

Abstract
Recent studies employing Ca2+ indicators and confocal microscopy demonstrate substantial local Ca2+ release beneath the cell plasma membrane (subspace) of sinoatrial node cells (SANCs) occurring during diastolic depolarization. Pharmacological and biophysical experiments have suggested that the released Ca2+ interacts with the plasma membrane via the ion current (INaCa) produced by the Na+/Ca2+ exchanger and constitutes an important determinant of the pacemaker rate. This study provides a numerical validation of the functional importance of diastolic Ca2+ release for rate control. The subspace Ca2+ signals in rabbit SANCs were measured by laser confocal microscopy, averaged, and calibrated. The time course of the subspace [Ca2+] displayed both diastolic and systolic components. The diastolic component was mainly due to the local Ca2+ releases; it was numerically approximated and incorporated into a SANC cellular electrophysiology model. The model predicts that the diastolic Ca2+ release strongly interacts with plasma membrane via INaCa and thus controls the phase of the action potential upstroke and ultimately the final action potential rate.
INTRODUCTION
Recent studies employing Ca2+ indicators together with confocal microscopy demonstrate substantial cyclic intracellular Ca2+ changes beneath the cell plasma membrane of sinoatrial node cells (SANCs) (Bogdanov et al., 2001; Vinogradova et al., 2002). Pharmacological and biophysical experiments have suggested that those cyclic variations of submembrane Ca2+ are an important determinant of the pacemaker firing rate, and thus represent a critical factor determining pacemaker cell dominance, as well as for achieving the maximum possible chronotropic effect under hormonal stimulation of the heart (Lakatta et al., 2003). An important experimental finding was that part of the cyclic Ca2+ release occurs in the form of localized events during diastolic depolarization (CRDD). The spontaneous (voltage-independent) nature of the diastolic release has been recently demonstrated in voltage-clamp experiments and in experiments in saponin-permeabilized SANCs (Vinogradova et al., 2004). It was suggested that CRDD could accelerate the diastolic depolarization (DD) via inward current (INaCa) produced by Na+/Ca2+ exchanger (NCX) and thus control the SANC rate.
Until recently numerical SANC models available in the literature have not considered local [Ca2+] changes. Accordingly, they were unable to reproduce many recent experimental findings such as chronotropic effects of intracellular Ca2+ buffering and sarcoplasmic reticulum (SR) function modulation. The role of [Ca2+] changes in the submembrane space has been introduced in a new SANC model by Kurata et al. (2002). In contrast to previous SANC models, this model can reproduce some Ca2+-related phenomena including different effects of BAPTA and EGTA on the SANC beating rate. However, the model still cannot explain many experimental findings such as spontaneous Ca2+ release during DD and the chronotropic effect of blocking SR calcium release with ryanodine (Bogdanov et al., 2001; Li et al., 1997; Rigg and Terrar, 1996). Furthermore, quantitative intracellular Ca2+ concentrations ([Ca2+]) in the subspace of SANCs have not previously been measured. The aim of this study was to measure [Ca2+] and to characterize the diastolic part of the Ca2+ release in the subspace so that it could be approximated numerically and included in a new primary pacemaker cell model to provide numerical validation to the implied functional importance of the cyclic spontaneous Ca2+ release during DD for the SANC pacemaker function.
MATERIALS AND METHODS
Sinus node cell preparations and electrophysiological recordings
Single sinoatrial nodal cells were isolated from rabbit hearts as previously described (Vinogradova et al., 2000). We studied spindle-shaped sinoatrial nodal cells that exhibited spontaneous contractions in normal Tyrode solution containing 1.8 mM Ca2+. Perforated patch-clamp technique with 50 μM β-escin (Fan and Palade, 1998) added to the pipette solution was employed to record spontaneous action potentials (AP) with an Axopatch-200 B patch-clamp amplifier (Axon Instruments, Foster City, CA). The bath solution had the following composition (in mM): NaCl, 140; KCl, 5.4; MgCl2, 1; HEPES, 5; CaCl2, 1.8; Glucose, 5.5; pH, 7.4. The pipette solution contained (in mM): K-gluconate, 120; NaCl, 10; MgATP, 5; HEPES, 5; KCl, 20; pH, 7.2. The bath temperature was maintained at 35° ± 0.5°C.
Confocal Ca2+ imaging
Cells were placed on the stage of a Zeiss LSM-410 inverted confocal microscope (Carl Zeiss, Mannheim, Germany) and loaded for 15 min with 20 μM fluo-3 AM. Cellular fluo-3 fluorescence was recorded either in x–y (two-dimensional) or line-scan mode. In the line-scan mode the lines are plotted every 2–5 ms, and each line is added to the right of the preceding line to form the line-scan image. Image processing was performed with IDL software (Ver. 5.2, Research Systems, Boulder, CO). To identify and measure local CRDD, custom software was employed which selected them on the basis of their statistical deviation from the background noise (Cheng et al., 1999).
