly reflecting the fact that the lysine at position 14 in the T14K variant is well exposed to the solvent.
It is perhaps worth noting here that the stabilization afforded by the T14→K mutation in the absence of salts, as measured by ΔΔG, is ∼1 kJ/mol, that is, less than half the value predicted by the Tanford-Kirkwood calculation (∼2.5 kJ/mol; see Fig. 1). This discrepancy is approximately by the same factor than that we described above between estimate of the total charge-charge contribution to denaturation ΔG and the corresponding Tanford-Kirkwood prediction, and, probably, the same reasons (except, of course, reason iii) can be adduced to explain it (see above).
Finally, it is interesting that a ΔΔG of ∼1 kJ/mol in the absence of salt translates into an increment in denaturation temperature (ΔTm) of ∼1° only. To a good degree of approximation, mutation effects on ΔG and Tm are related through the Schellman equation (Schellman, 1987) (see Materials and Methods),
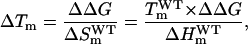 |
(15) |
where the superscript WT means wild-type value and the subscript m with ΔH and ΔS indicates that they correspond to the denaturation temperature. According to Eq. 15, the low ΔTm value is associated to comparatively high value for the denaturation enthalpy at the denaturation temperature (∼450 kJ/mol for thioredoxin). Indeed, it is well known that small proteins with low denaturation enthalpy values are more sensitive (in terms of denaturation temperature) to stabilizing effects of mutations and the environment, and often display high Tm values (Alexander et al., 1992; Ibarra-Molero et al., 2000). It is clear that the achievement of significant increments in Tm for not-so-small proteins via optimization of charge-charge interactions must rely on the cumulative effect of several mutations (for a discussion, see Sanchez-Ruiz and Makhatadze, 2001), that is, in the design of the surface charge distribution (see Ibarra-Molero and Sanchez-Ruiz, 2002).
Guanidinium thiocyanate m1/2 values for WT thioredoxin
As originally planned, this work was meant to consist in a detailed energetic characterization of the effect of sodium chloride and guanidinium chloride on thioredoxin denaturation addressed at determining the relative charge-screening efficiencies of these salts. However, since our results point to a relation between the screening efficiency and the denaturing strength, we deemed convenient to include some experimental data on the effect of guanidinium thiocyanate (a stronger denaturant than guanidinium chloride) on thioredoxin stability.
Guanidinium thiocyanate m1/2 values for WT thioredoxin denaturation were calculated in the same manner as the guanidinium chloride m1/2 values (although, in this case, a linear dependence sufficed to describe the denaturant-concentration dependence of Tm: see Fig. 5) and are shown in the upper panel of Fig. 6. Due to the small range of guanidinium thiocyanate concentration studied (see Results), we could not fully characterize the m versus C dependence. It is clear, however, that the m values for guanidinium thiocyanate are much larger than those for guanidinium chloride and that they decrease with concentration in a sharper manner, suggesting a higher charge-charge screening efficiency.
CONCLUDING REMARKS
Plots of denaturation Gibbs energy versus sodium chloride concentration (see, for instance, Perl and Schmid, 2001, and Dominy et al., 2002) often show an exponential-like dependence in the ∼0–1 M concentration range (attributed to screening of charge-charge interactions by the salt) and a gradual and almost linear dependence at higher concentrations. In the case of E. coli thioredoxin at pH 7, only a moderate linear increase in ΔG is observed upon increasing NaCl concentration; that is, our experimental data do not provide evidence for significant NaCl screening of charge-charge interactions. The simplest explanation for this is, of course, that screening does not take place in this case (since we do not find evidence for it). However, there is an alternative explanation that is, at least, plausible. Dominy et al. (2002) have pointed out that NaCl is expected to screen more efficiently long-range interactions over short-range ones, and that the former mainly correlate with the total charge of the protein (Dominy et al., 2002; Zhou and Dong, 2003), which, for thioredoxin at pH 7, is ∼−5 units. We might conceive then that unequal screening of short-range (predominantly stabilizing) interactions and long-range (predominantly destabilizing) interactions may perhaps yield a close-to-zero Gibbs energy balance in the low-NaCl concentration range, in such a way that screening would not be apparent in the NaCl dependence of the thermodynamic stability.
