

Abstract
Ion permeation through the gramicidin channel is studied using a model that circumvents two major difficulties inherent to standard simulational methods. It exploits the timescale separation between electronic and structural contributions to dielectric stabilization, accounting for the influence of electronic polarization by embedding the channel in a dielectric milieu that describes this polarization in a mean sense. The explicit mobile moieties are the ion, multipolar waters, and the carbonyls and amides of the peptide backbone. The model treats the influence of aromatic residues and the membrane dipole potential. A new electrical geometry is introduced that treats long-range electrostatics exactly and avoids problems related to periodic boundary conditions. It permits the translocating ion to make a seamless transition from nearby electrolyte to the channel interior. Other degrees of freedom (more distant bulk electrolyte and nonpolar lipid) are treated as dielectric continua. Reasonable permeation free energy profiles are obtained for potassium, rubidium, and cesium; binding wells are shallow and the central barrier is small. Estimated cationic single-channel conductances are smaller than experiment, but only by factors between 2 (rubidium) and 50 (potassium). When applied to chloride the internal barrier is large, with a corresponding miniscule single-channel conductance. The estimated relative single-channel conductances of gramicidin A, B, and C agree well with experiment.
INTRODUCTION
The semimicroscopic (SMC) approach to modeling permeation free energies for ion translocation through narrow channels (Dorman et al., 1996, 1999; Garofoli and Jordan, 2003; Garofoli et al., 2002; Partenskii et al., 1991a,b, 1994; Partenskii and Jordan, 1992a,b; Sancho et al., 1995) is a hybrid microscopic-mesoscopic method, developed to bridge the gap between continuum electrostatic modeling and molecular dynamics (MD). It is designed to provide a physically transparent way to model the most salient features of ion channels, while circumventing two limitations common to almost all MD studies: the input force fields ignore effects due to electronic polarization; long-range electrostatic contributions are sensitive to the use of periodic boundary conditions. In the most popular force fields such as AMBER (Cornell et al., 1995; Weiner et al., 1984), CHARMM (Brooks et al., 1983; MacKerell et al., 1998), and GROMOS (Hermans et al., 1984; van Gunsteren and Berendsen, 1987) the parameters for water and for various partially charged moieties used to describe peptides and lipids have been fitted to account for interaction with bulk aqueous solutions. Since water structure in the narrow confines of ion channels is typically single-file, vastly different from the hydrogen-bonded network characteristic of ambient water, it is quite conceivable that reliable error compensation, which is effectively demonstrated by the ability of these force fields to describe a wide range of problems, does not hold in the confines of a selectivity filter. In addition, for systems composed of dielectrically significantly dissimilar regions, ionic energetics is sensitive to the long-range artifacts imposed by periodic boundary conditions (Bostick and Berkowitz, 2003; Hunenberger and McCammon, 1999; Yeh and Berkowitz, 1999). These points were dramatically illustrated recently. The potential of mean force (PMF) for K+ permeation through a gramicidin A (gA) channel inserted in a model dimyristoyl phosphatidylcholine (DMPC) bilayer sandwiched between two bulk water boxes, using both the CHARMM and GROMACS force fields, was computed by MD (Allen et al., 2003b) and contrasted with profile parameters deduced using Brownian dynamics (BD) to deconvolute experimental data (Edwards et al., 2002). The results differ sharply. The computational predictions were essentially identical: binding wells of ∼6 kT located ∼10 Å from the channel midpoint and an internal barrier of ∼22 kT. Other than the binding site near ∼10 Å, the parameters bore scant resemblance to that deduced from experiment, in which wells were much deeper, ∼8 kT, and the internal barrier much smaller, ∼5 kT. This profile is qualitatively similar to deconvolutions based on electrodiffusion theories (EDT) (Chiu and Jakobsson, 1989; McGill and Schumaker, 1996), where wells are in the 5–9 kT range and barriers vary from 4 to 8 kT. Computing K+ conductance with the simulational PMFs leads to underestimates of six to seven orders of magnitude.
Gramicidin, although marvelous for studying aspects of single-file permeation, is structurally very different from physiological ion channels since its β-helical pore scaffolding is formed by the peptide backbone. Thus the inability of MD, based on employing standard force fields coupled with long-range Ewald methods to account for the energetics of cation permeation in this system, might be considered an annoying anomaly. However, with the recent striking successes in the determination of crystal structures for the pore domains of ion and water channels (Doyle et al., 1998; Dutzler et al., 2002; Fu et al., 2000; Jiang et al., 2002; Murata et al., 2000; Sui et al., 2001; Zhou et al., 2001) their theoretical study has become a miniature growth industry. These selective channels share two important features with gramicidin: narrow filter regions of variable length, where the translocating ions and waters move in single-file with sharp dielectric discontinuities between aqueous and lipid environments. In the potassium channel filter, there are four or five single-file sites, of overall length 12–15 Å (Bernèche and Roux, 2001; Doyle et al., 1998; Jiang et al., 2002; Morais-Cabral et al., 2001), and up to three ions may occupy single-file sites. In the ClC chloride pore, the transport pathway has not been as well characterized. However, modeling based on the crystal structure of a prokaryotic ClC (Dutzler et al., 2002), which is an antiporter, not a channel (Accardi and Miller, 2004), suggests an arched single-file ∼16 Å long (Cohen and Schulten, 2004; Miloshevsky and Jordan, 2004), long enough to comprise approximately four or five sites; inasmuch as there is clear coupling between ion conduction and gating (Chen and Miller, 1996; Pusch et al., 1995; Richard and Miller, 1990) two of these sites may well be simultaneously anion-occupied. In both the aquaporin (Murata et al., 2000; Sui et al., 2001) and the aquaglyceroporin (Fu et al., 2000) filters, the single-file extends for ∼10–12 Å. In all these cases water structure in the filter may well be more reminiscent of gramicidin, with its ∼20 Å single-file region, than of bulk water, and one can question whether the use of standard force fields to determine the PMF of the permeating species in these physiologically important systems could not be plagued by the problems that arise in gA. Both structural and electrophysiological evidence suggest that there is no long single-file in the acetylcholine receptor (Dani, 1989; Miyazawa et al., 2003). However, for constriction diameters as large as ∼9 Å, studies on water nanotubes give rise to ordered water alignments, differing sharply from those seen in bulk (Mashl et al., 2003). Thus even in this wider filter, standard force fields may conceivably yield unreliable energetic predictions. And in every instance periodic boundary conditions are used and long-range electrostatics corrections determined via Ewald methods.
One of the factors possibly contributing to the gramicidin dilemma is that standard force fields ignore electronic polarization. In the absence of a truly polarizable force field, reconsideration of gramicidin energetics from the SMC perspective is in order. Dielectric reorganization, which is the origin of the solvating ability of polar media, both uniform (like bulk water) and nonuniform (like the surroundings of ion channel selectivity filters), cleanly separates into structural and electronic components. The structural terms account for ligand rearrangement in response to the motion of the permeating ionic or polar moiety; the electronic terms describe the response of solvent (here waters, lipid, and peptide) molecules' charge distribution to the permeating species. The timescales for these processes are substantially different. Electronic relaxation occurs in femtoseconds, whereas structural relaxation takes place over wide-ranging timescales; however, even the fastest such process, translational relaxation, is at least 100 times slower. The behavior of water is representative (Hasted, 1973). In low frequency electric fields (<1 GHz) water exhibits bulk dielectric properties. As the frequency increases water molecules can no longer rotate or translate rapidly enough, and by ∼10 THz the solvent cannot respond to the electrical stimulus; the dielectric constant drops to its optical value, ɛ = 1.8. Structural reorganization takes place with the electrons relaxed. As a first approximation this electronic background is essentially uniform with a roughly universal polarizability; in effect molecular rearrangement takes place under conditions where ɛ = ∼2.
