

Abstract
The aim of this study was to use both a theoretical and experimental approach to determine the influence of the sarco-endoplasmic Ca2+-ATPase (SERCA) activity and mitochondria Ca2+ uptake on Ca2+ homeostasis in airway myocytes. Experimental studies were performed on myocytes freshly isolated from rat trachea. [Ca2+]i was measured by microspectrofluorimetry using indo-1. Stimulation by caffeine for 30 s induced a concentration-graded response characterized by a transient peak followed by a progressive decay to a plateau phase. The decay phase was accelerated for 1-s stimulation, indicating ryanodine receptor closure. In Na2+-Ca2+-free medium containing 0.5 mM La3+, the [Ca2+]i response pattern was not modified, indicating no involvement of transplasmalemmal Ca2+ fluxes. The mathematical model describing the mechanism of Ca2+ handling upon RyR stimulation predicts that after Ca2+ release from the sarcoplasmic reticulum, the Ca2+ is first sequestrated by cytosolic proteins and mitochondria, and pumped back into the sarcoplasmic reticulum after a time delay. Experimentally, we showed that the [Ca2+]i decay after Ca2+ increase was not altered by the SERCA inhibitor cyclopiazonic acid, but was slightly but significantly modified by the mitochondria uncoupler carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone. The experimental and theoretical results indicate that, although Ca2+ pumping back by SERCA is active, it is not primarily involved in [Ca2+]i decrease that is due, in part, to mitochondrial Ca2+ uptake.
INTRODUCTION
Cell calcium homeostasis and calcium signaling are the result of the dynamic interactions between the ON mechanisms that lead to cytosolic Ca2+ concentration increase via extracellular calcium influx and/or calcium release from intracellular Ca2+ stores and the OFF mechanisms that tend to decrease [Ca2+]i (Berridge et al., 2000). Although it is known that Ca2+ pumping back by sarco-endoplasmic Ca2+-ATPase (SERCA) and pumping out by plasma membrane Ca2+-ATPase (PMCA) and Na+-Ca2+ exchanger are not the unique processes involved in the OFF mechanisms and that mitochondrial Ca2+ uptake and Ca2+ binding to cytosolic proteins may also participate in the maintenance of a low [Ca2+]i, SERCA and PMCA are generally considered as the major and the first structures implicated in [Ca2+]i decrease after [Ca2+]i increase due to cell stimulation (Berridge et al., 2000; Sanders, 2001). However, although theoretical studies have pointed out that Ca2+ binding to cytosolic proteins and mitochondrial uptake may greatly influence the Ca2+ response pattern (Marhl et al., 2000), few studies have been performed to investigate the relative participation of SERCA activity and mitochondrial Ca2+ uptake in Ca2+ signaling of airway smooth muscle cells.
Caffeine is a cell-permeant agonist of ryanodine receptors (RyR) that is largely used to investigate calcium signaling. In particular, pharmacological stimulation of RyRs by caffeine can be used to trigger calcium release from the sarcoplasmic reticulum and investigate the mechanisms activated by the initial [Ca2+]i increase (Guibert et al., 1996; Roux et al., 1998; Drummond and Tuft, 1999; Pacher et al., 2000; Vallot et al., 2001; Kamishima and Quayle, 2002). On the other hand, RyRs are expressed in a large variety of cells, including airway myocytes, and may be implicated in their physiological response (Kannan et al., 1997; Prakash et al., 1998). The understanding of the effect of pharmacological stimulation of ryanodine receptors by caffeine as well as their physiological involvement in cell stimulation would be greatly improved by use of a theoretical model of calcium handling upon RyR stimulation.
The aim of the present study was first to characterize the calcium response to caffeine stimulation in rat freshly isolated cells and to build a theoretical model of calcium handling upon caffeine stimulation based on these experimental results. The model included calcium release from the sarcoplasmic reticulum (SR) through RyR stimulation and pumping back by SERCAs. In vascular smooth muscle cells, several recent studies have shown that mitochondria Ca2+ uptake occurs after SR Ca2+ release and [Ca2+]i increase (Drummond and Fay, 1996; Drummond and Tuft, 1999; Pacher et al., 2000; Vallot et al., 2001; Kamishima and Quayle, 2002; Szado et al., 2003). It is also known that Ca2+ may bind to two classes of protein binding sites in the cytosol. The first class represents the buffering proteins such as parvalbumin, calbindin, and also C-domains of calmodulin, which bind calcium relatively slowly but with a high affinity (Falke et al., 1994; Smith et al., 1996). The second class, which is referred to as signaling proteins, comprises binding sites like N-domains of calmodulin that have very high rate constants of binding and dissociation with respect to calcium, but low affinity. Hence the model also included, as additional OFF mechanisms, mitochondrial Ca2+ uptake and Ca2+ binding to buffering proteins.
This model was further used, in combination with experimental investigations, to evaluate the involvement of these various Ca2+ intracellular compartments in Ca2+ dynamics upon Ca2+ release from the SR. In particular, we determined the influence of SERCA activity and mitochondrial Ca2+ uptake in comparison with the other buffering mechanisms on Ca2+ homeostasis and on [Ca2+]i variations after caffeine-induced [Ca2+]i increase.
MATERIALS AND METHODS
Tissue preparation
Rat tracheae were obtained from male Wistar rats 10–15 weeks old, weighing 300–400 g. For each experiment, a rat was stunned and killed by cervical dissociation. Heart and lungs were removed en bloc, and the trachea was rapidly dissected out. The muscular strip located on the dorsal face of the trachea was further dissected under binocular control, as previously described (Roux et al., 2002). The epithelium-free muscular strip was cut into several pieces and the tissue was then incubated overnight (14 h) in low-Ca2+ (200 μM) physiological saline solution (PSS; composition given below) containing 0.5 mg ml−1 collagenase, 0.35 mg ml−1 pronase, 0.03 mg ml−1 elastase, and 3 mg ml−1 bovine serum albumin at 4°C. After this time, the muscle pieces were triturated in a fresh enzyme-free solution with a fire polished Pasteur pipette to release cells. Cells were stored for 1–3 h to attach on glass coverslips at 4°C in PSS containing 0.8 mM Ca2+ and used on the same day. In control experiments, immunocytochemistry was performed using monoclonal mouse anti-smooth muscle α-actin antibodies and fluorescein-5′-isothiocyanate-conjugated anti-mouse immunoglobulin G antibodies to verify that the isolated cells obtained by dissociation were smooth muscle cells (data not shown).
Fluorescence measurement and estimation of [Ca2+]i
Changes in [Ca2+]i were monitored fluorimetrically using the Ca2+-sensitive probe indo-1 as previously described (Roux et al., 2002). Briefly, freshly isolated cells were loaded with indo-1 by incubation in PSS containing 1 μM indo-1 acetoxymethylester for 25 min at room temperature and then washed in PSS for 25 min. Coverslips were then mounted in a perfusion chamber and continuously superfused at room temperature. A single cell was illuminated at 360 ± 10 nm. Emitted light from that cell was counted simultaneously at 405 nm and 480 nm by two photomultipliers (P100, Nikon, Tokyo, Japan). [Ca2+]i was estimated from the 405/480 ratio using a calibration for indo-1 determined within cells.
Caffeine was applied to the tested cell by a pressure ejection from a glass pipette located close to the cell. No changes in [Ca2+]i were observed during test ejections of PSS (data not shown). Generally, each record of [Ca2+]i response to caffeine was obtained from a different cell. Each type of experiment was repeated for the number of cells indicated in the text.
Solutions, chemicals, and drugs
Normal PSS contained (in mM): 130 NaCl, 5.6 KCl, 1 MgCl2, 2 CaCl2, 11 glucose, 10 Hepes, pH 7.4 with NaOH. In Ca2+-free solution, Ca2+ was removed and 0.4 mM EGTA was added. In “Ca2+-confining” solution, Ca2+ and Na+ were omitted and 130 mM N-methyl-d-glucamine, 0.4 mM EGTA, and 0.5 mM LaCl3 were added to the solution. Such a solution is Ca2+ confining, i.e., inhibits any Ca2+ fluxes throughout the plasma membrane (Tribe et al., 1994), because removal of external Ca2+ inhibits any Ca2+ influx, removal of external Na+ inhibits the Na+/Ca2+ exchanger, and lanthanum is a nonspecific inhibitor of Ca2+-dependent transport activities, in particular the PMCA (Herscher and Rega, 1996) and the Na+/Ca2+ exchanger (Iwamoto and Shigekawa, 1998).