Ca2+ calibration
To identify absolute Ca2+ concentrations, the fluo-3 signals were calibrated using a procedure that was a modification of that we previously reported (Maltsev et al., 1994). In short, absolute [Ca2+] was calculated using a well-known equation (see e.g., Grynkiewicz et al., 1985),
 |
where Kd is the dissociation constant for Ca2+-bound fluo-3 and is 864 nM at 37°C (Merritt et al., 1990). Maximum and minimum fluorescence (Fmax and Fmin) were determined offline in the images using the MetaMorph Imaging System (Universal Imaging, Downingtown, PA). Ca2+ ionophores such as ionomycin or 4-bromo A-23187 are usually used to obtain Fmax. However, in SANCs, with the bath [Ca2+] being 1.8 mM, we found that application of 5 μM ionomycin to the bath solution did not saturate fluo-3 with Ca2+. To reach the saturation, the bath [Ca2+] was increased to 10 mM. Fmin was calculated as described previously by Kao et al. (1989), taking into account that metal-free fluo-3 has 1/40 the fluorescence of the Ca2+ complex. Therefore,
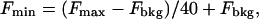 |
where Fbkg was background cell fluorescence (autofluorescence) measured after cell lysis by digitonin (30 μM) in the presence of 10 mM EGTA and no Ca2+.
We used the x–y scans instead of the line-scan images for Ca2+ calibration to precisely monitor the subspace signals. When maximum fluorescence signal Fmax was measured, ionomycin strongly increased intracellular [Ca2+] which, in turn, slightly changed the cell shape. With this change, in the line-scan mode it is difficult to maintain the precise close relative position (within 1–2 μm) of the scanning line with respect to the cell membrane. In other words, during calibration the scan line tends to miss the subspace and the signal becomes lost. The x–y scans obviously avoid this problem, inasmuch as the Ca2+ signals in subspace can be relatively easily identified and measured during the calibration.
Materials
All substances for Ca2+ calibration and for pipette and bath solutions were from Sigma (St. Louis, MO). Fluo-3-AM was purchased from Molecular Probes (Eugene, OR).
Computer simulations
Computer modeling studies were performed using Delphi-7 (Borland, Scotts Valley, CA) software. To model SANC electrophysiology we modified a recently published model of the primary pacemaker cell (Kurata et al., 2002) to include a phenomenological representation of CRDD, as described below. We received the original model text directly from Dr. Kurata.
RESULTS
The two phases of Ca2+ changes in the subspace
To illustrate the need for special consideration of Ca2+ signals in the subspace due to the inhomogeneity of Ca2+ changes within the cell, we oriented the scan line across the cell so that it crossed the longitudinal axis of the cell at approximately one-half of the cell depth (Fig. 1). It is clearly seen that during DD Ca2+ changes first occur in the subspace. We have previously shown that these Ca2+ changes are mainly contributed by local Ca2+ releases (Bogdanov et al., 2001; Vinogradova et al., 2002). The releases (shown by arrows) either terminate locally in the subspace or spread/propagate relatively slowly toward the cell center, inasmuch as SANCs lack t-tubules. The second part of the Ca2+ change, the systolic phase, comes simultaneously with the AP upstroke and occurs both in the subspace and in the cell center.
FIGURE 1.

Ca2+ signals across the cells as identified by line-scanning confocal microscopy. The scan line was oriented across the cell so that it crossed the longitudinal axis of the cell at approximately one-half of the cell depth (inset). Arrows on the confocal image show local Ca2+ releases in the submembrane space during diastolic depolarization. The blue line shows simultaneous recording of the membrane potential measured by the perforated patch-clamp technique.
Averaged Ca2+ signals in the subspace
To dissect the Ca2+ changes in subspace and to improve the signal/noise ratio, the details of the Ca2+ signals were further studied with the scanned line oriented along the longitudinal axis of the cell close to the plasma membrane. Examples of the line-scan images together with respective local Ca2+ signals in the subspace are shown in Fig. 2. The SANC model developed in the study describes the subspace [Ca2+] by a single variable, Casub. On the other hand, the Ca2+ releases and related Ca2+ changes in the subspace occur as local stochastic events. To avoid the stochastic-related noise and to approximate Casub for the SANC model, we averaged the local Ca2+ signals using a custom-made computer program. Spatial inhomogeneity was found to have negligible effect on the contribution of CRDD to whole-cell cellular electrophysiology, as discussed in detail in the Appendix. The SANC membrane almost instantly integrates any change in local electrical charge throughout its length. For example, for distances up to 100 μm, it needs <0.1 ms for almost complete charge integration by the cell capacitance. Thus, for determining membrane potential (Vm) changes, the spatial complexity of the diastolic Casub changes does not seem to be important. Our computer program splits experimental raw Casub traces into fragments and then averages them for further numeric approximation. Each fragment consists of two cycles identified by their peak positions. To avoid the phase shift due to various cycle lengths, we assign the same phase as t = 0 for the moment of maximum peak in the middle of the fragment. Fig. 3 shows Casub signals recorded from four cells along with their respective averages, which reveal a characteristic bump-and-spike morphology reflecting the diastolic and systolic components (marked by D and S in Fig. 3), respectively. In the absence of hormonal stimulation, the amplitude of the average diastolic Ca2+ component varied from 23% to 30% of the systolic component (26.3 ± 1.75%, mean ± SE, n = 4). The duration of the diastolic Ca2+ component varied from 69 ms to 160 ms (100 ± 24 ms), remaining ∼18% of the total cycle length (18.0 ± 1.2%, n = 4).
FIGURE 2.

Ca2+ signaling identified by confocal line-scan imaging of SANCs in a subsarcolemmal space. The scanned line was oriented along to the longitudinal axis of the cell close to the plasma membrane (inset). The diastolic Ca2+ releases are indicated by arrows on the image. The respective trace of the average Ca2+ signal (in F/F0 scale) processed from the whole image width is shown on the bottom panel.
FIGURE 3.

