

Abstract
The deactivation of the bovine G-protein-coupled receptor, rhodopsin, is a two-step process consisting of the phosphorylation of specific serine and threonine residues in the cytoplasmic tail of rhodopsin by rhodopsin kinase. Subsequent binding of the regulatory protein arrestin follows this phosphorylation. Previous results find that at least three phosphorylatable sites on the rhodopsin tail (T340) and at least two of the S338, S334, or S343 sites are needed for complete arrestin-mediated deactivation. Thus, to elucidate the details of the interaction between rhodopsin with arrestin, we have employed both a computational and an in vitro experimental approach. In this work, we first simulated the interaction of the carboxy tail of rhodopsin with arrestin using a Monte Carlo simulated annealing method. Since at this time phosphorylation of specific serines and threonines is not possible in our simulations, we substitute either aspartic or glutamic acid residues for the negatively charged phosphorylated residues required for binding. A total of 17 simulations were performed and analysis of this shows specific charge-charge interactions of the carboxy tail of rhodopsin with arrestin. We then confirmed these computational results with assays of comparable constructed rhodopsin mutations using our in vitro assay. This dual computational/experimental approach indicates that sites S334, S338, and T340 in rhodopsin and K14 and K15 on arrestin are indeed important in the interaction of rhodopsin with arrestin, with a possible weaker S343 (rhodopsin)/K15 (arrestin) interaction.
INTRODUCTION
The extracellular signaling of hormones, light, and odorants is facilitated by a large family of G-protein-coupled receptors. Specifically, for phototransduction in mammalian rod photoreceptors, this process involves the visual pigment apoprotein, rod opsin, and the chromophore, 11-cis-retinal. Initiation of the phototransduction cascade occurs by isomerization of the chromophore from 11-cis to all-trans leading to activation of the receptor (Molday, 1998; Jacobs, 1998; Sakmar et al., 2002). Once the signaling cascade has been activated, the receptor must then be quickly inactivated. Specifically for the photopigment rhodopsin, deactivation is initiated by a receptor-specific serine/threonine kinase, rhodopsin kinase (GRK1), which phosphorylates residues on the cytoplasmic tail and hence initiates partial deactivation. Complete deactivation of the receptor does not occur until a second regulatory protein, arrestin, binds. The interaction between receptor and arrestin results in the fast quenching of the signal transduction by decreasing the coupling of the receptor to the G-protein, transducin (Alloway and Dolph, 1999; Wilden et al., 1986; Lohse et al., 1990, 1992).
Phosphorhodopsin induces a large conformational change in arrestin which shifts this protein from an inactive, low-affinity binding state to an active high-affinity binding state, where there is a fivefold increase of the affinity of arrestin for phosphorhodopsin (Kuhn et al., 1986). Mutagenic studies have elucidated three regions of arrestin that respond to different states of rhodopsin (inactive, active, and phosphorylated; Gurevich, 1998). It is the interaction between phosphorylated rhodopsin residues and the arrestin protein that is of interest to our studies.
The cytoplasmic tail of rhodopsin contains seven possible phosphorylation sites. In vivo, the identity and number of phosphate groups incorporated into the tail is still under investigation. Several studies have found evidence that up to three phosphates are incorporated into the cytoplasmic tail of rhodopsin (Zhang et al., 1997; Kennedy et al., 2001a; Ablonczy et al., 2002; McDowell et al., 2001), and site-specific mutagenesis indicates that at least two rhodopsin phosphorylation sites are required for full desensitization (McDowell et al., 2001), whereas studies with transgenic mice indicate at least three phosphorylated resides are required (Mendez et al., 2000). Candidate phosphorylation sites include serine 334, serine 338, threonine 340, and serine 343 (Brannock et al., 1999; McDowell et al., 2001). The mechanism that these phosphorylated residues play in the desensitization process is still under investigation; however, it is believed that they will initially interact with arrestin via a charge-charge mechanism (Vishnivetskiy et al., 2000).
Therefore, our study aims to determine specific site-site interactions between the cytoplasmic tail of rhodopsin and arrestin using both computational methods and in vitro biochemical assays. For our approach, we draw on previous studies that have examined the regions necessary for this interaction. Specific crystallographic studies of bovine arrestin have elucidated regions that may respond to the phosphorylated residues on rhodopsin. The crystal structure of the inactive conformation of arrestin suggest that it is composed of two β-sheets oriented in such a way as to create a hydrophobic core between them (Gray-Keller et al., 1997). Within this hydrophobic core there exist at least two salt bridges which act to stabilize the low-affinity binding state (Hirsch et al., 1999). During activation, the phosphorylated residues are believed to penetrate into the hydrophobic core and thereby disrupt these salt bridges which then induces the conformational change in arrestin to achieve an active high-affinity binding state (Vishnivetskiy et al., 2000, 1999; Gurevich, 1998; Palczewski et al., 2001).
The initial interaction between rhodopsin and arrestin, however, is not well understood. Vishnivetskiy et al. (2000) proposed that it might be a charge-charge interaction between two key positively charged lysine residues (Lys14 and Lys15) on arrestin, interacting with the negatively charged phosphorylated residues of rhodopsin. This charge-charge interaction is thought to guide the rhodopsin into the polar core of the arrestin protein and thus allow for arrestin's conformational change. At present, this mechanism is still hypothetical; however, it is based on site-specific mutation studies of arrestin (K14A and K15A) that resulted in a nearly eightfold reduction of rhodopsin binding (Vishnivetskiy et al., 2000).
It is the aim of this study to investigate the desensitization process in G-protein-coupled receptors, from both a computational and experimental prospective. In particular, we will study the rod-specific visual pigment, rhodopsin, in complex with visual arrestin as a model system for this process. We should point out that any simulation of phosphorylated rhodopsin must contend with the fact that a molecular force field to describe specific phosphorylated residues does not exist at this time. To address this issue, we will perform key mutation studies to model the negatively charged phosphorylated residues as either aspartic or glutamic acid. Therefore, our simulations are only directly comparable to the experimental work of McDowell et al. (2001) and the current experiments. We stress that although this computational work is of peptide binding to arrestin, it is believed that the larger rhodopsin-arrestin complex will interact in a similar fashion as the peptide-arrestin complex (McDowell et al., 2001; Puig et al., 1995). To the best of our knowledge, this investigations represent the first such simulation of the desensitization process. Recently, Orry and co-workers have begun a study of identifying ligand binding sites in G-protein-coupled receptors using a novel docking approach (Cavasotto et al., 2003; Orry and Wallace, 2000). Our approach is slightly different in that we are looking at specific protein-protein interactions, allowing for full conformational flexibility of both proteins. Therefore, our approach is not suitable for a full screening of drug assays, but is meant to simulate possible binding events in the desensitization process.
