

Abstract
Numerous investigations in the last years focused on chromosome arrangements in interphase nuclei. Recent experiments concerning the radial positioning of chromosomes in the nuclear volume of human and primate lymphocyte cells suggest a relationship between the gene density of a chromosome territory (CT) and its distance to the nuclear center. To relate chromosome positioning and gene density in a quantitative way, computer simulations of whole human cell nuclear genomes of normal karyotype were performed on the basis of the spherical 1 Mbp chromatin domain model and the latest data about sequence length and gene density of chromosomes. Three different basic assumptions about the initial distribution of chromosomes were used: a statistical, a deterministic, and a probabilistic initial distribution. After a simulated decondensation in early G1, a comparison of the radial distributions of simulated and experimentally obtained data for CTs Nos. 12, 18, 19, and 20 was made. It was shown that the experimentally observed distributions can be fitted better assuming an initial probabilistic distribution. This supports the concept of a probabilistic global gene positioning code depending on CT sequence length and gene density.
INTRODUCTION
The compartmentalization of the nucleus in several well-defined subregions such as nucleoli, nuclear bodies, chromosome territories (CTs), and their higher compartmentalization levels into subchromosomal domains as well as the spatial arrangements of these compartments may have a profound impact on functional processes inside the nucleus (for review, see Dundr and Misteli, 2001; Cremer and Cremer, 2001; Parada and Misteli, 2002; O'Brien et al., 2003). For example, it has been shown that chromosome territories are compartmentalized into domains of early and later replicating chromatin (Visser et al., 1998, Zink et al., 1999): early replicating chromatin domains are found throughout the nucleus except for the utmost nuclear periphery and the perinucleolar space, whereas midreplicating chromatin domains form typical rims both along the nuclear periphery and around the nucleoli (Dimitrova and Berezney, 2002). This specific arrangement of differently replicating chromatin may mirror the results of recent investigations, regarding the positioning of whole CTs inside the nuclear volume. Chromosome painting experiments of single CTs and groups of CTs in different species suggest a relationship between the gene density of a chromosome and its radial positioning (distance to the nuclear center) in the nuclear volume. This was first shown by Croft et al. in 1999 for the different positions of CTs Nos. 18 and 19 in human lymphocytes in a two-dimensional semiquantitative analysis: both chromosomes are of similar DNA content, but the gene-poor CT No. 18 was found at the nuclear periphery, whereas the gene-dense CT No. 19 was found in the nuclear interior. A quantitative three-dimensional (3D) evaluation confirmed the positioning of the gene-dense CTs No. 19 toward the nuclear center and of the gene-poor CTs No. 18 and Y toward the nuclear periphery in morphologically preserved spherical nuclei of lymphocytes, which have an average diameter of ∼10 μm (Cremer et al., 2001). A gene density-correlated radial CT position for almost all chromosomes was described by Boyle et al. in 2001. Additionally, it could be shown that the distinct localization of the chromatin homologous to human chromosome No. 18 and of chromatin homologous to human chromosome No. 19, respectively, was maintained in lymphocytes during the evolution of higher primates, irrespective of major karyotype rearrangements that occurred in these phylogenetic lineages during their evolution, suggesting a functional significance for such an order (Tanabe et al., 2002).
However, the different positioning of a gene density-related radial dependence of chromatin obviously does not apply for all human cell types. In nuclei of human diploid fibroblasts, the CTs of small CTs were found in the nuclear center irrespective of the gene density, while large chromosomes were positioned toward the nuclear periphery, arguing for a chromosome size rather than a gene density-correlated radial arrangement (Cremer et al., 2003; A. Bolzer et al., unpublished results). In contrast to the size-correlated positioning found for chromosomes in these flat nuclei with a thickness of 3–4 μm, model calculations assuming a linear correlation between DNA content and CT volume revealed an inverse distribution of small and large chromosomes with small chromosomes in the nuclear periphery and large chromosomes in the nuclear center in flat ellipsoid nuclei (Habermann et al., 2001). Similarly, in simulated spherical nuclei, the same behavior for small and large chromosomes was predicted (Cremer et al., 2001).
This indicates that the applied geometrical constraints alone are not sufficient to explain the observed radial arrangements. In this contribution, model calculations based on the existing “spherical 1 Mbp chromatin domain (SCD)” model were extended to estimate the influence of gene density (number of genes per Mbp) as an additional geometrical constraint in the initial distribution of “mitotic-like” chromosomes. In this model, each chromosome was described as a linear chain of elastic spherical 1 Mbp-sized domains that are linked together by entropic spring potentials. Starting from such mitotic-like chromosome configurations, assumed to exist shortly after cell division, Metropolis Monte Carlo relaxation runs were applied to calculate relaxed interphase configurations for all the chromosome territories simultaneously. This relaxation process made it possible that during the decondensation, the dynamical spreading of CTs can change their positions and thus is not fixed by the initial distribution. In the case of spheres, for example, this latter case is realized by the modeling procedure performed in Holley et al. (2002).