Collagenase (type CLS1) was from Worthington Biochemical Corp. (Freehold, NJ). Bovine serum albumin, cyclopiazonic acid (CPA), thapsigargin, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), and caffeine (CAF) were purchased from Sigma (Saint Quentin Fallavier, France). Indo-1 AM was from Calbiochem (France Biochem, Meudon, France). Indo-1 AM, CPA, and FCCP were dissolved in dimethyl sulphoxide, for which the maximal concentration used in our experiments was <0.1% and had no effect on the resting value of the [Ca2+]i nor on the variation of the [Ca2+]i induced by caffeine (data not shown).
Data analysis and statistics
Cytosolic calcium concentrations ([Ca2+]i) in control conditions are expressed as mean ± SE. For each protocol, experiments were repeated on two or more rats, control cells were obtained in each rat, and [Ca2+]i values recorded for each protocol are expressed as a percentage of the corresponding values obtained in control cells from the rats used for that protocol. [Ca2+]i values that characterize the Ca2+ response, i. e., baseline, peak, and plateau values, were compared between control and each specific experimental condition using unpaired student t-tests (peak and plateau values were calculated above baseline). Time-dependent decrease in [Ca2+]i induced by SERCA inhibition was fitted by a sigmoidal equation using the Levenberg-Marquardt algorithm with ORIGIN software (Microcal Software Inc., Northhampton, MA). Parameters derived form curve fitting are given with SE. Comparison of the decay phase following the maximal [Ca2+]i value in control versus specific conditions (i.e., Ca2+ confining solution, CPA, and FCCP) were performed as follows: for each cell tested, [Ca2+]i measurements were repeated every 0.2 s for 10 s, time 0 corresponding to the maximal [Ca2+]i. Comparison between the points obtained in control versus specific condition was done by analysis of variance (ANOVA) for repeated measurements, using SPSS software (SPSS, Chicago, IL).
Computational
Simulations were performed using the Runge-Kutta routine for numerical solving of differential equations. The software used was MADONNA (University of Berkeley, Berkeley, CA).
RESULTS
Characterization of caffeine-induced [Ca2+]i response
[Ca2+]i response to caffeine stimulation
In this series of experiments, cells were stimulated with caffeine for 30 s in a concentration range from 0.01 to 5 mM. Stimulation with caffeine induced a [Ca2+]i response characterized by a first transient peak followed by a [Ca2+]i decay to a plateau slightly above the resting [Ca2+]i value. At the end of the stimulation the [Ca2+]i rapidly returned to baseline. Both the amplitude of the peak and the plateau depended on caffeine concentration. Typical traces of [Ca2+]i response to 0.1 and 5 mM caffeine are shown in Fig. 1 A. Fig. 1 B shows the mean peak (left panel) and plateau (right panel) values for the various caffeine concentrations are given in (n = 10–35 cells for each concentration). Log IC50, i.e., the logarithm of the half-maximal response concentration determined from curve fitting, was −3.51 ± 0.07 logM and −3.85 ± 0.19 logM for the peak and the plateau, respectively.
FIGURE 1.

Effect of various caffeine concentrations on [Ca2+]i responses. (A) Original traces of calcium response to 0.1 mM (left trace) and 5 mM (right trace) CAF (30-s stimulation). (B) Amplitude of the calcium peak (•, left panel) and plateau (♦, right panel) for CAF concentrations from 0.01 mM to 5 mM. Each point is a mean value from 10 to 35 cells. Vertical bars are SE.
The peak and plateau values in response to 30-s stimulation by 5 mM caffeine were 697 ± 59 nM and 43 ± 4.0 nM, respectively (n = 19). When the stimulation was stopped after 1 s, the amplitude of the Ca2+ peak was not significantly modified (681 ± 66 nM; n = 24), but the [Ca2+]i decreased more rapidly and returned to baseline. When cell stimulation was stopped after 5 s, i.e., during the decay phase, the [Ca2+]i decrease rate augmented at the end of caffeine exposure and became similar to that observed for 1-s stimulation. Typical traces are shown in Fig. 2 A.
FIGURE 2.

Effect of caffeine stimulation for various durations on [Ca2+]i. (A) Original traces of cells stimulated by 5 mM CAF for 30 s, 5 s, and 1 s. (B) Mean [Ca2+]i decay upon stimulation with 5 mM CAF for 30 s (▪), 5 s (□), and 1 s (shaded squares). Time 0 corresponds to max [Ca2+]i increase (peak). Values are expressed as percent of the peak obtained when cells were stimulated for 30 s (reference peak), 0 corresponding to baseline. Each curve is the mean value from 10 to 12 cells. (C) Effect of CAF ejection on fluorescence intensities and ratio. CAF was ejected for 5 s on a cell preincubated for 11 min with 10 μM CPA. During CAF ejection, fluorescence quenching at both wavelengths is observed, without alteration in the fluorescence ratio. Fluorescence intensities and ratio are in arbitrary units.
Fig. 2 B shows specifically the [Ca2+]i decay phase following the peak, when cells were stimulated with caffeine for 1 s, 5 s, and 30 s. Values are mean values calculated from 10 to 12 cells for each condition, and are expressed as the percent of the mean peak obtained for 30-s caffeine stimulation. Time 0 corresponds to peak, i. e., the maximal [Ca2+]i increase. The fact that the [Ca2+]i decay was quicker for 1-s ejection than for 5-s or 30-s ejection indicates that caffeine was rapidly washed out at the end of the ejection, inducing RyR closure. Caffeine has the property to decrease the indo-1 fluorescence intensity at both wavelengths, without altering the fluorescence ratio (O'Neill et al., 1990). The analysis of fluorescent intensity confirmed the quick penetration and washout of caffeine, as shown in Fig. 2 C.
Role of transmembrane Ca2+ fluxes in the response to caffeine stimulation
The possible implication of calcium fluxes through the plasma membrane was assessed by a series of experiments in Ca2+-free medium and in Ca2+-confining solution. In normal PSS, baseline value was 139.7 ± 4.2 nM (n = 42). Removal of external Ca2+ or incubation in Ca2+-confining solution did not significantly modify the resting [Ca2+]i value (88.0 ± 3.2% control, n = 23 and 99.9 ± 3.9% control, n = 23, respectively). When cells in Ca2+-free or Ca2+-confining medium were stimulated with 5 mM caffeine for 30 s, neither the peak nor the plateau was significantly different from control. Fig. 3 A shows the mean results, expressed as percent to control conditions, for the peak (left panel) and the plateau (right panel).
FIGURE 3.

Effect of Ca2+-free and Ca2+-confining solutions on response to 5 mM caffeine. (A) Peak and plateau values in response to 30-s CAF stimulation, in Ca2+-free medium (open columns, n = 21) and Ca2+-confining medium (shaded columns, n = 11). (B) Original trace of Ca2+ response to two successive stimulations by CAF for 1 s, at a 15-s interval in the presence of external Ca2+ (left), in Ca2+-free medium (middle), and in Ca2+-confining solution (right). (C) Left panel, mean values of the Ca2+ response to two successive stimulations by CAF for 1 s, at a 15-s interval in Ca2+-free medium (open columns, n = 13) and Ca2+-confining solution (shaded columns, n = 23). (C) Right panel, mean [Ca2+]i decay after the first CAF-induced peak in control (•, n = 22) and Ca2+-confining solution (○, n = 23), as percentage of the control peak, 0 corresponding to baseline. Vertical bars are SE.
Due to the superficial SR that may accumulate Ca2+ that enters through the plasmalemma, the so-called buffer barrier, Ca2+ influx may reload the SR without increasing the [Ca2+]i (van Breemen et al., 1995; Marin et al., 1999; Sanders, 2001). To test whether such a mechanism was triggered by caffeine stimulation, cells were stimulated by two successive 1-s ejections of 5 mM caffeine at a 15-s interval. The first stimulation induced a Ca2+ peak followed by a quick decay, the RyRs being closed. The second stimulation 15 s after the first one induced a second Ca2+ peak, the amplitude of which was used as an estimate of Ca2+ loading of the SR after the first stimulation. In control experiments, the mean [Ca2+]i response to the first and the second stimulation was 695 ± 82 nM and 206 ± 26 nM, respectively (n = 35). The amplitude of the first peak was not significantly modified in the absence of extracellular calcium; neither was the amplitude of the second one (n = 13). Original traces are shown in Fig. 3 B, and mean values in Fig. 3 C (left panel). A similar stimulation protocol was used in Ca2+-confining solution. As in Ca2+-free medium, neither the amplitude of the first response nor that of the second one was altered compared to normal medium (n = 23), Fig. 3, B (left trace) and C (left panel). To see whether Ca2+ fluxes through the plasma membrane may be significantly involved during cytosolic Ca2+ removal upon cell stimulation, we analyzed the [Ca2+]i phase after 1-s caffeine ejection in control versus Ca2+-confining solution. Mean values obtained in control condition (n = 22, filled circles) and in Ca2+-confining solution (n = 23, open circles) are shown in Fig. 3 C (right panel). ANOVA analysis of the decay phase showed no significant difference in normal and Ca2+-confining solution.