Our results support a relation between screening and the accessibility to solvent of the charged groups. Thus, 1 M NaCl (as well as 1 M guanidinium chloride) does appear to screen significantly the interactions of a well-exposed Lys group in a T14K variant of thioredoxin. It must be noted that Lys-14 in this variant has an accessibility to solvent (0.81, taking a Gly-Lys-Gly peptide as reference) higher than that for the charged groups involved in strongly stabilizing interactions in WT thioredoxin (the accessibilities of K3, K18, K36, K57, K69, K82, K90, K96, and K100 in the native structure of WT thioredoxin are 0.34, 0.67, 0.59, 0.19, 0.58, 0.39, 0.41, 0.37, and 0.41, respectively).
Finally, but most important, this work suggest that efficiency of the studied salts to screen charge interactions follows the order guanidinium thiocyanate > guanidinium chloride > sodium chloride, which matches the order of denaturing strength for these salts. A higher screening efficiency for denaturing salts (as compared with nondenaturing or stabilizing salts) is to be expected from the fact that denaturants tend to interact preferentially with protein surfaces, whereas stabilizers tend to be preferentially excluded. (We would like to emphasize that we are not proposing that the denaturing effect of certain salts is due to its charge-screening character but, rather, that the screening efficiency is higher for denaturing salts. Whether the charge-screening effect enhances or reduces the denaturing effect may depend on the balance of the screened charge-charge interactions: predominantly stabilizing (dominated by interactions between unlike charges in the native state, for instance) or predominantly destabilizing (dominated by interactions between like charges in the native state, for instance)). In fact, it has been proposed (Courtenay et al., 2001) that the Hofmeister ranking of cations and anions originate in the extent of accumulation or exclusion of the solute from the protein surface (note, for instance, that SCN− is higher than Cl− in the Hofmeister ranking for anions). This suggests, therefore, that the efficiency of different salts to screen charge-charge interactions in proteins may also reflect the position of the constituent ions in the Hofmeister series.
Acknowledgments
We thank Dr. Maria Luisa Tasayco for advice regarding thioredoxin expression and purification.
We acknowledge financial support from the Spanish Ministry of Science and Technology (grant BIO2000-1437). Raul Perez-Jimenez is a recipient of a predoctoral fellowship from the Spanish Ministry of Science and Technology.
APPENDIX 1: ON THE DETERMINATION OF THE EFFECT OF DENATURANT CONCENTRATION ON m VALUES
The m values determined in this work (see section “Denaturant m values from DSC experiments”) are m1/2 values that belong to the C-T equilibrium line (defined by the {C,T} couples for which the denaturation change in Gibbs energy is zero). Therefore, there could be some concern that the denaturant-concentration effect on the m values shown in Fig. 6 might actually reflect a temperature effect. In the main text (see section “Guanidinium chloride m1/2 values for WT thioredoxin and the T14K variant”), we have carried out a simple and intuitive analysis that suggests that the temperature effect on m values can be neglected in this case. In this appendix, we approach the issue in a more formal and rigorous way.
The effect of denaturant concentration on calorimetrically determined m values can be described by the following derivative:
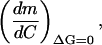 |
(A1,1) |
where the subscript ΔG = 0 means that the effect of C on m is computed along the C-T equilibrium line (so that T is changing with C to keep ΔG = 0). Actually we are interested in the derivative,
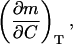 |
(A1,2) |
which describes the deviations from the linear ΔG versus C dependence at a given temperature. That is, if ΔG changes linearly with C, m is constant (independent of C) and the derivative A1,2 is zero. Consequently, deviations from linearity are signaled by values of (∂m/∂C)T significantly different from zero.
The two derivatives (A1,1 and A1,2) can be easily related using the well-known mathematics of partial differentiation (Blinder, 1966):
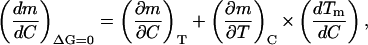 |
(A1,3) |
where we have used that the slope of the equilibrium line [(∂T/∂C)ΔG = 0] is, in fact, the denaturant-concentration effect on denaturation temperature ((dTm/dC)).
