

Abstract
The endoplasmic reticulum (ER) and Golgi have robust bidirectional traffic between them and yet form distinct membrane compartments. Membrane tubules are pulled from large aggregates of ER or Golgi by microtubule motors to form ER tubulovesicular networks or Golgi tubules both in vivo and in vitro. The physical properties of membranes are critical for membrane traffic and organelle morphology. For example, tension applied to membranes can create tethers, drive membrane flow, and set the diameter of the tubules. Here, we formed ER and Golgi membrane networks in vitro and used optical tweezers to measure directly, for the first time, the membrane tensions of these organelles to clarify the possible role of tension in membrane flow. We report that higher forces are needed to form tethers from ER (18.6 ± 2.8 pN) than from Golgi (11.4 ± 1.4 pN) membrane tubules in vitro. Since ER tubules are smaller in diameter than Golgi tubules, it follows that Golgi networks have a lower tension than ER. The lower tension of the ER could be an explanation of how Golgi tubules can be rapidly drawn into the ER by tension-driven flow after fusion, as is observed in vivo.
INTRODUCTION
Although membrane traffic among ER, Golgi, and plasma membrane clearly involves fission and fusion of membrane vesicles, there is an increasing body of evidence that membrane can flow through a compartment or from one compartment to another. Flow of membrane can be driven by tension drawing material from a region of low tension to a region of higher tension, similar to what has been observed for the movement of membrane on growing axons (Dai and Sheetz, 1995a). Because biological membranes are fluid, the energy needed to move large amounts of membrane from one region to another is quite small (Dai and Sheetz, 1995b). Small tension gradients in the plane of the membrane are sufficient to power very rapid movements. We provide evidence here that the physical properties of Golgi and ER membrane tubules are particularly different and could provide a simple means for driving some of the movements from one compartment to another.
In the processes of protein synthesis and maturation, the initial events in the ER are followed by considerable bidirectional traffic between the ER and Golgi, the Golgi and plasma membrane, or the Golgi and lysosomes. Once a compartment is formed, communication with other compartments is limited and Golgi tubules going to the plasma membrane rarely fuse with ER or other internal membranes. The limited fusion of compartments enables them to maintain their specialized functions and to only modify the specific proteins that traffic through them. When the barriers to fusion do break down, the compartments can mix rapidly. For example, the drug Brefeldin A (BFA) causes Golgi membrane to fuse and co-mingle with ER, and has been used extensively to study mechanisms regulating membrane traffic. When a cell is treated with BFA, the Golgi membrane starts tubulating along microtubules and is rapidly drawn into the ER (Lippincott-Schwartz et al., 1989, 1990). Although intact Golgi tubules can extend through the cytoplasm for 5–10 min normally, the fusion of Golgi tubules with ER causes the Golgi contents to mix into the ER within 15–30 s (Sciaky et al., 1997). Sciaky and co-workers interpret their results as an indication of tension-driven flow between the two membrane compartments. The ER is thought to provide a lower energy environment for membrane protein and lipid than the Golgi system, and flow rather than diffusion could cause mixing of components. The first step in verifying this hypothesis is to measure the tensions in the ER and Golgi tubules directly.
A well-characterized measure of tension within membrane bilayers is the force on membrane tethers pulled from those membranes (reviewed in Sheetz, 2001; Morris and Homann, 2001). Tether force can be rapidly measured with laser tweezers using beads attached to membranes to form tethers. In the case of plasma membranes, tether force is inversely related to the rates of endocytosis, membrane resealing, and lamellipodial extension. Internal membranes are also under tension and differences in tension could drive movements in a manner consistent with observed events in vivo. By bringing membrane tubules into contact with each other using laser tweezers, it is possible to measure the difference between homotypic and heterotypic fusion.