The 3D mapping of CTs Nos. 18 and 19 described in Cremer et al. (2001) and of CTs Nos. 12 and 20 performed in Weierich et al. (2003) was used as an experimental basis for the comparison with the radial arrangements of simulated CTs.
MATERIAL AND METHODS
Visualization and 3D mapping of individual CTs in 3D FISH experiments
For the experimentally obtained data sets, 3D fluorescence in situ hybridization (FISH) was performed on morphologically preserved lymphocytes that have a spherical shape with mean diameters of ∼10 μm. CTs of chromosomes Nos. 18, 19, 20, and 12 were visualized after chromosome painting with labeled fluorochromes. In these experiments, the CTs of No. 18 and No. 19 were visualized simultaneously by painting these territories with differently labeled fluorochromes. The CTs No. 12 and No. 20 were hybridized in two different experiments. The shape of the nucleus was visualized using a DNA counterstain in all experiments. For details see Cremer et al. (2001) and Weierich et al. (2003). Nuclei were scanned with an axial distance of 200 nm between light optical sections using a three-channel laser scanning confocal microscope (Zeiss LSM 410, Carl Zeiss, Jena, Germany). For each optical section, images were collected sequentially for all three fluorochromes. Stacks of eight-bit gray scale two-dimensional images were obtained with a pixel size of 66–80 nm and used for the quantitative evaluation (see below).
A detailed description of the quantitative radial 3D evaluation of light optical serial sections by a voxel- (volume element) based algorithm was published elsewhere (Cremer et al., 2001). Briefly, as a first step, the geometrical center and the border of the nucleus were determined using the 3D data set of the DNA-counterstain fluorescence. For segmentation, we defined all voxels not belonging to the nuclear interior as image background. For comparison of nuclei with different shape and size, the distance between the nuclear center and each point located on the segmented nuclear border was given as the relative radius (r0 = 100). A decline of the curve for the nuclear counterstain in the most peripheral shells observed by this approach results in part from the Gaussian filtering of the data and in part from irregularities of the nuclear border. In the second step, segmentation of CTs was performed in each 3D stack representing the color channels for painted CTs. All voxel intensities below an automatically set threshold were set to zero. Using an iterative procedure, a threshold value was estimated for each 3D data set for CT thresholding. The segmented nuclear space was divided into 25 equidistant shells with a thickness of Δr = 1/25 r0. For each voxel located in the nuclear interior, the relative distance r from the nuclear center was calculated as a fraction of r0. A shell at a given r contains all nuclear voxels with a distance between r − Δr/2 and r + Δr/2. For each shell, all voxels assigned to a given CT were identified and the fluorescence intensities derived from the respective emission spectrum were summed up. This procedure yielded the individual relative DNA content (differential DNA content) within each shell for painted CTs as well as the overall DNA content as reflected by the DNA counterstain. The sum of the voxel intensities measured in each nucleus was set to 100% for each fluorochrome. Using this normalization, the differential DNA content within a nuclear shell as a function of the relative distance r from the 3D center in the entire set of evaluated nuclei was plotted as a graph.
Virtual microscopy of simulated CTs
To allow a comparison between the experimentally observed and the simulated distributions of CTs inside the nuclear volume, the influence of the limited light optical resolution was simulated by “virtual microscopy”. For this purpose, from the simulated nuclear configurations, virtual image data stacks were calculated. This virtual microscopy approach consisted of a digitization of the spherical domains with diameters of 500 nm by a grid of 39 × 39 × 156 nm voxel spacing and a convolution of the digitized stacks with a measured confocal point spread function (with a full width at half-maximum (FWHM): FWHMx = 279 nm, FWHMy = 254 nm, FWHMz = 642 nm). By this procedure, the mapping of simulated nuclei can be made in the same way as for the experimental one (see method described above).