Our problem here is the evaluation of the second term on the right-hand side of A1,3, since we do not know the value of the derivative (∂m/∂T)C. However, the denaturation Gibbs energy change is function of both C and T (Eq. 2) and its two second cross-derivatives must be equal:
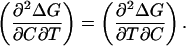 |
(A1,4) |
Now, since m and the denaturation entropy change are first derivatives of ΔG (ΔS = −(∂ΔG/∂T)C and m is defined by Eq. 3), Eq. A1,4 reduces to the following linkage relationship,
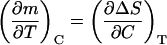 |
(A1,5) |
and Eq. A1,3 becomes,
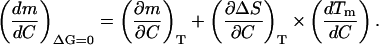 |
(A1,6) |
Values of ΔS can be easily calculated as ΔHm/Tm, but they will correspond to the equilibrium line. That is, the slope of a plot of ΔS (=ΔHm/Tm) versus Tm (with the different Tm values achieved by changing denaturant concentration) is actually the derivative,
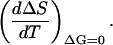 |
(A1,7) |
Again, a convenient expression for this derivative can be obtained using the known mathematics of partial differentiation (Blinder, 1966):
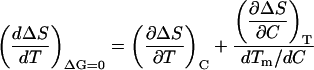 |
(A1,8) |
or, solving for (∂ΔS/∂C)T,
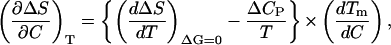 |
(A1,9) |
where we have already used the thermodynamic relation between heat capacity changes and temperature effects on entropy changes: (∂ΔS/∂T)C = ΔCP/T.
Substituting A1,9 into A1,6 and solving for (∂m/∂C)T, we obtain,
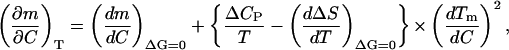 |
(A1,10) |
which, as we elaborate below, allows us to calculate the desired derivative, (∂m/∂C)T, in the limit of low denaturant concentration (strictly, in the C→0 limit).
The second term on the right-hand side of A1,10 is evaluated as follows. First, a plot of ΔS (calculated as ΔHm/Tm) versus Tm, including data corresponding to the 0–1 M guanidinium chloride concentration range, is linear within the experimental scatter (results not shown); the slope of this plot (1.71 × 10−2 ± 0.06 × 10−2 kJ·K−2·mol−1) provides then an estimate of the derivative (dΔS/dT)ΔG = 0 in the low denaturant concentration region. Several determinations of ΔCP for thioredoxin denaturation in the absence of denaturant (the C→0 limit) were reported by Georgescu et al. (2001); the average value is 6.1 ± 0.4 kJ·K−1/mol−1, and using as temperature value the Tm in the absence of denaturant, we get 1.69 × 10−2 ± 0.11 × 10−2 kJ·K−2·mol−1 for ΔCP/T. Therefore, the bracket 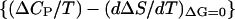 in A1,10 turns out to be zero within an uncertainty of ∼±0.13 × 10−2 kJ·K−2·mol−1. Finally, the derivative dTm/dC at low denaturant concentration can be calculated from the data shown in Fig. 5. The result is −18.3 ± 2.7 K·M−1 from the linear fitting of Tm versus C in the 0–1 M denaturant concentration range. Using these values, the second term on the right-hand side of A1,10 can be easily estimated to be zero within an approximate uncertainty of ±0.6 kJ·mol−1·M−2.
in A1,10 turns out to be zero within an uncertainty of ∼±0.13 × 10−2 kJ·K−2·mol−1. Finally, the derivative dTm/dC at low denaturant concentration can be calculated from the data shown in Fig. 5. The result is −18.3 ± 2.7 K·M−1 from the linear fitting of Tm versus C in the 0–1 M denaturant concentration range. Using these values, the second term on the right-hand side of A1,10 can be easily estimated to be zero within an approximate uncertainty of ±0.6 kJ·mol−1·M−2.