MATERIALS AND METHODS
Preparation of beads
For the purposes of optical trapping we used 0.5-μm-diameter protein-coated carboxylate microspheres from Polysciences (Polysciences, Inc., Warrington PA). The beads were activated using standard protocol (Kuo and Sheetz, 1993) with 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC). Briefly, 0.5 ml of a 2.5% suspension of carboxylate beads were washed in carbonate and phosphate buffers and incubated with a 2% EDC solution for 3–4 h at room temperature. The carbodiimide was washed with a phosphate buffer and the beads were resuspended in a borate buffer. After activation, the beads were coated with 1), anti-kinectin antibody, which has a preference for ER membranes; or 2), Wheat Germ Agglutinin (WGA), which binds preferentially to the Golgi membrane; or 3), Bovine Serum Albumin (BSA) as a control. 200–300 micrograms of the protein was added to the bead suspension and incubated overnight at 4°C with gentle end-to-end mixing. Beads were centrifuged and mixed with first ethanolamine and then BSA to block nonspecific protein binding sites. The beads were suspended in a storage buffer (containing sodium phosphate and sodium chloride) and stored at 4°C.
Formation of networks from membrane extracts
Internal membranes and cytosol rich in motors were obtained from chicken embryo fibroblasts following standard protocol (McIlvain et al., 1993). To separate the Golgi from the ER, the supernatant from a 1000-g spin of the cell homogenate was diluted with PMEE′ + (standard buffer, McIlvain et al., 1993) to 1 ml and centrifuged at low speed (10,000 g) to obtain a pellet of the heavier membranes (H-fraction). The resulting supernatant was spun at high speed (100,000 g) to get a pellet of lighter vesicles (L-fraction). H- and L-fractions were then resuspended in PMEE′ + and used for network formation and fluorescence assays. Ten-microliter capacity flow chambers were assembled from two coverslips separated by parallel strips of 70-μm-thick double-stick tape. The chambers were perfused with taxol-stabilized, bovine brain microtubules (10 μl, at 0.1–0.5 mg/ml), and incubated in a humid chamber for 15–20 min. Unbound microtubules were removed with 40 μl of washing buffer (PMEE′ +, 1 mM GTP, 20 μM taxol). Membrane fractions (5 μl) with motor supernatant (3 μl) and Mg-ATP (2 μl) were introduced into the flow chambers and network formation was assayed after ∼60 min incubation at 37°C.
Microscopy and force measurement
Network formation was imaged using video-enhanced differential interference contrast (DIC) microscopy as described before in Dabora and Sheetz (1988). Fluorescent images were taken using a cooled charge-coupled device camera (Princeton Instruments, Princeton, NJ). The force of the optical trap was calculated by computing the viscous drag of a bead through the aqueous medium as described before in Dai and Sheetz (1995a). A linear force-displacement graph was obtained to calculate the calibration constant for the trap stiffness. Carboxylate beads of 0.5-μm diameter (Polysciences) were covalently coupled (using user-supplied protocol) with either anti-kinectin antibody (which has a preference for ER membranes) or WGA (which binds preferentially to the Golgi membrane). Protein-coated beads were flowed into the chamber along with motor-containing supernatant with GTP, taxol, and Mg-ATP. Force measurement is described in Fig. 3. The recorded sequences of tether pulling events were digitized and analyzed using the tracking software ISEE (Inovision, Durham, NC)—a nanometer-level particle-tracking program that calculates the centroid of beads with a maximum precision of a few nanometers.
FIGURE 3.

Force measurement using optical tweezers. (a) DIC image of a typical tether-pull sequence. The bead is held in the optical trap and pulled orthogonal to the membrane tubule. The bead was trapped with the laser, placed onto one branch of the network, and allowed to bind by holding for a few seconds. Then, it was pulled at a constant velocity perpendicular to the network branch. The straight branch first bent into a V and then a Y as a tether was pulled out. The tether was of the same radius as the surrounding network and the triple point at the Y relaxed to a 120° angle. The tether was held static for 30–60 s. This whole sequence was video-recorded and later digitized for analysis. The scale bar is 3 μm. (b) Schematic of a tether-pull showing the displacement ΔR of a trapped bead from the trap center. The tether force Ft balances the trap force Fb pulling the bead toward the trap center. (c and d) Typical curves showing the displacement of a trapped bead after pulling a membrane tether from a branch for networks from the Golgi (c) and the ER (d). On the x axis, 30 frames correspond to 1 s.
