

Abstract
Imaging of fluorescence resonance energy transfer (FRET) between suitable fluorophores is increasingly being used to study cellular processes with high spatiotemporal resolution. The genetically encoded Cyan (CFP) and Yellow (YFP) variants of Green Fluorescent Protein have become the most popular donor and acceptor pair in cell biology. FRET between these fluorophores can be imaged by detecting sensitized emission. This technique, for which CFP is excited and transfer is detected as emission of YFP, is sensitive, fast, and straightforward, provided that proper corrections are made. In this study, the detection of sensitized emission between CFP and YFP by confocal microscopy is optimized. It is shown that this FRET pair is best excited at 430 nm. We identify major sources of error and variability in confocal FRET acquisition including chromatic aberrations and instability of the excitation sources. We demonstrate that a novel correction algorithm that employs online corrective measurements yields reliable estimates of FRET efficiency, and it is also shown how the effect of other error sources can be minimized.
INTRODUCTION
Fluorescence resonance energy transfer (FRET), the radiationless transfer of energy from a donor fluorophore to a closeby acceptor fluorophore, is rapidly gaining importance as a means to study molecular interactions in single cells. FRET is apparent as quenching of the donor and increased acceptor emission. Its main applications include the study of interactions between different proteins tagged with either a donor or an acceptor fluorophore (intermolecular FRET), following sterical alterations within a single protein labeled with both a donor and an acceptor (intramolecular FRET), and as the readout signal for biochemical sensors. In the latter case, constructs are engineered to respond to changes in a cellular signal (e.g., cAMP, Ca2+, or protein phosphorylation) by altering FRET. Depending on these different applications, very different design considerations may apply to the detection method. For FRET to occur, the fluorescent dipoles of donor and acceptor must be properly aligned, and there must be overlap between the donor emission spectrum and the acceptor excitation spectrum (Lakowicz, 1999). Furthermore, resonance energy transfer is steeply dependent on the distance between the fluorophores, decreasing with the sixth power of the distance. Characteristic half-maximal distances (Förster radii) for a number of biologically important fluorophores are ∼4–5 nm, and thus the distance range over which FRET changes (∼2–10 nm) is well-matched to the dimensions of individual proteins.
The recent introduction of color mutants of the Green Fluorescent Protein as donor and acceptor labels for FRET has fuelled interest in this technique. Because Green Fluorescent Proteins are genetically encoded, laborious in vitro conjugation of fluorophores to proteins as well as the introduction into the cell by microinjection or other means are no longer necessary. By far the most popular variants for FRET are the Cyan and Yellow variants, CFP and YFP, respectively (Tsien, 1998). First used to demonstrate a genetically encoded calcium sensor (cameleon; Miyawaki et al., 1997), this FRET pair has been the basis for several interesting sensors developed over the last few years, including those for cAMP, cGMP, PIP2, phosphorylation, and protein activation status (Zaccolo and Pozzan, 2002; Honda et al., 2001; van der Wal et al., 2001; Nagai et al., 2000; Mochizuki et al., 2001). Despite their bulkiness, CFP and YFP are also successfully applied to study protein-to-protein interactions and conformational changes. Concomitantly, several approaches to image FRET with this pair from single (living) cells have been exploited (for review, see Wouters et al., 2001). These include acceptor photobleaching, a technique whereby the fluorescent acceptor is destroyed and which therefore is not suited for timelapse imaging, and fluorescence lifetime imaging of the donor. Fluorescence lifetime imaging requires dedicated and expensive equipment, and CFP is not particularly suited for this technique because it intrinsically possesses several fluorescence lifetimes (Pepperkok et al., 1999). The most widely employed approach therefore is to calculate sensitized emission (i.e., the acceptor fluorescence resulting from energy transfer from excited donor molecules) from separately acquired donor and acceptor images. Because the spectra of CFP and YFP show considerable overlap, the detected sensitized acceptor emission must be corrected for leakthrough of the donor emission into the acceptor emission channel and for direct excitation of the acceptor during donor excitation. The latter correction requires that an additional image is captured from the acceptor, directly excited at its own wavelength. Several correction schemes were worked out for images that were acquired with wide-field fluorescence microscopes equipped with charge-coupled-device (CCD) cameras (Gordon et al., 1998; Nagy et al., 1998; Hoppe et al., 2002).
In this study, we focus on CFP/YFP FRET imaging by confocal microscopy. Confocal imaging has a number of advantages over wide-field imaging, the most important of which is that it produces crisp optical sections of the preparation. However, detecting sensitized emission by multiexcitation confocal acquisition raises a number of complications in the correction scheme. Unlike FRET imaging with digital camera systems, which have a single detector and a fixed ratio of excitation intensities that is determined by the filter sets, during (conventional) confocal imaging at least two individual detectors (photomultiplier tubes, PMT) are used, as well as two independent excitation laser lines. Dependent on the design of the deployed confocal instrument the spectral response of the detectors may even be tuned individually. Both donor and acceptor excitation intensities and PMT gain provide additional degrees of freedom and can be independently controlled by the user. Therefore, relative sensitivity for given fluorophores and leakthrough coefficients are not necessarily constant and need in all cases to be determined for each set of experimental conditions. Furthermore, errors stem from temporal variations in the relative intensities of the excitation lines for CFP and YFP, from slight misalignment between laser lines, and from the axial chromatic aberrations of the optical system. We quantified these effects and describe methods to correct for them. We also show that CFP is optimally excited at 430 nm to detect FRET, and we demonstrate suitability of a frequency-doubled diode laser for this application. These improvements result in a significant increase in quality of confocal sensitized emission images.
MATERIALS AND METHODS
Materials
Ionomycin was from Calbiochem-Novabiochem (La Jolla, CA), BAPTA was from Sigma Chemical (St. Louis, MO), and 0.17-μm, yellow-green fluorescent beads (490/515, component B from the PS-Speck Microscope Point Source Kit P-7220) were from Molecular Probes (Eugene, OR).