The derivative (dm/dC)ΔG = 0 at low guanidinium chloride concentration can be easily estimated from the m1/2 data shown in Fig. 6 as being of the order of −30 kJ·mol−1·M−2 . It is clear then that the second term on the right-hand side of A1,10 (0 ± 0.6 kJ·mol−1·M−2) is not significant compared with (dm/dC)ΔG = 0. We conclude that
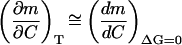 |
(A1,11) |
at low guanidinium chloride concentration and, therefore, that the sharp decrease in m value observed at low C (Fig. 6) reflects the actual denaturant-concentration dependence of m and reveals that the ΔG versus C dependence is clearly nonlinear at low denaturant concentrations.
The above illustrative calculations correspond to guanidinium chloride-induced denaturation. However, the same general conclusion (Eq. A1,11) also holds for guanidinium thiocyanate denaturation (calculations not shown).
APPENDIX 2: ON THE DEVIATIONS FROM THE LINEAR ΔG VERSUS DENATURANT-CONCENTRATION DEPENDENCE CAUSED BY SPECIFIC DENATURANT BINDING TO THE NATIVE STATE
Specific binding of denaturant molecules (or ions) to the native state is expected to cause deviations from the linear ΔG versus C dependence, in such a way that linear extrapolation from high denaturant concentration will overestimate the value of the denaturation Gibbs energy change at zero denaturant concentration. The reason is that, regardless of its structural consequences, specific binding to the native state will always have stabilizing effect (i.e., it will shift the denaturation equilibrium toward the native state). Thus, at low denaturant concentration, the stabilizing effect of the specific denaturant binding will cancel to some extent the denaturing effect, giving rise to a smaller value of m. In this appendix, we demonstrate these ideas with the analysis of a simple binding model.
We assume that the native protein can exist as a ligated form (NL) and a nonligated form (N), where the “ligand” (L) is actually a denaturant molecule or ion. We further assume that the binding,
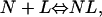 |
(A2,1) |
being comparatively strong, specific, and stoichiometric, can be described by an equilibrium constant of the following form:
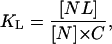 |
(A2,2) |
where C is the ligand (i.e., denaturant) concentration.
Since we are interested in the deviations from linearity that result from binding (Eq. A2,1) we take, as a starting point of our analysis, that the Gibbs energy change for the denaturation of the nonligated protein does change linearly with denaturant concentration:
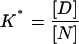 |
(A2,3) |
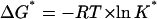 |
(A2,4) |
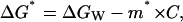 |
(A2,5) |
where we are using asterisks to designate parameters belonging to the denaturation of the nonligated protein, ΔGW is the denaturation Gibbs energy change in the absence of denaturant, and m* is taken to be constant (i.e., independent of denaturant concentration).
However, the denaturation equilibrium constant we can determine from experiments is not K*, but,
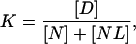 |
(A2,6) |
which includes the total concentration of native protein (ligated plus nonligated). Accordingly, the experimentally accessible denaturation Gibbs energy change is
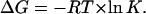 |
(A2,7) |
The relation between ΔG and ΔG* can be easily found as follows. From Eq. A2,2 we have that [NL] = KL × [N] × C, so that K (Eq. A2,6) can be written as K*/(1 + KL × C) and ΔG (Eq. A2,7) as
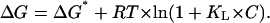 |
(A2,8) |
The expression for m is then obtained easily from its definition (Eq. 3):
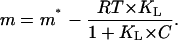 |
(A2,9) |
For high C, the second term on the right-hand side of A2,9 approaches zero and the m value for high denaturant concentration (strictly, in the C→∞ limit) is equal to m*. For C = 0, on the other hand, m becomes equal to m* − RT × KL, which is smaller than the value at high denaturant concentration. In general, Eq. A2,9 predicts that m increases with increasing denaturant concentration; accordingly, deviations from the linear ΔG versus C dependence are such that linear extrapolation will overestimate the value of the denaturation Gibbs energy in the absence of denaturant. This conclusion can also be directly verified as follows:
Let C′ be the denaturant concentration at which ΔG is zero at the temperature of interest. Assume that we have carried out a “traditional” chemical denaturation experiment from which we can determine the value of C′ and the effect of denaturant concentration on ΔG in the neighborhood of C′; that is, we determine C′ and the value of m corresponding to C′: m(C′). The linear extrapolation estimate of ΔG in the absence of denaturant would then be (see Ibarra-Molero and Sanchez-Ruiz, 1996):
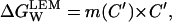 |
(A2,10) |
and using A2,9 for m(C′):
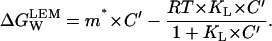 |
(A2,11) |
Applying Eq. A2,5 to the concentration C′ we have:
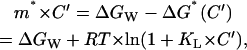 |
(A2,12) |
where we have also used for ΔG*(C′) the expression given by Eq. A2,8 (for C = C′, ΔG = 0, and ΔG*(C′) = −RT × ln(1+KL·C′)). Substituting A2,12 into A2,11 we obtain:
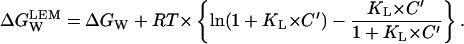 |
(A2,13) |
The difference in brackets in the above equation is necessarily a positive number (ln(1 + x) is always larger than x/(1 + x) for any positive value of x; here x = KL × C′). We conclude, therefore that,
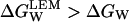 |
(A2,14) |
and linear extrapolation overestimates the value of the denaturation Gibbs energy change in the absence of denaturant.