The SMC method was designed to exploit this timescale separation. By placing the mobile polar permeant species in a dielectric background of ɛ = ∼2, electronic polarization is taken into consideration. Structural polarization is treated by explicitly including those charged features of the peptide-lipid ensemble that interact strongly with the permeating moiety. Of crucial importance is that a sufficient number of these be mobile, since dielectric shielding arises from charge rearrangements in the solvent in response to motion of the permeant species (Partenskii and Jordan, 1992b; Schutz and Warshel, 2001).
In addition to problems attributable to putative force field deficiencies, MD simulations of ion channel systems impose severe computational loads. To account for the influence of cytoplasmic and extracellular water, a computational membrane with its embedded channel is sandwiched between two large water boxes, generally the bulk of the computational system. Due to boundary effects, even simulations involving ∼105 atomic centers are not representative of bulk; errors are minimized by employing periodic boundaries in directions perpendicular to the channel protein. Since electrostatic forces are long-range, the behavior of charged groups in the membrane-peptide milieu is significantly affected by interaction with the surrounding bulk water. To avoid the ambiguity introduced by electrical cutoffs, reaction field effects are treated by particle mesh Ewald methods (Deserno and Holm, 1998; Essman et al., 1995). However, for ion channel geometries the standard approaches must be modified in the nonperiodic direction (the channel axis), lest spurious correlations (Bostick and Berkowitz, 2003; Yeh and Berkowitz, 1999) and free energy anomalies (Hunenberger and McCammon, 1999) be introduced. In the SMC treatment, embedding the membrane-peptide-ion channel ensemble in electrical geometries for which the reaction field can be rigorously computed (Dorman et al., 1996; Dorman and Jordan, 2003; Garofoli and Jordan, 2003; Miloshevsky and Jordan, 2004) finesses the difficulties associated with long-range electrostatics (Jackson, 1962; Smythe, 1968).
Our SMC approach differs sharply from most other mesoscopic-microscopic hybrids. BD descriptions, pioneered by Jakobsson and co-workers for gramicidin (Chiu and Jakobsson, 1989; Cooper et al., 1985; Jakobsson and Chiu, 1987) and exploited by Chung and co-workers for a range of physiological systems (Chung et al., 1999, 2002a,b; Edwards et al., 2002), all treat single-file pores as dielectric continua, assigning them artificially high dielectric constants. In the case of gramicidin, this compensates for the large image energy contribution due to interaction with nearby nonpermittive lipid domains. For physiological channels, this is analogous to the familiar use of a high ɛ in Poisson-Boltzmann descriptions to describe protein interiors; in this way dielectric shielding due to peptide reorganization is accommodated (Antosiewicz et al., 1994). The same device has been used recently (Mamonov et al., 2003) in modifications of the Poisson-Nernst-Planck approach (Eisenberg, 1999; Nonner et al., 1999) specifically tailored to circumvent the image energy issue. Only the surface-constrained all atom solvent (SCAAS) approach (King and Warshel, 1989) appears to treat both dielectric shielding and the associated reaction field in a completely natural fashion. However, it is as computationally intensive as MD methods generally.
In previous applications of the SMC method (Dorman et al., 1996, 1999; Garofoli and Jordan, 2003; Garofoli et al., 2002), the only mobile entities explicitly treated were single-file waters, ions, and carbonyl groups bounding the filter. In modeling KcsA, the midchannel aqueous cavity and various (fixed) charged moieties were also included. These simplified systems were sandwiched between planar conducting continua (ɛ ≡ ∞), introducing a discontinuous transition between the single-file ion-water chain in the model filter and bulk water, possibly creating spurious correlations. Here we introduce a new model geometry. It explicitly includes a few solvation shells of bulk water at the channel mouth, permitting continuous ion movement from bulk water into the channel. It is reminiscent of Warshel's surface-constrained soft spherical dipole and SCAAS methods (King and Warshel, 1989; Warshel, 1979); the parallels and differences have been fully discussed previously (Dorman and Jordan, 2003). Our earlier description of gramicidin only included the peptide carbonyls. Here, we treat the whole backbone, take into account the aromatic residues near the channel mouth, and also model effects attributable to the membrane dipole potential. Finally, instead of the dipolar water model used previously (Dorman et al., 1996) we use an improvement, an SPC-like multipolar model that reproduces water's dipole moment (Dorman and Jordan, 2003; Garofoli and Jordan, 2003). As in the SCAAS method (King and Warshel, 1989), which also explicitly accounts for electronic polarization, albeit quite differently, water is assigned its gas-phase dipole moment, μ0 = 1.86 D, not an effective μ > μ0.
We first describe our new electrical geometry, and develop a rigorous representation of its reaction field. We then outline the extended model for gramicidin, and demonstrate how to use it to determine ionic PMFs for the three naturally occurring gramicidins. We consider four representative ions: the three largest alkali cations and Cl−, and demonstrate that the SMC method, in which electronic polarizability is taken into account from the outset and long-range electrostatics is treated exactly, recovers important structural features of experimental potential profiles determined from data inversion via BD (Edwards et al., 2002) or EDT deconvolutions (Chiu and Jakobsson, 1989; McGill and Schumaker, 1996). The approach separates the influence that the backbone, the aromatic residues, and the membrane dipole potential have on the PMF. Unlike our earlier study (Dorman et al., 1996), where the source of cationic stabilization in the channel was limited to interaction among the ion, the single-file waters and the peptide backbone, we find that interplay between all electrical features is required to mimic the experimental profile.
COMPUTATIONAL MODEL
The computational model is illustrated in Fig. 1 A. It is an idealization of the physical system, but it incorporates the most significant features. Since the gA channel is symmetric and the single-file region is long, ionic energetics on one side of the channel is basically independent of how the other side is modeled as long as the pore is singly occupied. A clear instance of this principle was observed when modeling KcsA. There the selectivity filter is separated from the cytoplasm by the aqueous midchannel cavity and the inner pore. Filter energetics is essentially unaltered if the system is artificially truncated and these features replaced by three or more single-file waters abutting the cytoplasm (Garofoli et al., 2002). Thus, for a singly occupied pore, ionic energetics on the channel's left-hand side can be reliably determined using the model illustrated, if a correction is introduced to account for the reaction field due to interaction with the missing bulk water phase on the right-hand side.
FIGURE 1.

(A) Modified electrical geometry of gramicidin, permitting continuous transition from single-file to bulk water. All explicit sources are in the low ɛ-region. The backbone is tethered, with mobile CO-Os and NH-Hs. Only 20 COs are shown. The dotted line is the physical boundary of the truncated system. The solid line is the electrical boundary. The Helmholtz layer is an electrical buffer region between the low ɛ-background and the high ɛ-bulk electrolyte. The coordinate arrows beneath the central water indicate the location of the channel midpoint. (B) For comparison, the full gramicidin dimer is sited relative to the water bubble with the regions encompassing the aromatic moieties indicated.