RESULTS AND DISCUSSION
Several studies in the past have successfully formed networks in vitro both from ER (Dabora and Sheetz, 1988; Lane and Allan, 1999) and Golgi (Allan and Vale, 1991; Fullerton et al., 1998) membranes on microtubules. The dynamics and morphology of these reconstituted networks is remarkably similar to that in living cells (Lee and Chen 1988; Terasaki et al., 1986). They, therefore, provide an extremely useful cell-free system to conduct mechanical measurements on intracellular membranes.
In the process of forming networks, the membrane tubules were extremely dynamic on microtubules. An amorphous aggregate of membrane, which adhered to a microtubule meshwork on the glass coverslip, was the precursor of the network. Microtubule-dependent motors (kinesin and dynein) attached to regions of the membrane and moved along stationary microtubules, providing the force to draw out tubular branches. In the absence of motors or microtubules, we did not observe any tubular extensions, suggesting that motor force itself created tubules from a membrane with no preferred curvature, as opposed to motor proteins simply guiding pre-existing membrane extensions. The growing tubule was frequently observed to retract to its point of origin (possibly due to detachment from the microtubule). Membrane branches formed when another active motor contacted and moved along an intersecting microtubule, pulling a new membrane tubule from a pre-existing branch. A growing membrane branch fused with another branch if they overlapped. After fusion, the branches relaxed to a configuration connected by trigonal vertices with 120° angles between branches to minimize the local energy. This resulted in a reticular network of long membrane tubules on a microtubule mesh. In some instances, pre-existing polygons shrank in size due to movement of one of the branches and even disappeared, causing a local rearrangement of the network. After a few hours, the dynamics of tubules ceased and the entire structure stabilized. The network was interconnected and could stretch unbroken over hundreds of microns similar to the structures found in vivo (Terasaki et al., 1986). The tubulovesicular structure was attached to the underlying bed of microtubules at discrete points. The cause of attachment is yet unknown and could be due to inactive motors or some other attachment proteins. A typical example of a tubulovesicular network is shown in Fig. 1.
FIGURE 1.

DIC image of a typical membrane network. Hollow cylindrical tubules of lipid bilayer membranes are interconnected into a branched network. The underlying mesh of microtubules is not visible. Branches typically meet at trigonal vertices with 120° angles between two sides. The bright object in the center is a bead of 500-nm diameter. The scale bar corresponds to 3 μm.
Differential sedimentation of membrane fractions from chick embryo fibroblasts gave heavy (H) and light (L) fractions that formed networks of primarily Golgi and ER membranes, respectively. Networks formed from the L-fraction labeled primarily with an ER-specific antibody (Fig. 2 a) and networks formed from the H-fraction were primarily Golgi as determined by rhodamine WGA staining (Fig. 2 b). Additionally, differences in size of the network strands enabled us to reliably differentiate ER from Golgi for the tether measurements. We also determined that endocytotic membranes from the trans-Golgi network were not involved in the network formation process by determining that endocytosed Lucifer yellow did not appear in the tubulovesicular networks.
FIGURE 2.

Identification of ER and Golgi networks by fluorescence labeling. (a) Double-labeling of networks from the L-fraction. (Left) Ribosome receptor-Texas Red staining shows extensive labeling of network branches and hence enrichment of the L-fraction in ER membrane. A typical strand from the ER network is indicated by the arrow. (Right) Staining with WGA-FITC indicating the lack of Golgi membrane in the L-fraction. The scale bar is 5 μm in length. (b) Double-labeling of networks from the H-fraction. (Left) Staining with WGA-FITC indicating the presence of Golgi membrane. The arrow shows a typical strand from the Golgi network. (Right) Ribosome receptor-TR staining shows negligible staining of network branches and hence lack of ER membrane in the H-fraction. In some cases there was a small degree of contamination; the arrow indicates an ER branch in the H-fraction.