References
- Acevedo, O., M. Guzman-Casado, M. M. Garcia-Mira, B. Ibarra-Molero, and J. M. Sanchez-Ruiz. 2002. pH corrections in chemical denaturant solutions. Anal. Biochem. 306:158–161. [DOI] [PubMed] [Google Scholar]
- Alexander, P., S. Fahnestock, T. Lee, J. Orban, and P. Bryan. 1992. Thermodynamic analysis of the folding of the streptococcal protein G IgG-binding domains B1 and B2: why small proteins tend to have high denaturation temperatures. Biochemistry. 31:3597–3603. [DOI] [PubMed] [Google Scholar]
- Arakawa, T., and S. N. Timasheff. 1984. Protein stabilization and destabilization by guanidinium salts. Biochemistry. 23:5924–5929. [DOI] [PubMed] [Google Scholar]
- Arakawa, T., and S. N. Timasheff. 1985. The stabilization of proteins by osmolytes. Biophys. J. 47:411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin, R. L. 1996. How Hofmeister ions affect protein stability. Biophys. J. 71:2056–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinder, S. M. 1966. Mathematical methods in elementary thermodynamics. J. Chem. Ed. 43:85–92. [Google Scholar]
- Bolen, D. W., and I. V. Baskarov. 2001. The osmophobic effect: natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 310:955–963. [DOI] [PubMed] [Google Scholar]
- Bolen, D. W., and M. Yang. 2000. Effects of guanidine hydrochloride on the proton inventory of proteins: implications for the interpretations of protein stability. Biochemistry. 39:15208–15216. [DOI] [PubMed] [Google Scholar]
- Chothia, C. 1976. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 105:1–12. [DOI] [PubMed] [Google Scholar]
- Courtenay, E. S., M. W. Capp, and M. T. Record Jr. 2001. Thermodynamics of interactions of urea and guanidinium salts with protein surface: relationship between solute effects on protein stability and changes in water-accessible surface area. Protein Sci. 10:2484–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy, B. N., D. Perl, F. X. Schmid, and C. L. Brooks 3rd. 2002. The effects of ionic strength on protein stability: the cold-shock protein family. J. Mol. Biol. 319:541–554. [DOI] [PubMed] [Google Scholar]
- Garcia-Mira, M. M., and J. M. Sanchez-Ruiz. 2001. pH corrections and protein ionization in water/guanidinium chloride. Biophys. J. 81:3489–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu, R. E., M. M. Garcia-Mira, M. L. Tasayco, and J. M. Sanchez-Ruiz. 2001. Heat capacity of oxidized Escherichia coli thioredoxin fragments (1–73, 74–108) and their noncovalent complex. Evidence for burial of apolar surface in protein unfolded states. Eur. J. Biochem. 268:1477–1485. [DOI] [PubMed] [Google Scholar]
- Greene, R. F., and C. N. Pace. 1974. Urea and guanidinium chloride denaturation of ribonuclease, α-chymotrypsin, and β-lactoglobulin. J. Biol. Chem. 249:5388–5393. [PubMed] [Google Scholar]
- Grimsley, G. R., K. L. Shaw, L. R. Fee, R. W. Alston, B. M. Huyghues-Despointes, R. L. Thurlkill, J. M. Scholtz, and C. N. Pace. 1999. Increasing protein stability by altering long-range coulombic interactions. Protein Sci. 8:1843–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman-Casado, M., A. Parody-Morreale, S. Robric, S. Marqusee, and J. M. Sanchez-Ruiz. 2003. Energetic evidence for formation of a pH-dependent hydrophobic cluster in the denatured state of thermus thermophilus ribonuclease H. J. Mol. Biol. 329:731–743. [DOI] [PubMed] [Google Scholar]
- Hagihara, Y., S. Aimoto, A. L. Fink, and Y. Goto. 1993. Guanidine hydrochloride-induced folding of proteins. J. Mol. Biol. 231:180–184. [DOI] [PubMed] [Google Scholar]