The mobile moieties are the ion, explicit waters in the single-file and the left-hand hemispherical bubble, carbonyl oxygens, and amide hydrogens. To provide a sense of the truncations involved, the half-hydrated full channel is shown in Fig. 1 B; the influence of the immobilized aromatic side chains is accounted for in what follows. The water model is that developed for use in SMC modeling (Dorman and Jordan, 2003), which reliably reproduces hydration free energies of both the three smallest halides and the three larger alkali cations. As done previously (Dorman and Jordan, 2003; Garofoli and Jordan, 2003) ions are modeled as charged hard spheres, with crystallographic radii (Pauling, 1960). The peptide backbone is rigid, with coordinates taken from a symmetrized, equilibrated gramicidin structure, PDB code 1MAG (Ketchem et al., 1997). COs and NHs are treated as rigid dipoles, with bond lengths 1.24 and 0.96 Å, respectively. Carbonyl oxygens, amide nitrogens, and α-carbons were all assigned hard-sphere radii of 1.4 Å. Results were insensitive to varying N and Cα radii by as much as ±0.1 Å. As in earlier work (Dorman et al., 1996; Garofoli and Jordan, 2003), CO and NH bending are described by harmonic restoring forces, Ubend = kbend (θ–θo)2/2. The bending force constants differ slightly, kCO = 7.5 × 10−20 J and kNH = 7.1 × 10−20 J, both values representative of low energy torsional modes involving these subunits of the peptides (see e.g., Hermans et al., 1984); just as in the SMC analysis of cation interaction with the KcsA potassium channel (Garofoli and Jordan, 2003), results are insensitive to moderate force constant variation. Because electronic polarization is accounted for by the choice of background dielectric, dipolar moieties are assigned charges consistent with their permanent dipole moments. For CO carbons the charge is +0.38 eo (Garofoli and Jordan, 2003; Lee and Jordan, 1984; Pethig, 1979), identical with the GROMOS assignment (Hermans et al., 1984; van Gunsteren and Berendsen, 1987). We thus use the GROMOS charges, −0.28 eo for NH nitrogens, with uncharged α-carbons. To assess the influence of the charge assignment, we also considered the noticeably different CHARMM19 parameters: CO carbon and oxygen, +0.55 eo and −0.55 eo; NH nitrogen and hydrogen −0.35 eo and +0.25 eo; and α-carbon +0.10 eo (MacKerell et al., 1998).
The left-hand hemispherical bubble permits continuous changes of solvation as an ion moves from bulk water into the channel. The bubble is centered 15.4 Å from the channel midpoint, which is also the location of the planar surface separating the two electrical domains. The SMC model for ion solvation converged if the ion was embedded in a water-filled sphere with a background ɛ of 2 and a mechanical radius of 7.9 Å, inside an intermediate spherical 1.3 Å electrical buffer shell (also with ɛ = 2) and all surrounded by high ɛ-dielectric (Dorman and Jordan, 2003). The same parameters are used here; the hemisphere contains ∼20 explicit waters. The region to the right is the electrical background, ɛbackground = 2. The region to the left is bulk electrolyte, treated in a continuum approximation. For computational convenience ɛbulk is assigned the value ∞ rather than its true value, ∼80; earlier work showed that the associated error in the reaction field contribution to system energetics is negligible, ∼1 kJ mol−1. This perfect conductor description of aqueous systems has been discussed previously (Partenskii et al., 1991b, 1994; Partenskii and Jordan, 1992a); it becomes ever more reliable as the bulk electrolyte's ionic strength increases (Partenskii et al., 1994, 1998).
In addition to the mobile groups, we include the electrical influence of the aromatic moieties of gramicidins A (gA), B (gB), and C (gC), with tryptophan and tyrosine coordinates taken from structural data, PDB files 1JNO (gA), 1JO3 (gB), and 1JO4 (gC) (Townsley et al., 2001). We used 1JNO to model gA because, based on analysis of NMR data, its tryptophan orientations more closely resemble those in gA solvated by membrane (Allen et al., 2003a). Moreover, comparisons are more reliable since the three structures were determined under similar conditions, and structural details are environmentally sensitive (Allen et al., 2003a). It should be noted that this backbone switch is reasonable since, after equilibration, the RMS deviations between 1MAG and 1JNO backbones is small, ∼0.21 Å (Allen et al., 2003a). Depending upon the backbone charge assignment used, either GROMOS or CHARMM19 charge conventions are used for the Trp and Tyr residues. Ethanolamine headgroups only matter at the channel-bulk water interface; as we focus on the channel interior, they are ignored. Formyl groups at midchannel are treated like carbonyls.
Due to the influence of the lipid headgroups, the interiors of neutral lipids are significantly positive with respect to bulk water (Pickar and Benz, 1978), giving rise to membrane dipole potentials in the 300–400 mV range. The headgroups have complex charge distributions, partially bathed by aqueous solvent; it is not obvious how to treat these reliably in our electrical model, with its sharp delineation of the background-bulk boundary. A point dipole approximation is surely inadequate. Thus, rather than explicitly modeling the lipid headgroups, we account for them heuristically, relying on electrophysiological measurements on gramicidin to determine their energetic influence within the channel (Thompson et al., 2001).
Electrical potential and reaction field
The great advantage of the SMC approach is that the reaction field problem is exactly soluble for a felicitous choice of electrical geometry. Previously a planar low ɛ-slab (containing explicit moieties) was sandwiched between high ɛ-surroundings (continuum bulk electrolyte). With this geometry the reaction field generated by explicit charges (those in the low ɛ-domain) were readily treated by the image method resulting in a closed form expression for the electrical potential. The geometry of Fig. 1 A is more difficult. The solution to the electrical mapping problem is not self-evident; we describe it briefly.
Fig. 2 is a confocal map transforming the hemispherical bubble that separates low and high ɛ-regions into a right-angled wedge, for which the electrical potential (including its reaction field) can be expressed in integral form. Relec is the electrical radius of the bubble; the transformation is based on the invariant point where the dashed circle is tangent to the bubble. For points on or below the dividing surface, their electrical equivalents are found by inverting them through the surface defined by the dashed circle of radius 2Relec with its center at point I. As shown in the upper panel, a generic point r = (x, y, z) is translated along x by –Relec; the translated vector, rT = (x + Relec, y, z) is then inverted, a procedure which requires charge and potential rescaling (Dorman and Jordan, 2003; Jackson, 1962). The inverted vector is
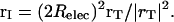 |
(1) |
FIGURE 2.

Confocal mapping of the bubble geometry of Fig. 1 A into a wedge. The point I, translated by x = −Relec from the bubble center, is the center of the inversion sphere of radius 2Relec. In the transformed geometry, points on the bubble surface lie on the plane at x = 2Relec. The invariant point in the transformation is the intersection of the bubble and the inversion sphere.
Charge and potential rescaling yields
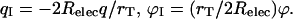 |
(2) |
The lower panel illustrates the x–z projection of the new geometry. The hemisphere and the original plane become semi-infinite planes, and the new surfaces are (xI = 2Relec, −∞< yI <+∞, zI > 0) and (xI < 2Relec, −∞< yI <+∞, zI = 0), respectively. Representative transformed points within the region containing all explicit electrical sources, the hemisphere (z > 0) and cylinder (−Relec < ρ < Relec, z < 0), are shown in the upper panel. The four points within the low ɛ-region map into the exterior points. Points on the bubble map to points on the surface x = 2 Relec. In this planar projection the high ɛ-region is the second quadrant. The original bubble geometry is thus equivalent to a wedge, a solved electrical problem (Smythe, 1968).