EXPERIMENTAL MATERIALS AND METHODS
Construction, expression, and reconstitution of rhodopsin mutants
A synthetic bovine rhodopsin gene was cloned into a bacterial eukaryotic expression vector (pMT3), which is a modified form of the pMT2 vector. Mutations were introduced in the synthetic bovine rhodopsin gene using the QuickChange site-directed mutagenesis protocol (Stratagene, La Jolla, CA) and were verified by sequencing. Rhodopsin mutants were transiently expressed in COS-7 mammalian cells through transfection using LipofectAMINE (Invitrogen, Carlsbad, CA) reagent. Cells were harvested 48 h post-transfection and the cell membranes were isolated by sucrose density-gradient centrifugation as described previously in Weiss et al. (1994). Rhodopsin concentrations of membrane preparations were approximated by Western blot using mAb 4D2, an antibody against the N-terminus of bovine rhodopsin (antibody was a gift from R. Molday). Blots were quantitated using ImageQuant (Molecular Dynamics, Sunnyvale, CA) software. The concentration of rhodopsin in membrane preparations varied between 50 and 100 nM. The samples were stored at −80°C until ready for use. The 50-μL rhodopsin samples were reconstituted with 360 μM 11-cis retinal at 4°C for 1 h on a rotating plate.
Purification of arrestin from bovine retinas
Arrestin was purified from dark-adapted bovine retinas according to the method described by Hargrave (1986) and the samples were stored at −80°C.
Purification of transducin from bovine rod outer segments
Transducin was purified as described by Wessling-Resnick and Johnson (1987), and the protein was stored in 50% glycerol at −20°C.
Expression and purification of rhodopsin kinase
Rhodopsin kinase was expressed in High-Five insect cells (Invitrogen). A viral construct containing the rhodopsin kinase gene was a gift from K. Palczewski. High-Five cells were grown as a suspension culture to a cell density of 1 × 106 cells/mL. High-Five cells were infected with baculovirus containing RK gene at a multiplicity of infection = 5. The cells were incubated at 27°C for 72 h and were harvested by centrifugation (1000 × g) of 50 mL aliquots and subsequently stored at −80°C until ready for purification. For RK purification, a single aliquot was thawed in ice water for 30 min and resuspended into 3 mL of homogenization buffer (10 mM BTP, 0.04% Tween 20). The cells were homogenized with a Dounce homogenizer and the homogenate was mixed with 10 mL volume of regenerated DE52 resin (Whatman, Maidstone, Kent, UK). The kinase was eluted from the column with 100 mM NaCl in the homogenization buffer. Rhodopsin kinase positive fractions were verified using a filter-binding assay as described previously by Brannock et al. (1999).
GTPγS35-binding filter-binding assays
The ability of arrestin to bind to rhodopsin was indirectly measured through measurement of transducin activation by rhodopsin. A filter-binding assay was employed for this purpose. 2.5–5 picomoles of rhodopsin were reconstituted in the presence of 360 μM 11-cis retinal in a 50 μL volume for 1 h on a vertically rotating platform at 30 rpm. 12 μL of the reconstituted membranes were incubated in a buffer containing 30 mM BTP (pH 7.5), 3 mM MgCl2, 0.4 mM ATP, 0.3 mM DTT, and 2–4 μL kinase or kinase elution buffer (see Arrestin-Mediated Rhodopsin Deactivation Assays, below) to generate a final volume of 35 μL. The mixture was incubated at 30°C for 15 min under bright light. After the 15-min incubation, 15 μL of arrestin or arrestin buffer were added to generate 60% of the final arrestin concentration. The mixture was incubated for another 15 min at 30°C under bright light. The reaction was terminated by placing the tubes on ice for 2 min, at which time reaction volumes were increased to generate 100% of the final arrestin concentration of 5 μM, 2 μM transducin, 100 mM NaCl, 10 mM Tris-Cl (pH 7.5), 5 mM MgCl2, and 1 mM DTT to a final volume of 90 μL. The reactions were started by adding 10 μL of 30 μM GTPγS35. 10 μL of the reaction mixture were added to nitrocellulose filters on a Millipore vacuum manifold at 30-s intervals. The filters were washed with 15 mL of reaction buffer. Then the filters were added to 5 mL of scintillation fluid (Amersham Biosciences, Piscataway, NJ) and allowed to shake vigorously overnight. Reactions rates were expressed as picomoles of GTPγS35 bound per min. The linear regressions were calculated using SigmaPlot software.
Arrestin-mediated rhodopsin deactivation assays
Arrestin-mediated rhodopsin deactivation was indirectly measured by measuring transducin activation. Deactivation was expressed as percentage of inhibition of transducin activation. For experiments requiring phosphorylation of rhodopsin, three reactions were run simultaneously and the rates of transducin activation (v) were calculated for each. Reaction rates were as follows: v1 = (–) kinase, (–) arrestin; v2 = (+) kinase, (–) arrestin; and v3 = (+) kinase, (+) arrestin. Enough kinase was added to the reaction so v2 = 50% for Wt (v1). The decrease in rate of transducin activation due to arrestin-mediated deactivation was given as: % inhibition = [1 − (v3/v2)]100. For the experiments with rhodopsin mutants that had glutamic acid and aspartic acid substitutions, only two reactions were run. Reaction rates are as follows: v1 = (–) kinase, (–) arrestin, and v2 = (–) kinase, (+) arrestin. For these reactions, the decrease in rate of transducin activation due to arrestin-mediated deactivation is given as: % inhibition = [1 − (v2/v1)]100, and is reported as an arrestin effect.