Simulation of human cell nuclei
For a simulation of the overall structure of CTs in human cell nuclei, the SCD model was applied (Kreth et al., 2001; Cremer et al., 2000). According to this model, each chromosome of the diploid human genome is approximated by a linear chain of spherical 1 Mbp-sized chromatin domains (with a diameter of 500 nm each). The number of 1 Mbp domains is given directly by the DNA content of a given chromosome (according to National Center for Biotechnology Information (NCBI) data, http://www.ncbi.nlm.nih.gov/genome/guide/human/; September 2003). To relate these domains in a linear sequence, adjacent domains are linked together by entropic spring potentials (Fig. 1), which describe the rigidity of “real” 120 kbp linker connections. These latter are assumed to connect adjacent rosettes (of ∼10 120 kbp loops) according to the multi-loop subcompartment model (Münkel and Langowski, 1998). For a description of the stiffness of flexible polymers, usually the worm-like chain model is used that correlates the mean-squared distance 〈R2〉 with the persistence length LP and the contour length LC to:
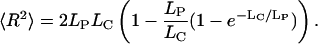 |
(1) |
In the limit case LC ≫ LP, the simple relation
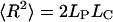 |
(2) |
is obtained, which corresponds directly to the mean-squared displacement of a random walk for a chain of N segments with the Kuhn segment length LK according to the freely jointed chain model:
 |
(3) |
In this case, the Kuhn segment length is equal to the double of the persistence length. Taking into account a Kuhn segment length of 300 nm in the case considered for the SCD model, the 120 kbp chromatin linker has a contour length of ∼1200 nm. This corresponds to the limit case mentioned above where the linker flexibility can be described by an ideal Gaussian chain (random walk). The connection between adjacent domains in the SCD model is therefore described by the potential energy (entropic spring energy) of such a chain:
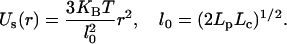 |
(4) |
With the Boltzmann factor kB and the absolute temperature T (= 310 K), in the thermodynamic equilibrium case, this entropic spring energy results in a mean distance of l0 = 600 nm between adjacent domains; r describes the actual distance. Furthermore, the exclusive structure of chromatin has to be regarded. Although Debye-type electrostatic interactions are expected to be limited to a range <10 nm (Cremer et al., 2000), an excluded volume potential might have a far larger range, e.g., due to protein/RNA complexes between domains. For different domains, a slightly increasing potential is assumed to exist that corresponds on a larger scale to the empiric excluded volume potential introduced in Münkel and Langowski, 1998:
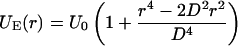 |
(5) |
Here, r describes the distance between the centers of the domains; U0 is the height of the potential and will be set to a value of 1.5 kBT to prevent an intermingling of different domains during the relaxation process. For all distances <D = 500 nm, the potential energy is positive and otherwise zero. These two model assumptions, however, are not sufficient to maintain the experimentally observed compactness of chromosomes in territories. The packaging of all 46 polymer chains (for the diploid human genome) in a spherical volume represents the typical case of a “semidilute” polymer solution that is affected by a chain interpenetration. This could also be shown by long-term Monte Carlo relaxation runs of simulated nuclei (not presented in this study). We therefore introduced a weak potential barrier around each simulated chromosome chain:
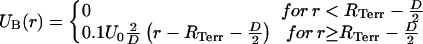 |
(6) |
with
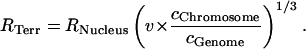 |
(7) |
Here, r describes the distance of a given domain from the gravity center of the simulated chromosome chain. In this way, only domains moving beyond this barrier experience an attractive force back to the center of the simulated CT chain. The radius RTerr of the potential barrier is given by the radius RNucleus of the simulated nucleus, the DNA content of the respective CT cChromosome, and the DNA content of the whole genome cGenome. In this case, the factor v was set equal to 1.
FIGURE 1.

Schematic drawing of the approximation of a chromosome by a linear chain of spherical 1 Mbp-sized domains, which are linked together by entropic spring potentials according to the spherical 1 Mbp chromatin domain (SCD) model.
This potential accounts in a drastically simplified way for forces, which in real nuclei may arise from a combination of parameters, including the rigidity of higher order chromatin segments, or the effects of chromosome territory anchoring proteins.
Relaxation process
To obtain thermodynamic equilibrium configurations with respect to the energies, the Metropolis Monte Carlo method was applied. For this purpose, in a first-start configuration, the spherical domains of each simulated chromosome were placed side by side in a “mitosis-like” arrangement (“start cylinders”, compare Fig. 2 a) with a distance of 14 nm from each other. Random displacements of the domains resulted in relaxed interphase-like configurations using the Metropolis Monte Carlo procedure. According to this procedure, consecutive states in the relaxation process were generated by a Markov process (see, e.g., Binder and Heermann, 1997). This process implicates the principle of the “microscopic reversibility”; this means that the relation between the transition probabilities from an old to a new state and vice versa depend only on the energy difference of the two states. In this way, the procedure can be performed by the following: beginning from an arbitrary state, a new state (generated by a random displacement of a domain) is accepted when the potential energy difference between the new and the old state is ΔH ≤ 0. When the energy difference ΔH is larger than zero, the new state is accepted with the probability exp(−ΔH/kBT). In this way, the energies of the states are distributed according to a Boltzmann distribution in the equilibrium.
FIGURE 2.

Visualization of a modeled human nuclear genome according to the SCD model. In a, the “initial” configuration with the 46 “start cylinders” is shown. Here, the 1 Mbp domains were placed side by side within the start cylinders. (b) Relaxed interphase state after 400,000 Monte Carlo steps. The simulated CTs are visualized in 24 pseudocolors. The visualization was done using the Persistence of Vision Ray-Tracer Pov-Ray (POV-Team, Williamstown, Victoria, Australia). Bar, 5 μm.
For the relaxation of each start configuration, ∼400,000 Monte Carlo steps (in one Monte Carlo step for each CT a randomly chosen domain was displaced) were used (Fig. 2 b). The achievement of an equilibrium state was controlled by the calculation of different geometrical modes during the relaxation process, like the gyration radius (the slowest increasing mode for this system). When this mode showed no further increase, the equilibrium state was assumed to be reached (∼200,000 Monte Carlo steps). Further 200,000 Monte Carlo steps were then executed; these end configurations were used for the quantitative evaluations.