The two distinct components of Ca2+ signaling in submembrane space: D, the diastolic Ca2+ release and S, the systolic Ca2+ release (right panels), as identified by averaging the raw F/F0 traces (superimposed, left panels) recorded by line-scanning confocal microscopy in four cells. The traces were split into two-cycle fragments by custom-made software and centered, assigning time = 0 for the peak in the middle (see dashed line marked as t = 0). Numbers of superimposed traces are 15, 9, 8, and 14 (left panels, from top to bottom).
Measurements of systolic and diastolic absolute Ca2+ levels
In addition to the Ca2+ signal shape, another important characteristic of our SANC model with the new diastolic Ca2+ component is a correct range of absolute [Ca2+] changes in the subspace. In this study we determined the systolic and diastolic absolute [Ca2+] levels in the subspace by calibrating the respective fluo-3 fluorescence signals. For this purpose, cellular fluo-3 fluorescence was recorded in x–y (two-dimensional) scan mode in a thin cell slice approximately in the middle of the cell height as confirmed by an additional scan image across the cell along the z axis (perpendicularly to the surface of the bath). The systolic and diastolic Ca2+ signals were identified on the x–y cell scans by appearance of bright and dark strips of fluo-3 fluorescence, respectively. We measured the signals in areas that were close to the image edge and thus presumed to reflect Ca2+ changes in the cell submembrane space. The absolute values of fluo-3 fluorescence were quantified offline using The MetaMorph Imaging System (Universal Imaging Corporation). In this set of Ca2+ calibration experiments, we acquired and processed three x–y images for each cell. The collected data were then calibrated using Ca2+ ionophore ionomycin (see Materials and Methods for calibration procedure details). We determined absolute [Ca2+] levels for diastolic and systolic phases in three cells. The average data for systolic and diastolic [Ca2+] levels were 1363 ± 394 nM and 204 ± 12 nM (mean ± SD, n = 3), respectively.
Approximation of the spontaneous Ca2+ release
The importance of the diastolic Ca2+ component for the DD phase and the SANC rate was then explored by means of computer modeling. Casub changes, which are important for cell membrane potential control, are considered in the most recent SANC model by Kurata et al. (2002) (Fig. 4 A). The model presumes that jrel, Ca2+ release flux from the junctional SR to subspace, is described by a simple quadratic function of Casub, as
 |
(1) |
where Carel is the Ca2+ concentration in the junctional SR, Prel is the rate constant for Ca2+ release from the junctional SR, and Krel is the half-maximal Casub for Ca2+ release from the junctional SR. However, jrel = f (Casub)—i.e., it is an instantaneous relation between jrel and Casub that is generally not true. During Ca2+-induced Ca2+ release (CICR) the ryanodine receptor (RyR) channels become inactivated, terminating the Ca2+ release. Multiple mechanisms have been suggested for the release termination, e.g., via RyR regulation by luminal Ca2+ (Gyorke et al., 2002), by time-dependent inactivation and/or thermodynamically irreversible gating of RyR (Wang et al., 2002), or by a cytosolic Ca2+/calmodulin mechanism (Fruen et al., 2000). Robust CICR termination is the central problem of local Ca2+ control of cardiac excitation-contraction coupling (Stern, 1992) and is not solved yet. As the aim of this study was only to demonstrate the importance of the diastolic Ca2+ release for the SANC rate, we did not test a particular mechanism for RyR inactivation but rather developed a SANC Ca2+ release formulation based on a phenomenological representation. To approximate the experimental pattern of Ca2+ signals in the subspace (Fig. 3), we added to the release flux in the model a “spontaneous” Ca2+ release component, jspont (Fig. 4 B),
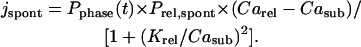 |
(2) |
Prel,spont is the rate constant for the spontaneous Ca2+ release from the junctional SR and it determines the amplitude of the Ca2+ release. In this study we considered a wide range of possible Prel variations from 0.5 ms−1 as in the original Kurata et al. (2002) model (this value was misprinted in the original article) up to 10 ms−1. Compared to Eq. 1, we also added the phasic factor (Fig. 4, blue line), which reflects the phase of Ca2+ release in the subspace and is described within each SANC cycle by one complete cycle of a cosine function, scaled by 0.5 and shifted by 0.5, that varies between 0 and 1 as
 |
(3) |
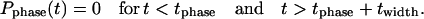 |
The variable t is time that is left after the previous Ca2+ transient peak. The release phase tphase describes the moment when the spontaneous release begins. The total duration of the spontaneous Ca2+ release is described by twidth. The physiological significance of these phenomenological variables is that twidth would reflect the half-width of the temporal distribution of the signal mass of CRDDs, whereas the moment t = tphase + twidth/2 would coincide with the maximum spontaneous release phase (when the signal mass from CRDDs reaches its maximum). The cosine function itself does not have any theoretical significance but was chosen as a phenomenological curve to model the Ca2+ cycling process in SANCs. The phasic factor, Pphase(t) described by Eq. 3, starts from 0 at the beginning of the spontaneous release phase (t = tphase) and after reaching its peak (Pphase = 1) returns to 0. We assume that Pphase(t) = 0 before the spontaneous release begins (t < tphase) and after the cycle is over (t > tphase + twidth). It is important to note that our new model is a recursive computer model inasmuch as its variable t is determined for each cycle by the Ca2+ release phase generated by the same model on the previous cycle, thereby modeling the “resetting” of SR oscillations by the systolic Ca2+ release. The final Ca2+ transient incorporating our phenomenological model of the diastolic Ca2+ release is close to that which we obtained for the averaged [Ca2+] signals in the subspace (compare Fig. 3, Averages, and Fig. 4 B, green line).