COMPUTATIONAL METHODS
Development of the Monte Carlo simulated annealing algorithm
In understanding the possible interactions between two proteins (in this case rhodopsin and arrestin) one needs to find low energy minima of the complex. This is similar, in spirit, to reaching the global energy minimum in ligand-protein docking simulations (Trosset and Scheraga, 1998; Ewing and Kuntz, 1997; Makino and Kuntz, 1997; Eisen et al., 1994). Recently Orry and Wallace utilized a docking approach to test their molecular model of the Endothelin G-protein-coupled receptor (Orry and Wallace, 2000; Cavasotto et al., 2003). Although this approach is highly successful for rigid-body (and semiflexible) ligands, it will not allow for gross conformational changes to occur in neither protein nor ligand. However, in our case, the entire complex must be allowed to change conformation during the docking procedure. This requires an effective search of all conformational space available to the complex, as a function of conformational changes in either, or both, docking proteins. In other words, both proteins must be able to refold upon interaction. Therefore, a rigid-body docking algorithm would not suffice for this particular study where there is a definite conformational change in the complex. Thus to effectively search the conformational space available to the rhodopsin-arrestin complex, and hence get a sense of the interactions between the two proteins, a torsion space, all-atom, Monte Carlo simulated annealing algorithm was developed. Our algorithm is based on the all-atom Monte Carlo algorithm of Avbelj and Moult developed for the folding of small peptide structures (independent folding units) of between 11 and 14 residues in length (Avbelj and Moult, 1995). Although we utilized the same free energy function (ΔG) and Monte Carlo sampling techniques as Avbelj and Moult, we have extended the original algorithm to include a Monte Carlo simulated annealing optimization procedure that will effectively handle the refolding of protein-protein complexes.
In accordance with the original Monte Carlo algorithm, all of the atoms were allowed to move in the simulation; however, only the bonding angles (φ, ψ, and χ) were allowed to vary. Thus, the length of all bonds was kept fixed throughout the simulation. As in many standard Monte Carlo techniques, an angle (backbone or side-chain) was selected and moved and then a Metropolis criterion (Metropolis et al., 1953) was used to evaluate the acceptance of the move as follows: if the free energy (ΔG) decreased, the new conformation was accepted; if the free energy increased, then the move was accepted if
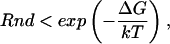 |
where Rnd is a random number, ΔG is the free energy of the complex, k is the Boltzmann constant, and T is the current simulation temperature (in K).
Angles were randomly chosen for a given move and the following move set of Avbelj and Moult was employed throughout the simulations:
40% of the angles chosen were backbone angles.
60% of the angles chosen were side-chain angles.
Of the angles chosen (backbone or side-chain) the following types of moves were allowed:
20% of all moves of angles were random.
60% of all moves of angles were taken from a precompiled library (Holm and Sander, 1994).
20% of all moves of angles were local steps of 5°.
If any given move resulted in a steric clash, then that move was immediately disregarded and a new angle was chosen. The initial temperature of the simulation was set at 300 K, the final temperature was set to 50 K and a cooling rate of 0.988 was employed. Thus the temperature decreased ∼0.1 K per every 10 simulation steps.
The free energy function (ΔG)
The MC-SA procedure optimizes geometry by optimizing an all-atom implicit solvent free energy function. The free energy function used in the MC-SA algorithm, defined as ΔG, was derived by Avbelj and Moult (1995). It is a potential of mean force based on a set of 114 nonhomologous protein crystal structures and has the form
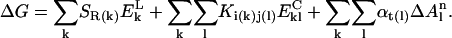 |
(1) |
The first term of the free energy function is defined as the local backbone electrostatic energy. The sum over k, is a sum over all residues in the peptide. SR(k) is a scaling dependent factor for a residue of type R at position k in the sequence. 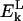 is the Coulomb energy of residue k, arising from interactions between the NH and C=0 groups with those of the flanking residues (k−1 and k+1). In general,
is the Coulomb energy of residue k, arising from interactions between the NH and C=0 groups with those of the flanking residues (k−1 and k+1). In general, 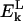 is unfavorable for residues in a helical conformation and favorable for extended conformations. The balance of this term and that of main-chain hydrogen bonding will establish the secondary structure of the protein.
is unfavorable for residues in a helical conformation and favorable for extended conformations. The balance of this term and that of main-chain hydrogen bonding will establish the secondary structure of the protein.
The second term in the free energy function is defined as the electrostatic energy for the other intramolecular interactions. The sums over k and l are over polar (k) and charged (l) residues of types i and j, for which the distance between the proton donor and acceptor is shorter than 6.5 Å. Ki(k)j(l) is a scaling factor that is dependent on the type of charged or polar groups involved. The equation for the Coulomb energy,  , is given as
, is given as
 |
(2) |
where the sum over m and n are over all atoms of groups k and l. Here qm and qn are the partial charges on the atoms, and rnm is the interatomic distance between the two atoms.
The third term in the free energy function (Eq. 1) is the solvation free energy, and is based on the average local solvent accessibility of the atoms in the observed conformation. For each set of atoms of the same type in any configuration,  can be expressed as in Avbelj and Moult (1995),
can be expressed as in Avbelj and Moult (1995),
 |
(3) |
The exponent n is equal to 1 for carbon and sulfur atoms and 3 for oxygen and nitrogen atoms. In Eq. 3, 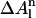 is the difference between the surface area for an estimated random coil conformation (At; Lee and Richards, 1971) and the exposed surface area for each atom, m, in the current conformation (Am).
is the difference between the surface area for an estimated random coil conformation (At; Lee and Richards, 1971) and the exposed surface area for each atom, m, in the current conformation (Am).
Taken together, the three terms in Eq. 1 represent the potential of mean force, which is optimized for the lowest energy conformation using the Monte Carlo simulated annealing algorithm.
COMPUTATIONAL RESULTS
In an attempt to understand the role of the phosphorylated residues in the desensitization process of G-protein-coupled receptors, we have studied the interaction of various methyl-capped rhodopsin cytoplasmic tail analogs (22 residues) with the basal state of the rod-specific arrestin protein (R-arrestin) (Protein Data Bank entry: 1CF1.pdb) (Hirsch et al., 1999).
Modeling the cytoplasmic tail of rhodopsin
Due to the size of rhodopsin (348-residue Protein Data Bank entry: 1HZX.pdb), we are not yet able to include all of the residues of the rhodopsin-arrestin complex in our simulations. This is because the number of degrees of freedom, or the number of movable dihedral angles, is too large for our current simulation to handle. However, at present we are only interested in studying the initial mechanism of the desensitization process, which involves only the tail portion of rhodopsin (residues 329–348) associating with the full arrestin protein. Thus the remaining 328 residues of rhodopsin are relatively unimportant for this initial study. It must be stressed, however, that the full 353 residues of arrestin are important for this study to ascertain if a conformational change of this protein occurs when associating with rhodopsin.
Because the cytoplasmic tail of rhodopsin is disordered in solution and thus the x-ray crystal structure is not available for these residues, we have modeled the tail as follows:
Methyl groups were added to both the N- and C-termini of the cytoplasmic tail to avoid artificial end-end interactions.