Constructs and transfection
The yellow cameleon (2.0 and 3.1) in pcDNA3 were a kind gift of Drs. R. Tsien and A. Miyawaki (Miyawaki et al., 1997). The GST-tagged yellow cameleon 2 proteins were purified from an Escherichia coli culture expressing the pGEX261 vector inserted with the yellow cameleon 2.0 into the HindIII and NotI sites. eYFP-PH(PLCδ1) and eCFP-PH(PLCδ1) in pcDNA3 expression vector were described elsewhere (van der Wal et al., 2001). Transfections were performed using calcium phosphate precipitate, at ∼0.8 μg DNA/well. After overnight transfection, cells were washed with fresh medium and incubated until usage.
Fluorometry
For fluorometry, a dual-emission channel Quantamaster fluorometer (Photon Technology International, Lawrenceville, NJ) was used. Purified fluorescent proteins were dissolved at a final concentration of ∼1 μM in HEPES-buffered intracellular solution. Free [Ca2+] of the solution was set to 50 nM using BAPTA. Fluorescence was detected from 2-ml aliquots of solution, kept at 37°C in a stirred cuvette.
Confocal microscopy
For registration of images, coverslips with transfected cells were transferred to a culture chamber and mounted on the inverted microscope. The cells were kept in bicarbonate-buffered saline (containing in mM: 140 NaCl, 5 KCl, 1 MgCl2, 10 glucose, 1 CaCl2, 23 NaHCO3, and 10 HEPES, pH 7.2), under 5% CO2 at 37°C. Imaging was with a DM-IRE2 inverted microscope fitted with TCS-SP2 scanhead (Leica, Mannheim, Germany). CFP was excited at 430 nm and detected from 460 to 490 nm, and YFP was excited at 514 nm, and detected from 528 to 603 nm. Excitation power was ∼100–400 μW.
Image processing
Image acquisition and specimen refocusing were automated from within a custom-made Visual Basic (v6.0) program by calling commands from the Leica macro tool package. To obtain FRET images, the following post-acquisition image processing steps were carried out. First the imported images were shading-corrected, and optionally smoothed. Then regions of interest (ROIs) were designed corresponding with cells expressing only CFP or YFP. From these ROIs, correction factors were measured and calculated. With these factors, sensitized emission was calculated as outlined in Results and Discussion. The sensitized emission image was ratioed to the excitation intensity-corrected MDirectAcceptor or FDonor (see Appendix) image to obtain the apparent FRET efficiency picture. Images were scaled appropriately for onscreen visualization. To suppress excessive noise in dim parts of the images, a mask was applied as follows. First, the FRET efficiency image was smoothed with a spatial filter to distinguish noise from signal. Then, a mask was created by setting a threshold equal to the background from this image. Subsequently, unwanted noise in dim areas was rejected by applying this mask to the original, unfiltered FRET image.
RESULTS AND DISCUSSION
Experimental setup and corrective terms for sensitized emission
In the most general case, proteins with CFP and YFP labels are independently expressed in living cells. Relative fluorescence levels are thus not fixed, and pixel-to-pixel intensities may differ widely for each fluorophore. To image sensitized emission, acceptor fluorescence is to be detected while exciting the donor. However, due to spectral overlap the recorded image in the acceptor emission channel contains components of leakthrough of donor emission into the acceptor channel and of direct excitation of the acceptor at the donor excitation wavelength (Gordon et al., 1998; Nagy et al., 1998; Hoppe et al., 2002). Estimation of the latter term requires information on the acceptor distribution, which is gained by taking an additional image at acceptor excitation and emission wavelength. In the following treatment, it is assumed that detector gain and offset are correctly adjusted, and that autofluorescence of the cells is either negligible, or properly subtracted (for an excellent correction method, see Nagy et al., 1998). In the more extensive treatment given in the Appendix the individual factors that influence brightness of the images (such as PMT gain, laser intensities, the CFP and YFP quantum yield, etc.) are factored out to allow clear assessment of the influence of these factors.
Thus, provided that independent estimates of cross-talk magnitude are present, straightforward corrections can be carried out from three acquired images (denoted M for Measured): donor excitation with donor emission, MDonor; donor excitation with acceptor emission, MIndirectAcceptor; and acceptor excitation with acceptor emission, MDirectAcceptor.
The measured images are composite images consisting of multiple terms as follows: MDonor is the sum of fluorescence of the donor diminished by donor fluorescence lost to energy transfer (FDonor − FSen), and of leakthrough components consisting of fractions of FDirectAcceptor (the actual acceptor fluorescence) and of FSen, as
 |
(1) |
where α is the correction factor for acceptor fluorescence excited and detected at donor wavelength, and δ that for leakthrough of sensitized emission back into the donor filters.
MIndirectAcceptor represents the sum of fluorescence of energy transfer (FSen), leakthrough of the donor minus the component lost to energy transfer (FDonor − FSen), and of the directly excited acceptor (FAcceptor),
 |
(2) |
where β is the leakthrough factor of the fluorescence of donor into acceptor filters, and γ is the excitation efficiency of the acceptor upon excitation at donor wavelength.
Finally, MDirectAcceptor represents the acceptor fluorescence. Formally, a component consisting of donor fluorescence, excited and emitting at acceptor wavelengths, is present. However, using the 514-nm argon ion laser line and the CFP/YFP pair, the magnitude of this component is essentially zero. Thus,
 |
(3) |
To derive the sensitized emission, Eqs. 1 and 3 are combined as
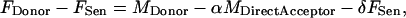 |
(4) |
and Eqs. 3 and 4 are substituted into Eq. 2, yielding
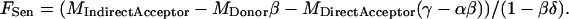 |
(5) |
For detailed derivation, see the Appendix (corresponding equation is Eq. A11). In Eq. 5, the parameters α, β, γ, and δ are effectively used as correction factors that must be determined independently. Estimates for α, γ, and δ can be obtained by imaging a sample with only acceptor molecules, and can then be calculated as
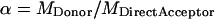 |
(6) |
 |
(7) |
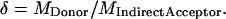 |
(8) |
Similarly, β is estimated from a sample with only donor molecules, as
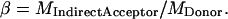 |
(9) |
To obtain an indication for apparent FRET efficiency, the derived expression for FSen (Eq. 5) can be related to the total acceptor levels as
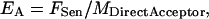 |
(10) |
or it can be related to the donor levels, which makes the calculated efficiency over time independent of laser fluctuations (see Appendix for further detail),
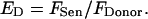 |
(11) |
It is evident that any changes in cell morphology (e.g., locomotion) that occur in between acquisition of the images will severely compromise the accuracy. Therefore, the images should be acquired in rapid succession by simultaneously detecting MDonor and MIndirectAcceptor, immediately followed by MDirectAcceptor at its own excitation line. When acquisition parameters are chosen with some care, the Leica TCS SP2 confocal system used here, controlled by our in-house developed macro program, is capable of grabbing a full-sized (512 × 512 pixels) set of images in two seconds.