- Holmgren, A., and P. Reichard. 1967. Thioredoxin 2: cleavage with cyanogen bromide. Eur. J. Biochem. 2:187–196. [DOI] [PubMed] [Google Scholar]
- Ibarra-Molero, B., V. V. Loladze, G. I. Makhatadze, and J. M. Sanchez-Ruiz. 1999a. Thermal versus guanidine-induced unfolding of ubiquitin. Analysis in terms of the contributions from charge-charge interactions to protein stability. Biochemistry. 38:8138–8149. [DOI] [PubMed] [Google Scholar]
- Ibarra-Molero, B., G. I. Makhatadze, and J. M. Sanchez-Ruiz. 1999b. Cold denaturation of ubiquitin. Biochim. Biophys. Acta. 1429:384–390. [DOI] [PubMed] [Google Scholar]
- Ibarra-Molero, B., R. Perez-Jimenez, R. Godoy-Ruiz, and J. M. Sanchez-Ruiz. 2004. Linkage between temperature and chemical denaturant effects on protein stability: the interpretation of calorimetrically determined m values. In Biocalorimetry II: Applications of Calorimetry in the Biological Sciences. M. Doyle and J. Ladbury, editors. John Wiley & Sons, New York. In press.
- Ibarra-Molero, B., I. M. Plaza del Pino, B. Souhail, H. O. Hammou, and J. M. Sanchez-Ruiz. 2000. The sarcosine effect on protein stability: a case of nonadditivity? Protein Sci. 9:820–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra-Molero, B., and J. M. Sanchez-Ruiz. 1996. A model-independent, nonlinear extrapolation procedure for the characterization of protein folding energetics from solvent-denaturation data. Biochemistry. 35:14689–14702. [DOI] [PubMed] [Google Scholar]
- Ibarra-Molero, B., and J. M. Sanchez-Ruiz. 1997. Are there equilibrium intermediate states in the urea-induced unfolding of hen egg-white lysozyme? Biochemistry. 36:9616–9624. [DOI] [PubMed] [Google Scholar]
- Ibarra-Molero, B., and J. M. Sanchez-Ruiz. 2002. A genetic algorithm to design stabilizing surface charge distributions in proteins. J. Phys. Chem. B. 106:6609–6613. [Google Scholar]
- Irun, M. P., M. M. Garcia-Mira, J. M. Sanchez-Ruiz, and J. Sancho. 2001. Native hydrogen bonds in a molten globule: the apoflavodoxin thermal intermediate. J. Mol. Biol. 306:877–888. [DOI] [PubMed] [Google Scholar]
- Kao, Y. H., C. A. Fitch, S. Bhattacharya, C. J. Sarkisian, J. T. Lecomte, and B. E. Garcia-Moreno. 2000. Salt effects on ionization equilibria of histidines in myoglobin. Biophys. J. 79:1637–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, R. F., W. Shalongo, M. V. Jagannadham, and E. Stellwagen. 1987. Equilibrium and kinetic measurements of the conformational transition of reduced thioredoxin. Biochemistry. 26:1406–1411. [DOI] [PubMed] [Google Scholar]
- Ladbury, J. E., R. Wynn, H. W. Hellinga, and J. M. Sturtevant. 1993. Stability of oxidized Escherichia coli thioredoxin and its dependence on protonation of the aspartic acid residue in the 26 position. Biochemistry. 32:7526–7530. [DOI] [PubMed] [Google Scholar]
- Laurents, D. V., B. M. Huyghues-Despointes, M. Bruix, R. L. Thurlkill, D. Schell, S. Newsom, G. R. Grimsley, K. L. Shaw, S. Trevino, M. Rico, J. M. Briggs, J. M. Antosiewicz, J. M. Scholtz, and C. N. Pace. 2003. Charge-charge interactions are key determinants of the pK values of ionizable groups in ribonuclease Sa (pI = 3.5) and a basic variant (pI = 10.2). J. Mol. Biol. 325:1077–1092. [DOI] [PubMed] [Google Scholar]
- Lee, L. P., and B. Tidor. 2001. Optimization of binding electrostatics: charge complementarity in the barnase-barstar complex. Protein Sci. 10:362–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loladze, V. V., B. Ibarra-Molero, J. M. Sanchez-Ruiz, and G. I. Makhatadze. 1999. Engineering a thermostable protein via optimization of charge-charge interactions on the protein surface. Biochemistry. 38:16419–16423. [DOI] [PubMed] [Google Scholar]