RESULTS
In this study, we compared the experimental results obtained for the radial distributions of single CTs Nos. 12, 18, 19, and 20 in spherical human lymphocytes of normal karyotype (Cremer et al., 2001; Weierich et al., 2003) with model calculations depending on gene density. Chromosome No. 12 (CT sequence length, 133 Mbp) and chromosome No. 20 (63 Mbp) represent chromosomes with intermediate gene densities (see Table 1), chromosome No. 18 (77 Mbp) represents a gene-poor chromosome, whereas chromosome No. 19 (63 Mbp) represents the human chromosome with the highest gene density. CT sequence length and gene density data used were based on the latest information available (NCBI data, September 2003, http://www.ncbi.nlm.nih.gov/genome/guide/human/).
TABLE 1.
Order of human chromosomes (normal karyotype) according to gene density
| No. 19 | No. 17 | No. 22 | No. 16 | No. 20 | No. 11 | No. 1 | No. 12 | No. 15 | No. 7 | No. 14 | No. 6 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 28 | 21 | 17 | 15 | 15 | 15 | 13 | 13 | 12 | 12 | 12 | 11 |
| No. 9 | No. 2 | No. 10 | No. 8 | No. 5 | No. 3 | No. 21 | No. X | No. 18 | No. 4 | No. 13 | No. Y |
| 11 | 11 | 11 | 10 | 10 | 10 | 10 | 10 | 9 | 9 | 7 | 4 |
The gene density values for each human chromosome are given by the number of genes per Mbp (NCBI data, September 2003: http://www.ncbi.nlm.nih.gov/genome/guide/human/).
According to the SCD model, the simulated chromosome chains consisting of a certain number of spherical 1 Mbp-domains (according to the DNA content of a chromosome) were arranged at the beginning (start configuration) in mitotic-like start cylinders (see Material and Methods). The model calculations were based on three different assumptions about the initial distribution of these start cylinders: a statistical, a deterministic, and a probabilistic distribution (see Fig. 3). In addition, two nucleoli were inserted in all three cases, simulated as additional CTs with a DNA content of 80 Mbp. The midpoints of the nucleoli in the start configuration were considered to maintain a minimal distance of 1.75 μm to the nuclear envelope and a minimal distance of 3.75 μm from each other. To regard a certain amount of the final volume (simulation procedure), the midpoints of the start cylinders were located first in so called “initial” CT spheres with radii according to Eq. 7 (Material and Methods). In the case of the statistical initial distribution of the CTs (see Fig. 3 a), the initial spheres were positioned randomly in the nuclear volume with the condition that overlapping with already existing initial spheres was forbidden. As a consequence, in case of an overlap of a randomly chosen position of a given initial CT sphere with another CT sphere, this position was discarded, and a new random position was chosen. This procedure was repeated, until a nonoverlap position was obtained. To ensure that the algorithm is not running in an endless loop (termed as “loop criteria” in the following, meaning that the algorithm finds for all initial CT spheres a nonoverlap position in a tolerable computing time (e.g., a few minutes)), the volumes of the initial spheres had to be reduced by a common factor v (v = 0.22, Eq. 7, Material and Methods).
FIGURE 3.

Schematic drawing of the localization of the initial CT spheres in the nuclear volume for the three simulated cases. In the statistical simulation case (a), the initial CT spheres were put in the nucleus in a random order without further assumptions. In the probabilistic simulation case (b), the initial CT spheres were put in the nucleus in the order of their gene densities, and the distances of the CT spheres to the nuclear center were weighted with the probability density function (Eq. 8) according to their gene densities. In the deterministic simulation case (c), the initial spheres were located on discrete shells in the order of their gene densities (see text for details). Starting with the initial spheres of CT No. 19 on the first shell in the interior, the next two CTs, No. 17 and No. 22, follow in the upper shells and so on. A constraint that has to be fulfilled in all three cases is that overlapping of the initial CT volumes is not allowed.
In the case of the deterministic and probabilistic initial distribution, a gene density correlated distribution of the initial CT spheres in the nuclear volume was performed. To create the deterministic start distribution (Fig. 3 c) after the incorporation of the two nucleoli, the initial spheres of the homologous CTs were located on discrete shells in the nuclear volume in the order of their gene densities as following: Nos. 19, 17, 22, 16, 20, 11, 1, 12, 15, 7, 14, 6, 9, 2, 10, 8, 5, 3, 21, X, 18, 4, 13, and Y (see Table 1; http://www.ncbi.nlm.nih.gov/genome/guide/human/). The simulation was started with the initial spheres of the CTs with the maximum gene density (CTs No. 19) with a shell radius of RTerr(19) (with v = 1, Eq. 7, Material and Methods); then the CTs No. 17 spheres with the second highest gene density were located with a distance of 0.11 × RTerr(17) from the first shell and so on. Here, the loop criteria (see above) enforced a size of v = 0.11 (Eq. 7, Material and Methods) for the initial CT spheres. In this deterministic start distribution, all probabilistic constraints were eliminated, except that on a given shell surface, an initial CT sphere was allowed any radial position not resulting in an overlap.