It should be stressed that whereas the derived expressions for FRET efficiency allow direct comparison of FRET between different preparations and for different laser intensity and PMT settings, information on either the fraction of acceptor in complex with donor or the characteristic maximum FRET efficiency between donor and acceptor in complex is lacking. Since we anticipate this to be the reality in the vast majority of experiments, estimates of the actual fraction of donors and acceptors that engage in FRET (such as presented in FRET stoichiometry; Hoppe et al., 2002) cannot be derived from our data.
The confocal acquisition parameters
Although corrective factors for emission leakthrough and indirect excitation are analogous to those described for wide-field CCD imaging of FRET (Gordon et al., 1998), confocal acquisition introduces a major complication in that relative sensitivities for donor and acceptor emission of the detector channels are no longer fixed. With CCD acquisition, weaker fluorescent cells are imaged with increased integration time, causing both direct signals from the fluorophores as well as leakthrough terms to increase proportionally. Thus, leakthrough factors are fixed for a particular combination of fluorophores and filters, and alterations in integration time can be easily compensated for. In contrast, during confocal imaging, sensitivity is adjusted by finetuning the individual excitation line intensities and by controlling PMT gain (high voltage) and offset settings for each channel separately. In addition, since excitation sources and PMT channels are physically separate, drift will have differential effects on sensitivity for donor and acceptor fluorophores. Taken together, these factors necessitate that new estimates for α, β, γ, and δ be determined for each experiment, even if identical filter and pinhole settings are used from experiment to experiment. The advantage—on the other hand—is that the added flexibility allows simultaneous optimized acquisition of the often weak FRET signals without compromising acquisition time.
In the Appendix, dependence of the parameters α, β, γ, and δ on instrument settings is derived (Eqs. A7–A10). Note that parameters β and δ depend on signal amplifications in the utilized detector (PMT), which normally operate nonlinearly, and elements in the optical path (optical filter, spectral detection bands) only, whereas α and γ are additionally influenced by relative laser line intensities. Furthermore, from Eqs. A7–A10, it is seen that δ = α/γ. This relationship, as well as the dependencies of correction factors on PMT and/or laser intensity settings were verified experimentally by imaging cells expressing CFP or YFP under a variety of settings (data not shown, but available on request).
For experiments with cells expressing CFP- and YFP-tagged constructs at ∼1:1 stoichiometry, spectral detection bandwidth of the SP2 channels were set up to balance minimal cross talk with optimal collection efficiency of CFP and YFP (460–490 nm and 528–603 nm, respectively). Under these conditions, typical ranges for the parameter values were 0.00001 < α < 0.0005; 0.2 < β < 1.5; 0.02 < γ < 0.5; and 0.0003 < δ < 0.003. However, at different expression stoichiometry, or when spatial distribution is very inhomogeneous for one of the fluorophores, selection of widely different instrument settings may be favorable, with consequent large changes in α, β, γ, and δ. Thus, optimized instrument settings for cells expressing low CFP and high YFP levels caused large δ- and α-values, whereas cells expressing high CFP and low YFP resulted in δ and α being negligibly small. In the latter case, Eq. 5 may be simplified to the numerator.
As outlined above, parameters α, β, γ, and δ are determined by imaging cells expressing either CFP or YFP alone. Stochastical errors in the calculated values for either of these parameters systematically bias the FRET efficiency results over the entire image, and should therefore be minimized. Thus, it is important to obtain the parameter values from extended image regions, averaging out statistic fluctuations over many pixels. The correction factor δ is particularly sensitive to noise because it is calculated by dividing MDonor by MIndirectAcceptor (Eq. 8) from a cell expressing only YFP. Both of these images are very dim, because they stem from acceptor molecules excited at donor wavelength (430 nm). Since δ depends on filter and PMT settings, but not on relative laser line intensities (see Eq. A9), δ may be acquired using increased laser power or with 514-nm excitation. In practical experiments, errors in calculated FRET efficiencies for each pixel are dominated by the rather large noise in the MDonor, MDirectAcceptor, and MIndirectAcceptor images, with stochastical variations in the parameter values contributing <1%, on average.
CFP excitation for sensitized emission is optimal at 430 nm
The 458-nm and 514-nm Argon ion laser lines have been used (He et al., 2003; Karpova et al., 2003) to excite CFP and YFP in FRET experiments. However, as deduced from the excitation spectra of these fluorophores (see Appendix Fig. 1), the 458-nm line overlaps considerably with the YFP excitation spectrum, resulting in direct acceptor excitation and poor discrimination. To determine the optimal wavelength for CFP excitation in sensitized emission experiments, we expressed and purified the CFP/YFP-based Ca2+ sensor yellow cameleon (Miyawaki et al., 1997) from bacteria. When dissolved at ∼1 μg/ml in a Ca2+-free intracellular buffer solution, this construct shows little FRET in the fluorometer. Upon addition of 1 mM Ca2+, a robust and reliable increase in FRET is detected. In a series of experiments, the excitation wavelength was varied in the range of 340–452 nm, and both the magnitude of the CFP emission, as well as the magnitude of the Ca2+-induced FRET change (defined as percent change in the ratio YFP/CFP induced by Ca2+) were recorded (Fig. 1, A and B). It is apparent from Fig. 1 A that FRET changes are most efficiently detected at excitation wavelength below 432 nm, whereas direct YFP excitation caused the Ca2+-induced change in ratio to drop dramatically at higher wavelength. Conversely, decreasing wavelength below ∼425 nm had little effect on Ca2+-induced ratio changes but significantly reduced CFP excitation. From Fig. 1, it can be concluded that optimal excitation to resolve FRET changes is at 432 nm.
FIGURE 1.