Mathematical modeling
Presentation of the model
In accordance with our experiments, the model takes into account the calcium exchange between the cytosol and the intracellular calcium stores, neglecting any transplasmalemmal Ca2+ fluxes. We focus on four different intracellular calcium stores: the SR, mitochondria, and two classes of calcium-binding proteins in the cytosol, i.e., signaling and buffering proteins. The signaling proteins characterize protein-binding sites with high affinity and low capacity, whereas the buffering proteins characterize protein-binding sites with low affinity and high capacity. Regarding the SR, three different calcium fluxes are included in the model: the ATP-dependent calcium uptake from the cytosol into the SR (JSERCA), the Ca2+ efflux from the SR through ryanodine sensitive Ca2+ channels following the calcium-induced calcium release (CICR) mechanism (JRyR), and an additional Ca2+ leak flux from the SR into the cytosol (Jleak). For the exchange of Ca2+ between the mitochondria and the cytosol we take into account active Ca2+ uptake by mitochondrial uniporters (Jin) and calcium release through Na+/Ca2+ and H+/Ca2+ exchangers (Jout). In the cytosol Ca2+ binding to signaling and buffering proteins is considered.
The concentration of free-Ca2+ binding sites on signaling proteins, SPr, can be calculated by applying a rapid-equilibrium approximation to the fast binding reactions (see, e.g., Wagner and Keizer, 1994; Heinrich and Schuster, 1996; Marhl et al., 1998a; Höfer et al., 2001). Together with the conservation relation for the total concentration of Ca2+ binding sites on signaling proteins, SPrtot, the concentration of free-Ca2+ binding sites on signaling proteins, SPr, is given by the following equation:
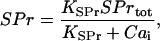 |
(1) |
where 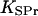 is the dissociation constant of binding sites on signaling proteins.
is the dissociation constant of binding sites on signaling proteins.
The rapid-equilibrium approximation for the fast Ca2+-binding to signaling proteins is justified in view of the very high values of the rate constants for signaling proteins (Wagner and Keizer, 1994; Smith et al., 1996). The rate constants for binding and dissociation of 90–500 μM−1s−1 and 300–500 s−1, respectively (Falke et al., 1994; Smith et al., 1996), imply a time constant of <0.01 s. Even under consideration of diffusional resistance and competition with Mg2+, this time constant is much smaller than the duration of any calcium pulse.
Using the conservation relation for the total concentration of Ca2+ binding sites on signaling proteins, 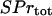 , the concentration of bound Ca2+ binding sites on signaling proteins can be calculated as:
, the concentration of bound Ca2+ binding sites on signaling proteins can be calculated as:
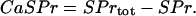 |
(2) |
Taking into account the conservation relations for the total cellular calcium, 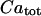 , and the total concentration of Ca2+ binding sites on buffering proteins,
, and the total concentration of Ca2+ binding sites on buffering proteins, 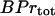 , we obtain equations for bound and free-Ca2+ binding sites on buffering proteins, CaBPr and BPr, respectively:
, we obtain equations for bound and free-Ca2+ binding sites on buffering proteins, CaBPr and BPr, respectively:
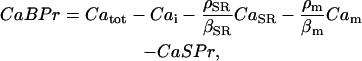 |
(3) |
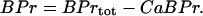 |
(4) |
Here 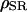 and
and 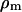 represent the volume ratio between the SR and the cytosol and between the mitochondria and the cytosol, respectively. Assuming very fast unsaturated buffering of Ca2+ in the SR and mitochondrial compartments, we use factors
represent the volume ratio between the SR and the cytosol and between the mitochondria and the cytosol, respectively. Assuming very fast unsaturated buffering of Ca2+ in the SR and mitochondrial compartments, we use factors 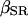 and
and 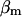 , which are the free/total calcium concentration ratios, in the SR and in the mitochondria, respectively (Marhl et al., 1998b; Haberichter et al., 2001). Applying the rapid-equilibrium approximation for relating the free-calcium concentration in each organelle,
, which are the free/total calcium concentration ratios, in the SR and in the mitochondria, respectively (Marhl et al., 1998b; Haberichter et al., 2001). Applying the rapid-equilibrium approximation for relating the free-calcium concentration in each organelle, 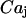 (j stands for SR and m), with the total concentration in the corresponding organelle,
(j stands for SR and m), with the total concentration in the corresponding organelle, 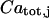 , leads to
, leads to 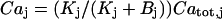 , where
, where 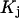 is the dissociation constant of the binding sites on buffering proteins
is the dissociation constant of the binding sites on buffering proteins 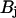 in the organelle. Taking into account that
in the organelle. Taking into account that 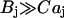 , we can assume that
, we can assume that 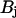 nearly equals the total buffer concentration,
nearly equals the total buffer concentration, 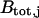 . Therefore, we consider
. Therefore, we consider 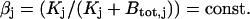 Because the values for
Because the values for 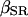 and
and 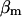 have not been experimentally determined for airway myocytes, we take values that are in the range of experimentally obtained values for other cell types. For
have not been experimentally determined for airway myocytes, we take values that are in the range of experimentally obtained values for other cell types. For 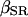 values usually span from 0.0025 (Li et al., 1995) to 0.01 (Smith et al., 1996), whereas
values usually span from 0.0025 (Li et al., 1995) to 0.01 (Smith et al., 1996), whereas 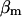 can be found in the range from 0.00001 to 0.01 (Babcock et al., 1997; Fall and Keizer, 2001; Chalmers and Nicholls, 2003).
can be found in the range from 0.00001 to 0.01 (Babcock et al., 1997; Fall and Keizer, 2001; Chalmers and Nicholls, 2003).
The time dependence of the free cytosolic calcium concentration, 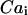 , is determined by Ca2+ fluxes across the SR membrane, by Ca2+ exchange between the cytosol and mitochondria, and by Ca2+ binding to signaling and buffering proteins in the cytosol:
, is determined by Ca2+ fluxes across the SR membrane, by Ca2+ exchange between the cytosol and mitochondria, and by Ca2+ binding to signaling and buffering proteins in the cytosol:
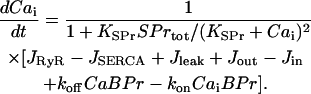 |
(5) |
Here 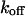 and
and 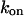 denote the off and on rate constants, respectively, of the Ca2+ binding to the buffering proteins. The first factor in Eq. 5, characterizing Ca2+ binding to signaling proteins, is obtained by the method of eliminating fast reactions (see Heinrich and Schuster, 1996; Marhl et al., 1998a).
denote the off and on rate constants, respectively, of the Ca2+ binding to the buffering proteins. The first factor in Eq. 5, characterizing Ca2+ binding to signaling proteins, is obtained by the method of eliminating fast reactions (see Heinrich and Schuster, 1996; Marhl et al., 1998a).
The equation for the free-calcium concentration in the SR, 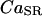 , is linked with the fluxes across the SR membrane as follows:
, is linked with the fluxes across the SR membrane as follows:
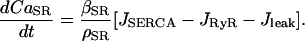 |
(6) |
The equation for the free-Ca2+ concentration in mitochondria, 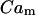 , reads:
, reads:
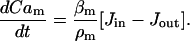 |
(7) |
Here, 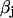 relates fluxes, i.e.,
relates fluxes, i.e., 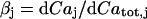 , like in previous papers by Wagner and Keizer (1994) and Smith et al. (1996). Under the condition of very fast unsaturated buffering of Ca2+ in the organelles used above, this is in agreement with using the constant
, like in previous papers by Wagner and Keizer (1994) and Smith et al. (1996). Under the condition of very fast unsaturated buffering of Ca2+ in the organelles used above, this is in agreement with using the constant 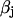 for relating the concentrations in Eq. 3. Differentiating
for relating the concentrations in Eq. 3. Differentiating 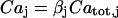 with respect to time gives
with respect to time gives 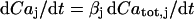 and dividing by
and dividing by 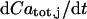 gives the above formula
gives the above formula 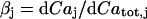 .