The 22-residue (methyl-capped) wild-type tail sequence was first modeled as an extended random coil.
This extended structure was then folded into a lowest free energy conformation (global minimum) using the Monte Carlo simulated annealing algorithm. We find that the lowest energy conformation of the wild-type (unphosphorylated) rhodopsin tail is ΔG = −174.68 Kcal/mol, and has a two-turn helix turn structure, as illustrated in Fig. 1.
FIGURE 1.

Lowest energy configuration of the cytoplasmic tail of wild-type rhodopsin, as found by the MC-SA method. The possible phosphorylation sites (Ser/Thr) are illustrated in red.
To ascertain that both the crystal structure of the basal state of the arrestin and the cytoplasmic tail of rhodopsin are in an energy minimum configuration (before docking), we also performed an energy minimization (MC-SA) on the crystal structure of R-arrestin. We find that the basal state of arrestin protein has low energy of ΔG = −3464.86 kcal/mol, with an RMS change of 0.539 Å as compared to the basal state crystal structure. This indicates that the crystal structure of arrestin is indeed at an energy minimum, hence any conformational changes observed during association with rhodopsin will be due to the interactions of the rhodopsin-arrestin complex and not an artifact of our simulation procedures.
The energy-minimized wild-type cytoplasmic tail of rhodopsin was manually docked into the binding pocket of arrestin using the program PSSHOW (Swanson, 1995). Care was taken that the distance between most residues of rhodopsin and arrestin were >9.0 Å apart, to begin our simulations from an unbiased starting configuration. Fig. 2 illustrates the wild-type complex, which is used as a starting structure for the simulations.
FIGURE 2.

The initial structure of the cytoplasmic tail of rhodopsin (parallel orientation) in association with R-arrestin.
Simulations and results of the rhodopsin-arrestin complex
Monte Carlo simulated annealing, as described in Computational Methods, was performed on the wild-type complex for 20,000 steps, beginning at 300 K and slowly reducing the simulation temperature to 50 K during the course of the simulation. We find that neither the wild-type cytoplasmic tail of rhodopsin nor arrestin changed conformation during the simulation. This is seen in Table 1, where the Cα RMS (root mean-square deviation) between the initial structure (Fig. 2) and the final structure (not shown) is 0.56 Å. In fact, the wild-type cytoplasmic tail began to leave the active site during the simulation. Furthermore, there did not appear to be any interaction between the two structures. This is seen in Table 2, where the residue-residue distance (in Å) is never <8 Å. This confirms the experimental evidence that unphosphorylated (wild-type) rhodopsin will not interact with basal state arrestin. As a check of our simulation, we mutated the seven serine/threonine residues to alanine in the cytoplasmic tail of rhodopsin. We then performed the same MC-SA simulation on this mutant complex and again observed neither conformational changes (Table 1) nor binding interactions in the complex (Table 2), as expected.
TABLE 1.
Monte Carlo simulated annealing of C-tail rhodopsin/arrestin: energetics and RMS compared to starting structures
| Simulation | ΔG (Kcal/mol), tail | ΔG (Kcal/mol), arrestin | CαRMS tail | CαRMS arrestin | CαRMS complex |
|---|---|---|---|---|---|
| Wild-type | −174.68 | −3591.63 | 0.694 | 0.157 | 0.566 |
| ALA mutant | −149.06 | −3507.95 | 0.461 | 0.090 | 0.290 |
| GLU mutant | −171.29 | −3501.23 | 5.58 | 0.56 | 1.67 |
| ASP mutant | −188.83 | −3523.45 | 5.269 | 0.133 | 1.902 |
ΔG = free energy of the final structure, relative to the unfolded state.
TABLE 2.
Monte Carlo simulated annealing of C-tail rhodopsin/arrestin: residue-residue interaction distances in Å
| Simulation | Structure | S334-K14 | S338-K14 | T340-K14 | S334-K15 | S338-K15 | T340-K15 |
|---|---|---|---|---|---|---|---|
| 1. Wild-type | in | 13.6 | 12.6 | 14.6 | 13.4 | 6.9 | 9.6 |
| 1. Wild-type | fn | 15.9 | 9.7 | 11.9 | 14.5 | 8.0 | 9.7 |
| 2. All-D mutant* | in | 14.3 | 13.1 | 14.2 | 13.3 | 8.2 | 9.4 |
| 2. All-D mutant* | fn | 10.1 | 18.6 | 18.5 | 3.7 | 10.3 | 10.8 |
| 3. All-D mutant† | in | 23.8 | 25.5 | 24.4 | 16.1 | 15.9 | 12.3 |
| 3. All-D mutant† | fn | 24.4 | 24.6 | 24.4 | 16.2 | 14.9 | 12.8 |
| 4. All-D mutant‡ | in | 15.16 | 7.3 | 8.0 | 14.68 | 9.03 | 16.9 |
| 4. All-D mutant‡ | fn | 14.83 | 7.41 | 6.2 | 14.42 | 8.7 | 15.96 |
| 5. S334D:T340D* | in | 14.3 | NA | 14.2 | 13.3 | NA | 9.36 |
| 5. S334D:T340D* | fn | 17.3 | NA | 17.1 | 16.1 | NA | 14.9 |
| 6. S334D:T340D† | in | 23.8 | NA | 24.4 | 16.1 | NA | 12.3 |
| 6. S334D:T340D† | fn | 24.5 | NA | 24.3 | 16.7 | NA | 11.9 |
| 7. S334D:T340D‡ | in | 20.3 | NA | 13.6 | 11.7 | NA | 9.1 |
| 7. S334D:T340D‡ | fn | 21.6 | NA | 8.9 | 13.1 | NA | 7.6 |
| 8. S334D:S338D:T340D* | in | 14.3 | 13.1 | 14.2 | 13.3 | 8.2 | 9.6 |
| 8. S334D:S338D:T340D* | fn | 16.8 | 8.4 | 19.2 | 15.6 | 11.9 | 21.3 |
| 9. S334D:S338D:T340D† | in | 25.1 | 23.6 | 16.9 | 15.2 | 13.2 | 5.2 |
| 9. S334D:S338D:T340D† | fn | 25.4 | 23.9 | 16.6 | 15.2 | 13.2 | 4.9 |
| 10. S334D:S338D:T340D‡ | in | 16.1 | 21.2 | 14.3 | 14.9 | 13.9 | 5.4 |
| 10. S334D:S338D:T340D‡ | fn | 15.6 | 20.7 | 15.8 | 14.9 | 13.2 | 4.7 |
| 11. All-E mutant* | in | 16.96 | 11.95 | 13.44 | 19.92 | 6.18 | 8.05 |
| 11. All-E mutant* | fn | 17.70 | 6.65 | 16.18 | 18.40 | 13.22 | 22.26 |
| 12. S334E:T340E* | in | 14.24 | NA | 16.29 | 15.56 | NA | 9.05 |
| 12. S334E:T340E* | fn | 17.76 | NA | 9.26 | 17.07 | NA | 17.12 |
| 13. S338E:T340E* | in | NA | 14.3 | 16.29 | NA | 6.2 | 9.0 |
| 13. S338E:T340E* | fn | NA | 14.2 | 19.09 | NA | 10.6 | 15.1 |
| 14. S334E* | in | 14.3 | NA | NA | 15.6 | NA | NA |
| 14. S334E* | fn | 17.4 | NA | NA | 16.9 | NA | NA |
| 15. T340E* | in | NA | NA | 16.3 | NA | NA | 9.0 |
| 15. T340E* | fn | NA | NA | 15.3 | NA | NA | 15.4 |