To measure the tension of these networks, we added beads coated with specific antibodies to the ER or lectins that bound to the Golgi fraction in preformed networks. Using an optical trap, beads were bound to network strands and then pulled to form tethers (Fig. 3, a). Bead binding specificity was controlled by determining that BSA-coated control beads did not bind to the networks. Tether formation was analogous to tubule branching during the network growth phase. As the new tether was formed, membrane flowed into the tether from the surrounding branches of the network. Since the network was interconnected, it essentially acted as an infinite reservoir of membrane material, particularly when large membrane aggregates were present. Beads with tethers could be pulled for long distances laterally across the branches indicating that the network was interconnected and fluid-like. Tethers were pulled at a constant speed from the membrane tubules and held stationary for 30–60 s to measure the static tether force. Tethers rapidly retracted when the bead was released from the laser trap, indicating that a significant force was pulling the membrane in the tether back into the network. The displacement of the bead from the trap center was used to measure the tether force (Fig. 3, b, c and d). Forces were measured from several different parts of the network for larger networks. For each of the fractions, we found that a fixed value of force was maintained throughout the entire network. The tether force for the Golgi was 11.4 ± 1.4 pN whereas the tether force for the ER was 18.6 ± 2.8 pN (standard deviations from the mean of ∼30 measurements for each case). The distribution of forces was normal. For all of these measurements, the identity of the ER and the Golgi membranes was confirmed by immunostaining.
The energy required to pull a tether is given by Bozic et al. (2001) and Bukman et al. (1996) as
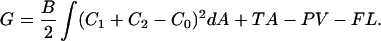 |
(1) |
Here B is the bending stiffness of the membrane, C1 and C2 are the principal curvatures, and C0 is the spontaneous curvature. A and V are the membrane surface area and volume, respectively, and L is the tether length. T is the membrane tension, P is the pressure, and F is the tether force. The contribution due to nonlocal bending that arises from area difference between monolayers is neglected because the rate at which lipids have been shown to move between monolayers in ER and Golgi is rapid compared to our experimental timescales (Buton et al., 1996). The pressure difference term can be neglected because the tether volume is not conserved. Both ER and Golgi membranes do not tubulate spontaneously and require the action of motors or cytosolic factors to form tubular networks. Further, purified ER membranes tend to aggregate into large micron-sized vesicles (Dreier and Rapoport, 2000). These observations suggest that we can neglect the effect of spontaneous curvature. Taking these simplifications into account, the free energy is minimized with respect to the membrane shape for a cylinder (Bozic et al., 2001). An inverse relationship between tether (tubule) diameter and membrane tension indicates that the Golgi tubules should be larger in diameter than ER tubules. The tether force F is related to the radius of the tether, Rt, the bending stiffness of the membrane, and the membrane tension by the following equations (Hochmuth et al., 1996; Waugh et al., 1992):
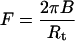 |
(2) |
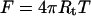 |
(3) |
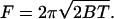 |
(4) |
A difference in tether force can arise either from a difference in bending stiffness or a difference in membrane tension. From the above equations, the tether forces and radii of the two membrane types ER (E) and Golgi (G) can be related by 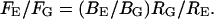 From the contrast of the tubules in the DIC images it was possible to estimate the relative diameters (Fig. 4). Golgi tubules had an average diameter of ∼180 nm whereas the ER network branches were ∼115 nm in diameter or a factor of RG/RE = 1.57 ± 0.2 (Fig. 4). The inverse ratio of tether forces is FE/FG = 1.63 ± 0.3. We find that the two ratios (FE/FG and RG/RE) are the same within experimental error. This implies that both membrane types have approximately the same bending stiffness: BE ≈ BG ≈ 3.3 × 10−19 N/m, similar to that of growth cone membranes and phospholipid bilayers (Dai and Sheetz, 1995a, Evans and Rawicz, 1990; Song and Waugh, 1993). Calculating the tensions explicitly using Eq. 3, we find that the tension in the ER membrane networks is TE ≈ 0.013 dyn/cm and the tension in the Golgi membrane is TG ≈ 0.005 dyn/cm. To put these tensions in perspective, membrane lysis requires a tension of 5–10 dyn/cm. As a further indication that the Golgi networks were at a lower tension, the Golgi tubules were more “floppy” (had larger thermal fluctuations) than the ER tubules. For this analysis we have assumed that the spontaneous curvature of the membranes is negligible. However, it is possible that both ER and Golgi have nonzero spontaneous curvatures. Therefore, the numerical values of tension and bending stiffness could be different from that reported here. Further work measuring the spontaneous curvatures of the two membrane types is required to determine more accurately the relative contributions of spontaneous curvature and tension to the tether force.