.
There are three genetically distinct isoforms of RyR in Mammals, RyR1, RyR2, and RyR3. In smooth muscle, several isoforms may be expressed (Marin et al., 1999; Ogawa et al., 2000). In airway smooth muscle, RT-PCR in porcine trachea has revealed that both RyR2 and RyR3 mRNA were present (Kannan et al., 1997). However, in human bronchial smooth muscle, we have previously showed using RT-PCR and RNase protection assay that RyR3 was the unique isoform expressed (Hyvelin et al., 2000). On this basis, our model was built on RyR3 activity. The Ca2+ flux through ryanodine channels, 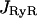 , is caffeine-activated. The open probability depends on caffeine concentration, Caff, and follows the Hill kinetics with
, is caffeine-activated. The open probability depends on caffeine concentration, Caff, and follows the Hill kinetics with  . As driving force for the Ca2+ channel flux the concentration gradient across the SR membrane is taken into account and the equation for
. As driving force for the Ca2+ channel flux the concentration gradient across the SR membrane is taken into account and the equation for 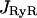 reads:
reads:
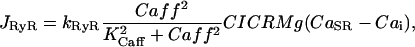 |
(8) |
where 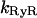 is the rate constant of the ryanodine channels and
is the rate constant of the ryanodine channels and 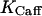 represents the half-saturation constant of the ryanodine channels for caffeine. The kinetics for caffeine activation was obtained by fitting our experimental data (see Fig. 1 B). This is also fully in accordance with previous experimental studies in airway smooth muscle cells (Roux et al., 1998). On the basis of experimental investigations (Ogawa et al., 2000) on the Ca2+-induced Ca2+ release (CICR) mechanism of ryanodine channels, CICRMg, the effect of Mg2+ on the activity of ryanodine receptors is taken into account as follows:
represents the half-saturation constant of the ryanodine channels for caffeine. The kinetics for caffeine activation was obtained by fitting our experimental data (see Fig. 1 B). This is also fully in accordance with previous experimental studies in airway smooth muscle cells (Roux et al., 1998). On the basis of experimental investigations (Ogawa et al., 2000) on the Ca2+-induced Ca2+ release (CICR) mechanism of ryanodine channels, CICRMg, the effect of Mg2+ on the activity of ryanodine receptors is taken into account as follows:
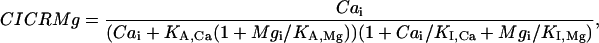 |
(9) |
where 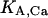 and
and 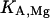 represent the half-saturation constants of activate sites for Ca2+ and Mg2+, respectively, and
represent the half-saturation constants of activate sites for Ca2+ and Mg2+, respectively, and 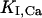 and
and 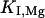 represent the half-saturation constants of inactivate sites for Ca2+ and Mg2+, respectively.
represent the half-saturation constants of inactivate sites for Ca2+ and Mg2+, respectively.
For the SERCA flux into the SR lumen, 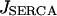 , the Hill kinetics with
, the Hill kinetics with 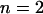 is taken:
is taken:
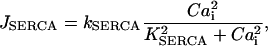 |
(10) |
where 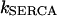 is the rate constant of the SERCAs and
is the rate constant of the SERCAs and 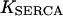 stands for the half-saturation constant of SERCAs.
stands for the half-saturation constant of SERCAs.
The leak flux 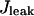 depends on the concentration gradient across the SR membrane and a simple relation is taken:
depends on the concentration gradient across the SR membrane and a simple relation is taken:
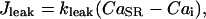 |
(11) |
where 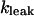 is the rate constant for Ca2+ leak flux through the SR membrane and has been estimated on the basis of our experimental data.
is the rate constant for Ca2+ leak flux through the SR membrane and has been estimated on the basis of our experimental data.
There is experimental evidence of a very fast and effective calcium sequestration by mitochondria through a specific uniporter (Hehl et al., 1996; Applegate et al., 1997; Babcock et al., 1997). In some cases the Ca2+ uptake by mitochondria can be extremely fast due to a mechanism called the rapid mode (RaM) (Gunter et al., 2000; Rizzuto et al., 2000). Based on the experimental results that Ca2+ sequestration takes place at free cytosolic calcium levels of >∼0.5–1.0 μM (Jouaville et al., 1995; Bernardi and Petronilli, 1996; Hehl et al., 1996; Herrington et al., 1996; Hoth et al., 1997; Ricken et al., 1998) a step-like kinetics is considered for the mitochondrial Ca2+ uptake by uniporters, 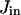 (Marhl et al., 1998a; Grubelnik et al., 2001; Haberichter et al., 2001).
(Marhl et al., 1998a; Grubelnik et al., 2001; Haberichter et al., 2001).
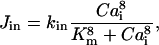 |
(12) |
where 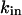 represents the maximal permeability of the uniporters in the mitochondrial membrane, and
represents the maximal permeability of the uniporters in the mitochondrial membrane, and 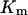 represents the half-saturation for Ca2+ (Marhl et al., 2000). As in our previous publication (Marhl et al., 1998a), in factor
represents the half-saturation for Ca2+ (Marhl et al., 2000). As in our previous publication (Marhl et al., 1998a), in factor 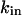 the constant value of the mitochondrial transmembrane potential,
the constant value of the mitochondrial transmembrane potential, 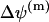 , is implicitly included. The potential difference
, is implicitly included. The potential difference 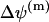 is usually strongly changed only by fast release of calcium through the permeability transient pore (PTP). Note that under normal physiological conditions just a slow release of calcium from the mitochondria takes place (Bernardi and Petronilli, 1996; Eriksson et al., 1999; Svichar et al., 1999).
is usually strongly changed only by fast release of calcium through the permeability transient pore (PTP). Note that under normal physiological conditions just a slow release of calcium from the mitochondria takes place (Bernardi and Petronilli, 1996; Eriksson et al., 1999; Svichar et al., 1999).
For the mitochondrial Ca2+ release through Na+/Ca2+ and H+/Ca2+ exchangers we consider a simple linear dependency on 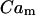 (Marhl et al., 1998b; Grubelnik et al., 2001):
(Marhl et al., 1998b; Grubelnik et al., 2001):
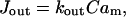 |
(13) |
where 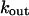 is the maximal rate for calcium efflux from mitochondria.
is the maximal rate for calcium efflux from mitochondria.
Model parameters used in our calculations are given in figure captions. The parameter values for Ca2+ fluxes across the SR membrane were estimated in accordance with our own experimental results. Data regarding the RyR activity were taken from the experiments done by Ogawa et al. (2000). The parameter values regarding Ca2+ binding to signaling and buffering proteins were taken from the experiments in the literature and were extensively discussed in our previous paper (Marhl et al., 1998a). The parameter values regarding the mitochondrial Ca2+ handling were also taken from the experimental results published in the literature and were exhaustively discussed in our previous papers (Marhl et al., 1998b, 2000; Haberichter et al., 2001).
Modeling of caffeine-induced [Ca2+]i response
Cell stimulation with different concentrations of caffeine was simulated by varying the model parameter Caff. The calculated traces for Caff = 0.1 mM and 5 mM for 30 s are shown in Fig. 4 A, and correspond to the experimental results presented in Fig. 1 A. Fig. 4 B represents the calculated concentration-dependent RyR activity. The sigmoidal relationship between caffeine concentration and RyR activity is similar to that between caffeine concentration and [Ca2+]i response showed in Fig. 1 B. Model calculations of [Ca2+]i responses to 5-mM caffeine exposures for 1 s, 5 s, and 30 s are shown in Fig. 4 C (dotted line, dashed line, and solid line, respectively). They correspond to the experimental traces shown in Fig. 2 A, and reflect fast closure of RyR at the end of caffeine exposure.
FIGURE 4.

Simulation of Ca2+ response to caffeine. (A) Simulated response to 0.1 (left) and 5 (right) mM CAF for 30 s. (B) Concentration-activity curve of RyR. (C) Simulated responses to 30-s (solid line), 5-s (dashed line), and 1-s (dotted line) caffeine (5 mM). Parameter values are ρSR = 0.01; ρm = 0.01; βSR = 0.0025; βm = 0.0025; Catot = 50 μM; SPrtot = 90 μM; BPrtot = 120 μM; Mgi = 500 μM; kRyR = 2000 s−1; kSERCA = 1 μM s−1; kleak = 0.02 s−1; kin = 20 μM s−1; kout = 0.1 s−1; kon = 0.1 μM−1 s−1; koff = 0.01 s−1; KCaff = 250 μM; KA,Ca = 2.5 μM; KA,Mg = 75 μM; KI,Ca = 400 μM; KI,Mg = 300 μM; KSERCA = 0.1 μM; Km = 1 μM; and KSPr = 5 μM.