To ascertain if a possible charge-charge interaction occurs between the cytoplasmic tail of rhodopsin and the basal state of arrestin, we would ultimately need to phosphorylate the serine/threonine residues in the cytoplasmic tail. However, at this time such a simulation is not feasible. Thus we performed two additional rhodopsin cytoplasmic tail mutation studies. In one study, the serine/threonine residues were mutated to aspartic acid, and in another study the serine/threonine residues were mutated to glutamic acid. Although this may seem like a drastic mutation, our simulations will be directly compared to the aspartic and glutamic peptide tail analogous studies of McDowell et al. (2001) as well as our current experimental work. In the investigations of McDowell and co-workers, the authors found evidence for an interaction between the all-aspartic and all-glutamic peptide tail analogous with arrestin. These interactions are not necessarily the same as in the phosphorylated case but it is exactly this interaction which we will elucidate as a foundation for future studies.
Simulations of the rhodopsin aspartic tail analog
In an attempt to elucidate the charge-charge interaction of rhodopsin with R-arrestin we have performed computational site-directed mutagenic studies of this system. Our mutation studies include both aspartic acid and glutamic acid mutations for the serine/threonine residues of the cytoplasmic tail of rhodopsin. We have also looked at different initial orientations of the rhodopsin tail interacting and refolding within the binding pocket of R-arrestin. We define one orientation as parallel to the β-sheet of the Lys14/Lys15 pair (Fig. 2), and the other initial orientation of the rhodopsin is defined as perpendicular (Fig. 3). Based on our Monte Carlo simulated annealing simulations, we find that only the parallel orientation of the rhodopsin tail analog is capable of interacting with the Lys14/Lys15 residues on arrestin. This finding is best illustrated in Tables 1–3 and Fig. 4. In Table 1 we see that the CαRMS distance between the initial and final structures of the aspartic acid mutant complex is 1.9 Å. The change in this distance is due mostly to the refolding of the aspartic mutant tail (CαRMS 5.3 Å). In previous simulations of both the wild-type and the alanine mutant, the tail did not refold in the binding pocket of arrestin, indicating that the charge-charge interaction is necessary for this refolding process to occur.
FIGURE 3.

The initial structure of the cytoplasmic tail of rhodopsin (perpendicular orientation) in association with R-arrestin.
TABLE 3.
Monte Carlo simulated annealing of C-tail rhodopsin/arrestin: residue-residue interaction distances in Å
FIGURE 4.

The final structure of the all-aspartic acid mutant of the C-terminus tail of rod-specific visual pigment rhodopsin (blue) in complex with rod-specific arrestin protein (red). The charge-charge interactions between the Asp334/Lys15 and Asp340/His301 are shown in stick representation. The final structure was found by a Monte Carlo simulated annealing algorithm and this figure was generated using RASMOL (Sayle and Milner-White, 1995).
Table 2 is a measure of the interaction distance between key residues on rhodopsin and arrestin before (in) and after (fn) the simulation. We find that in a perpendicular orientation this interaction distance is typically >12 Å, indicating no significant interaction (simulation 3, Table 2). However, when the initial orientation of the all-aspartic tail mutant is parallel with the arrestin binding pocket, we find that after the simulation the interaction distance between residues D334 (rhodopsin) and K15 (arrestin) is 3.7 Å. We believe this indicates a definite charge-charge interaction (simulation 2, Table 2). We also find three additional interactions between residues D340 (rhodopsin) and K300 (6.48 Å), H301 (3.51 Å), and R29 (4.95 Å) on the arrestin protein. These additional interactions were not proposed in the original model of Vishnivetskiy et al. (2000) and came as a surprise to us. The final structure of the all-aspartic acid tail analog interacting with arrestin (simulation 2) is illustrated in Fig. 4. The key residues are drawn in stick representation to highlight these strong interactions.
We also determined if different parallel conformations would interact in the same way with the R-arrestin protein. To address this issue, we created a new initial parallel conformation by refolding the all-aspartic tail analog separately into a new configuration. We then placed this new structure into the binding pocket of R-arrestin in a parallel conformation (Fig. 5). This new structure was also tested for all the constitutive mutation studies we performed. We find that during the course of the simulation the rhodopsin residues D338 and D340 (parallel structure 2) interact with the R-arrestin protein residues K14 and K15. However, this interaction must be somewhat weaker as the interaction distance is 6.2 Å and 8.7 Å, respectively (simulation 4, Table 2). In a recent article by Kumar and Nussinov on the relationship between ion pair geometry and electrostatic strengths in proteins, the authors find that if the ion pair is within 4 Å this tends to be a stabilizing interaction, whereas if the ion pair is >4 Å apart this tends to be a destabilizing interaction (Kumar and Nussinov, 2002). This work is done by exploring 11 nonhomologous protein NMR structures and is based on a continuum electrostatic calculation. Because we have not calculated the electrostatic strength of our ion pairs separately, we cannot say if the short-range 3.7 Å distance is stabilizing or destabilizing to the complex. We are now in the process of investigating this effect for the ion pairs formed in the complex.
FIGURE 5.

The initial structure of the cytoplasmic tail of rhodopsin (parallel orientation, Table 2‡) in association with R-arrestin.