For a wedge with β = π/p, the angle subtended by the low ɛ-region, the potential at (ρ, α, z) due to an electrical source at (ρ0, α0, z0) is
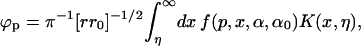 |
(3) |
where η, the kernel K(x,η), and the function f(p,x,α,α0) are, respectively,
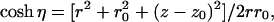 |
(4) |
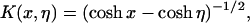 |
(5) |
and
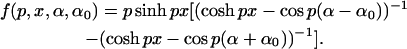 |
(6) |
Here β = 3π/2 and p = 2/3. Eq. 3 is analytically well defined, but the integrand has singularities requiring careful handling. In determining the source's self-energy, [r, z, α] → [r0, z0, α0], and the denominator of the first term in Eq. 6 can approach zero. This cannot be handled by simply superposing the two points; the putative singularity must be integrated before carrying out the limiting process [r, z, α] → [r0, z0, α0]. The denominator of the second term approaches zero as both η → 0 and α → α0 → 3π/2 and in Eq. 5 the kernel, K(x,η), is singular as both η → 0 and x → 0. These singularities are integrable, and are handled by a reformulation that treats them analytically, all remaining contributions to the reaction field being computed numerically. It should be noted that the dimple geometry (a wedge angle β = π/2) is much simpler; it can be treated by extensions of standard image approaches since, for all β < π, a point within the low ɛ-region has a unique image. This is not the case for the bubble treated here.
Thermodynamic cycle
The basic computational problem is removing an ion from a solvation sphere and exchanging it for a water in the channel. The solvation sphere is that used previously, which accounts for ionic hydration energetics (Dorman and Jordan, 2003). The interior of the channel and the solvation sphere are electrical background regions (ɛbackground ≡ 2); respectively, they abut or are surrounded by bulk electrolyte (ɛbulk ≡ ∞). The transfer free energy is computed by gradually mutating an ion in the sphere to water while simultaneously transforming a water in the channel to an ion. During this procedure the channel moiety's center-of-mass is fixed axially; it remains free to rotate and to translate radially. The solvation sphere's mutating moiety is held at the sphere center, but rotates freely. The associated thermodynamic process is simple:
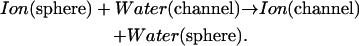 |
(7) |
The entity being altered in the sphere has the charge distribution
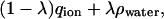 |
(8) |
with ρwater the charge distribution in the SPC-like water molecule; λ varies from 0 to 1 in the ion to water transformation. In the channel, the mutating unit's charge distribution is complementary, λqion + (1 − λ)ρwater. The total system Hamiltonian is H = Hsphere(λ) + Hchannel(λ) where
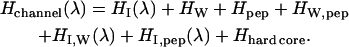 |
(9) |
HI(λ) describes the interaction of the variable species with its reaction field, HW, Hpep, and HW,pep the water-water, peptide-peptide, and peptide-water interactions, respectively, including reaction field effects HI,W(λ) and HI,pep(λ), the interactions between the variable species and water and peptide, respectively, again including reaction field influences. The hard-core term keeps the various species apart. The sphere Hamiltonian is analogous, with peptide terms deleted.
During this transformation the mutating groups' interaction with their surroundings changes in two distinct ways, the details of which have been discussed previously (Dorman and Jordan, 2003). First, there is dielectric shielding due to solvent reorganization at each alteration locus; the associated free energy change is ΔGreorg. In addition, the mutating ion (or water) is surrounded by a Born solvation cavity, which need not be exactly the same size in the two environments; the free energy change is ΔGBorn. The contribution from transferring the ion from the aqueous sphere to the channel is simply a Born energy,
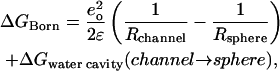 |
(10) |
where Rsphere and Rchannel are the cavity radii for the ion in the two regions; the second term in Eq. 10 accounts for the energetic consequences of any changes in water's solvation cavity in the complementary process (Beveridge and Schnuelle, 1975). The reorganizational free energy, ΔGreorg, is determined by standard thermodynamic perturbation,
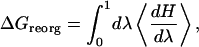 |
(11) |
where
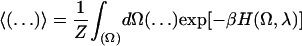 |
(12) |
is the canonical statistical average, Ω the complete set of phase space variables, and Z the corresponding partition function with β ≡ 1/kT. The λ-integral, Eq. 11, is broken up into 20 discrete intervals and each contribution to ΔGreorg calculated from 105 Monte Carlo steps using the Metropolis algorithm (Metropolis et al., 1953). At each λ the system is re-equilibrated for 5 × 105 steps before data collection is started. The cavity radii needed for computing the Born terms of Eq. 10 are determined from the relevant ion-solvent and water-solvent radial distribution functions, i.e., when λ = 0 or 1.
Residual terms in the free energy
The geometry of Fig. 1 A is incomplete since the channel only abuts one aqueous phase. As long as the focus is on ions fairly distant from the excluded water, correcting for this omission is simply done by recognizing that the bubble geometry really only matters for ions near the channel mouth where explicit solvation by bulk water is important. The deeper the ion in the channel, the more reliable is the planar geometry used previously (Dorman et al., 1996). Interaction between an ion on one side of the channel and water on the other can be treated assuming a planar interface. We assume that explicit bubble water does not significantly alter this interaction and estimate the long-range correction as the difference between the image energy (the reaction field contribution) for an ion sandwiched between two high ɛ-domains and that for an ion in a single-interface geometry. For a low ɛ-region of width W with an ion a distance z from the left-hand surface, the correction is
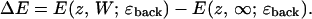 |
(13) |
This substitute geometry is generated from the geometry of Fig. 1 A by shrinking the radius of the bubble to zero; W is 30.8 Å because the Helmholtz layer is 1.4 Å thick.
Interaction with gramicidin's aromatic side chains are treated in the same spirit. Their locations are determined from symmetrized crystallographic coordinates, and only their influence on the ion is considered; the reaction field contribution is calculated assuming the same planar, two-interface geometry used in estimating long-range contributions to the ion's reaction field.
The membrane dipole potential is treated heuristically. Experiments indicate that, at the channel midpoint, the influence of this source at the channel midpoint is ∼38% of that in the membrane itself (Thompson et al., 2001). For a DMPC-like membrane the membrane dipole potential, relative to water is ∼390 mV (Pickar and Benz, 1978), leading to a cationic contribution of +5.7 kT at the channel midpoint. The energy profile is determined by scaling the potential computed for a conductive channel embedded in a membrane (Jordan, 1983). The energy change from the three contributions just described is equated to the free energy change.
RESULTS
Fig. 3 presents backbone contributions to the permeation free energy for three alkali cations and chloride in gA (symmetrized, equilibrated 1MAG structure) based on the GROMOS charge assignment, including interaction with all explicit waters, with bulk electrolyte and correcting for the distant water phase. Two properties are immediately apparent. Backbone interaction with the cations is quite similar; all exhibit a gradual rise in energy as the ion enters deeper into the pore with a maximum ∼0.4 nm from the channel midpoint. The cations differ distinctly from chloride, where the energy rises much more steeply, with an internal barrier typically ∼10 kT larger than for the cations. Just as in our previous study (Dorman et al., 1996), backbone interactions do not create a well-articulated binding site in the channel. Here and in the remaining figures, results based on the CHARMM19 charge parameterization exhibit no qualitative differences and only minor quantitative ones.
FIGURE 3.

Backbone contributions to the permeation free energy in gA for the three larger cations and chloride: Cs+, ▪; Rb+, ▴; K+, •; and Cl−, □.