Modeling of Ca2+ dynamics upon calcium release from the SR
The model was used to predict a scenario of how Ca2+ is distributed among the calcium stores that may be involved in the OFF mechanisms after Ca2+ release from the SR. Ca2+ release was triggered by simulating 1-s caffeine stimulation (5 mM), and Fig. 5 presents the predicted scenario. Time 0 corresponds to the beginning of caffeine stimulation. During the [Ca2+]i decay phase, the RyRs are closed, allowing effective Ca2+ pumping back by the SERCAs without further Ca2+ release through open RyRs. However, the increase in luminal Ca2+ concentration is slower than [Ca2+]i decrease. The first process that takes place is Ca2+ binding to fast protein binding sites quickly followed by Ca2+ sequestration into mitochondria. After a short time delay, Ca2+ is shifted to the slow protein binding sites, and finally, after a considerably longer time delay, it is pumped back into the SR. The predictions of the model for the first 5 s after 1-s stimulation are shown in Fig. 5, A and B. Fig. 5 C represents the predicted long time variations in SR Ca2+ concentration. As in Fig. 5, A and B, stimulation runs up for 1 s. Simulations were performed for 0.1 and 5 mM caffeine. It can be seen that complete Ca2+ reuptake into the SR after maximal Ca2+ release is a slow process, that takes ∼10 min. Caffeine (0.1 mM) triggers a smaller Ca2+ release from the SR, and faster recovery.
FIGURE 5.

Model prediction of Ca2+ distribution after Ca2+ release from the SR. (A and B) Model predictions on Ca2+ handling upon 5-mM CAF stimulation. Stimulation begins at time 0 s and runs up to 1 s (double arrow). Parameter values are the same as in Fig. 4. Concentration in the cytosol (Cai) is indicated by the dashed line. (A) Concentration in the SR (CaSR) and mitochondria (Cam). (B) Ca2+ bound to fast-kinetics (CaSPr) and to slow-kinetics (CaBPr) cytosolic proteins. (C) Predicted long-time variations in SR Ca2+ concentration for 0.1 (dashed line) and 5 mM CAF (solid line). Stimulation begins at time 0 s and runs up to 1 s (arrow). Parameter values are the same as in Fig. 4.
Involvement of SERCA in cytosolic Ca2+ clearance
Model prediction of SERCA blockade
According to the predictions of the model presented above, SERCA activity is not necessarily the key mechanism in the [Ca2+]i decrease after Ca2+ release from the SR. To test this hypothesis, we compared model predictions of Ca2+ dynamics with active versus blocked SERCAs. We simulated two successive stimulations by 5 mM caffeine of 1 s at a 15-s interval, with and without active SERCAs. Fig. 6 shows the predicted calcium response and the decay phase after the first Ca2+ peak. There is no significant difference in [Ca2+]i decay upon Ca2+ release from the SR in case of active versus blocked SERCAs, which indicates that in our model Ca2+ pumping back into the SR is not primarily involved in Ca2+ clearance from the cytosol.
FIGURE 6.

Model prediction of the effect of SERCA blockade on the Ca2+ response to two successive stimulations by 5 mM caffeine (1 s). (A) Predicted [Ca+]i response to two successive 1-s stimulations by 5 mM CAF at a 15-s interval, with active (left trace) and inactivated (right trace) SERCA. (B) Predicted values of the decay phase after the first [Ca+]i rise, with active (•) and inactivated (○) SERCA. Parameter values are the same as in Fig. 4.
Experimental effects of SERCA blockade
To test this prediction, experiments were performed using the same protocol as for simulated experiments, i.e., each cell was stimulated by two successive stimulations of 5 mM caffeine of 1 s at a 15-s interval, in the absence and in the presence of CPA, a reversible SERCA inhibitor. For each cell tested, exposure to 10 μM CPA was initiated 30 s before the first caffeine stimulation. Exposure to 10 μM CPA alone (n = 14) for 1 min induced no change or, in some cells, a slight increase in [Ca2+]i 5–15 s after the beginning of exposure. This CPA-induced [Ca2+]i increase, for which the mean value was 28.4 ± 7.4 nM, was transient and the [Ca2+]i at the end of CPA exposure was not significantly altered compared to baseline before CPA exposure (99.3 ± 0.9% control). When cells were stimulated by caffeine 30 s after the beginning of exposure to CPA, the first peak was not significantly modified versus control (96.4 ± 6.9% control; n = 21), in accordance with the predicted results, but the Ca2+ response to the second stimulation was greatly decreased (49.2 ± 8.1% control; p < 0.05), indicating that the SERCA was blocked by CPA and that, in the absence of SERCA inhibition, the Ca2+ response to the second caffeine stimulation was mainly due to effective Ca2+ pumping back into the SR. Typical traces and mean values are given in Fig. 7.
FIGURE 7.

Effect of CPA (10 μM) on [Ca2+]i response to successive 1-s stimulations with 5 mM caffeine. (A) Original traces of cells stimulated in the absence (left trace) and in the presence of CPA (right trace). (B) Effect of CPA on the calcium response to two successive CAF stimulations. Values were obtained from 21 cells, and are expressed as a percentage of control. Vertical bars are SE. *p < 0.05. (C) Mean [Ca2+]i decay after the first CAF stimulation in the absence (•) and in the presence (○) of 10 μM CPA. Each curve in the mean value of 21 cells in each experimental conditions, expressed as percentage of the control peak, 0 corresponding to baseline.
Under these conditions, we analyzed the [Ca2+]i decay phase after the first caffeine stimulation for 12 s, i.e., a time sufficient for the [Ca2+]i to return to a steady-state value. Mean values calculated from 21 cells in each condition are shown in Fig. 8. Statistical comparison showed no significant difference in the absence versus in the presence of 10 μM CPA.
FIGURE 8.

Effect of SERCA blockade on SR Ca2+ load. (A) Typical traces of [Ca2+]i response to two successive 5-s CAF stimulations in cells preincubated with 10 μM CPA for 7 min (left trace), 10 min (middle trace), and 13 min (right trace). (B) Individual Ca2+ response to 5 mM CAF of cells incubated in Ca2+-free medium (crosses) and incubated in Ca2+-free medium with 10 μM CPA (•) or with 1 μM TG (○). Abscissa: duration of incubation. Ordinate: first peak amplitude in percent of control, i.e., response of cells in normal PSS. [Ca2+]i in Ca2+-free medium are fitted by linear regression; [Ca2+]i responses in the presence of CPA and TG are fitted by a sigmoidal equation.
To verify that, because of low passive Ca2+ leak, short-time SERCA blockade does not significantly deplete the SR, we assessed the time-dependent effect of the SERCA inhibitors CPA and thapsigargin (TG) on Ca2+ homeostasis. Experiments were performed in Ca2+-free medium to avoid any calcium influx that may be involved in long-term SERCA blockade. Each cell tested was stimulated by two successive ejections of 5 mM caffeine for 5 s at a 15-s interval. The Ca2+ response to the first stimulation was used as an estimate of SR Ca2+ load. The loss of response to the second stimulation ensured that Ca2+ pumping back by SERCAs was actually inhibited. [Ca2+]i responses in Ca2+-free medium without SERCA inhibition were fitted by linear regression and the slope did not significantly differ from zero (Fig. 8 B, crosses fitted by solid straight line). Exposure to 10 μM CPA up to 25 min in the absence of external Ca2+ did not significantly alter the resting [Ca2+]i (100.3 ± 4.2% control, n = 20). Decrease in the calcium response to caffeine was observed several minutes after the beginning of exposure to CPA, and complete abolishment of the response needed >10 min. Typical traces are shown in Fig. 8 A. Similar results were obtained with the irreversible inhibitor TG (1 μM) (n = 14). The time-dependent decrease of the amplitude of the first Ca2+ peak was sigmoidal with both SERCA inhibitors, and the duration of exposure to CPA (10 μM) and TG (1 μM) needed to induce 50% inhibition was 10.8 ± 1.0 min and 8.0 ± 0.5 min, respectively. Fig. 8 B shows the experimental values obtained with CPA (solid circles) and TG (open circles). These results indicate that spontaneous Ca2+ leak from the SR at rest was small.