FIGURE 4.

Increase in SANC beating rate on introduction of the spontaneous Ca2+ release component, jspont, described by Eq. 2 (see text), into the model. (A) Subspace [Ca2+], Casub, and membrane potential, Vm, generated by the original model of Kurata et al. (2002) from Eq. 1. (B) Casub and Vm simulations of the same model but with introduced spontaneous Ca2+ release. The time course for Pphase(t), described by Eq. 3, is also shown (blue line). The beating rate increased from 195.4 bpm to 246.7 bpm with the introduction of jspont with the following parameters: Prel,spont = 5 ms−1, twidth = 150 ms, and tphase = 150 ms.
Electrophysiological effects of spontaneous Ca2+ release
Introduction of the new diastolic spontaneous Ca2+ release component (jspont, Eq. 2) into the model resulted in significant increase in the AP firing rate. Fig. 4 shows an example of the strong chronotropic effect produced by the spontaneous Ca2+ release. The beating rate increased from 195.4 to 246.7 beats per min (bpm). With this new spontaneous release component, the AP firing rate becomes too high for a SANC in the absence of hormonal stimulation. Also, the maximum systolic [Ca2+] level decreased from 1815 nM (original model from Kurata et al., 2002) to 1450 nM because the diastolic Ca2+ release partially emptied SR before AP upstroke in the new model.
Our version of a SANC model with the diastolic Ca2+ release
We adjusted some of the conductances and Ca2+ handling parameters in the Kurata et al. (2002) model for the beating rate and the range of [Ca2+] changes to be in line with experimental data. Our variant of the SANC model (Fig. 5) has the spontaneous Ca2+ release given by Prel,spont = 2.5 ms−1, twidth = 150 ms, and tphase = 180 ms; and the following ion conductances (in nS/pF): maximum ICaL conductance, gCaL = 0.464; background Na+ conductance, gbNa = 0.00216; maximum ICaT conductance, gCaT = 0.4; and maximum Ist conductance, gst = 0.0075. The assigned conductance values are close to those in the original model and/or to those reported in the literature. Since with these new conductances the minimum diastolic Ca2+ level significantly dropped (to ∼120 nM), we compensated this effect with a slight decrease of the rate constant for Ca2+ uptake by the Ca2+ pump in the network SR, Pup = 0.004 M/s, and we also included the background Ca2+ current IbCa with a normalized conductance of gbCa = 1.2 pS/pF. This current was incorporated in the previous SANC models to balance the Ca2+ extrusion via NCX during diastole but it was assumed to be negligible in the Kurata et al. (2002) model. The adjusted SANC model has a beating rate of 201 bpm and the Ca2+ systolic and diastolic levels 1384 nM and 186 nM, respectively, which fit our experimentally measured values well (see above). Other important parameters of the AP cycle produced by the model were: maximum diastolic potential = −63.4 mV; peak overshoot potential = 12.3 mV; AP amplitude = 75.7 mV; maximum dVm/dt = 4.2 V/s; Cycle length = 298 ms; and AP duration at 50% repolarization = 102 ms.
FIGURE 5.

Our variant of SANC model with the diastolic Ca2+ release and adjusted membrane conductance and Ca2+ handling parameters to be in line with experimental data (left side marked as + Spontaneous Ca2+ Release) and its negative chronotropic response on the complete blockade of the spontaneous release (jspont = 0) simulating ryanodine effect. Shown are simulated traces for subspace [Ca2+], Casub, and membrane potential, Vm, changes. For model parameters see text. T is the action potential period; bpm is beats per min.
The parameters were basically similar to those of the previously published SANC models as well as those measured experimentally.
The phase of spontaneous Ca2+ release controls the phase of AP upstroke and the firing rate
In this SANC model, the blockade of spontaneous SR release results in an almost instant reduction of AP rate from 201 bpm to 148 bpm (reduction by ≈26%) that simulates ryanodine effect (jspont = 0, Fig. 5) reported previously (Bogdanov et al., 2001; Vinogradova et al., 2002; Li et al., 1997; Rigg and Terrar, 1996). This substantial ryanodine effect was missing in the two available primary SANC models by Kurata et al. (2002) and by Zhang et al. (2000) (the latter considered intracellular [Ca2+] = const). To examine the importance of the phase of spontaneous Ca2+ release occurrence for AP period (T), we generated a family of plasma membrane responses upon an instant introduction of the jspont component characterized by various phase, tphase (Fig. 6). We found that the AP upstroke phase closely followed (from 280 ms to 405 ms) the phase of the Ca2+ release flux (jrel) in the subspace within a wide range of tphase from 180 ms to 400 ms (see plot T vs. tphase in Fig. 6, inset). The next set of simulations shown in Fig. 7 illustrates the model prediction that the phase of Ca2+ spontaneous release can control the steady-state beating rate. The ability of the spontaneous Ca2+ release to control the SANC beating rate is increased as the amplitude of the release increased (see set of plots in Fig. 7 for various Prel,spont varying from 0.5 ms−1 to 10 ms−1). The control becomes almost saturated for Prel,spont > 5 ms−1 (compare plots for Prel,spont = 5 ms−1 and Prel,spont = 10 ms−1). The control is lost if the spontaneous release occurs too early (tphase < 180 ms). This happens because the junctional SR is not refilled with Ca2+ yet and the corresponding Ca2+ release is small. Interestingly, the premature Ca2+ release can also result in a cycle span increase. The SR leak (produced by that release) delays normal SR refilling with Ca2+, ultimately resulting in the prolongation of the complete SANC duty cycle.