Fig. 4 distinguishes some of the terms contributing to ionic stabilization in the channel. It illustrates backbone contributions to the permeation free energy of Cs+ for different electronic and structural scenarios. The behavior of the other cations is similar. As in Fig. 3, results account for interaction with the explicit waters, with bulk electrolyte and with the distant water phase. Both backbone immobilization and electrical background permittivity reduction destabilize an ion in the channel, but structural relaxation is clearly more significant energetically. Stabilization can be viewed in dielectric terms as the Born transfer of an ionic sphere of fixed radius from bulk water (ɛ = 80) to a milieu with a local, position-dependent, effective permittivity, ɛeff (Jordan et al., 1997; Partenskii et al., 1994). From this perspective, for a background ɛ of 2, the ɛeff at the center of the rigid channel is ∼10 whereas that of the relaxed channel is ∼20, this latter a relatively common choice for an effective protein dielectric constant (Antosiewicz et al., 1994; Schutz and Warshel, 2001). If the background ɛ is dropped to 1, both ɛeff decrease, to ∼5 for the rigid backbone and ∼12 for the mobile backbone.
FIGURE 4.

Effect of structural relaxation and mean background electronic polarizability on backbone contributions to the permeation free energy for Cs+: ɛbackground = 2, mobile backbone ▪; ɛbackground = 1, mobile backbone, •; and ɛbackground = 2, rigid backbone, ▴.
Fig. 5 compares the influence of the three separable contributions to the PMF of gA (backbone, aromatic residues, and membrane dipole potential) for the two similarly sized ions, chloride and cesium. Ionic interaction with the Trp side chains leads to a sharp separation of the cesium and chloride profiles. There is now a well-articulated binding site for the cation in roughly the proper location, ∼1.0 nm from the midpoint of the channel. The peak in the anion profile, at ∼0.6 nm, is even more pronounced. The anion-cation free energy difference is ∼30 kT. When the influence of the membrane dipole potential is included, the position of the cation binding site is shifted slightly and the anion-cation free energy difference is noticeably reduced, to ∼20 kT. Since, in accounting for side-chain and membrane dipole contributions to the permeation free energy profiles, no further structural relaxation is permitted, results are essentially the same for all three cations; consequently only cesium data are presented.
FIGURE 5.

Comparison of the cumulative contributions to the permeation free energy in gA for both cesium and chloride. Backbone only: Cs+ (+), Cl− (−); full gA (backbone and tryptophan residues): Cs+, (•), Cl− (○); all sources (backbone, tryptophan residues and membrane dipole potential): Cs+ (▪), Cl− (□).
Fig. 6 plots the total energy, for the three cations and for chloride, and contrasts these with the simulational and experimental free energy profiles. The striking feature is the large difference between the simulational profile and the experimental one, a point stressed by Allen et al. (2003b), and in agreement with familiar EDT results (Chiu and Jakobsson, 1989; McGill and Schumaker, 1996). The SMC profiles for the larger cations, even given the approximate nature of the modeling procedure, far more closely approach the experimental data than does the simulational profile, although in all instances the energy is somewhat high, typically by 5–10 kT. The shape of the SMC profile is quite similar to that deduced from data deconvolution, although displaced to somewhat higher energy (∼5 kT). The well is shallow and the translocational profile is flat; there is no hint of the very steep rise predicted by (in this case) the GROMOS force field. However, in each case the well depth is underestimated and the translocational barrier overestimated.
FIGURE 6.

Total permeation free energy profiles for the three larger cations and chloride. Symbols are those of Fig. 3. Solid line is an experimental profile for K+ (Edwards et al., 2002); dashed line is the simulational profile for K+ based on the GROMOS force field (Allen et al., 2003b).
As indicated at the outset, the differences between the simulational and experimental profiles may arise from neglect of electronic polarization or be due to long-range electrostatic artifacts attributable to the use of Ewald methods with periodic boundary conditions. The SMC approach circumvents both of these pitfalls, but not in a manner that isolates their influence. However, insight can be gained by comparing four profiles for K+, as shown in Fig. 7, which presents simulational, experimental, SMC (ɛbackground = 2, mobile backbone), and SMC (ɛbackground = 1, mobile backbone) results. Reducing ɛbackground eliminates the mean contribution of electronic polarizability. Comparison of the ɛbackground = 1 and simulational curves indicates that there is an important additional contribution. Recent simulational results for gramicidin (Allen et al., 2004) have shown that the correction for the Ewald artifact is ∼6.5 kT at midchannel, approximately the residual difference. The polarization and Ewald periodicity corrections are thus roughly comparable.
FIGURE 7.

Effect of mean background electronic polarizability on the total permeation free energy for K+: ɛbackground = 2, mobile backbone ▪; ɛbackground = 1, mobile backbone, •. As in Fig. 6, solid and dashed lines are experimental and simulational profiles for K+.
Fig. 8 contrasts the free energy profiles for Cs+ permeation of gA (1JNO), gB (1JO3), and gC (1JO4). As expected there is a clear separation, with permeation through gA substantially favored. In agreement with experiment there is little difference between gB and gC, with gC the more conductive (Sawyer et al., 1990). Simple diffusive modeling (Levitt, 1986) can be used to estimate the ratios of single-channel conductance. Table 1 compares the experimental ratios with those determined based on both GROMOS and CHARMM19 charge assignments. The qualitative pattern is fully consistent with experiment, although the computed ratios clearly overestimate the relative conductance of gA. The associated energy barrier discrepancies are small, ∼1 and ∼2 kT, for the CHARMM19 and GROMOS charge parameterizations, respectively. The gC-gB energy barrier discrepancies are trivial, in both cases, <0.2 kT.
FIGURE 8.

Comparison of total permeation free energy profiles for Cs+ permeation through gA, ▴; gB, ▪; and gC, •.
TABLE 1.
Comparisons of computed and experimental relative conductances of the three naturally occurring gramicidins
| GROMOS | CHARMM19 | Experiment* | |
|---|---|---|---|
| gA (1JNO) | ≡ 1 | ≡ 1 | ≡ 1 |
| gB (1JO3) | 0.079 | 0.23 | 0.59 |
| gC (1JO4) | 0.14 | 0.30 | 0.87 |
Data from Sawyer et al. (1990).
The free energy profiles can also be used to estimate absolute conductances, again utilizing Levitt's (1986) diffusive model for single-file conductance. The expression requires three parameters: the ionic diffusion coefficients, the channel radius, and the channel length. We assume that an ion's intrinsic friction in the channel (in the absence of the energy barrier) would be the same as in bulk water, a not-unreasonable assumption since water and cationic fluid dynamic diffusion coefficients in gramicidin differ little from their diffusion coefficients in aqueous media (Chiu et al., 1991, 1993; Smith and Sansom, 1999); thus we assign bulk values to ionic diffusion coefficients in the channel. The channel's length and radius are taken to be 2.8 nm and 0.25 nm, respectively. Based on these assumptions the absolute conductances in 0.1 M electrolyte are listed in Table 2. Two features stand out. Conductances determined from the simulational profiles bear no resemblance to the experimental values, being some seven or eight orders-of-magnitude too small. They are more reminiscent of the chloride conductances determined from the SMC procedure. Regardless of the charge parameters used, the trends are the same, with the ordering Rb+ > Cs+ > K+ ⋙ Cl−, in agreement with experiment, although the computed conductance ratios are much larger than those observed. Nonetheless, computed cation conductances are within a factor of ∼100 of the experimental values. The spread in computed cation conductances is substantially greater than that observed. Instead of the experimental, approximately twofold, K/Rb difference, we find factors of ∼50 (GROMOS) and ∼10 (CHARMM19); and for K+ the absolute barrier overestimates are ∼4 and ∼6 kT using GROMOS and CHARMM19, respectively.
TABLE 2.