For the probabilistic initial case (Fig. 3 b), after the incorporation of the two nucleoli, the CTs were put into the nuclear volume in the same order according to gene density as realized for the deterministic initial case. Here, however, in contrast to the deterministic case, the initial CT spheres were not located on discrete shells, but the distance from the center of the initial spheres to the nuclear center was weighted with an exponential probability density function that depends on the gene density of a given chromosome i and the distance d of the initial sphere to the nuclear center in units of the nuclear radius (equal to d = 1.0):
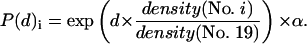 |
(8) |
Density(No. i) is the number of genes per Mbp in CT (No. i). The actual position of an initial CT sphere (i) was confirmed when P(d)i was equal to or smaller than a randomly chosen number between zero and 1: [0;1[ ≥ P(d)i, according to the Monte Carlo procedure. This means: a given initial sphere for a CT (i) = A (e.g., No. 22) was first placed into a nonoverlapping position into the nucleus, according to the rules described above. Then the distance d to the nuclear center was determined for this special position, and P(d)A was calculated using Eq. 8, with the gene density of CT A (e.g., No. 22). Then the calculated P(d)A value (e.g., P(d)A = 0.37) was compared with a random number between zero and 1. If the random number turned out to be equal to or larger than the calculated P(d)A value, then the position of the initial (CT A) sphere was accepted. If the random number turned out to be smaller than the calculated P(d)A value, then again a new randomly chosen position for CT A was tested for nonoverlap; the d value of the new nonoverlapping position was again inserted in Eq. 8 and tested as described above. The procedure was continued until a nonoverlap position was obtained with a random number equal to or larger than the P(d)A value tested. Besides a reduction of the volumes (v = 0.22; Eq. 7) of the initial CT spheres, an acceptance factor α is required (Eq. 8) to ensure the loop criteria (see above) for this procedure. With α = 0.774, typically 3 min on the personal computer used were needed.
After the start configuration with the initial spheres of the diploid human chromosome set (22, X, Y) and the two nucleoli had been created as described above, the midpoints of the start cylinders were placed in these spheres (in all three simulation cases the distance of adjacent domains in the start cylinders was the same). To create relaxed interphase configurations, in the next step, the start cylinders were relaxed into an equilibrium state (this can be interpreted as the dynamic spreading out early in G1); the initial spheres were then discarded and played no further role in the relaxation process. For all three cases, 50 nuclei each were calculated. The relaxation of one simulated nucleus took ∼1 day of computing time on the personal computer (1 GHZ Intel Pentium III) used.
To investigate the differences in the localization of CTs between the initial start arrangement and after the relaxation process, in Fig. 4 the radial distances (distances to the nuclear center) of the gravity centers are plotted for all CTs in the order of the gene density for the three simulation cases. The error bars denote the standard deviations determined by averaging over the 50 simulated nuclei and the both homologous CTs for each case.
FIGURE 4.

Mean radial distances of gravity centers of CTs of the start configurations (open rectangles) and after the completed relaxation (solid rectangles). On the abscissa, the CT numbers are given in the order of the gene density (genes per Mbp). The distances were determined for the statistical (a), probabilistic (b), and the deterministic (c) simulation case (i.e., statistical, probabilistic, and deterministic start configurations, respectively). The mean values were obtained by averaging over all 50 nuclei for each simulation case. Error bars denote the standard deviations.
Whereas in the case of a statistical simulation (i.e., a statistical initial distribution), there was no remarkable difference visible between start and relaxed configuration of a given CT type; for the probabilistic simulation case (i.e., probabilistic initial distribution) for some gene-dense CTs that are located initially in the interior, a slight movement toward the periphery after the relaxation process was predicted. The quite large standard deviations indicate that this process can take place for both homologous CTs in a different way. This can be ascribed to the limited space in the interior and to the fact that no fixing points were assumed for the simulated CTs. This behavior was still more pronounced for CTs in the deterministic simulation case (i.e., deterministic initial distribution) that were initially arranged on discrete shells.
For comparison of the experimentally observed and the simulated radial arrangements of the reconstructed CTs Nos. 12, 18, 19, and 20, the simulated nuclear genome configurations were virtually labeled using the virtual microscopy approach (see Material and Methods). Fig. 5 visualizes 3D reconstructions of painted CTs No. 18 and No. 19 in a nucleus of a human lymphocyte (Fig. 5 d) as well as for the three simulated model assumptions (Fig. 5, a–c). The quantitative 3D evaluation of the nuclear positions of the (virtually) painted territories was made by the assessment of the 3D relative radial distribution of each voxel assigned to the respective territory (Material and Methods). Fig. 6 shows the voxel distributions (differential DNA content) for the respective painted CTs plotted against the relative radius in lymphocyte nuclei (Fig. 6, g and h, experimental data described in Cremer et al. (2001) and Weierich et al. (2003)) and in simulated nuclei (Fig. 6, a–f), where Fig. 6, a and b, represents the quantitative distribution of the statistical, Fig. 6, c and d, of the probabilistic and Fig. 6, e and f, of the deterministic model assumptions. For each given relative radius, the respective differential DNA content was determined as the mean over the single distribution curves for each nucleus for this radius. The error bars represent the standard deviations of the mean. In Table 2, the mean differential DNA contents for all relative radii averaged over all nuclei with the respective standard deviations are given.