FIGURE 6.

The AP upstroke phase follows the phase of spontaneous Ca2+ release (tphase) upon introduction of spontaneous Ca2+ release into the model. Shown are superimposed simulated traces of [Ca2+] changes in subspace (Casub) along with respective APs generated with varying phase of the diastolic release (tphase, shown at peaks of Casub traces). (Inset) Plot of the first AP period (T) as a function, tphase.
FIGURE 7.

The phase and the amplitude of the diastolic Ca2+ release control the steady-state rate of AP firing. The family of plots shows relationships of cycle period (or respective SANC rate, right scale) versus the phase of the diastolic release (tphase in Eq. 3) for various Ca2+ release amplitude determined by Prel,spont (in ms−1) in Eq. 2.
Since the spontaneous beating rate of isolated rabbit SANCs varies from ∼2–3 Hz, we also explored whether the control of the SANC rate by diastolic release was robust and could be reproduced with various membrane conductance parameter sets simulating cells with various spontaneous activity. The cell model illustrated in Fig. 7 has the range of regulation from 2.5 Hz to 3.8 Hz that covers the upper range of SANC rates. We tested if the regulation is preserved for a SANC that has an overall slower DD. To achieve this, the following parameters were changed compared to the model set illustrated in Fig. 7 (in nS/pF): gCaL = 0.58; gbNa = 0.00162; gCaT = 0.229; gbCa = 0.0006; and maximum current produced by Na+/K+ ATPase INaK,max = 4.32 pA/pF. We found that with Prel,spont = 5 ms−1, Pup = 0.005 M/s, and with tphase varying from 475 ms to 225 ms, this model set generated spontaneous action potentials with the rate from 2.08 Hz to 3.18 Hz, respectively (not shown), covering essentially the entire range of spontaneous rabbit SANC activity.
Another issue related to the ryanodine effect is that ryanodine might affect sarcolemmal ion channels in some experimental conditions. However, the available data are limited and the reported effects were marginal and somewhat contradictory. In one report by Satoh (1997), ryanodine in a relatively high concentration of 10 μM produced a 45% decrease of hyperpolarization-activated current (If) at −90 mV, whereas others reported no significant ryanodine effect on If (Li et al., 1997). Satoh (1997) also observed a 10% increase of ICaL with 1 μM ryanodine and 8% decrease with 10 μM ryanodine, whereas others found no significant effect of ryanodine on ICaL (Li et al., 1997; Bogdanov et al., 2001). In the same report, the delayed rectifier K+ current (IK) was decreased by 6% and by 26% at 1 μM and 10 μM of ryanodine, respectively. In another report by Li et al. (1997), ryanodine (10 μM) slightly decreased ICaT by ∼17% in cultured pacemaker cells with no effect on the “background” inward current (Ibg) (Li et al., 1997). The authors did not separate whether the reported ICaT change was due to [Ca2+] or a direct effect of ryanodine on T-type Ca2+ channels. We examined the effect of these “extreme” reductions of ICaT (by 17%) and IK (IKr + IKs) (by 26%) and found that they resulted in minor AP rate decreases of ∼5% and 4%, respectively (or 10%, if combined). Although the ICaT and IK changes modulate the AP rate in the very limited range, the diastolic Ca2+ release controls the rate in a wide range from 150 to 226 bpm (Fig. 7) that corresponds to the rate reductions up to 34% when the release is blocked or reduced. Thus it appears that any direct effect of ryanodine on sarcolemmal currents is a minor issue.
The key role of NCX in transduction of Casub signals to the plasma membrane
The steady-state beating rate reflects the change in the phase of AP upstroke in each cycle. An important question is what is the mechanism of the AP upstroke phase change? The missing link in the mechanism is obviously a plasma membrane ion current(s) that would connect subspace Ca2+ changes with plasma membrane potential. The SANC model used in this study has only two ion current components, which are Ca2+-dependent—namely ICaL inactivation and INaCa. Occurrence of CRDD are associated with inward current produced by NCX (Bogdanov et al., 2001) and thus can explain the chronotropic effects of the Ca2+ release phase change. On the other hand, Ca2+-dependent ICaL inactivation cannot provide the link in the observed effects, inasmuch as the opposite effect is expected. Increase in Casub would inactivate the Ca2+ channels and thus decrease the whole cell (inward) ICaL. Also, ICaL is very small during the initial part of DD as the pacemaker potential starts below the level of the Ca2+ channel activation threshold. Other Ca2+-dependent mechanisms of ICaL regulation, such as modulation of ICaL inactivation and reactivation by CaMKII (Vinogradova et al., 2000), merit examination in future studies.
Fig. 8 A shows a complete AP cycle together with simulated ion currents that have inward direction during DD. It is clearly seen that INaCa remains the predominant source of membrane depolarization during DD until its very late phase. Compared to other currents during DD, it is always at least two times larger than ICaT and three times larger than If. The only current that becomes equal to INaCa is ICaL, when the membrane potential reaches −32.8 mV (i.e., close to ICaL activation threshold). Importantly, INaCa behaves very dynamically during DD: it significantly increases as NCX becomes stimulated by Ca2+ from spontaneous release. This INaCa increase results in a significantly faster DD rate (Fig. 8 B, Fast DD) and a higher frequency of AP firing. On the other hand, when spontaneous release is delayed, INaCa remains relatively small during the early and middle phases of DD, so that the DD rate is significantly slower inasmuch as other inward currents provide only a modest rate of depolarization of the plasma membrane (Fig. 8 B, Slow DD).