Ionic conductances (pS) of gA in 0.1 M electrolyte
| GROMOS | CHARMM19 | Experiment* | |
|---|---|---|---|
| K+ | 0.24 | 0.025 | 11 |
| Rb+ | 12 | 0.40 | 20 |
| Cs+ | 1 | 0.074 | 18 |
| Cl− | 2.4 × 10−8 | 6.0 × 10−6 | 0 |
| Simulation, K+ | 1.8 × 10−7 | 4.2 × 10−7 | 11 |
Based on the total permeation free energy profiles of Fig. 6 (and analogous results using CHARMM19 charges), and computed using Levitt's (1986) single-file algorithm. Simulational values are estimated from the free energy profiles of Allen et al. (2003a).
Data extrapolated from Fig. 5 of Andersen (1983).
DISCUSSION
Figs. 6 and 7 and Table 2 summarize the main results. The SMC protocol leads to reasonable permeation free energy profiles and single-channel conductances for alkali cation permeation through the gramicidin channel. That this blunt, highly approximate tool can successfully account for the low internal free energy barrier is attributable to two methodological features. By exploiting the timescale separation between electronic and structural contributions to dielectric shielding, the polarization dilemma is finessed, and electronic reorganization is dealt with in a mean sense. In addition, by judicious choice of electrical geometry, long-range electrostatics are treated exactly and artifacts due to Ewald corrections for periodic boundary conditions avoided. The results underscore an observation, important to simulating electrical behavior in single-file (or narrow) environments, where water structure is very different from that in bulk. In such instances water's mean charge distribution is in all likelihood quite different from that in bulk water. As a result computed ion-water and water-mediated ion-channel interactions are substantially altered from what would be expected on the basis of standard force fields. Given that standard simulational protocols account quite well for most local details of ion binding to gramicidin (Roux, 2002), comparison of Figs. 6 and 7 suggest that the problems are due both to differences in the channel's long-range water ordering and to electrostatic artifacts introduced by the periodic boundary conditions (Allen et al., 2004; Hunenberger and McCammon, 1999). The effect that polarization has on the persistence of orientational correlation provides evidence for the influence of long-range order. MD studies of ionic interaction with gramicidin based on a polarizable water model (Duca and Jordan, 1997; Jordan, 1990), exhibited much shorter correlation lengths than those using standard simulational waters (Chiu et al., 1989; Mackay et al., 1984), with their artificially large dipole moments. Since the polarizable water used had both a dipole and a quadrupole moment, it accommodates hydrogen bonding, and the differences cannot be ascribed to this source (Chiu et al., 1991). A more likely reason is the large difference in interaction energy between neighboring waters as computed from the SMC model and from standard force fields. Assuming perfect alignment, this would be −μ2/(ɛR3), with μ the water dipole moment and R the interwater distance. With standard force fields, this corresponds to an energy of ∼−6 kT; for the model used here it is one-third as much, ∼−2 kT, consistent with a reduction in correlation lengths. Calculations based on the model parameters used here for a channel embedded between two image planes yielded similar outcomes. With a cation located at one channel mouth, water-chain ordering propagated to midchannel, and then fell off rapidly. This differs from the behavior of an ion-dipolar water, single-file solvated by a primitive model of the gramicidin backbone, with immobile carbonyl oxygens; there dipolar ordering persisted (Jordan et al., 1997).
The effect of the membrane dipole potential is of interest as well. Based on the results of Thompson et al. (2001) it adds ∼5.7 kT to the central cationic barrier, a substantial energy penalty. Were it ignored, predicted conductances would increase dramatically, ∼300-fold, and the estimated single-channel conductances of Table 2 would be substantially in excess of the experimental values. The surrounding lipid quite clearly significantly influences ionic flow through gramicidin. This is a consequence of the small spatial extent of the polypeptide. Its outer radius is ∼1 nm, much smaller than that of typical physiological channels, where outer radii are more likely in the 3–5-nm range. In such systems any effect of the membrane dipole potential would be greatly attenuated, with associated energy barriers in the range of 1–2 kT.
The use of GROMOS or CHARMM19 charge parameters might be questioned. However, these quantities are basically determined by fitting to ab initio calculations (MacKerell et al., 1998), under conditions where the dielectric background is unquestionably a vacuum, with ɛ = 1. It is only in the condensed state that polarization becomes a consideration.
Although it is qualitatively similar to the free energy profiles of the larger cations, we did not present the PMF for Na+ interaction with gramicidin because the primitive hard-sphere SMC water model we use, when applied to Na+, fails to reproduce this ion's hydration free energy. With a Pauling radius of 0.095 nm, the associated hydration energy error is ∼35 kJ mol−1, which indicates that the approach would require significant refinement to be applicable to Na+; consequently we felt the computed PMF was unreliable. It would also have been interesting to model interaction between gramicidin and the smaller divalent ions, Mg+2 and Ca+2. Here too the SMC parameterization protocol for ionic hydration is inadequate. The SMC approach is, by its very nature, approximate. The basic assumption is that electronic dielectric response is everywhere uniform. This is clearly inappropriate. Background modulation, due to local differences in charge density perturbation, is certainly to be expected. As a result, expending substantial effort to refine the model's structural parameters and develop a 6–12 formalism suitable for SMC computation seems unwarranted.
We now contrast this study with our earlier analysis of gramicidin (Dorman et al., 1996). There are major model differences. Previously structural relaxation was due to carbonyl oxygen and water reorientation only. Water was a simple dipole, centered in a rigid sphere. Results were sensitive to the positioning of the image plane, which was a major motivation for developing the model geometry used here. Since the earlier study focused only on the backbone and ignored effects due to aromatic residues, the proper comparison is between Fig. 3 and Fig. 7 of Dorman et al. (1996). Previously the cationic free energy profiles were flat, with minima near the channel midpoint. Here there is a steady free energy increase as a positively charged ion moves more deeply into the channel. Energy orderings are reversed, with the larger cations now being (correctly) the more stable. The appearance of a Cl− binding site near the channel mouth, seen earlier, is eliminated. The changes all reflect the new, more realistic model. Here water is multipolar and NH groups are included. Anomalies due to abrupt transition from bulk water to the channel interior are eliminated by the bubble geometry. The new approach is internally consistent. All transmutations occur within the same uniform dielectric background; passage through a vacuum intermediate is not required. The cation binding site, absent in the earlier study, arises here as a direct consequence of interaction with the channel's aromatic residues, consistent with standard modeling approaches (Allen et al., 2003a,b; Roux and Karplus, 1993; Woolf and Roux, 1997).
This microscopic-mesoscopic approach, although highly approximate, finesses two serious problems with standard approaches to determining permeation free energy profiles. Long-range electrostatic corrections required by the imposition of periodic boundary conditions are not needed (Allen et al., 2004; Bostick and Berkowitz, 2003; Hunenberger and McCammon, 1999). Bulk solvent is realistically modeled as a dielectric continuum; the perfect conductor approximation, with ɛ ≡ ∞, is ever more reliable as ionic strength increases and the electrical geometry used permits exact solution of the reaction field. By exploiting the timescale separation between electronic and structural relaxation modes, it is possible, in an average sense, to account for electronic polarization without having to explicitly address the quantum problem. The dramatically lowered permeation free energy barrier highlights the effect that these two limitations of standard modeling approaches have on ionic energetics in channels, and suggest that care should be taken in relying on computed free energy profiles for ion translocation through narrow pores.
Acknowledgments
We thank the referees and G. V. Miloshevsky for valuable comments, and are indebted to Dr. Miloshevsky for his analysis of dipolar ordering.
Work was supported by the National Institutes of Health under grant #GM-28643.