Model prediction of mitochondrial Ca2+uptake blockade
The theoretical and experimental results presented above show that Ca2+ pumping back by SERCA is not significantly responsible for the [Ca2+]i decrease after Ca2+ release from the SR and, according to the model, Ca2+ uptake into mitochondria may be more important. To test this hypothesis, we compared model predictions of Ca2+ dynamics with active versus blocked mitochondrial Ca2+ uptake. We simulated the [Ca2+]i response to 1-s stimulation by 5 mM caffeine, with and without active Ca2+ uniporter, and determined the respective [Ca2+]i decays, as shown Fig. 9 A. According to the model, mitochondrial inhibition results in a slight change in the slope of the Ca2+ decay, indicating that mitochondrial Ca2+ uptake is actually involved in cytosolic Ca2+ clearance after Ca2+ release from the SR, but has a slight influence on the shape of the [Ca2+]i decay.
FIGURE 9.

Predicted and experimental effect of mitochondrial Ca2+ uptake inhibition on the Ca2+ response to 5 mM caffeine (1 s). (A) Predicted values of the decay phase after CAF stimulation, with active (solid line) and inactivated (dashed line) mitochondrial Ca2+ uptake. Parameter values are the same as in Fig. 4. (B) Mean [Ca2+]i decay after CAF-induced peak in the absence (n = 21, •) and in the presence (n = 25, ○) of 5 μM FCCP, as a percentage of the control peak, 0 corresponding to baseline.
Experimental effects of mitochondrial Ca2+uptake blockade
To test this prediction, experiments were performed in the presence of 5 μM FCCP, an uncoupler of mitochondrial function usually used at 1–5 μM (Gurney et al., 2000; Albrecht et al., 2002; Kang et al., 2003; Szado et al., 2003). FCCP (5 μM) assures a quick mitochondrial inhibition (Kang et al., 2003). For each cell tested, exposure to 5 μM FCCP was initiated 30 s before 1-s caffeine stimulation. Exposure to 5 μM FCCP alone for 30 s induced no change in [Ca2+]i (data not shown). When cells were stimulated by caffeine 30 s after the beginning of exposure to FCCP, the first peak was not significantly modified versus control (102.4 ± 6.2% control; n = 25). We hence analyzed the [Ca2+]i decay phase after the caffeine-induced peak. Mean values calculated in control condition (n = 21) and in the presencve of FCCP (n = 25) are shown in Fig. 9 C. In the presence of FCCP, the [Ca2+]i decay was slower than that in control condition, and ANOVA analysis showed that the difference was statistically significant. However, the subsequent resting [Ca2+]i value was not modified compared to control.
DISCUSSION
Our experimental study has shown that calcium signaling upon short caffeine stimulation results in Ca2+ dynamics within the cell without significant involvement of Ca2+ fluxes through the plasma membrane. On the basis on these experimental data, we build a relevant theoretical model of RyR stimulation and subsequent Ca2+ handling. Using this model in combination with the experimental approach, we used short-time stimulation by caffeine to investigate the OFF mechanisms, i.e., the mechanisms implicated in the return of [Ca2+]i to baseline after [Ca2+]i increase. Our experimental results, in accordance with our theoretical model, indicate that although Ca2+ pumping back by SERCA is active after Ca2+ release from SR upon RyR stimulation, it is not primarily involved in [Ca2+]i decrease. Ca2+ uptake by mitochondria slightly but significantly shapes the [Ca2+]i decay that may also depend on additional intracellular buffering processes.
Our cell model of RyR stimulation is a “closed cell” one, i.e., in which Ca2+ fluxes through the plasma membrane are not considered and the total Ca2+ concentration within the cell is kept constant. Although transmembrane Ca2+ fluxes are likely to be present in these cells, our results showed that such an approximation is relevant in our experimental conditions. Indeed, Ca2+ influx was not triggered by caffeine-induced Ca2+ release, because the [Ca2+]i response was not altered in Ca2+-free medium. Store-operated channels, activated by the depletion of intracellular Ca2+ stores, have been described in a variety of smooth muscle cells. Some authors have speculated that the main physiological role of store-operated channels may be the refilling of the SR (Marin et al., 1999; Ng and Gurney, 2001). The fact that the second Ca2+ response to two successive caffeine stimulations, which is actually due to SR refilling because it is abolished in the presence of the SERCA blocker CPA, was not modified in Ca2+-free medium indicates that such a mechanism is not involved in our experimental conditions. Usually, Ca2+ extrusion by PMCA and/or Na+/Ca2+ exchanger is considered as an important OFF mechanism (Berridge et al., 2000; Sanders, 2001). In Na+- and Ca2+-free solution containing 0.5 mM lanthanum, that inhibits any Ca2+ influx or efflux (Tribe et al., 1994), the decay phase of the Ca2+ response was not modified, indicating that Ca2+ extrusion is not implicated in the quick [Ca2+]i return to baseline. This is in accordance with previous studies that showed that the [Ca2+]i response to contractile agonists like acetylcholine is not altered by removal of extracellular Ca2+ (Roux et al., 1996, 1997; Prakash et al., 1998; Bergner and Sanderson, 2002). In the absence of compensatory Ca2+ influx, fast Ca2+ extrusion would rapidly induce a complete loss of intracellular Ca2+ pool and hence loss of Ca2+ response.
The predicted values for the resting Ca2+ levels, that are ∼40 nM, are lower than the experimental values, that were ∼130 nM, in accordance with our previous results (Roux et al., 1997, 1998). However, due to relative imprecision of the calculation of absolute [Ca2+]i values and interindividual variations so that values measured in some cells from our experiments were similar to that predicted but the model, the predicted values, though lower than the mean estimated [Ca2+]i, can be considered as in a physiological range.
We analyzed the OFF mechanisms after [Ca2+]i increase using RyR stimulation. Single-channel activity measurement in lipid bilayer indicates that RyR2 and RyR3 differ in their sensitivity to Ca2+ inactivation and Mg2+ inhibition, so that for RyR3 Ca2+-induced Ca2+ release may be reduced in the presence of physiological Mg2+ concentration (Ogawa et al., 2000). In accordance with the simulated Ca2+ response, stimulation by various caffeine concentrations showed a sigmoidal relationship between the amplitude of the Ca2+ response and the logarithmic concentration of caffeine. The fact that the response is not an all-or-none phenomenon suggests that Ca2+-induced Ca2+ release is limited, which is consistent with a predominant RyR3 expression.
To investigate the mechanisms responsible for Ca2+ removal in smooth muscle cells, some authors have used voltage pulse to induce Ca2+ influx through voltage-dependent L-type Ca2+ channels (Kamishima and McCarron, 1998; Shmigol et al., 1999). In such a protocol, initial [Ca2+]i increase is due to additional Ca2+ influx into the cell without SR Ca2+ depletion. However, in airway smooth muscle, although L-type Ca2+ channels may be activated during stimulation (Tomasic et al., 1992), contractile agonists such as acetylcholine act primarily via Ca2+ release from the SR (Roux et al., 1996, 1997; Prakash et al., 1998; Bergner and Sanderson, 2002). Hence, in this type of cells, it seems physiologically relevant to induce Ca2+ release from the SR rather than extracellular Ca2+ influx to investigate the subsequent OFF mechanisms. Analysis of fluorescence quenching and comparison of the decay phase after 1-, 5-, and 30-s caffeine stimulation indicate that caffeine washout and RyR closure occurred ∼2–3 s after the beginning of caffeine ejection. Because the maximal [Ca2+]i increase occurred approximately in the same time (see Fig. 2), we assumed that during the decay phase, the duration of which was ∼10 s after maximal [Ca2+]i increase, the RyRs were closed. Hence the mechanisms involved in cytosolic clearance in our experiments do not seem to be specific to RyR stimulation and are likely to be also implicated upon airway smooth muscle stimulation by physiological agonists that induce Ca2+ release from the SR.