FIGURE 8.

NCX transmits Ca2+ signals to the plasma membrane. (A) Complete AP cycle (Vm) along with simulated ion currents that have inward direction during diastolic depolarization (DD), namely, current produced by the Na+/Ca2+ exchanger, INaCa; L-type Ca2+ current, ICaL; T-type Ca2+ current, ICaT; background Na+ and Ca2+ currents IbNa and IbCa; hyperpolarization-activated If current; and sustained inward current, Ist. INaCa remains the largest inward current during DD until the membrane potential reaches −32.8 mV (i.e., ICaL activation threshold). ICaL is shown truncated. (B) Two sets of superimposed simulations generated for normal and late spontaneous release occurrence (red and blue lines, respectively). When spontaneous release is late, INaCa is relatively small and DD significantly slows (Slow DD). Each set displays simulated traces of INaCa, membrane potential, Vm, and the phase of spontaneous Ca2+ release, Pphase(t), as defined by Eq. 3.
DISCUSSION
The present study for the first time provides a numerical approximation of diastolic Ca2+ release in SANCs. In contrast to previous models, the new SANC model that includes our Ca2+ release scheme reproduces the diastolic Ca2+ release (Figs. 3 and 4), ryanodine effect (Fig. 5), and the range of Ca2+ changes in the subspace. To our knowledge, this is the first report of absolute Ca2+ concentration measurements in the subspace of SANCs. An important result of the study is that the model predicts that NCX can provide a powerful functional link between SR and plasma membrane. Specifically, via NCX the diastolic Ca2+ release is able to control the AP upstroke timing and ultimately the final SANC rate in a wide and physiologically important range from 2.5 Hz to 3.8 Hz and from 2.08 Hz to 3.18 Hz, respectively, for the two model parameter sets explored in the study.
At present there is a general consensus about the importance of Ca2+ release for SANC rate (Lipsius and Bers, 2003). However, reports vary regarding its significance when compared to other factors contributing to pacemaking, and particularly regarding the magnitude of the ryanodine effect. Importantly, all research groups report some chronotropic effect of ryanodine in all of experimental conditions and preparations including isolated SANC and sinus-node preparations (see Lakatta et al., 2003). The extent of the ryanodine effect varies from a 20% rate decrease to a complete halt of pacemaking. Therefore, an important discussion still remains regarding intracellular Ca2+ as a dominant or critical factor for cardiac pacemaker cell dominance (Honjo et al., 2003 versus Lakatta et al., 2003). Part of this variability may be related to cellular heterogeneity within the sinus node, as ryanodine effect might be different within the primary and peripheral SANCs. However, our model is designed to represent the ionic currents present in primary SANCs, and the magnitude of diastolic calcium release was determined directly from localized imaging in the subspace; whole-cell measurements of Ca2+ release proteins may not reflect the true activity of the release machinery in this critical location. The subspace imaging combined with numerical modeling here is the first study that directly tested the hypothesis that local subspace Ca2+ variations could control the AP rate of a primary sinus node cell that leads the heart beat. We show here that CRDDs can control the SANC rate in a broad physiologically important range, indicating that this Ca2+-related mechanism is viable and can significantly contribute to the final heart rate.
Another important question concerns the pathway of β-adrenergic stimulation in SANCs via either If (Bucchi et al., 2003) or NCX (Vinogradova et al., 2002). One concept is that during DD the initial membrane depolarization via If and/or other membrane currents reaches the level of T-type (and then L-type) Ca2+ channel activation. The Ca2+ ions entering the Ca2+ channels induce CICR that, in turn, via NCX accelerates DD. Another concept is that the Ca2+ release is intrinsically spontaneous (Lakatta et al., 2003; Vinogradova et al., 2004) and under the control of β-adrenergic receptors; it has its own clock and does not need an initial push from the membrane. Accordingly, the two oscillators (one is in membrane and another is in SR) dynamically interact during the SANC duty cycle producing a variety of chronotropic effects (Lakatta et al., 2003). The present study provides theoretical support for the concept of the interacting oscillators: if such spontaneous (and cyclic) releases occur in SANCs they can effectively control the SANC rate via NCX. It was previously shown that the β-adrenergic stimulation of SANCs increased both the frequency and the amplitude of spontaneous Ca2+ release in the subspace (Vinogradova et al., 2002). The stimulatory effect can be analyzed using our series of plots shown in Fig. 7 depicting the model prediction for SANC rate as a function of tphase for various Prel,spont. As tphase is linked to the Ca2+ release frequency and Prel,spont is linked to the Ca2+ release amplitude, the model explains/predicts a substantial chronotropic effect of β-adrenergic stimulation apparently via an NCX effect. A further, more detailed analysis of Fig. 7 allows an additional important prediction: that increased amplitude of the release (such as under β-adrenergic stimulation) results in a wider range of rate regulation—for example, from 150 to 226 bpm for Prel,spont = 10 ms−1 but from 150 to only 185 bpm for Prel,spont = 1.25 ms−1. In other words, regulation via the phase shift of CRDD has limited capacity to alter action potential timing until the release amplitude becomes sufficiently large.