To ascertain if residues S334 and T340 are indeed important interaction sites in rhodopsin we carried out a series of site-directed computational mutations studies. We studied two different mutant rhodopsin tail analogs, S334D:T340D and S334D:S338D:T340D in all three starting conformations (Figs. 2, 3, and 5). We find that both the double and triple mutant tail analogs can interact with R-arrestin, although it is a somewhat weaker interaction than with the full all-aspartic tail analog. This is seen in Table 2, where the interaction distances are on the order of 7–8 Å for the double and triple mutant analogs, whereas it is 3.7 Å for the all-aspartic tail analog. Based on these findings we made a prediction that the key residues for interaction of the aspartic tail analog of rhodopsin with arrestin were Ser334 and Thr340, with a secondary and somewhat weaker interaction of Ser338 with arrestin.
Simulations of the rhodopsin glutamic tail analog
To address the issue of the effect of the side-chain size (Brannock et al., 1999) on the charge-charge interaction, we performed a glutamic acid mutation study. We begin with the same initial parallel structure as in the all-aspartic mutation study (Fig. 2). However, in this case, we mutated all-rhodopsin tail serine and threonine residues to glutamic acid. We then performed the same Monte Carlo simulated annealing algorithm as before. The results are presented in Tables 1–3. We see from Table 1 that the all-glutamic tail analog has refolded in the complex (Cα RMS is 5.58 Å) as in the case of the all-aspartic acid tail analog. However, in this particular case, we find that residue E338 on the rhodopsin tail analog will interact with the K14 residue on R-arrestin (Fig. 6). This interaction was not seen in the all-aspartic acid analog. Moreover, the all-glutamic acid analog did not show any evidence of a E334-K14 interaction or E340-K15 interaction, as in the all-aspartic acid case. We also created a two additional mutants in a parallel orientation: S334E:T340E and S338E:T340E. Although we know from experimental evidence that the S334E:T340E mutant should interact with the R-arrestin (as well as the S334D:T340D mutant), we could find no evidence of this interaction for the glutamic acid analogue. The bulky side chains of the glutamic acid mutant proved to be difficult to simulate. Thus we preformed only one simulation for each glutamic acid mutant. Based on our limited simulations, we find a somewhat weaker interaction between the glutamic acid tail analog and the R-arrestin protein, compared to the aspartic acid analog.
FIGURE 6.

The final structure of the all-glutamic acid mutant of the C-terminus tail of rod-specific visual pigment rhodopsin (blue) in complex with rod-specific arrestin protein (red). The charge-charge interactions between the Glu338/Lys14 are shown in stick representation. The final structure was found by a Monte Carlo simulated annealing algorithm and this figure was generated using RASMOL.
EXPERIMENTAL IN VITRO RESULTS
Given the results of the computational study, we generated rhodopsin mutants with aspartic acid residues substitutions at specific sites on the cytoplasmic tail. We constructed eight mutant rhodopsin proteins and studied the effect of arrestin-mediated deactivation. The results are presented in Fig. 7. Most strikingly we find that the double mutant with aspartic acid substituted at positions 334 and 340 (D334/D340) shows an arrestin effect that is comparable with all phosphorylatable residues replaced with aspartate (D-tail). This result confirms that the presence of a negative charge at these specific positions is sufficient to induce an arrestin effect. However, it is also clear from Fig. 7 that the aspartic D-tail mutant does not produce as strong an effect as phosphorylation of the cytoplasmic tail. It is also evident that a single negative charge present at either position 334 or 340 is not sufficient, although a negative charge at residue 334 does increase the arrestin effect relative to the unphosphorylated wild-type rhodopsin. To test whether the interactions seen were indeed site-specific, we mutated residues 335 and 343 to aspartic acid and subsequently tested the construct using our in vitro assay. We find that this mutant showed no interaction with the arrestin protein, as predicted from the computer simulations. Taken all together, these results suggest that the computer simulations can guide specific in vitro experiments in this system.
FIGURE 7.

This figure illustrates the effect of arrestin binding with various aspartic acid (D) rhodopsin mutants. Arrestin blocks transducin binding and this is measured as an arrestin effect. The largest effect is seen for the D334/D340 mutant, second column. The experiments were performed after the simulations and were design based on our computational results. The error bars are mean ± SE.
CONCLUSIONS
In total 17 simulations were performed, including a simulation to predict a putative structure for the cytoplasmic tail of rhodopsin. We find that the peptide analog of the rhodopsin cytoplasmic tail begins with an α-helix until residue V337, and thereafter the tail adopts a more random loop structure. This result is somewhat consistent with recent EPR spectroscopy that indicates the cytoplasmic tail is part of a short H-VII helix (residues 328–333; Altenback et al., 2001); however, solution NMR studies indicate that the tail is dynamically disordered beyond residue 327 (Langen et al., 1999).
Tables 1–3 summarize the conformational energetics, Cα root mean-square deviation (RMSD) (Table 1) and the residue-residue interaction distance (Table 2) between the initial and final structures of various mutated rhodopsin-arrestin complexes which we have simulated. We note that the alanine mutant is not represented in Tables 1 and 2, inasmuch as it is similar to the wild-type simulation. We find that both the glutamic acid and aspartic acid mutated tail analogs produced a slight conformational change in R-arrestin (0.56 and 0.133 Å). However, both of the negatively charged tail mutants refolded into completely new conformations when introduced in complex with R-arrestin (5.58 and 5.269 Å). Moreover, both the aspartic and glutamic acid tail analogs showed potential charge-charge interactions with R-arrestin, although at different sites. For example, in the aspartic tail analog we see specific interactions of S334D (tail) with K15, K300, H301, and R29 (R-arrestin) and also T340D (tail) with K15 (R-arrestin) (Fig. 4), whereas the glutamic tail analog mainly interacts at S338E (tail) with K14 (R-arrestin) (Fig. 6).
What about the role of the serine 343 residue in the rhodopsin cytoplasmic tail? There is compelling in vivo and in vitro work to suggest that this serine residue is indeed phosphorylated, and as such, the question is, does it play a role in desensitization (Ohguro et al., 1995, 1996; Zhang et al., 1997; Kennedy et al., 2001b). To address this issue, we have analyzed our simulation results for the interaction of residue 343 with the arrestin lysine residues (K14, K15) of interest (Table 3). We find, in one simulation (the all-glutamic acid mutant, conformation 1), an interaction between E343 (rhodopsin) and K15 (arrestin). Moreover, this is an obvious interaction as the interaction distance changes from 17.22 Å to 7.35 Å. The question is how to interpret these results, since we find only one simulation with an obvious rhodopsin S343 interaction with arrestin. In all other simulations studied, we consistently see interactions of rhodopsin residues S338, T340, and strikingly S334 with the arrestin protein. One possibility could be that we have simply not run enough simulations to sample all possible interactions of rhodopsin with arrestin. We are currently running multiple mutant glutamic acid simulations to test this hypothesis.