Optimization of CFP excitation wavelength to resolve FRET from CFP/YFP. Yellow cameleon 2.0 was expressed and purified from bacteria, and introduced at ∼1 μM in a 2-ml cuvette in a spectrofluorometer. FRET changes were measured upon increasing the Ca2+ concentration from 50 nM to 1 mM. (A) Excitation efficiency of CFP (black line) and Ca2+-induced change in YFP/CFP emission intensity (shaded line) are plotted as a function of wavelength. (B) The efficacy of various excitation wavelengths in resolving FRET changes was approximated by multiplying the excitation efficiency with the efficiency to resolve Ca2+-induced ratio changes. Note the considerable decline at wavelengths longer than 432 nm. (C) Sensitized emission of yellow cameleon was imaged using either 430-nm (left panel) or 458-nm (right panel) CFP excitation, and 514-nm YFP excitation. The average sensitized emission in the cytosol was 111 ± 40 with 430-nm excitation and 48 ± 41 with 458-nm excitation (8-bits grayscale).
We employ a 10-mW Melles Griot (Irvine, CA) type 58-BTL-008 frequency-doubled diode laser to excite CFP at 430 nm on our Leica TCS-SP2 confocal microscope. YFP excitation is by the 514-nm Argon laser line. The use of 430-nm CFP excitation, rather than the more commonly used 458-nm excitation, also allows collection of a larger part of the CFP emission spectrum, resulting in brighter CFP images. Together with the aforementioned optimal discrimination between CFP and YFP, this significantly increases the signal/noise ratio. Fig. 1 C shows FRET images of a cell that expresses yellow cameleon using either 430-nm or 458-nm excitation.
Correcting misfocusing deviations
As the calculation of FSen involves mathematical operations based on three raw images, it is of the utmost importance that these channels spatially overlap tightly, both in lateral and in axial direction. Compared with wide-field microscopy, the focusing deviations—i.e., deviations that occur if donor and acceptor images are offset in the axial direction—are emphasized by the confocals' inherent optical sectioning. The CFP and YFP images are effectively taken from slightly different planes in the cell (Fig. 2 A), causing erroneous results during calculation of the sensitized emission, resulting in pixels with extreme high or low FRET efficiencies (Fig. 2 B). Two main sources for this type of deviation exist: chromatic aberrations within the objective and other optics, and slight differences in the collimation of the laser beams. Chromatic aberrations are due to the wavelength dependency of the refractive index of optical glasses, which causes axial misregistration of images taken at different wavelengths (Cogswell and Larkin, 1995). Depending on the objective used, chromatic aberrations may be several micrometers (worst case). Chromatically corrected objectives are available, but it should be stressed that these are optimized only for a limited spectral range, typically in the midvisible range. Therefore, significant chromatic aberration may still be present at 430 and 458 nm.
FIGURE 2.

Axial misregistration of images using 430- and 514-nm laser lines. (A) A confocal X/Z image of the green emission (∼525 nm) of a 0.17-μm bead was registered using a HCX PL APO CS 63× objective upon 430-nm (blue line) and 514-nm (red line) excitation. The profiles of fluorescence intensities, detected at 525 nm, demonstrate the axial misregistration. (B) Confocal images were acquired from a cell expressing CFP- and YFP-tagged pleckstrin homology (PH) domains, with or without using the refocusing macro routine, and FRET efficiency images were determined (lower and upper photomicrograph, respectively). The intensity profiles plotted along the indicated line (red, uncorrected routine; blue, refocusing routine) show the extreme FRET values in the profile from the uncorrected FRET image (arrows).
Using a good, standard corrected 63× magnification, 1.32-NA oil immersion objective (HCX PL APO CS, #506180, Leica), we noticed focusing deviations of ∼400 nm (Fig. 2 A). Use of a UV-corrected 63× magnification objective (HCX PL APO lbd.BL, #506192, Leica) significantly, but not completely, remedied this chromatic aberration. Chromatic focusing deviations are not limited to violet wavelengths because significant deviations exist for dye pairs excited throughout the visible spectrum (Table 1).
TABLE 1.
Differences in focus distance between commonly used laser line pairs using a standard 63×, 1.32 NA oil immersion objective
| Laser line pair (nm) | Distance (μm) |
|---|---|
| 458/514 | 0.3 |
| 488/568 | 0.17 |
| 514/633 | 0.04 |
Slight collimation differences between the laser beams are the second source of focusing inaccuracies, in particular if donor and acceptor excitation wavelength are derived from separate lasers. Lasers which are coupled via separate collimation lenses are normally optimized for three-dimensional resolution. This causes the focal plane of excitation to vary depending on excitation wavelength, resulting in an offset between the images and also in inefficient excitation with consequent unnecessary specimen bleaching. In principle, slight adjustments in collimation of one beam could be used to correct the objective chromatic aberration, at least partially. The lower-wavelength beam can be adjusted to be a little bit more divergent, which compromises three-dimensional resolution, but brings the different focus planes nearer to each other. However, this is not a practical solution, as chromatic aberrations vary with lens types, and even for different objectives of the same type (L. Oomen and K. Jalink, unpublished; Zucker and Price, 2001).
To provide a more generic approach to overcome focusing deviations, we used the fine focusing capacity of the Z-galvanometer of the microscope stage. First, MDonor and MIndirectAcceptor images were recorded using 430-nm excitation. Then, before taking the MDirectAcceptor image with 514-nm excitation, the preparation is refocused to minimize chromatic aberration. Because for a given combination of objective and excitation lines the focus deviation is constant, the correction distance needs to be determined only once. We used x/z-scanning of fixed cells or fluorescent beads for this goal (Fig. 2 A). Applying this focus correction in an automated acquisition routine (macro), MDonor, MIndirectAcceptor, and MDirectAcceptor images are collected from the same focal plane in the biological sample. Thus, the FRET efficiency calculated from images acquired in this manner is effectively corrected for misfocusing as shown in Fig. 2 B.
Lateral image errors
Lateral image errors occur when raw images do not overlap precisely in the image plane (x/y direction). Both geometric and intensity errors may occur. Geometric errors are most apparent at the borders of the image, and errors of this type can best be avoided by zooming in slightly. Lateral intensity errors may be present over the entire image and occur on CCD and confocal systems alike. For CCD systems, a standard correction algorithm exists: corrections are carried out by normalizing pixel intensities using a reference image, a procedure called shading correction (Tomazevic et al., 2002). On the confocal system with independent excitation lines, these corrections are slightly more complex because spatial excitation intensities may vary independently (L. Oomen, L. Brocks, and K. Jalink, unpublished; Zucker and Price, 2001) and similar effects also occur in the detection path. This necessitates that both channels be normalized by shading correction.