From the contrast of the tubules in the DIC images it was possible to estimate the relative diameters (Fig. 4). Golgi tubules had an average diameter of ∼180 nm whereas the ER network branches were ∼115 nm in diameter or a factor of RG/RE = 1.57 ± 0.2 (Fig. 4). The inverse ratio of tether forces is FE/FG = 1.63 ± 0.3. We find that the two ratios (FE/FG and RG/RE) are the same within experimental error. This implies that both membrane types have approximately the same bending stiffness: BE ≈ BG ≈ 3.3 × 10−19 N/m, similar to that of growth cone membranes and phospholipid bilayers (Dai and Sheetz, 1995a, Evans and Rawicz, 1990; Song and Waugh, 1993). Calculating the tensions explicitly using Eq. 3, we find that the tension in the ER membrane networks is TE ≈ 0.013 dyn/cm and the tension in the Golgi membrane is TG ≈ 0.005 dyn/cm. To put these tensions in perspective, membrane lysis requires a tension of 5–10 dyn/cm. As a further indication that the Golgi networks were at a lower tension, the Golgi tubules were more “floppy” (had larger thermal fluctuations) than the ER tubules. For this analysis we have assumed that the spontaneous curvature of the membranes is negligible. However, it is possible that both ER and Golgi have nonzero spontaneous curvatures. Therefore, the numerical values of tension and bending stiffness could be different from that reported here. Further work measuring the spontaneous curvatures of the two membrane types is required to determine more accurately the relative contributions of spontaneous curvature and tension to the tether force.
FIGURE 4.

Determination of tubule diameter. (a) DIC image of a membrane network from the Golgi. The scale bar is 3 μm. (b) DIC image of a network from the ER. The network tubules from the Golgi show higher contrast than network tubules from the ER. To measure the diameter of the network branches, orthogonal scans were taken across the membrane tubules oriented along the DIC shear axis to give maximal contrast and across beads of known diameter (500 nm). The relative intensities were determined as the area under the intensity profile curve (Schnapp et al., 1988). Since the intensity is proportional to the square of the diameter, the radius of each type of tubule (Rt) was calculated by multiplying the radius of the bead (Rb) by the square-root of the ratio of the intensity of the tubule (It) to that of the bead (Ib): 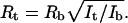 (c and d) Normalized intensity profile across a network branch from the Golgi (c) and ER (d). The lower contrast of the ER branch is indicated by the smaller area under the intensity profile curve.
(c and d) Normalized intensity profile across a network branch from the Golgi (c) and ER (d). The lower contrast of the ER branch is indicated by the smaller area under the intensity profile curve.
Previous studies showed that the networks did not form in the absence of microtubule motors, but that once the networks were formed, active motors were no longer needed to remain statically spread. If there was a large reserve area in the network as it appeared, we imagined that the motors were not needed to maintain the network tension. Therefore, we incubated network samples with kinesin-inhibitor (2 mM AMP-PNP) and dynein-inhibitor (0.5 μM sodium vanadate) before measuring tether forces. Neither drug had an effect on the tether force for ER or Golgi membranes. Thus, active motors are needed to generate the force needed to pull out the membrane tubules but not for maintenance of the force.
Because the membrane fractions were not pure we worried that fusion between ER and Golgi tubules could produce hybrid networks in vitro although the two compartments do not normally fuse in vivo. To study fusion, tethers were pulled from tubules, placed over a nearby network branch, and allowed to fuse for several seconds. For homologous membrane types (i.e., ER-ER or Golgi-Golgi), fusion was observed ∼70% of the time. For heterologous membrane types, ER to Golgi or vice versa, no fusion event was observed (25 trials). The behavior of membrane tubules in vitro is consistent with the properties of ER and Golgi in vivo. The much lower rate of heterologous versus homologous fusion of the networks in vitro helps to explain how separate compartments could be maintained in vitro as well as within the cell. Homologous fusion would explain how a reticular network of ER could form. If agents such as BFA would cause heterologous fusion, then the rapid movement of the Golgi into the ER would be explained by the greater tension in the ER.