FIGURE 5.

Visualization of reconstructed CTs of simulated human cell nuclei (a–c) according to the SCD model and of an experimental human lymphocyte cell nucleus with FISH-painted CTs (d). The simulated virtual microscopy data stacks are reconstructions from the three simulation cases of the relaxed configurations: statistical simulation case (a), probabilistic simulation case (b), and deterministic simulation case (c). In all cases, CTs No. 18 were visualized in red and CTs No. 19 in green. The visualization tool was kindly provided by Dr. R. Heintzmann, MPI Göttingen, Germany.
FIGURE 6.

Radial distribution curves of experimental (compare Cremer et al., 2001; Weierich et al., 2003) and virtual chromosome painting experiments applying a 3D mapping algorithm (see Material and Methods). The radial arrangements were evaluated for CTs No. 12 and No. 20 (left column) and CTs No. 18 and No. 19 (right column). The counterstain distribution results from the mapping of all chromosomes. The different simulated cases and the experimental distribution curves are arranged in the same way as in Fig. 5: statistical simulation case (a and b), probabilistic simulation case (c and d), deterministic simulation case (e and f), and the experimental case observed in human lymphocytes after CT painting (g and h). The relative radius determines the relative position of a shell with respect to the nuclear border. E.g., a shell at the relative radius 0 is located at the nuclear center, whereas the shell 98 is positioned at the nuclear periphery. Error bars represent the standard deviations of the mean. The mean value for each relative radius was obtained by the average of the single distribution curves for each nucleus.
TABLE 2.
The mean relative radii (plus/minus standard deviations) of the radial distributions shown in Fig. 6*
| Mean/Std | Simulation (statistical) | Simulation (probabilistic) | Simulation (deterministic) | Experiment |
|---|---|---|---|---|
| No. 12 | 73.65 ± 11.40 | 73.58 ± 6.33 | 70.24 ± 7.63 | 75.41 ± 3.15 |
| No. 18 | 75.30 ± 9.28 | 80.17 ± 3.09 | 81.68 ± 2.89 | 73.95 ± 4.09 |
| No. 19 | 77.03 ± 8.23 | 36.5 ± 6.86 | 49.93 ± 12.31 | 42.99 ± 11.08 |
| No. 20 | 77.33 ± 11.29 | 63.77 ± 10.96 | 68.32 ± 12.83 | 68.95 ± 7.15 |
These were determined as the average of the mean relative radii of the single radial distributions for each nucleus. The last column gives the experimentally observed mean relative radii (evaluations from Cremer et al., 2001; Weierich et al., 2003).
The radial distribution curves for the statistical simulation case (Fig. 6, a and b) showed a peripheral position and an almost identical distribution pattern of the average radial arrangements for all CTs with a peak content at ∼76% of the relative radius. A small shift of the larger CTs Nos. 12 and 18 toward the interior results here from the larger DNA amount (volume) of the CTs Nos. 12 and 18 in comparison to the CTs Nos. 20 and 19, which enforces larger distances to the nuclear envelope (compare Cremer et al., 2001; Habermann et al., 2001). These distributions obtained by the statistical model assumption are not compatible with the experimentally evaluated radial arrangements of CTs No. 19 (Fig. 6 h). For the CTs Nos. 18, 12, and 20, the null hypothesis of equality cannot be rejected without further statistical evaluations. The comparison of CT positions obtained by the experimental data with the data obtained by the deterministic simulation case showed an agreement for all CTs if only the mean relative radii values are regarded (Table 2). However, the radial distributions for CTs Nos. 19 and 20 showed clear differences (Fig. 6, e and f, and g and h). This difference results from the effect mentioned above that CTs located initially around the center of the nucleus have the tendency to move toward the periphery (Fig. 6 c), which results in a broadening of the radial distribution.
For the probabilistic simulation case, the distribution patterns (Fig. 6, c and d) are in good agreement with the experimentally obtained data (Fig. 6, g and h) for CTs Nos. 18, 19, and 12: Here, the CTs No. 19 with the highest gene density are localized in the interior whereas the gene-poor No. 18 CTs are arranged close to the nuclear envelope; CTs No. 12 shows an intermediate position. For the CTs No. 20, the slight movement of a homologous CT in some model nuclei (data not shown) revealed a quite broad distribution compatible with the experimentally observed distribution. Here also a broad distribution was observed. Furthermore, the mean relative radii of the radial distribution values (given in Table 2) also agreed fairly well with the experimental data.
DISCUSSION
In this study, we applied the SCD computer model for large-scale chromatin organization in spherical human cell nuclei to interpret the experimentally observed specific arrangement pattern of CTs in the nuclear volume. Chromosome painting experiments have suggested a close relationship between the localization of CTs in the nuclear volume and their gene densities in a variety of cell types with a spherical nuclear shape (Cremer et al., 2003). To relate gene density and CT positioning, we tested three model assumptions for the initial arrangement of mitotic-like chromosomes in the nucleus: a statistical, a deterministic, and a probabilistic initial distribution.