For 430- and 514-nm lines, excitation inhomogeneities were measured by registration of reference images of a solution of the FRET calcium sensor yellow cameleon (Miyawaki et al., 1997). We observed significant deviations from unity flatness: 430-nm excitation intensity dropped by as much as 50% at the image corners, whereas 514-nm deviated by ∼15% (Fig. 3 A, left panels). Importantly, significant differences (up to 20%) may also occur over the center of the images. Deviations of this magnitude are not uncommon in confocal systems (Zucker and Price, 2001), although they can be diminished by increasing the zoom factor. Therefore, shading correction was routinely applied to MDonor and MIndirectAcceptor by normalizing to the 430-nm reference, and to MDirectAcceptor by normalizing to the 514-nm reference image. This completely corrects for lateral fluorescence inhomogeneities (Fig. 3, A and B, right panels). To illustrate the impact of shading correction on practical experiments, the γ-values calculated for two cells expressing YFP-PH are also indicated in Fig. 3 B. Whereas in the uncorrected images (left panels) these values differ by as much as 50%, shading correction (right panels) effectively canceled out the differences. Consequently, we used shading-corrected images to determine FSen as well as the correction factors throughout this study.
FIGURE 3.

Lateral image errors. (A) Shown are parts of reference images that were acquired by averaging eight confocal images of a solution of yellow cameleon at 430-nm excitation (upper left panel) and at 514-nm excitation (lower left panel). The right panels demonstrate shade correction of (single-pass) confocal images through division by the respective normalized reference images. (B) Two cells expressing YFP-PH were registered with 430- and 514-nm excitation (upper and lower left panels, respectively). Using the shade correction reference image, fluorescence inhomogeneities were corrected (right panels). The γ-values, calculated according to Eq. 7, are indicated for the two cells. Note that the differences in γ-values in the uncorrected images are remedied in the right panel.
Temporal errors: laser intensity fluctuation
Unstable excitation sources generate temporal intensity variations. Excitation stability is extremely important because the correction factors α and γ depend on relative laser line intensities. We observed considerable drift and slow oscillations (at a timescale of one to several minutes) in excitation line intensity on several different confocal systems (Fig. 4 A). Changes of several percent are common, whereas worst-case variations of up to 20% are detected in poorly aligned systems. Importantly, individual laser line intensity variations are independent, even for different lines from the same laser. Although intensity variations may also occur in arc lamps from wide-field fluorescence microscopes, these changes are often much smaller (compare Fig. 4, A and B). Furthermore, slow arc lamp intensity variations affect the three raw images to the same degree if images are gathered in rapid succession, and thus have no effect on the apparent FRET image (Eqs. 10 and 11).
FIGURE 4.

Temporal intensity variations in excitation sources. The intensity of a 514-nm argon ion laser line (A) and a mercury arc lamp (B) were measured every 20 s for a 3-h time period and plotted after normalization. (C) A mixed population of cells expressing yellow cameleon, YFP-PH, or CFP-PH was imaged and analyzed for sensitized emission. The FRET efficiency (ED) and the correction factor γ (shaded line) are plotted versus time. FRET efficiency was calculated using a single fixed (red line) γ-factor and the online-updated (black line) γ-factor. After 15 min a large intensity fluctuation in the 514-nm laser line was simulated by manually diminishing laser power with ∼60%. (D) To correct for variations in excitation intensity, the leakthrough factors were determined in every pair of images from regions of interest (ROIs) corresponding to cells expressing either CFP-PH (blue ROI) or YFP-PH (red ROI), as detailed in the text. The calcium ionophore ionomycin and 2 mM extra Ca2+ were added to the medium to increase the FRET signal, and to translocate the PH-chimeras to the cytosol. Factors α, β, γ, and δ in the first acquired images were 0.00005, 0.47, 0.04, and 0.0013, respectively.
The independent variations in laser line intensity on confocal systems pose a major problem for timelapse FRET measurements. This is illustrated in Fig. 4 C, where the FRET efficiency (ED) of Yellow cameleon was followed over time (red line). Although in these unstimulated cells the FRET efficiency remains constant over time, the independent intensity variation of the 430 and 514-nm laser line cause fluctuations in ED. A supplier-installed stabilization system improved the excitation stability considerably, but not completely. In particular when expected FRET signals are a small fraction of the total fluorescence, the realized stability of ∼3% hampers acquisition of meaningful results. We therefore implemented an online correction scheme by recalculating the leakthrough factors for each image. To this goal, the cells under study were plated together with a mix of cells expressing either CFP or YFP on the same coverslip. In an image taken at low zoom factor, regions of interest (ROIs) were assigned to single CFP- or YFP-transfected cells (Fig. 4 D). From these ROIs, correction factors were determined as detailed in Eqs. 6–9. Sensitized emission was than calculated using these correction factors from cells expressing both CFP and YFP within the same image, or from a separate image collected at higher zoom factor. Provided that proper shading correction is carried out (see Lateral Image Errors), this procedure completely removed the effect of laser fluctuations, resulting in superior registration of FRET during acquisition of timelapse series (Fig. 4 C, black line).
Post-acquisition analysis
Having compensated for the most important sources of confocal acquisition deviations, significant improvements in image quality may still be obtained by post-acquisition procedures. The prime consideration is noise present in the images. Since photon noise is Poison-distributed (with the standard error being the square root of the number of photons), its effects will be most evident in low intensity regions of the image (see Fig. 5). In these regions, noise will be emphasized by image arithmetic, because it leads to extreme values in ratios as well as subzero intensity values in subtractions. This causes pixels with “false” high FRET values to appear in dim image regions (Fig. 5, upper right panel). Therefore, care must be taken to acquire MDonor, MIndirectAcceptor, and MDirectAcceptor images with a good signal/noise ratio. This can be accomplished in a number of ways on the confocal, including increasing the laser power which allows using lower PMT voltage, averaging of acquired images, and opening the pinhole. However, these measures come at the expense of increased fluorophore bleaching, prolonged imaging time, and degraded resolution.
FIGURE 5.