Voltage-independent Ca2+ release via RyRs has also been reported in other cardiac cell types (see Lakatta, 1992). For example, steady Ca2+ oscillations and Ca2+ waves occur in Ca2+-overloaded ventricular myocytes. In cells with increased intracellular [Ca2+], RyRs stay close to their activation threshold and their activation can presumably be easily provoked, leading to autooscillatory patterns, although the quantitative dynamics of RyR oscillations are by no means understood. In this study we measured a minimum diastolic [Ca2+] level in SANCs of ∼200 nM, twice the diastolic [Ca2+] level in resting ventricular myocytes. This new finding gives us a basis to speculate that the normal pacemaker SANC activity may have some similar features to the abnormal Ca2+ automaticity in Ca2+-overloaded ventricular myocytes.
Acknowledgments
The authors are grateful to Dr. Heping Cheng for the helpful comments and to Dr. Harold A. Spurgeon and Bruce Ziman for help and technical support.
APPENDIX
Consideration of possible spatial limitations
To approximate Casub, we simply averaged local Ca2+ signals in subspace (Fig. 3). To justify the additive effect of individual CRDDs with respect to their effect on the plasma membrane potential, we considered possibilities for a nonuniform Vm distribution along the cell surface. The potential spatial heterogeneity may arise from local changes in the membrane ion permeability produced by individual CRDDs (via Ca2+-dependent membrane channels and transporters). Theoretically, on the one hand, the local electrical charge change (e.g., generated by NCX) would passively propagate toward cell ends (propagation effect). On the other hand, the local electric charge can be neutralized by the membrane conductance in its vicinity and thus may never reach the cell ends (space constant effect).
Propagation effect
We approximated a SANC as an infinite cable with a diameter d = 10 μm and considered its response upon an instant introduction of an excessive electrical charge at x = 0 described by a delta function, δ(t). The solution for the membrane potential Vm(x,t) is an “expanding” and decrementing Gaussian function,
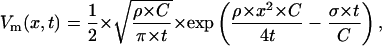 |
with the width w(t) of the “expanding” Gaussian given as
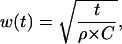 |
where ρ is the resistance of intracellular medium per unit length (Ω/cm), σ is the specific membrane conductance (Ω−1 × cm−1), and C is the membrane capacitance per unit length (F/cm). The well-known estimates for specific membrane capacitance and resistivity of intracellular medium are Cs = 1 μF/cm2 = 10−6 F/cm2 and Ri = 200 Ω × cm, respectively. Accordingly,
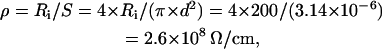 |
where S is the cell cross-section area, and
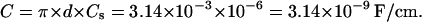 |
Finally,
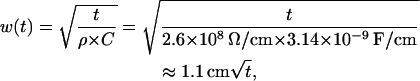 |
where t is given in seconds. It means that after 1 ms the “expanding” Gaussian width w could reach the distance of 1.1 cm × 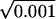 = 0.0348 cm = 348 μm. On the other hand, the Gaussian width will reach the cell ends (for a cell length Lcell = 100 μm) from its center after t = 0.83 w2 = 0.83 s/cm2 × (5 × 10−3 cm)2 ≈ 20.8 μs. This means that the passive Vm propagation occurs almost instantly along the electric cable approximating the cell membrane.
= 0.0348 cm = 348 μm. On the other hand, the Gaussian width will reach the cell ends (for a cell length Lcell = 100 μm) from its center after t = 0.83 w2 = 0.83 s/cm2 × (5 × 10−3 cm)2 ≈ 20.8 μs. This means that the passive Vm propagation occurs almost instantly along the electric cable approximating the cell membrane.
Spatial effect: estimation for the space constant, λ
We used a well-known formula for a cable space constant λ = 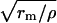 , where rm is membrane resistance per unit length in the voltage range of DD. We estimated the total cell membrane resistance Rtot from the Kurata et al. (2002) model as the ratio δVm/δItotal, where δVm = 1 mV is an instant change of Vm induced by a switch to the voltage-clamp during DD and δItotal is the total membrane current change as the response to that change. We found that minimum Rtot is 0.25 GΩ in the range of DD from −40 mV to −20 mV. Accordingly,
, where rm is membrane resistance per unit length in the voltage range of DD. We estimated the total cell membrane resistance Rtot from the Kurata et al. (2002) model as the ratio δVm/δItotal, where δVm = 1 mV is an instant change of Vm induced by a switch to the voltage-clamp during DD and δItotal is the total membrane current change as the response to that change. We found that minimum Rtot is 0.25 GΩ in the range of DD from −40 mV to −20 mV. Accordingly,
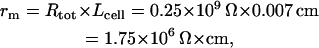 |
and
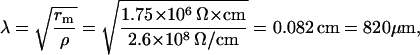 |
which is still much longer than the SANC length (Lcell = 70 μm in the Kurata model). We conclude that the electrical effect of individual CRDD will be distributed instantaneously and uniformly over the surface membrane.