Another possibility is, if phosphorylated, residue S343 will have a weak, but viable interaction with the arrestin protein. We tested this hypothesis with our in vitro assay (Fig. 7). We find that single mutants of aspartic acid on any phosphorylated rhodopsin residue produces a relatively insignificant arrestin effect, with the possible exception of residue D334. Moreover, the double mutant D335/D343 also produces an insignificant arrestin effect. This suggest that the role of residue S343 in desensitization is secondary at best. However, a polar (negative) charge at this position may facilitate the formation/stabilization of the rhodopsin-arrestin complex. We stress that we have not tested all possible mutant combinations involving residue S343; we merely suggest that, if one relates the distance between the ion pair residues to the strength of this interaction, that the order of affinity would be S334 > T340 > S338 > S343. This would imply that mutation of the S334 residue could have a significant effect on the binding of the rhodopsin-arrestin complex. Our results are consistent with the dephosphorylation results of Palczewski and co-workers, who find that dephosphorylation occurs in the following order (slowest to fastest): S334 < S338 < S343 (Ohguro et al., 1996).
We believe that our findings may be significant, inasmuch as it is thought that the rhodopsin residues Ser334, Ser338, and Thr340 are key phosphorylation sites (McDowell et al., 2001). Our results also indicate the relative importance of the R-arrestin Lys14 and Lys15 as a possible charge-charge interaction site (Vishnivetskiy et al., 2000). Moreover, we find that the orientation of the cytoplasmic tail of rhodopsin in the binding pocket of arrestin is important. That is to say, only a parallel tail orientation facilitates binding, whereas a perpendicular orientation did not seem to show any interaction between the two proteins. We also note that our simulations do not show any evidence of a significant role of the other serine/threonine residues in this binding process. We must stress that this is the first computational study of such a system.
Based on our computational studies, we predicted that the S334D/T340D aspartic acid mutant analog of rhodopsin would bind to arrestin in a comparable way to phosphorylated rhodopsin. This hypothesis was tested with our in vitro assays (Fig. 7) and results indicate this to be the case. We are now in the process of studying the effect of this ionic interaction, both experimentally and computationally.
Acknowledgments
S.K.G. thanks Professor John Moult of Center for Advanced Research in Biotechnology for his insightful discussions and use of his force field. P.R.R. thanks R. K. Crouch and the National Eye Institute for the 11-cis-retinal.
S.K.G. acknowledges funding from the University of Maryland, Baltimore County in the form of a Special Research Initiative Award, and P.R.R. acknowledges the National Science Foundation for continued support (grant #0119102).
References
- Ablonczy, Z., R. K. Crouch, P. W. Goletz, T. M. Redmond, D. R. Knapp, J. X. Ma, and B. Rohrer. 2002. 11-cis-retinal reduces constitutive opsin phosphorylation and improves quantum catch in retinoid-deficient mouse rod photoreceptors. J. Biol. Chem. 277:40491–40498. [DOI] [PubMed] [Google Scholar]
- Alloway, P. G., and P. J. Dolph. 1999. A role for the light-dependent phosphorylation of visual arrestin. Proc. Natl. Acad. Sci. USA. 96:6072–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenback, C., K. Cai, J. Klein-Seetharaman, H. G. Khorana, and W. L. Hubbell. 2001. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 65 in helix tm1 and residues in the sequence 306–319 at the cytoplasmic end of helix tm7 and in helix h8. Biochemistry. 40:15483–15492. [DOI] [PubMed] [Google Scholar]
- Avbelj, F., and J. Moult. 1995. The role of electrostatic screening in determining protein main chain conformational preferences. Biochemistry. 34:755–764. [DOI] [PubMed] [Google Scholar]
- Brannock, M., K. Weng, and P. R. Robinson. 1999. Rhodopsin's carboxyl terminal threonines are required for the arrestin mediated quench of transducin activation in vitro. Biochemistry. 38:3770–3777. [DOI] [PubMed] [Google Scholar]
- Cavasotto, C. N., A. J. W. Orry, and R. A. Abagyan. 2003. Structure-based identification of binding sites, native ligands and potential inhibitors for G-protein-coupled receptors. Protein Struc. Func. Gen. 51:423–433. [DOI] [PubMed] [Google Scholar]
- Eisen, M. E., D. C. Wiley, M. Karplus, and R. Hubbard. 1994. HOOK: a program for finding novel molecular architectures that satisfy the chemical and steric requirements for a macromolecule binding site. Protein Struc. Func. Gen. 19:199–221. [DOI] [PubMed] [Google Scholar]
- Ewing, T., and I. Kuntz. 1997. Critical evaluation of search algorithms for automated molecular docking and database screening. J. Comp. Chem. 18:1175–1189. [Google Scholar]
- Gray-Keller, M. P., P. B. Detwiler, J. L. Benovic, and V. V. Gurevich. 1997. Arrestin with a single amino acid substitution quenches light-activated rhodopsin in a phosphorylation-independent fashion. Biochemistry. 36:7058–7063. [DOI] [PubMed] [Google Scholar]
- Gurevich, V. V. 1998. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J. Biol. Chem. 273:15501–15506. [DOI] [PubMed] [Google Scholar]
- Hargrave, P. A. 1986. Molecular Dynamics of the Rod Cell, Vol. 1. Academic Press, Orlando, FL.