- Luisi, D. L., C. D. Snow, J. J. Lin, Z. S. Hendsch, B. Tidor, and D. P. Raleigh. 2003. Surface salt bridges, double-mutant cycles, and protein stability: an experimental and computational analysis of the interaction of the Asp 23 side chain with the N-terminus of the N-terminal domain of the ribosomal protein L9. Biochemistry. 42:7050–7060. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G. I., V. V. Loladze, D. M. Ermolenko, X. Chen, and S. T. Thomas. 2003. Contributions of surface salt bridges to protein stability: guidelines for protein engineering. J. Mol. Biol. 327:1135–1148. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G. I., M. M. Lopez, J. M. Richardson 3rd, and S. T. Thomas. 1998. Anion binding to the ubiquitin molecule. Protein Sci. 7:689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhatadze, G. I., and P. L. Privalov. 1990. Heat capacity of proteins. I. Partial molar heat capacity of individual amino acid residues in aqueous solution: hydration effect. J. Mol. Biol. 213:375–384. [DOI] [PubMed] [Google Scholar]
- Makhatadze, G. I., and P. L. Privalov. 1992. Protein interactions with urea and guanidinium chloride. A calorimetric study. J. Mol. Biol. 226:491–505. [DOI] [PubMed] [Google Scholar]
- Marshall, S. A., C. S. Morgan, and S. L. Mayo. 2002. Electrostatics significantly affect the stability of designed homeodomain variants. J. Mol. Biol. 316:189–199. [DOI] [PubMed] [Google Scholar]
- Martin, A., I. Kather, and F. X. Schmid. 2002. Origins of the high stability of an in vitro-selected cold-shock protein. J. Mol. Biol. 318:1341–1349. [DOI] [PubMed] [Google Scholar]
- Mayr, L. M., and F. X. Schmid. 1993. Stabilization of a protein by guanidinium chloride. Biochemistry. 32:7994–7998. [DOI] [PubMed] [Google Scholar]
- Monera, O. D., C. M. Kay, and R. S. Hodges. 1994. Protein denaturation with guanidine hydrochloride or urea provides a different estimate of stability depending on the contributions of electrostatic interactions. Protein Sci. 3:1984–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, J. K., C. N. Pace, and J. M. Scholtz. 1995. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4:2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohaile, M. J., Z. S. Hendsch, B. Tidor, and R. T. Sauer. 2001. Altering dimerization specificity by changes in surface electrostatics. Proc. Natl. Acad. Sci. USA. 98:3109–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C. N. 2000. Single surface stabilizer. Nat. Struct. Biol. 7:345–346. [DOI] [PubMed] [Google Scholar]
- Pace, C. N., R. W. Alston, and K. L. Shaw. 2000. Charge-charge interactions influence the denatured state ensemble and contribute to protein stability. Protein Sci. 9:1395–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C. N., B. M. Huyghues-Despointes, J. M. Briggs, G. R. Grimsley, and J. M. Scholtz. 2002. Charge-charge interactions are the primary determinants of the pK values of the ionizable groups in ribonuclease T1. Biophys. Chem. 101–102:211–219. [DOI] [PubMed] [Google Scholar]
- Pace, C. N., D. V. Laurents, and J. A. Thomson. 1990. pH dependence of the urea and guanidine hydrochloride denaturation of ribonuclease A and ribonuclease T1. Biochemistry. 29:2564–2572. [DOI] [PubMed] [Google Scholar]
- Pace, C. N., B. A. Shirley, and J. A. Thomson. 1989. Measuring the conformational stability of a protein. In Protein Structure, a Practical Approach. T. E. Creighton, editor. IRL Press at Oxford University Press, Oxford. 311–330.