For the deterministic simulation case, initial CT spheres (representing a certain start volume of the CTs according to their DNA content) were located on discrete shells in the nuclear volume in the order of their gene densities; for the probabilistic simulation case, the distances of the initial CT spheres to the center of the nuclear volume were weighted with the respective gene densities (derived from the latest sequence data). This weighting was executed in a probabilistic way using a Monte Carlo procedure. In the case of the statistical simulation case, the initial CT spheres were located randomly in the nuclear volume. After the location of the initial CT spheres, Metropolis Monte Carlo relaxation runs were performed to calculate relaxed interphase genome configurations. Using the same quantitative 3D mapping algorithm for experimental and simulated data, the evaluated radial distributions of single CTs Nos. 12, 18, 19, and 20 in experiment and simulation were compared.
In the statistical simulation case, large differences between predicted and experimental values were found for the mean relative radii for CTs No. 19. The radial distributions were fairly different for all evaluated CTs. In the probabilistic simulation case, the evaluated more interior arrangement (in the nuclear volume) of the CTs No. 19, the more peripheral arrangement of the CTs No. 18, the intermediate arrangement of the CTs No. 12, and the quite broad intermediate arrangement of the CTs No. 20 fitted quite well the experimental data (with respect to the broadness, the mean values (Table 2), and the height of the radial distribution curves); for CT No. 20 in some simulated nuclei, a slight movement of one of the homologous CTs to a more peripheral position was predicted during the relaxation process, which caused the determined broad distribution. This may be also a reason for the experimentally observed broad distribution. In the deterministic simulation case, the mean relative radii (Table 2) for all CTs evaluated were in quite good agreement with the experimental values. For the CTs Nos. 19 and 20, however, quite large movements from the interior of the nucleus to a peripheral position was predicted during the relaxation process. At least for CT No. 19, this was not compatible with the experimental values. The reason here is the quite dense packaging of the CTs on discrete shells in the initial start configuration.
Recent experimental investigations indicated that global chromosome positions may be maintained through the cell cycle in mammalian cells (Gerlich et al., 2003; Walter et al., 2003). This may suggest that chromosomal localization might be controlled by a global chromosome positioning code. However, precise radial (e.g., Tanabe et al., 2002) or relative positioning is not found in all cells in a population, and relatively large variations in the positioning of a chromosome can be observed when single cells are compared (A. Bolzer et al., unpublished results). E.g., when analyzing the radial positioning of all human CTs, statistically significant patterns are evident, although every CT can be found at variable radial positions in a cell population. These findings are also in good agreement with the study of Cornforth et al. (2002): here, frequencies between all possible heterologous pairs of CTs with 24-color whole-chromosome painting after damage to interphase lymphocytes by sparsely ionizing radiation in vitro were performed to test the influence of nonrandom CT-CT associations on aberration frequencies between specific CTs. It was found that only a group of five chromosomes (Nos. 1, 16, 17, 19, and 22), previously observed to be preferentially located close to the center of the nucleus (suggested by Boyle et al., 2001), showed a statistically significant deviation of a random CT-CT association. According to Cornforth et al. (2002), these findings suggest a predominantly random location of CTs with respect to each other in interphase lymphocyte cells.
The results obtained in this report by computer simulations using the SCD model indicated that the idea of an appropriately designed global chromosome positioning code is compatible with such experimentally observed variations if an uncertainty condition is introduced in the initial distribution of CTs.
The computer simulations of nuclear genome structure presented here allowed first quantitative predictions about the possible influence of sequence length and gene density of a chromosome on its spatial positioning in the nuclear volume of lymphocyte cells. Besides some general constants and procedural rules, only linear sequence-derived data (chromosomal DNA content and gene density) were included as first parameters in the model. However, other constraints (not yet realized) also have to be regarded, like the arrangement of specific CTs around the nucleoli, the specific R-/G- band pattern, and other still unknown factors, e.g., specific attachment sites. The simulations presented here may help to determine the influence of such constraints on the arrangement of CTs in the nucleus and may provide a quantitatively testable model system for further experimental investigations. As a biophysically important application of such simulations, effects of ionizing irradiation and other clastogenic agents on specific chromosomal rearrangements (e.g., relative frequencies of translocations, dicentrics, deletions, and inversions) can be predicted.
Acknowledgments
For stimulating discussions, we thank T. Cremer and M. Hausmann. The chromosome painting experiments used for the comparison with simulated data were performed in the group of Prof. T. Cremer (University of Munich). Special thanks to I. Solovei, C. Weierich, and A. Brero.
These studies were supported financially from the Deutsche Forschungsgemeinschaft (grant CR 60/19-1) and the European Commission (grant FIGH-CT1999-00011).
References
- Binder, K., and D. W. Heermann. 1997. Monte Carlo Simulation in Statistical Physics, 3rd ed. Springer Verlag, Berlin, Heidelberg, and New York.