Sensitized emission calculated from noisy confocal images. Confocal images of cells expressing CFP-PH, YFP-PH, or both were registered as in Fig. 4. (Upper left panel, CFP image; middle left panel, YFP image; and lower left panel, corrected sensitized emission.) Apparent FRET efficiency (upper right panel) was calculated as detailed in the text. Note the appearance of excessive noise in dim areas of the FRET image. The calculated FRET efficiency image was smoothed with a spatial filter to distinguish noise from signal and a threshold equal to background intensity (dotted line) was applied to this image to reject pixels without FRET signal (middle right panel). Applying this mask to the original nonsmoothed FRET image effectively rejects the noise (lower right panel; compare to the upper right panel to assess the noise rejection introduced by this step). Factors α, β, γ, and δ were 0.0004, 0.65, 0.46, and 0.001, respectively.
Post-acquisition spatial filtering (smoothing) can also be applied to reduce noise, but this will equally degrade the resolution and blur fine details. To abolish the incidence of false high FRET values in dim image regions, while simultaneously circumventing image blurring in the other regions, a masking technique was applied (Fig. 5, middle right panel). In the apparent FRET image, resonance can be distinguished from noise by smoothing the image with a spatial filter. Isolated noise pixels are averaged out, whereas consecutive adjacent pixels with positive FRET remain visible. Setting a threshold to just above background intensity, a mask is then generated from this image that contains only regions of true FRET. This mask is subsequently applied to the original, unfiltered FRET image. Fig. 5 (middle and lower right panels) illustrates that this approach results in near-complete rejection of noise pixels.
Final remarks
Confocal microscopy has a number of advantages over wide-field fluorescence microscopy, the most important of which is that it records thin optical sections from the preparation. We therefore aimed to optimize confocal imaging of FRET between CFP and YFP by detecting sensitized emission. Earlier confocal studies focused on acceptor photobleaching (Karpova et al., 2003), and main confocal suppliers now support this application with dedicated software. However, acceptor photobleaching is a destructive technique that cannot be used for timelapse studies, and we therefore focused on detecting sensitized emission. In this study, it was demonstrated that a 430-/514-nm excitation line pair outperforms the more commonly used 458-/514-nm lines (Karpova et al., 2003) by discriminating better between CFP and YFP, resulting in a marked decrease in noise of the FRET image. We also identified a number of confocal-specific error sources that complicate the leakthrough correction schemes commonly applied in wide-field fluorescence microscopy.
In this study, an approach was introduced to compensate for individual laser line intensity fluctuations, leakthrough, and detector gain by simultaneous imaging of cells expressing either CFP or YFP alone, present within the same image field. This complicates the experimental design because it requires the user to establish co-cultures with CFP- and YFP-expressing cell lines, unless in the cell under study reliable regions can be identified that contain either CFP or YFP fluorescence only. However, the advantages are numerous, because the online calibration procedure not only compensates for excitation intensity fluctuations, but also allows semiquantitative assessment of FRET efficiency, independent of system settings such as PMT gain. Importantly, this enables direct comparison of FRET values from experiment to experiment, even when detector gain and laser intensities have been adjusted by the user.
Careful consideration of the practical implementation of automated acquisition and analysis steps in the macro is necessary. For example, within a timelapse series, the correction factor δ, that is updated along with the other parameters, is deduced from division of MDonor by MIndirectAcceptor from a cell expressing YFP only. Both images are very dim, because they stem from acceptor molecules excited at donor wavelength, and this may result in some noise in consecutive determinations of δ. The independence of this parameter on relative laser line intensities (see Appendix) allows δ to be determined just once, e.g., at the onset, for the whole timelapse series, if needed at increased laser power or using the 514-nm laser line, as long as no further adjustments are made to the instrument during the experiment.
In summary, online corrected confocal imaging is a fast, sensitive, and straightforward approach to detect sensitized emission from the CFP/YFP pair. The speed is particularly important for live cell imaging, since it minimizes artifacts due to movement of organelles during acquisition. Furthermore, the introduced corrective approaches can be readily adapted to other FRET pairs.
Acknowledgments
We thank Drs. B. Ponsioen (Department of Biochemistry), A. Griekspoor, and L. Oomen (Department of Tumor Biology), and members of the Department of Cell Biology for stimulating discussions and critical reading of the manuscript. Drs. L. Oomen and L. Brocks (Department of Tumor Biology) are acknowledged for sharing unpublished data and Dr. T. W. Gadella (University of Amsterdam, Amsterdam, The Netherlands) for inspiring discussions. We also thank Drs. F. Olschewski and R. Borlinghaus (Leica Microsystems, Mannheim, Germany) for continued support and discussions and Drs. A. Miyawaki and R. Y. Tsien for the kind gift of plasmids.
This work was supported by Netherlands Organization for the Advancement of Pure Research grant 901-02-236.
APPENDIX: IMAGING FRET BY SENSITIZED EMISSION
In this Appendix, we will assume 430-nm donor and 514-nm acceptor excitation to image FRET from the CFP/YFP pair. As detailed in the text, three images are collected that allow independent estimates of cross-talk magnitude to perform correction of leakthrough: 430-nm excitation with CFP emission, MDonor; 430-nm excitation with YFP emission, MIndirectAcceptor; and 514-nm excitation with YFP emission, MDirectAcceptor.
The acquired images are composite images that consist of multiple terms (see Appendix Fig. 1; for symbols, see Appendix Table 1) as described in the following equations.
APPENDIX FIGURE 1.

Spectral overlap of CFP and YFP. Emission spectra of CFP and YFP were recorded on a spectrofluorometer from bacterially expressed purified protein. Note that the two fluorophores have considerable spectral overlap. The graph also illustrates the difference in excitation efficiency of YFP depending on the method of excitation; 514 nm (dark shaded line), 430 nm (black line), and FRET (light shaded line); not to-scale.
APPENDIX TABLE 1.