Usually, Ca2+ uptake by SERCAs is considered as the main OFF mechanism (Marin et al., 1999; Berridge et al., 2000; Sanders, 2001). The analysis of the decay phase after short caffeine stimulation in vitro and in silico demonstrated that, although Ca2+ pumping back is actually active and does reload the SR, SERCA activity does not significantly modulate the shape of the [Ca2+]i decay. Several authors have used CPA to inhibit the SERCA activity, with significant effect on Ca2+ decrease, including in airway myocytes (Sims et al., 1996, 1997; Yoshikawa et al., 1996; Shmigol et al., 1999). Global comparison of these different studies is difficult because the experimental protocols used to trigger [Ca2+]i increase as well as to analyze [Ca2+]i decay differed between studies. However, it appears in these studies that even if the [Ca2+]i decay rate was altered in the presence of CPA, the general pattern was not deeply modified. Hence, [Ca2+]i return to baseline should be attributed to mechanisms other than SERCA activity.
The model prediction about Ca2+ sequestration in mitochondria is in agreement with experimental results in some other systems, showing that mitochondria indeed sequester a large amount of Ca2+ released from the sarco/endoplasmic reticulum (for review see Schuster et al., 2002). For example, in chromaffin cells, around 80% of the Ca2+ released from the ER is cleared first into mitochondria (Babcock and Hille, 1998). In vascular smooth muscle cells, several recent studies have shown that mitochondria Ca2+ uptake occurs after SR Ca2+ release and [Ca2+]i increase (Drummond and Fay, 1996; Drummond and Tuft, 1999; Pacher et al., 2000; Vallot et al., 2001; Kamishima and Quayle, 2002; Szado et al., 2003) but, to the best of our knowledge, this issue has not been previously addressed in airway smooth muscle cells. It should be noted that although our model predicts that mitochondrial Ca2+ uptake is effective after [Ca2+]i increase, simulated inhibition of mitochondria uniporter does not greatly alter the shape of the [Ca2+]i decay after 1-s caffeine stimulation, and this was confirmed by our experimental results. In particular, the [Ca2+]i decay phase itself was slowed in the presence of FCCP, but the subsequent resting value did not differ from control.
In conclusion, we have built a relevant theoretical model of Ca2+ handling upon RyR stimulation in airway myocytes. We have experimentally confirmed the model predictions that Ca2+ pumping back into the SR by SERCA, though effective, is not primarily involved in [Ca2+]i decay upon Ca2+ release from the SR, and that Ca2+ uptake by mitochondria slightly but significantly shapes the [Ca2+]i decay. The model provides a possible scenario for Ca2+ handling in which cytosolic Ca2+-binding proteins play a key role in cytosolic Ca2+ clearance. Further studies are needed to check the model predictions about the role of these buffering processes in [Ca2+]i modulation.
Acknowledgments
The authors thank Professors J. P. Mazat and R. Marthan from Bordeaux for fruitful discussions, Dr. C. Guibert from Bordeaux for reading the manuscript, and Professor S. Schuster from Jena and Professors R. Heinrich and T. Höfer from Berlin for checking the mathematical expressions used in the model.
This work was financially supported by the French Ministère des Affaires Étrangères and the Slovenian Ministry of Education, Science and Sport (bilateral project PROTEUS, F-No. 03811NH/SI-No. FR-2002-04), and INSERM.
References
- Albrecht, M. A., S. L. Colegrove, and D. D. Friel. 2002. Differential regulation of ER Ca2+ uptake and release rates accounts for multiple modes of Ca2+-induced Ca2+ release. J. Gen. Physiol. 119:211–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegate, T. L., A. Karjalainen, and F. L. Bygrave. 1997. Rapid Ca2+ influx induced by the action of dibutylhydroquinone and glucagon in the perfused rat liver. Biochem. J. 323:463–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock, D. F., J. Herrington, P. C. Goodwin, Y. B. Park, and B. Hille. 1997. Mitochondrial participation in the intracellular Ca2+ network. J. Cell Biol. 136:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock, D. F., and B. Hille. 1998. Mitochondrial oversight of cellular Ca2+ signaling. Curr. Opin. Neurobiol. 8:398–404. [DOI] [PubMed] [Google Scholar]
- Bergner, A., and M. J. Sanderson. 2002. Acetylcholine-induced calcium signaling and contraction of airway smooth muscle cells in lung slices. J. Gen. Physiol. 119:187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi, P., and V. Petronilli. 1996. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J. Bioenerg. Biomembr. 28:131–138. [DOI] [PubMed] [Google Scholar]
- Berridge, M. J., P. Lipp, and M. D. Bootman. 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1:11–21. [DOI] [PubMed] [Google Scholar]
- Chalmers, S., and D. G. Nicholls. 2003. The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278:19062–19070. [DOI] [PubMed] [Google Scholar]
- Drummond, R. M., and F. S. Fay. 1996. Mitochondria contribute to Ca2+ removal in smooth muscle cells. Pflugers Arch. 431:473–482. [DOI] [PubMed] [Google Scholar]
- Drummond, R. M., and R. A. Tuft. 1999. Release of Ca2+ from the sarcoplasmic reticulum increases mitochondrial [Ca2+] in rat pulmonary artery smooth muscle cells. J. Physiol. 516:139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson, O., P. Pollesello, and E. Geimonen. 1999. Regulation of total mitochondrial Ca2+ in perfused liver is independent of the permeability transition pore. Am. J. Physiol. 276:C1297–C1302. [DOI] [PubMed] [Google Scholar]
- Falke, J. J., S. K. Drake, A. L. Hazard, and O. B. Peersen. 1994. Molecular tuning of ion binding to calcium signaling proteins. Q. Rev. Biophys. 27:219–290. [DOI] [PubMed] [Google Scholar]
- Fall, C. P., and J. E. Keizer. 2001. Mitochondrial modulation of intracellular Ca2+ signaling. J. Theor. Biol. 210:151–165. [DOI] [PubMed] [Google Scholar]
- Grubelnik, V., A. Z. Larsen, U. Kummer, L. F. Olsen, and M. Marhl. 2001. Mitochondria regulate the amplitude of simple and complex calcium oscillations. Biophys. Chem. 94:59–74. [DOI] [PubMed] [Google Scholar]
- Guibert, C., R. Marthan, and J. P. Savineau. 1996. Angiotensin II-induced Ca2+-oscillations in vascular myocytes from the rat pulmonary artery. Am. J. Physiol. 270:L637–L642. [DOI] [PubMed] [Google Scholar]
- Gunter, T. E., L. Buntinas, G. Sparagna, R. Eliseev, and K. Gunter. 2000. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 28:285–296. [DOI] [PubMed] [Google Scholar]
- Gurney, A. M., R. M. Drummond, and F. S. Fay. 2000. Calcium signalling in sarcoplasmic reticulum, cytoplasm and mitochondria during activation of rabbit aorta myocytes. Cell Calcium. 27:339–351.11013464 [Google Scholar]
- Haberichter, T., M. Marhl, and R. Heinrich. 2001. Birhythmicity, trirhythmicity and chaos in bursting calcium oscillations. Biophys. Chem. 90:17–30. [DOI] [PubMed] [Google Scholar]
- Hehl, S., A. Golard, and B. Hille. 1996. Involvement of mitochondria in intracellular calcium sequestration by rat gonadotropes. Cell Calcium. 20:515–524. [DOI] [PubMed] [Google Scholar]
- Heinrich, R., and S. Schuster. 1996. The Regulation of Cellular Systems. Chapman Hall, New York.