- Boyle, S., S. Gilchrist, J. M. Bridger, N. L. Mahy, J. A. Ellis, and W. A. Bickmore. 2001. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 10:211–219. [DOI] [PubMed] [Google Scholar]
- Cornforth, M. N., K. M. Greulich-Bode, B. D. Loucas, J. Arsuaga, M. Vazquez, R. K. Sachs, M. Brückner, M. Molls, P. Hahnfeldt, L. Hlatky, and D. J. Brenner. 2002. Chromosomes are predominantly located randomly with respect to each other in interphase human cells. J. Cell Biol. 159:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer, T., and C. Cremer. 2001. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2:292–301. [DOI] [PubMed] [Google Scholar]
- Cremer, T., G. Kreth, H. Koester, R. H. A. Fink, R. Heintzmann, M. Cremer, I. Solovei, D. Zink, and C. Cremer. 2000. Chromosome territories, interchromatin domain compartment and nuclear matrix: an integrated view of the functional nuclear architecture. Crit. Rev. Eukaryot. Gene Expr. 10:179–212. [PubMed] [Google Scholar]
- Cremer, M., K. Küpper, B. Wagler, L. Wizelmann, J. von Hase, Y. Weiland, L. Kreja, J. Diebold, M. R. Speicher, and T. Cremer. 2003. Inheritance of gene density-related higher order chromatin arrangements in normal and tumor cell nuclei. J. Cell Biol. 162:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer, M., J. von Hase, T. Volm, A. Brero, G. Kreth, J. Walter, C. Fischer, I. Solovei, C. Cremer, and T. Cremer. 2001. Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome. Res. 9:541–567. [DOI] [PubMed] [Google Scholar]
- Croft, J. A., J. M. Bridger, S. Boyle, P. Perry, P. Teague, and W. A. Bickmore. 1999. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145:1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova, D. S., and R. Berezney. 2002. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell Sci. 115:4037–4051. [DOI] [PubMed] [Google Scholar]
- Dundr, M., and T. Misteli. 2001. Functional architecture in the cell nucleus. Biochem. J. 356:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlich, D., J. Beaudouin, B. Kalbfuss, N. Daigle, R. Eils, and J. Ellenberg. 2003. Global chromosome positions are transmitted through mitosis in mammalian cells. Cell. 112:751–764. [DOI] [PubMed] [Google Scholar]
- Habermann, F. A., M. Cremer, J. Walter, G. Kreth, J. von Hase, K. Bauer, J. Wienberg, C. Cremer, T. Cremer, and I. Solovei. 2001. Arrangements of macro- and microchromosomes in chicken cells. Chromosome Res. 9:569–584. [DOI] [PubMed] [Google Scholar]
- Holley, W. R., I. S. Mian, S. J. Park, B. Rydberg, and A. Chatterjee. 2002. A model for interphase chromosomes and evaluation of radiation-induced aberrations. Radiat. Res. 158:568–580. [DOI] [PubMed] [Google Scholar]
- Kreth, G., P. Edelmann, and C. Cremer. 2001. Towards a dynamical approach for the simulation of large scale, cancer correlated chromatin structures. Ital. J. Anat. Embryol. 106(Suppl.):21–30. [PubMed] [Google Scholar]
- Münkel, C., and J. Langowski. 1998. Chromosome structure predicted by a polymer model. Phys. Rev. E. 57:5888–5896. [Google Scholar]
- O'Brien, T. P., C. J. Bult, C. Cremer, M. Grunze, B. B. Knowles, J. Langowski, J. McNally, T. Pederson, J. C. Politz, A. Pombo, G. Schmahl, J. P. Spatz, and R. van Driel. 2003. Genome function and nuclear architecture: from gene expression to nanoscience. Genome Res. 13:1029–1041. [DOI] [PubMed] [Google Scholar]
- Parada, L., and T. Misteli. 2002. Chromosome positioning in the interphase nucleus. Trends Cell Biol. 12:425–432. [DOI] [PubMed] [Google Scholar]
- Tanabe, H., S. Müller, M. Neusser, J. von Hase, E. Calcagno, M. Cremer, I. Solovei, C. Cremer, and T. Cremer. 2002. Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc. Natl. Acad. Sci. USA. 99:4424–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser, A. E., R. Eils, A. Jauch, G. Little, P. J. M. Bakker, T. Cremer, and J. A. Aten. 1998. Spatial distribution of early and late replicating chromatin in interphase chromosome territories. Exp. Cell Res. 243:398–407. [DOI] [PubMed] [Google Scholar]
- Walter, J., L. Schermelleh, M. Cremer, S. Tashiro, and T. Cremer. 2003. Chromosome order in HeLa cells changes during mitosis and early G1, but is stably maintained during subsequent interphase stages. J. Cell Biol. 160:685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weierich, C., A. Brero, S. Stein, J. von Hase, C. Cremer, T. Cremer, and I. Solovei. 2003. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res. 11:485–502. [DOI] [PubMed] [Google Scholar]
- Zink, D., H. Bornfleth, A. E. Visser, C. Cremer, and T. Cremer. 1999. Organization of early and late replicating DNA in human chromosome territories. Exp. Cell Res. 247:176–188. [DOI] [PubMed] [Google Scholar]