References
- Accardi, A., and C. Miller. 2004. Secondary active transport mediated by a prokaryotic homologue of CIC CI− channels. Nature. 427:803–807. [DOI] [PubMed] [Google Scholar]
- Allen, T. W., O. S. Andersen, and B. Roux. 2003a. Structure of gramicidin A in a lipid bilayer environment determined using molecular dynamics simulations and solid-state NMR. J. Am. Chem. Soc. 125:9868–9877. [DOI] [PubMed] [Google Scholar]
- Allen, T. W., O. S. Andersen, and B. Roux. 2004. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA. 101:117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, T. W., T. Bastug, S. Kuyucak, and S. H. Chung. 2003b. Gramicidin A channel as a test ground for molecular dynamics force fields. Biophys. J. 84:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, O. S. 1983. Ion movement through gramicidin A channels. Single-channel measurements at very high potentials. Biophys. J. 41:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antosiewicz, J., J. A. McCammon, and M. K. Gilson. 1994. Prediction of pH-dependent properties of proteins. J. Mol. Biol. 238:415–436. [DOI] [PubMed] [Google Scholar]
- Bernèche, S., and B. Roux. 2001. Energetics of ion conduction through the K+ channel. Nature. 414:73–77. [DOI] [PubMed] [Google Scholar]
- Beveridge, D. L., and G. W. Schnuelle. 1975. Free energy of a charge distribution in concentric dielectric continua. J. Phys. Chem. 79:2562–2566. [Google Scholar]
- Bostick, D., and M. L. Berkowitz. 2003. The implementation of slab geometry for membrane-channel molecular dynamics simulations. Biophys. J. 85:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM—a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Chen, T. Y., and C. Miller. 1996. Nonequilibrium gating and voltage dependence of the ClC-0 Cl− channel. J. Gen. Physiol. 108:237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S. W., and E. Jakobsson. 1989. Stochastic theory of singly occupied ion channels. II. Effects of access resistance and potential gradients extending into the bath. Biophys. J. 55:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S. W., E. Jakobsson, S. Subramaniam, and J. A. McCammon. 1991. Time-correlation analysis of simulated water motion in flexible and rigid gramicidin channels. Biophys. J. 60:273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S. W., J. A. Novotny, and E. Jakobsson. 1993. The nature of ion and water barrier crossings in a simulated ion channel. Biophys. J. 64:98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu, S. W., S. Subramaniam, E. Jakobsson, and J. A. McCammon. 1989. Water and polypeptide conformations in the gramicidin channel. A molecular dynamics study. Biophys. J. 56:253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, S. H., T. W. Allen, M. Hoyles, and S. Kuyucak. 1999. Permeation of ions across the potassium channel: Brownian dynamics studies. Biophys. J. 77:2517–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, S. H., T. W. Allen, and S. Kuyucak. 2002a. Conducting-state properties of the KcsA potassium channel from molecular and Brownian dynamics simulations. Biophys. J. 82:628–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, S. H., T. W. Allen, and S. Kuyucak. 2002b. Modeling diverse range of potassium channels with Brownian dynamics. Biophys. J. 83:263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen, J., and K. Schulten. 2004. Mechanism of anionic conduction across ClC. Biophys. J. 86:836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, K., E. Jakobsson, and P. Wolynes. 1985. The theory of ion transport through membrane channels. Prog. Biophys. Mol. Biol. 46:51–96. [DOI] [PubMed] [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second-generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Dani, J. A. 1989. Open channel structure and ion binding sites of the nicotinic acetylcholine receptor channel. J. Neurosci. 9:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deserno, M., and C. Holm. 1998. How to mesh up Ewald sums. J. Chem. Phys. 109:7678–7701. [Google Scholar]
- Dorman, V., M. B. Partenskii, and P. C. Jordan. 1996. A semi-microscopic Monte Carlo study of permeation energetics in a gramicidin-like channel: the origin of cation selectivity. Biophys. J. 70:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman, V. L., S. Garofoli, and P. C. Jordan. 1999. Ionic interactions in multiply occupied channels. Novartis Found. Symp. 225:153–167. [DOI] [PubMed] [Google Scholar]
- Dorman, V. L., and P. C. Jordan. 2003. Ion-water interaction potentials in the semi-microscopic model. J. Chem. Phys. 118:1333–1340. [Google Scholar]
- Doyle, D. A., J. Morais-Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Duca, K. A., and P. C. Jordan. 1997. Ion-water and water-water interactions in a gramicidin-like channel: effects due to group polarizability and backbone flexibility. Biophys. Chem. 65:123–141. [DOI] [PubMed] [Google Scholar]
- Dutzler, R., E. B. Campbell, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity. Nature. 415:287–294. [DOI] [PubMed] [Google Scholar]
- Edwards, S., B. Corry, S. Kuyucak, and S. H. Chung. 2002. Continuum electrostatics fails to describe ion permeation in the gramicidin channel. Biophys. J. 83:1348–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg, R. S. 1999. From structure to function in open ionic channels. J. Membr. Biol. 171:1–24. [DOI] [PubMed] [Google Scholar]
- Essman, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- Fu, D., A. Libson, L. J. Miercke, C. Weitzman, P. Nollert, J. Krucinski, and R. M. Stroud. 2000. Structure of a glycerol-conducting channel and the basis for its selectivity. Science. 290:481–486. [DOI] [PubMed] [Google Scholar]
- Garofoli, S., and P. C. Jordan. 2003. Modeling permeation energetics in the KcsA potassium channel. Biophys. J. 84:2814–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofoli, S., G. Miloshevsky, V. L. Dorman, and P. C. Jordan. 2002. Permeation energetics in a model potassium channel. Novartis Found. Symp. 245:109–122. [PubMed] [Google Scholar]
- Hasted, J. B. 1973. Water, A Comprehensive Treatise, Vol. 1. F. Franks, editor. Plenum, New York. 405–458.
- Hermans, J., H. J. C. Berendsen, W. F. van Gunsteren, and J. P. M. Postma. 1984. A consistent empirical potential for water-protein interactions. Biopolymers. 23:1513–1518. [Google Scholar]
- Hunenberger, P. H., and J. A. McCammon. 1999. Effect of artificial periodicity in simulations of biomolecules under Ewald boundary conditions: a continuum electrostatics study. Biophys. Chem. 78:69–88. [DOI] [PubMed] [Google Scholar]
- Jackson, J. D. 1962. Classical Electrodynamics. John Wiley, New York.