- Hirsch, J. A., C. Schubert, V. V. Gurevich, and P. B. Sigler. 1999. A model for arrestin's regulation: the 2.8 Å crystal structure of visual arrestin. Cell. 97:257–269. [DOI] [PubMed] [Google Scholar]
- Holm, L., and C. Sander. 1994. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233:123–138. [DOI] [PubMed] [Google Scholar]
- Jacobs, G. H. 1998. Photopigments and seeing: lessons from natural experiments. Invest. Opth. Vis. Sci. 39:2204–2216. [PubMed] [Google Scholar]
- Kennedy, M., K. A. Lee, G. A. Niemi, D. B. Craven, G. G. Garwin, J. C. Saari, and J. B. Hurley. 2001a. Multiple phosphorylation of rhodopsin and the in vivo chemistry underlying rod photoreceptor dark adaptation. Neuron. 31:87–101. [DOI] [PubMed] [Google Scholar]
- Kennedy, M. J., K. A. Lee, G. A. Niemi, K. B. Craven, G. G. Garwin, J. C. Saari, and J. B. Hurley. 2001b. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 32:451–461. [DOI] [PubMed] [Google Scholar]
- Kuhn, H., S. W. Hall, and U. Wilden. 1986. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 176:473–478. [DOI] [PubMed] [Google Scholar]
- Kumar, S., and R. Nussinov. 2002. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys. J. 83:1595–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langen, R., K. Cai, C. Altenbach, H. G. Khorana, and W. L. Hubbell. 1999. Structural features of the C-terminal domain of bovine rhodopsin: a site-directed spin-labeling study. Biochemistry. 38:7918–7924. [DOI] [PubMed] [Google Scholar]
- Lee, B., and F. M. Richards. 1971. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55:379–400. [DOI] [PubMed] [Google Scholar]
- Lohse, M. J., S. Andexinger, J. Pitcher, S. Trukawinski, J. Codina, J. Faure, M. G. Caron, and R. J. Lefkowitz. 1992. Receptor-specific desensitization with purified proteins. Kinase dependence and receptor specificity of β-arrestin and arrestin in the β2-adrenergic receptor and rhodopsin systems. J. Biol. Chem. 267:8558–8564. [PubMed] [Google Scholar]
- Lohse, M. J., J. L. Benovic, J. Codina, M. G. Caron, and R. J. Lefkowitz. 1990. β-arrestin: a protein that regulates β-adrenergic receptor function. Science. 248:1547–1550. [DOI] [PubMed] [Google Scholar]
- Makino, S., and I. Kuntz. 1997. Automated flexible ligand docking method and its application for database search. J. Comp. Chem. 18:1812–1825. [Google Scholar]
- McDowell, J. H., P. R. Robinson, R. L. Miller, M. T. Branock, A. Arendt, W. C. Smith, and P. A. Hargrave. 2001. Activation of arrestin: requirement of phosphorylation as the negative charge on residues in synthetic peptides from the carboxyl-terminal region of rhodopsin. Invest. Opth. Vis. Sci. 42:1439–1443. [PubMed] [Google Scholar]
- Mendez, A., M. E. Burns, A. Roca, J. Lem, L. W. Wu, M. I. Simon, D. A. Baylor, and J. Chen. 2000. Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites. Neuron. 28:153–164. [DOI] [PubMed] [Google Scholar]
- Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. Teller, and E. Teller. 1953. Equations of state calculations by fast computing machine. J. Chem. Phys. 21:1087–1091. [Google Scholar]
- Molday, R. S. 1998. Photoreceptor outer segment proteins, phototransduction and retinal degenerative diseases: the Friedenwald Lecture. Invest. Opth. Vis. Sci. 39:2493–2513. [PubMed] [Google Scholar]
- Ohguro, H., J. P. V. Hooser, A. H. Milam, and K. Palczewski. 1995. Rhodopsin phosphorylation and dephosphorylation in vivo. J. Biol. Chem. 270:14259–14264. [DOI] [PubMed] [Google Scholar]
- Ohguro, H., M. Rudnicka Nawrot, J. Buczylko, X. Zhao, J. A. Taylor, K. A. Walsh, and K. Palczewski. 1996. Structural and enzymatic aspects of rhodopsin phosphorylation. J. Biol. Chem. 271:5215–5224. [DOI] [PubMed] [Google Scholar]
- Orry, A. J. W., and B. A. Wallace. 2000. Modeling and docking the endothelin G-protein-coupled receptor. Biophys. J. 79:3083–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski, K., Z. Pulvermuller, J. Buczylko, and K. P. Hofmann. 1991. Phosphorylated rhodopsin and heparin induce similar conformational changes in arrestin. J. Biol. Chem. 266:18649–18654. [PubMed] [Google Scholar]
- Puig, J., A. Arendt, F. L. Tomson, G. Abdulaeva, R. Miller, P. A. Hargrave, and J. H. McDowell. 1995. Synthetic phosphopeptide from rhodopsin sequence induces retinal arrestin binding to photoactivated unphosphorylated rhodopsin. FEBS Lett. 362:185–188. [DOI] [PubMed] [Google Scholar]
- Sakmar, T. P., S. T. Menton, E. P. Marin, and E. W. Awad. 2002. Rhodopsin: insights from recent structural studies. Annu. Rev. Biophys. Biomol. Struct. 31:443–484. [DOI] [PubMed]
- Sayle, R. A., and E. J. Milner-White. 1995. RASMOL: biomolecular graphics for all. Trends Biochem. Sci. 20:374–376. [DOI] [PubMed] [Google Scholar]
- Swanson, E. 1995. PSSHOW, Vers. 1.9. University of Washington, Seattle, WA.
- Trosset, J. Y., and H. Scheraga. 1998. Reaching the global minimum in docking simulations: a Monte Carlo energy minimization approach using Bezier splines. Proc. Natl. Acad. Sci. USA. 95:8011–8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy, S. A., C. L. Paz, C. Schubert, J. A. Hirsch, P. B. Sigler, and V. V. Gurevich. 1999. How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 274:11451–11454. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy, S. A., C. Schubert, G. C. Climaco, Y. V. Gurevich, M. G. Velez, and V. V. Gurevich. 2000. An additional phosphate-binding element in arrestin molecule. Implications for the mechanism of arrestin activation. J. Biol. Chem. 275:41049–41057. [DOI] [PubMed] [Google Scholar]
- Weiss, E. R., S. Osawa, W. Shi, and C. D. Dickerson. 1994. Effects of carboxy-terminal truncation on the stability and G-protein-coupling activity of bovine rhodopsin. Biochemistry. 33:7587–7593. [DOI] [PubMed] [Google Scholar]
- Wessling-Resnick, M., and G. L. Johnson. 1987. Allosteric behavior in transducin activation mediated by rhodopsin. Initial rate analysis of guanine nucleotide exchange. J. Biol. Chem. 262:3697–3705. [PubMed] [Google Scholar]
- Wilden, U., S. Hall, and H. Kuuhn. 1986. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48 kDa protein of rod outer segments. Proc. Natl. Acad. Sci. USA. 83:1174–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L., C. D. Sports, S. Osawa, and E. R. Weiss. 1997. Rhodopsin phosphorylation sites and their role in arrestin binding. J. Biol. Chem. 272:14762–14768. [DOI] [PubMed] [Google Scholar]