References
- Bogdanov, K. Y., T. M. Vinogradova, and E. G. Lakatta. 2001. Sinoatrial nodal cell ryanodine receptor and Na-Ca exchanger: molecular partners in pacemaker regulation. Circ. Res. 88:1254–1258. [DOI] [PubMed] [Google Scholar]
- Bucchi, A., M. Baruscotti, R. B. Robinson, and D. DiFrancesco. 2003. If-dependent modulation of pacemaker rate mediated by cAMP in the presence of ryanodine in rabbit sino-atrial node cells. J. Mol. Cell. Cardiol. 35:905–913. [DOI] [PubMed] [Google Scholar]
- Cheng, H., L. S. Song, N. Shirokova, A. Gonzales, E. G. Lakatta, E. Rios, and M. Stern. 1999. Amplitude distribution of calcium sparks in confocal images: theory and studies with an automatic detection method. Biophys. J. 76:606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, J.-S., and P. Palade. 1998. Perforated patch recording with β-escin. Pflugers Arch. Eur. J. Physiol. 436:1021–1023. [DOI] [PubMed] [Google Scholar]
- Fruen, B. R., J. M. Bardy, T. M. Byrem, G. M. Strasburg, and C. F. Louis. 2000. Differential Ca2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. Cell Physiol. 279:C724–C733. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz, G., M. Poenie, and R. Y. Tsien. 1985. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260:3440–3450. [PubMed] [Google Scholar]
- Gyorke, S., I. Gyorke, V. Lukyanenko, D. Terentyev, S. Viatchenko-Karpinski, and T. F. Wiesner. 2002. Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle. Front. Biosci. 7:d1354–d1363. [DOI] [PubMed] [Google Scholar]
- Honjo, H., S. Inada, M. K. Lancaster, M. Yamamoto, R. Niwa, S. A. Jones, N. Shibata, K. Mitsui, T. Horiuchi, K. Kamiya, I. Kodama, and M. R. Boyett. 2003. Sarcoplasmic reticulum Ca2+ release is not a dominating factor in sinoatrial node pacemaker activity. Circ. Res. 92:e41–e44. [DOI] [PubMed] [Google Scholar]
- Kao, J. P. Y., A. T. Harootunian, and R. Y. Tsien. 1989. Photochemically generated cytosolic calcium pulses and their detection by fluo-3. J. Biol. Chem. 264:8179–8184. [PubMed] [Google Scholar]
- Kurata, Y., I. Hisatome, S. Imanishi, and T. Shibamoto. 2002. Dynamical description of sinoatrial node pacemaking: improved mathematical model for primary pacemaker cell. Am. J. Physiol. Heart. Circ. Physiol. 283:H2074–H2101. [DOI] [PubMed] [Google Scholar]
- Lakatta, E. G. 1992. Functional implications of spontaneous sarcoplasmic reticulum Ca2+ release in the heart. Cardiovasc. Res. 26:193–214. [DOI] [PubMed] [Google Scholar]
- Lakatta, E. G., V. A. Maltsev, K. Y. Bogdanov, M. D. Stern, and T. M. Vinogradova. 2003. Cyclic variation of intracellular calcium: a critical factor for cardiac pacemaker cell dominance. Circ. Res. 92:e45–e50. [DOI] [PubMed] [Google Scholar]
- Li, J., J. Qu, and R. D. Nathan. 1997. Ionic basis of ryanodine's negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am. J. Physiol. 273:H2481–H2489. [DOI] [PubMed] [Google Scholar]
- Lipsius, S. L., and D. M. Bers. 2003. Cardiac pacemaking: If vs. Ca2+, is it really that simple? J. Mol. Cell. Cardiol. 35:891–893. [DOI] [PubMed] [Google Scholar]
- Maltsev, V. A., B. Wolff, J. Hess, and G. Werner. 1994. Calcium signalling in individual T-cells measured by confocal microscopy. Immunol. Lett. 42:41–47. [DOI] [PubMed] [Google Scholar]
- Merritt, J. E., S. A. McCarthy, M. P. Davies, and K. E. Moores. 1990. Use of fluo-3 to measure cytosolic Ca2+ in platelets and neutrophils. Loading cells with the dye, calibration of traces, measurements in the presence of plasma, and buffering of cytosolic Ca2+. Biochem. J. 269:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigg, L., and D. A. Terrar. 1996. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 81:877–880. [DOI] [PubMed] [Google Scholar]
- Satoh, H. 1997. Electrophysiological actions of ryanodine on single rabbit sinoatrial nodal cells. Gen. Pharmacol. 28:31–38. [DOI] [PubMed] [Google Scholar]
- Stern, M. D. 1992. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63:497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova, T. M., Y.-Y. Zhou, K. Y. Bogdanov, D. Yang, M. Kuschel, H. Cheng, and R.-P. Xiao. 2000. Sinoatrial node pacemaker activity requires Ca2+/calmodulin-dependent protein kinase II activation. Circ. Res. 87:760–767. [DOI] [PubMed] [Google Scholar]
- Vinogradova, T. M., K. Y. Bogdanov, and E. G. Lakatta. 2002. β-adrenergic stimulation modulates ryanodine receptor Ca2+ release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ. Res. 90:73–79. [DOI] [PubMed] [Google Scholar]
- Vinogradova, T. M., Y.-Y. Zhou, V. Maltsev, A. Lyashkov, M. Stern, and E. G. Lakatta. 2004. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. In press. [DOI] [PubMed]
- Wang, S. Q., L. S. Song, L. Xu, G. Meissner, E. G. Lakatta, E. Rios, M. D. Stern, and H. Cheng. 2002. Thermodynamically irreversible gating of ryanodine receptors in situ revealed by stereotyped duration of release in Ca2+ sparks. Biophys. J. 83:242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, H., A. V. Holden, I. Kodama, H. Honjo, M. Lei, T. Varghese, and M. R. Boyett. 2000. Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am. J. Physiol. 279:H397–H421. [DOI] [PubMed] [Google Scholar]