- Jakobsson, E., and S. W. Chiu. 1987. Stochastic theory of ion movement in channels with single-ion occupancy. Application to sodium permeation of gramicidin channels. Biophys. J. 52:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Jordan, P. C. 1983. Electrostatic modeling of ion pores. II. Effects attributable to the membrane dipole potential. Biophys. J. 41:189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, P. C. 1990. Ion-water and ion-polypeptide correlations in a gramicidin-like channel. A molecular dynamics study. Biophys. J. 58:1133–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, P. C., M. B. Partenskii, and V. Dorman. 1997. Electrostatic influences on ion-water correlation in ion channels. Prog. Cell Res. 6:279–293. [Google Scholar]
- Ketchem, R., B. Roux, and T. Cross. 1997. High-resolution polypeptide structure in a lamellar phase lipid environment from solid-state NMR-derived orientational constraints. Structure. 5:1655–1669. [DOI] [PubMed] [Google Scholar]
- King, G., and A. Warshel. 1989. A surface constrained all-atom solvent model for effective simulations of polar solutions. J. Chem. Phys. 91:3647–3661. [Google Scholar]
- Lee, W. K., and P. C. Jordan. 1984. Molecular dynamics simulation of cation motion in water-filled gramicidin-like pores. Biophys. J. 46:805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt, D. G. 1986. Interpretation of biological ion channel flux data—reaction-rate versus continuum theory. Annu. Rev. Biophys. Biophys. Chem. 15:29–57. [DOI] [PubMed] [Google Scholar]
- Mackay, D. H. J., P. H. Berens, K. R. Wilson, and A. T. Hagler. 1984. Structure and dynamics of ion transport through gramicidin A. Biophys. J. 46:229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, A. D., D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Mamonov, A. B., R. D. Coalson, A. Nitzan, and M. G. Kurnikova. 2003. The role of the dielectric barrier in narrow biological channels: a novel composite approach to modeling single-channel currents. Biophys. J. 84:3646–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashl, R. J., S. Joseph, N. Aluro, and E. Jakobsson. 2003. Effect of hydrophobic channels on water structure and dynamics. Biophys. J. 84:2385A. [Google Scholar]
- McGill, P., and M. F. Schumaker. 1996. Boundary conditions for single-ion diffusion. Biophys. J. 71:1723–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metropolis, N., A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller, and E. Teller. 1953. Equation of state calculations by fast computing machines. J. Chem. Phys. 21:1087–1092. [Google Scholar]
- Miloshevsky, G. V., and P. C. Jordan. 2004. Anion pathway and potential energy profiles along curvilinear bacterial ClC Cl− pores: electrostatic effects of charged residues. Biophys. J. 86:825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa, A., Y. Fujiyoshi, and N. Unwin. 2003. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 424:949–955. [DOI] [PubMed] [Google Scholar]
- Morais-Cabral, J. H., Y. Zhou, and R. MacKinnon. 2001. Energetic optimization of ion conduction rate by the K+ selectivity filter. Nature. 414:37–42. [DOI] [PubMed] [Google Scholar]
- Murata, K., K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J. B. Heymann, A. Engel, and Y. Fujiyoshi. 2000. Structural determinants of water permeation through aquaporin-1. Nature. 407:599–605. [DOI] [PubMed] [Google Scholar]
- Nonner, W., D. P. Chen, and B. Eisenberg. 1999. Progress and prospects in permeation. J. Gen. Physiol. 113:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partenskii, M. B., M. Cai, and P. C. Jordan. 1991a. A dipolar chain model for the electrostatics of transmembrane ion channels. Chem. Phys. 153:125–131. [Google Scholar]
- Partenskii, M. B., M. Cai, and P. C. Jordan. 1991b. Influence of pore-former charge distribution on the electrostatic properties of dipolar water chains in transmembrane ion channels. Electrochim. Acta. 36:1753–1756. [Google Scholar]
- Partenskii, M. B., V. Dorman, and P. C. Jordan. 1994. Influence of a channel-forming peptide on energy barriers to ion permeation, viewed from a continuum dielectric perspective. Biophys. J. 67:1429–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partenskii, M. B., V. L. Dorman, and P. C. Jordan. 1998. Membrane stability under electrical stress: a nonlocal electroelastic treatment. J. Chem. Phys. 109:10361–10371. [Google Scholar]
- Partenskii, M. B., and P. C. Jordan. 1992a. Nonlinear dielectric behavior of water in transmembrane ion channels: ion energy barriers and the channel dielectric constant. J. Phys. Chem. 96:3906–3910. [Google Scholar]
- Partenskii, M. B., and P. C. Jordan. 1992b. Theoretical perspectives on ion-channel electrostatics: continuum and microscopic approaches. Q. Rev. Biophys. 25:477–510. [DOI] [PubMed] [Google Scholar]
- Pauling, L. 1960. The Nature of the Chemical Bond, 3rd Ed. Cornell University Press, Ithaca, NY.
- Pethig, R. 1979. Dielectric and Electronic Properties of Biological Materials. John Wiley & Sons, Chichester, England.
- Pickar, A. D., and R. Benz. 1978. Transport of oppositely charged lipophilic probe ions in lipid bilayer membranes having various structures. J. Membr. Biol. 44:353–376. [Google Scholar]
- Pusch, M., U. Ludewig, A. Rehfeldt, and T. J. Jentsch. 1995. Gating of the voltage-dependent chloride channel CIC−0 by the permeant anion. Nature. 373:527–531. [DOI] [PubMed] [Google Scholar]
- Richard, E. A., and C. Miller. 1990. Steady-state coupling of ion-channel conformations to a transmembrane ion gradient. Science. 247:1208–1210. [DOI] [PubMed] [Google Scholar]
- Roux, B. 2002. Computational studies of the gramicidin channel. Acc. Chem. Res. 35:366–375. [DOI] [PubMed] [Google Scholar]
- Roux, B., and M. Karplus. 1993. Ion transport in the gramicidin channel: free energy of the solvated right-handed dimer in a model membrane. J. Am. Chem. Soc. 115:3250–3262. [Google Scholar]
- Sancho, M., M. B. Partenskii, V. Dorman, and P. C. Jordan. 1995. Extended dipolar chain model for ion channels: electrostriction effects and the translocational energy barrier. Biophys. J. 68:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer, D. B., L. P. Williams, W. L. Whaley, R. E. Koeppe II, and O. S. Andersen. 1990. Gramicidins A, B, and C form structurally equivalent ion channels. Biophys. J. 58:1207–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutz, C. N., and A. Warshel. 2001. What are the dielectric “constants” of proteins and how to validate electrostatic models. Proteins. 44:400–417. [DOI] [PubMed] [Google Scholar]
- Smith, G. R., and M. S. P. Sansom. 1999. Effective diffusion coefficients of K+ and Cl− ions in ion channel models. Biophys. Chem. 79:129–151. [DOI] [PubMed] [Google Scholar]
- Smythe, W. R. 1968. Static and Dynamic Electricity, 3rd Ed. McGraw-Hill, New York.
- Sui, H., B. G. Han, J. K. Lee, P. Walian, and B. K. Jap. 2001. Structural basis of water-specific transport through the AQP1 water channel. Nature. 414:872–878. [DOI] [PubMed] [Google Scholar]
- Thompson, N., G. Thompson, C. D. Cole, M. Cotten, T. A. Cross, and D. D. Busath. 2001. Noncontact dipole effects on channel permeation. IV. Kinetic model of 5F-Trp13 gramicidin A currents. Biophys. J. 81:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsley, L. E., W. A. Tucker, S. Sham, and J. F. Hinton. 2001. Structures of gramicidins A, B, and C incorporated into sodium dodecyl sulfate micelles. Biochemistry. 40:11676–11686. [DOI] [PubMed] [Google Scholar]
- van Gunsteren, W. F., and H. J. C. Berendsen. 1987. Groningen Molecular Simulations (GROMOS) Library Manual. BIOMOS, Groningen, The Netherlands.
- Warshel, A. 1979. Calculations of chemical processes in solutions. J. Phys. Chem. 83:1640–1652. [Google Scholar]
- Weiner, S. J., P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta, and P. Weiner. 1984. A new force field for molecular mechanical simulation of nucleic-acids and proteins. J. Am. Chem. Soc. 106:765–784. [Google Scholar]
- Woolf, T. B., and B. Roux. 1997. The binding of sodium in the gramicidin A channel: comparison of molecular dynamics with solid-state NMR data. Biophys. J. 72:1930–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh, I. C., and M. L. Berkowitz. 1999. Ewald summation for systems with slab geometry. J. Chem. Phys. 111:3155–3162. [Google Scholar]
- Zhou, Y., J. H. Morais-Cabral, A. Kaufman, and R. MacKinnon. 2001. Chemistry of ion coordination and hydration revealed by a K+ channel-FAB complex at 2.0-Å resolution. Nature. 414:43–48. [DOI] [PubMed] [Google Scholar]