- Perl, D., U. Mueller, U. Heinemann, and F. X. Schmid. 2000. Two exposed amino acid residues confer thermostability on a cold shock protein. Nat. Struct. Biol. 7:380–383. [DOI] [PubMed] [Google Scholar]
- Perl, D., and F. X. Schmid. 2001. Electrostatic stabilization of a thermophilic cold shock protein. J. Mol. Biol. 313:343–357. [DOI] [PubMed] [Google Scholar]
- Perutz, M. F., A. M. Gronenborn, G. M. Clore, J. H. Fogg, and D. T. Shih. 1985. The pKa values of two histidine residues in human haemoglobin, the Bohr effect, and the dipole moments of α-helices. J. Mol. Biol. 183:491–498. [DOI] [PubMed] [Google Scholar]
- Plaza del Pino, I. M., and J. M. Sanchez-Ruiz. 1995. An osmolyte effect on the heat capacity change for protein folding. Biochemistry. 34:8621–8630. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ruiz, J. M., and G. I. Makhatadze. 2001. To charge or not to charge? Trends Biotechnol. 19:132–135. [DOI] [PubMed] [Google Scholar]
- Santoro, M. M., and D. W. Bolen. 1988. Unfolding free energy changes determined by the linear extrapolation method. I. Unfolding of phenyl-methanesulfonyl α-chymotypsin using different denaturants. Biochemistry. 27:8063–8068. [DOI] [PubMed] [Google Scholar]
- Santoro, M. M., and D. W. Bolen. 1992. A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry. 31:4901–4907. [DOI] [PubMed] [Google Scholar]
- Schellman, J. A. 1987. The thermodynamic stability of proteins. Annu. Rev. Biophys. Biophys. Chem. 16:115–137. [DOI] [PubMed] [Google Scholar]
- Shrake, A., and J. A. Rupley. 1973. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 79:351–372. [DOI] [PubMed] [Google Scholar]
- Spector, S., M. Wang, S. A. Carp, J. Robblee, Z. S. Hendsch, R. Fairman, B. Tidor, and D. P. Raleigh. 2000. Rational modification of protein stability by the mutation of charged surface residues. Biochemistry. 39:872–879. [DOI] [PubMed] [Google Scholar]
- Sundd, M., N. Iverson, B. Ibarra-Molero, J. M. Sanchez-Ruiz, and A. D. Robertson. 2002. Electrostatic interactions in ubiquitin: stabilization of carboxylates by lysine amino groups. Biochemistry. 41:7586–7596. [DOI] [PubMed] [Google Scholar]
- Tanford, C. N., and J. G. Kirkwood. 1957. Theory of protein titration curves. I. General equations for impenetrable spheres. J. Am. Chem. Soc. 79:5333–5339. [Google Scholar]
- Yu, Y., O. D. Monera, R. S. Hodges, and P. L. Privalov. 1996. Ion pairs significantly stabilize coiled-coils in the absence of electrolyte. J. Mol. Biol. 255:367–372. [DOI] [PubMed] [Google Scholar]
- Zhou, H. X., and F. Dong. 2003. Electrostatic contributions to the stability of a thermophilic cold shock protein. Biophys. J. 84:2216–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]