Is the observed tension difference sufficient to cause membrane flow from Golgi to ER as observed after addition of BFA (Lippincott-Schwartz et al., 1989, 1990)? From the measured redistribution of a fluorescent Golgi protein into the ER, Sciaky et al. (1997) have concluded that movement of membrane protein between Golgi and ER is due to convective flow rather than diffusive movement, with velocities on the order of 10 μm/s. Chizmadzhev et al. (1999) have calculated the velocity v of lipid transfer between fusing membranes at different tensions to be v = C(Δσ/η), where Δσ is the tension difference, η is the surface viscosity of the membrane, and C is a factor depending on the pore geometry. Making reasonable assumptions about the pore geometry and viscosity of these membranes, η = ∼10−5−10−6 g/s (Evans and Hochmuth, 1978; Saffman, 1976), we find that the measured tension difference (∼0.01 dyn/cm) is sufficient to produce the observed lipid flow velocities. Experiments on plasma membranes show that even the presence of a cytoskeleton does not increase the surface viscosity sufficiently to block this effect (Hochmuth et al., 1996). One possible origin of the tension is the action of microtubule motors that extend the membrane. Since motor inhibitors block network spreading but have no effect on the tether force of existing networks, it appears that there is a large reservoir of membrane at a constant tension. Further, throughout a single type of network we observe the same tubule radius, suggesting that the equilibrium radius is not set by the motor force, which could vary across the network, but by a constant chemical potential. The difference in tension could arise from a difference in chemical potential of the two membrane compartments. The cell may maintain the surface tension of its different membrane compartments at fixed levels by keeping lipid reservoirs at a fixed chemical potential. The normal block to heterologous fusion would maintain the separation between the different membrane types. Thus, the in vitro behavior of the tubulovesicular networks formed from ER and Golgi membrane provides important insights into aspects of function in vivo. Even though there could be differences between in vitro and in vivo situations, our experiments are a first step in identifying the physical properties of intracellular organelles. Experiments with BFA in vivo (Sciaky et al., 1997) show rapid movement of Golgi into the ER, ruling out diffusional mixing of the two compartments, which would be the case if the membranes were at the same tension. This is important evidence in favor of our hypothesis that tension differences measured in vitro could in fact reflect the in vivo situation. Differences in membrane tension between ER and Golgi provide a simple explanation for the rapid transfer of Golgi to ER.
References
- Allan, V. J., and R. D. Vale. 1991. Membrane formation and tubule transport in vitro. J. Cell Biol. 113:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozic, B., V. Heinrich, S. Svetina, and B. Zeks. 2001. Shapes of nearly cylindrical, asymmetric bilayer membranes. Eur. Phys. J. E. 6:91–98. [Google Scholar]
- Bukman, D. J., J. H. Yao, and M. Wortis. 1996. Stability of cylindrical vesicles under axial tension. Phys. Rev. E. 54:5463–5468. [DOI] [PubMed] [Google Scholar]
- Buton, X., G. Morrot, P. Fellmann, and M. Seigneuret. 1996. Ultrafast glycerophospholipid-selective transbilayer motion mediated by a protein in the endoplasmic reticulum membrane. J. Biol. Chem. 271:6651–6657. [DOI] [PubMed] [Google Scholar]
- Chizmadzhev, Y. A., D. A. Kumenko, P. I. Kuzmin, L. V. Chernomordik, J. Zimmerberg, and F. S. Cohen. 1999. Lipid flow through fusion pores connecting membranes of different tensions. Biophys. J. 76:2951–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabora, S. L., and M. P. Sheetz. 1988. The microtubule-dependent formation of a tubulovesicular network with characteristics of the ER from cultured cell extracts. Cell. 54:27–35. [DOI] [PubMed] [Google Scholar]