- Herrington, J., Y. B. Park, D. F. Babcock, and B. Hille. 1996. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 16:219–228. [DOI] [PubMed] [Google Scholar]
- Herscher, C. J., and A. F. Rega. 1996. Pre-steady-state kinetic study of the mechanism of inhibition of the plasma membrane Ca2+-ATPase by lanthanum. Biochemistry. 35:14917–14922. [DOI] [PubMed] [Google Scholar]
- Höfer, T., A. Politi, and R. Heinrich. 2001. Intercellular Ca2+ wave propagation through gap-junctional Ca2+ diffusion: a theoretical study. Biophys J. 80:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth, M., C. M. Fanger, and R. S. Lewis. 1997. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J. Cell Biol. 137:633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvelin, J. M., C. Martin, E. Roux, R. Marthan, and J. P. Savineau. 2000. Human isolated bronchial smooth muscle contains functional ryanodine/caffeine-sensitive Ca-release channels. Am. J. Respir. Crit. Care Med. 162:687–694. [DOI] [PubMed] [Google Scholar]
- Iwamoto, T., and M. Shigekawa. 1998. Differential inhibition of Na+/Ca2+ exchanger isoforms by divalent cations and isothiourea derivative. Am. J. Physiol. 275:C423–C430. [DOI] [PubMed] [Google Scholar]
- Jouaville, L. S., F. Ichas, E. L. Holmuhamedov, P. Camacho, and J. D. Lechleiter. 1995. Synchronization of calcium waves by mitochondrial substrates in Xenopus laevis oocytes. Nature. 377:438–441. [DOI] [PubMed] [Google Scholar]
- Kamishima, T., and J. G. McCarron. 1998. Ca2+ removal mechanisms in rat cerebral resistance size arteries. Biophys. J. 75:1767–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamishima, T., and J. M. Quayle. 2002. Mitochondrial Ca2+ uptake is important over low [Ca2+]i range in arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 283:H2431–H2439. [DOI] [PubMed] [Google Scholar]
- Kang, T. M., M. K. Park, and D. Y. Uhm. 2003. Effects of hypoxia and mitochondrial inhibition on the capacitative calcium entry in rabbit pulmonary arterial smooth muscle cells. Life Sci. 72:1467–1479. [DOI] [PubMed] [Google Scholar]
- Kannan, M. S., Y. S. Prakash, T. Brenner, J. R. Mickelson, and G. C. Sieck. 1997. Role of ryanodine receptor channels in Ca2+ oscillations of porcine tracheal smooth muscle. Am. J. Physiol. 272:L659–L664. [DOI] [PubMed] [Google Scholar]
- Li, Y. X., J. Keizer, S. S. Stojilkovic, and J. Rinzel. 1995. Ca2+ excitability of the ER membrane: an explanation for IP3-induced Ca2+ oscillations. Am. J. Physiol. 269:C1079–C1092. [DOI] [PubMed] [Google Scholar]
- Marhl, M., T. Haberichter, M. Brumen, and R. Heinrich. 2000. Complex calcium oscillations and the role of mitochondria and cytosolic proteins. Biosystems. 57:75–86. [DOI] [PubMed] [Google Scholar]
- Marhl, M., S. Schuster, and M. Brumen. 1998a. Mitochondria as an important factor in the maintenance of constant amplitudes of cytosolic calcium oscillations. Biophys. Chem. 2:125–132. [PubMed] [Google Scholar]
- Marhl, M., S. Schuster, M. Brumen, and R. Heinrich. 1998b. Modelling oscillations of calcium and endoplasmic reticulum transmembrane potential; role of the signalling and buffering proteins and of the size of the Ca2+ sequestering ER subcompartments. Bioelectrochem. Bioenerg. 46:79–90. [Google Scholar]
- Marin, J., A. Encabo, A. Briones, E. C. Garcia-Cohen, and M. J. Alonso. 1999. Mechanisms involved in the cellular calcium homeostasis in vascular smooth muscle: calcium pumps. Life Sci. 64:279–303. [DOI] [PubMed] [Google Scholar]
- Ng, L. C., and A. M. Gurney. 2001. Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circ. Res. 89:923–929. [DOI] [PubMed] [Google Scholar]
- Ogawa, Y., N. Kurebayashi, and T. Murayama. 2000. Putative roles of type 3 ryanodine receptor isoforms (RyR3). Trends Cardiovasc. Med. 10:65–70. [DOI] [PubMed] [Google Scholar]
- O'Neill, S. C., P. Donoso, and D. A. Eisner. 1990. The role of [Ca2+]i and [Ca2+] sensitization in the caffeine contracture of rat myocytes: measurement of [Ca2+]i and [caffeine]i. J. Physiol. 425:55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher, P., P. Csordas, T. Schneider, and G. Hajnoczky. 2000. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J. Physiol. 529:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash, Y. S., M. S. Kannan, T. F. Walseth, and G. C. Sieck. 1998. Role of cyclic ADP-ribose in the regulation of [Ca2+]i in porcine tracheal smooth muscle. Am. J. Physiol. 274:C1653–C1660. [DOI] [PubMed] [Google Scholar]
- Ricken, S., J. Leipziger, R. Greger, and R. Nitschke. 1998. Simultaneous measurements of cytosolic and mitochondrial Ca2+ transients in HT29 cells. J. Biol. Chem. 273:34961–34969. [DOI] [PubMed] [Google Scholar]
- Rizzuto, R., P. Bernardi, and T. Pozzan. 2000. Mitochondria as all-round players of the calcium game. J. Physiol. 529:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, E., M. Duvert, and R. Marthan. 2002. Combined effect of chronic hypoxia and in vitro exposure to gas pollutants on airway reactivity. Am. J. Physiol. Lung Cell. Mol. Physiol. 283:L628–L635. [DOI] [PubMed] [Google Scholar]
- Roux, E., C. Guibert, H. Crevel, J. P. Savineau, and R. Marthan. 1996. Human and rat airway smooth muscle responsiveness after ozone exposure in vitro. Am. J. Physiol. 271:L631–L636. [DOI] [PubMed] [Google Scholar]
- Roux, E., C. Guibert, J. P. Savineau, and R. Marthan. 1997. [Ca2+]i oscillations induced by muscarinic stimulation in airway smooth muscle cells: receptor subtypes and correlation with the mechanical activity. Br. J. Pharmacol. 120:1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, E., J. M. Hyvelin, J. P. Savineau, and R. Marthan. 1998. Calcium signaling in airway smooth muscle cells is altered by in vitro exposure to the aldehyde acrolein. Am. J. Respir. Cell Mol. Biol. 19:437–444. [DOI] [PubMed] [Google Scholar]
- Sanders, K. M. 2001. Invited review: mechanisms of calcium handling in smooth muscles. J. Appl. Physiol. 91:1438–1449. [DOI] [PubMed] [Google Scholar]
- Schuster, S., M. Marhl, and T. Hofer. 2002. Modelling of simple and complex calcium oscillations. From single-cell responses to intercellular signalling. Eur. J. Biochem. 269:1333–1355. [DOI] [PubMed] [Google Scholar]
- Shmigol, A. V., D. A. Eisner, and S. Wray. 1999. The role of the sarcoplasmic reticulum as a Ca2+ sink in rat uterine smooth muscle cells. J. Physiol. 520:153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims, S. M., Y. Jiao, and H. G. Preiksaitis. 1997. Regulation of intracellular calcium in human esophageal smooth muscles. Am. J. Physiol. 273:C1679–C1689. [DOI] [PubMed] [Google Scholar]
- Sims, S. M., Y. Jiao, and Z. G. Zheng. 1996. Intracellular calcium stores in isolated tracheal smooth muscle cells. Am. J. Physiol. 271:L300–L309. [DOI] [PubMed] [Google Scholar]
- Smith, G. D., J. Wagner, and J. Keizer. 1996. Validity of the rapid buffering approximation near a point source of calcium ions. Biophys. J. 70:2527–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svichar, N., V. Shishkin, and P. Kostyuk. 1999. Mitochondrial participation in modulation of calcium transients in DRG neurons. Neuroreport. 10:1257–1261. [DOI] [PubMed] [Google Scholar]
- Szado, T., K. H. Kuo, K. Bernard-Helary, D. Poburko, C. H. Lee, C. Seow, U. T. Ruegg, and C. van Breemen. 2003. Agonist-induced mitochondrial Ca2+ transients in smooth muscle. FASEB J. 17:28–37. [DOI] [PubMed] [Google Scholar]
- Tomasic, M., J. P. Boyle, J. F. Worley 3rd, and M. I. Kotlikoff. 1992. Contractile agonists activate voltage-dependent calcium channels in airway smooth muscle cells. Am. J. Physiol. 263:C106–C113. [DOI] [PubMed] [Google Scholar]
- Tribe, R. M., M. L. Borin, and M. P. Blaustein. 1994. Functionally and spatially distinct Ca2+ stores are revealed in cultured vascular smooth muscle cells. Proc. Natl. Acad. Sci. USA. 91:5908–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallot, O., L. Combettes, and A. M. Lompre. 2001. Functional coupling between the caffeine/ryanodine-sensitive Ca2+ store and mitochondria in rat aortic smooth muscle cells. Biochem. J. 357:363–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breemen, C., Q. Chen, and I. Laher. 1995. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharmacol. Sci. 16:98–105. [DOI] [PubMed] [Google Scholar]
- Wagner, J., and J. Keizer. 1994. Effects of rapid buffers on Ca2+ diffusion and Ca2+ oscillations. Biophys. J. 67:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa, A., C. van Breemen, and G. Isenberg. 1996. Buffering of plasmalemmal Ca2+ current by sarcoplasmic reticulum of guinea pig urinary bladder myocytes. Am. J. Physiol. 271:C833–C841. [DOI] [PubMed] [Google Scholar]