Glossary of used symbols
| Factor | Name | Description |
|---|---|---|
| Laser | ɛ430CFP | Excitation efficiency of CFP with 430 nm |
| ɛ430YFP | Excitation efficiency of YFP with 430 nm | |
| ɛ514YFP | Excitation efficiency of YFP with 514 nm | |
| Fraction (Spectral) | A | Fraction of CFP spectrum in the CFP channel |
| B | Fraction of YFP spectrum in the CFP channel | |
| C | Fraction of CFP spectrum in the YFP channel | |
| D | Fraction of YFP spectrum in the YFP channel | |
| PMT detector | g1 | Scaling factor relating fluorescence to donor channel grayscale value |
| g2 | Scaling factor relating fluorescence to indirect acceptor channel grayscale value | |
| g3 | Scaling factor relating fluorescence direct acceptor channel grayscale value | |
| Quantum yield | QCFP | The quantum yield of CFP is 0.40 (Tsien, 1998) |
| QYFP | The quantum yield of YFP (Citrine) is 0.76 (Griesbeck et al., 2001) |
MDonor is the output grayscale value after amplification by the PMT I detector (g1) of the sum of the fraction (A) of CFP fluorescence in the CFP channel and the fraction (B) of YFP fluorescence in the CFP channel. The fluorescence of CFP depends on the number of CFP molecules (NCFP), the 430-nm laser excitation (ɛ430CFP), the quantum yield (QCFP), diminished by the number of CFP molecules that lose their excited state energy due to FRET (NSen). Note that ɛ430CFP models both the laser intensity and the excitation efficiency of CFP at 430 nm. The fluorescence of YFP depends on it's quantum yield (QYFP), and the sum of the number of YFP molecules (NYFP) excited with 430-nm laser (ɛ430YFP) and those excited by FRET (NSen). Because the relaxation of excited CFP molecules by FRET results in equal amounts of excited YFP molecules, both pools are denoted by NSen. Since resonance is due to excited CFP, NSen is also dependent on the excitation efficiency of CFP at 430 nm (ɛ430CFP), as
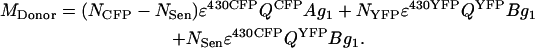 |
(A1) |
MIndirectAcceptor is the output grayscale value after the PMT II detector scaling (g2) of the sum of the fractions of CFP fluorescence in the YFP channel (C) and of YFP fluorescence in the YFP channel (D). The CFP fluorescence depends on QCFP, the 430-nm laser excitation efficiency (ɛ430CFP), the number of CFP molecules (NCFP), and the CFP molecules that lose their energy by FRET (NSen). The fluorescence of YFP depends on QYFP, the amount of YFP molecules (NYFP) excited with 430-nm laser (ɛ430YFP), and on the amount of YFP molecules excited by FRET (NSen, which is linear to ɛ430CFP), as
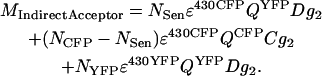 |
(A2) |
Finally, MDirectAcceptor is the output grayscale value after the PMT III detector scaling (g3) of the YFP fluorescence in the YFP channel (D), which depends on the quantum yield of YFP (QYFP) and the amount of YFP molecules (NYFP) excited with 514 nm (ɛ514YFP). Note that PMT III generally will be the same physical detector as PMT II, but operated at a different gain setting. Formally, donor fluorescence, excited with 514 nm is also present. However, using the 514-nm argon laserline, the magnitude of this component is essentially zero. Thus,
 |
(A3) |
To derive the sensitized emission, Eqs. A1 and A3 are combined as
 |
(A4) |
and Eqs. A3 and A4 are substituted into Eq. A2, yielding
 |
(A5) |
Relating back to Eq. 5 from Results and Discussion, the sensitized emission grayscale image FSen is composed of the emission from NSen, which depends on the quantum yield of YFP (QYFP), scaled by factors for PMT II gain (g2), fraction of YFP fluorescence in the YFP channel (D), and CFP excitation efficiency ɛ430CFP, as
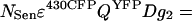 |
(A6) |
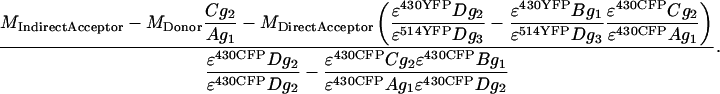 |
In Eq. A6, the constants α, β, γ, and δ (see Results and Discussion) are identified as detailed in Eqs. A7–A10. Values for α, γ, and δ can be deduced from imaging of a sample with only acceptor molecules,
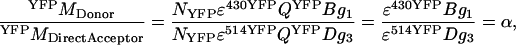 |
(A7) |
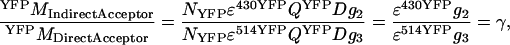 |
(A8) |
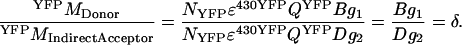 |
(A9) |
Similarly, β is calculated from a sample with only donor molecules, as
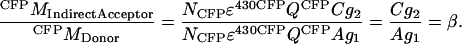 |
(A10) |
Note that in contrast to β and δ, α and γ depend on the relative laser line intensities.
Analogous to Eq. 5, we can thus rewrite Eq. A6 as
 |
(A11) |
To obtain an indication for FRET efficiency, the derived expression for FSen (Eq. A11) can be related to the total acceptor level, or to the total donor level. Depending on the biological application, either way may have specific advantages. Relating FSen to FDirectAcceptor, the expression for efficiency becomes
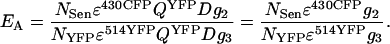 |
(A12) |
This corresponds to Eq. 10 from Results and Discussion. It is evident that EA depends on the excitation of YFP at both laser lines and on PMT II and III settings. Therefore, EA is useful to compare FRET efficiencies within a cell, or between different cells in the same image, but not to compare efficiencies when excitation intensities or PMT settings may have changed unless an additional correction is introduced to compensate for such changes. The magnitude of this corrective term is
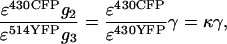 |
(A13) |
where κ is a constant relating the efficiency of CFP excitation by the 430-nm laser line to that of YFP (using our settings, κ = ∼15).