- Dai, J., and M. P. Sheetz. 1995a. Axon membrane flows from the growth cone to the cell body. Cell. 83:693–701. [DOI] [PubMed] [Google Scholar]
- Dai, J., and M. P. Sheetz. 1995b. Mechanical properties of neuronal growth cone membranes studied by tether formation with laser optical tweezers. Biophys. J. 68:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier, L., and T. A. Rapoport. 2000. In vitro formation of the endoplasmic reticulum occurs independently of microtubules by a controlled fusion reaction. J. Cell Biol. 148:883–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E. A., and R. M. Hochmuth. 1978. Mechanochemical properties of membranes. Curr. Top. Membr. Transp. 10:1–62. [Google Scholar]
- Evans, E. A., and W. Rawicz. 1990. Entropy-driven tension and bending elasticity in condensed fluid membranes. Phys. Rev. Lett. 64:2094–2097. [DOI] [PubMed] [Google Scholar]
- Fullerton, A. T., M. Y. Bau, P. A. Conrad, and G. S. Bloom. 1998. In vitro reconstitution of microtubule plus end-directed, GTPγS-sensitive motility of Golgi membranes. Mol. Biol. Cell. 9:2699–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochmuth, R. M., J. Y. Shao, J. Dai, and M. P. Sheetz. 1996. Deformation and flow of membranes into tethers extracted from neuronal growth cones. Biophys. J. 70:358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo, S. C., and M. P. Sheetz. 1993. Force of single kinesin molecules measured with optical tweezers. Science. 260:232–234. [DOI] [PubMed] [Google Scholar]
- Lane, J. D., and V. J. Allan. 1999. Microtubule-based endoplasmic reticulum motility in Xenopus laevis: activation of membrane-associated kinesin during development. Mol. Biol. Cell. 10:1909–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, C., and L. B. Chen. 1988. Dynamic behavior of endoplasmic reticulum in living cells. Cell. 54:37–46. [DOI] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., L. C. Yuan, J. S. Bonifacino, and R. D. Klausner. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with Brefeldin A: evidence for membrane cycling from Golgi to ER. Cell. 56:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz, J., J. G. Donaldson, A. Schweizer, E. G. Berger, H. Hauri, L. C. Yuan, and R. D. Klausner. 1990. Microtubule-dependent retrograde transport of proteins into the ER in the presence of Brefeldin A suggests an ER recycling pathway. Cell. 60:821–836. [DOI] [PubMed] [Google Scholar]
- McIlvain, J. M., C. Lamb, S. Dabora, and M. P. Sheetz. 1993. Microtubule motor-dependent formation of tubulovesicular networks from endoplasmic reticulum and Golgi membranes. Methods Cell Biol. 39:227–236. [DOI] [PubMed] [Google Scholar]
- Morris, C. E., and N. Homann. 2001. Cell surface area regulation and membrane tension. J. Membr. Biol. 179:79–102. [DOI] [PubMed] [Google Scholar]
- Saffman, P. G. 1976. Brownian motion in thin sheets of viscous fluid. J. Fluid. Mech. 73:593–602. [Google Scholar]
- Schnapp, B. J., J. Gelles, and M. P. Sheetz. 1988. Nanometer-scale measurements using video-light microscope. Cell Motil. Cytoskeleton. 10:47–53. [DOI] [PubMed] [Google Scholar]
- Sciaky, N., J. J. Presley, C. Smith, K. J. M. Zaal, N. Cole, J. E. Moreira, M. Terasaki, E. Siggia, and J. Lippincott-Schwartz. 1997. Golgi tubule traffic and the effects of Brefelden A visualized in living cells. J. Cell Biol. 139:1137–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheetz, M. P. 2001. Cell control by membrane-cytoskeleton adhesion. Nat. Rev. Mol. Cell Biol. 2:392–396. [DOI] [PubMed] [Google Scholar]
- Song, J., and R. E. Waugh. 1993. Bending rigidity of SOPC membrane containing cholesterol. Biophys. J. 64:1967–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaki, M., L. B. Chen, and K. Fujiwara. 1986. Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol. 103:1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh, R. E., J. Song, S. Svetina, and B. Zeks. 1992. Local and nonlocal curvature elasticity in bilayer membrane by tether formation from lecithin vesicles. Biophys. J. 61:974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]