Alternatively, when FRET efficiency is expressed by relating FSen to FDonor, the results become directly independent from excitation intensity and PMT settings. To arrive at an expression for ED, the loss of signal due to FRET from the grayscale image MDonor is related to the total (i.e., when no FRET occurs) donor grayscale image,
 |
(A14) |
where in the numerator, the emission lost (the CFP quantum yield times NSen, see Eq. A5) is scaled by factors for PMT I gain (g1), fraction of CFP fluorescence in the CFP channel (A), and CFP excitation efficiency ɛ430CFP. Analogous to Eq. 6,
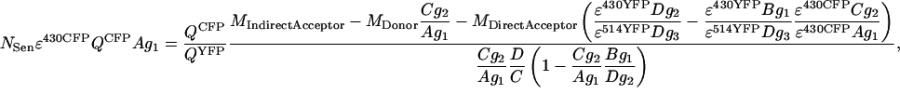 |
(A15) |
which, using Eqs. A7–A10, can be rewritten to
 |
(A16) |
In the divisor of Eq. A14, the expression for the grayscale image of the total donor fluorescence is
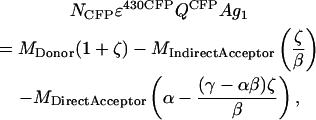 |
(A17) |
where
 |
Note that ζ does not depend on photomultiplier gain settings or laser intensity fluctuations, because βδ = BC/AD. For a given combination of confocal filter settings and fluorophores ζ is therefore a constant (for our settings, ζ = −0.248). It can be reliably determined by acquiring the MDonor, MIndirectAcceptor, and MDirectAcceptor images before and after complete acceptor photobleaching. Since postbleach MDonor is equal to NCFPɛ430CFPQCFPAg1 (see Eq. A1), ζ can be determined by rewriting Eq. A17 as
 |
(A18) |
Equation A17 is derived by combining Eqs. A1, A3, and A11,
 |
(A19) |
Equation A19 can be rewritten as
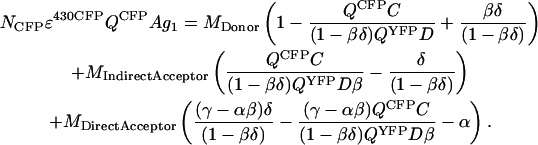 |
(A20) |
The constant ζ is brought outside the parentheses,
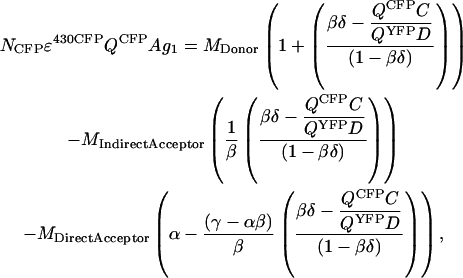 |
(A21) |
References
- Cogswell, C. J., and K. G. Larkin. 1995. The specimen illumination path and its effect on image quality. In Handbook of Biological Confocal Microscopy. J. B. Pawley, editor. Plenum Press, New York.
- Gordon, G. W., G. Berry, X. H. Liang, B. Levine, and B. Herman. 1998. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 74:2702–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck, O., G. S. Baird, R. E. Campbell, D. A. Zacharias, and R. Y. Tsien. 2001. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276:29188–29194. [DOI] [PubMed] [Google Scholar]
- He, L., T. D. Bradrick, T. S. Karpova, X. Wu, M. H. Fox, R. Fischer, J. G. McNally, J. R. Knutson, A. C. Grammer, and P. E. Lipsky. 2003. Flow cytometric measurement of fluorescence (Förster) resonance energy transfer from cyan fluorescent protein to yellow fluorescent protein using single-laser excitation at 458 nm. Cytometry. 53A:39–54. [DOI] [PubMed] [Google Scholar]
- Honda, A., S. R. Adams, C. L. Sawyer, V. Lev-Ram, R. Y. Tsien, and W. R. Dostmann. 2001. Spatiotemporal dynamics of guanosine 3′,5′-cyclic monophosphate revealed by a genetically encoded, fluorescent indicator. Proc. Natl. Acad. Sci. USA. 98:2437–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe, A., K. Christensen, and J. A. Swanson. 2002. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys. J. 83:3652–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova, T. S., C. T. Baumann, L. He, X. Wu, A. Grammer, P. Lipsky, G. L. Hager, and J. G. McNally. 2003. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy. J. Microsc. 209:56–70. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum, New York.
- Miyawaki, A., J. Llopis, R. Heim, J. M. McCaffery, J. A. Adams, M. Ikura, and R. Y. Tsien. 1997. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 388:882–887. [DOI] [PubMed] [Google Scholar]
- Mochizuki, N., S. Yamashita, K. Kurokawa, Y. Ohba, T. Nagai, A. Miyawaki, and M. Matsuda. 2001. Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature. 411:1065–1068. [DOI] [PubMed] [Google Scholar]
- Nagai, Y., M. Miyazaki, R. Aoki, T. Zama, S. Inouye, K. Hirose, M. Iino, and M. Hagiwara. 2000. A fluorescent indicator for visualizing cAMP-induced phosphorylation in vivo. Nat. Biotechnol. 18:313–316. [DOI] [PubMed] [Google Scholar]
- Nagy, P., G. Vamosi, A. Bodnar, S. J. Lockett, and J. Szollosi. 1998. Intensity-based energy transfer measurements in digital imaging microscopy. Eur. Biophys. J. 27:377–389. [DOI] [PubMed] [Google Scholar]
- Pepperkok, R., A. Squire, S. Geley, and P. I. Bastiaens. 1999. Simultaneous detection of multiple green fluorescent proteins in live cells by fluorescence lifetime imaging microscopy. Curr. Biol. 9:269–272. [DOI] [PubMed] [Google Scholar]
- Tomazevic, D., B. Likar, and F. Pernus. 2002. Comparative evaluation of retrospective shading correction methods. J. Microsc. 208:212–223. [DOI] [PubMed] [Google Scholar]
- Tsien, R. Y. 1998. The green fluorescent protein. Annu. Rev. Biochem. 67:509–544. [DOI] [PubMed] [Google Scholar]
- van der Wal, J., R. Habets, P. Varnai, T. Balla, and K. Jalink. 2001. Monitoring agonist-induced phospholipase C activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 276:15337–15344. [DOI] [PubMed] [Google Scholar]
- Wouters, F. S., P. J. Verveer, and P. I. Bastiaens. 2001. Imaging biochemistry inside cells. Trends Cell Biol. 11:203–211. [DOI] [PubMed] [Google Scholar]
- Zaccolo, M., and T. Pozzan. 2002. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 295:1711–1715. [DOI] [PubMed] [Google Scholar]
- Zucker, R. M., and O. Price. 2001. Evaluation of confocal microscopy system performance. Cytometry. 44:273–294. [DOI] [PubMed] [Google Scholar]