

Abstract
The linear sequence KLAL (KLALKLALKALKAALKLA-NH2) and its corresponding d,l-isomers k9a10-KLAL (KLALKLALkaLKAALKLA-NH2) and l11k12-KLAL (KLALKLALKAlkAALKLA-NH2) are model compounds for potentially amphipathic α-helical peptides which are able to bind to membranes and to increase the membrane permeability in a structure- and target-dependent manner (Dathe and Wieprecht, 1999) We first studied the secondary structure of KLAL and its analogs bound to the air/water using infrared reflection absorption spectroscopy. For the peptide films the shape and position of the amide I and amide II bands indicate that the KLAL adopts at large areas per molecule an α-helical secondary structure, whereas at higher surface pressures or smaller areas it converts into a β-sheet structure. This transition could be observed in the compression isotherm as well as during the adsorption at the air/water interface from the subphase as a function of time. The secondary structures are essentially orientated parallel to the air/water interface. The analogs with d-amino acids in two different positions of the sequence, k9a10-KLAL and l11k12-KLAL, form only β-sheet structures at all surface pressures. The observed results are interpreted using a comparison of hydrophobic moments calculated for α-helices and β-sheets. The differences between the hydrophobic moments calculated using the consensus scale are not large. Using the optimal matching hydrophobicity scale or the whole-residue hydrophobicity scale the β-sheet even has the larger hydrophobic moment.
INTRODUCTION
Membrane active peptides with bacterial selectivity are known to be a key component in the plant and animal defense system against predators or microorganisms. In spite of the structural diversity, a common feature of the peptides is that they all have an amphipathic structure, which allows them to bind to the membrane interface (Epand and Vogel, 1999). They can be classified into linear peptides tending to adopt an α-helical amphipathic conformation and linear peptides of unusual composition, i.e., rich in amino acids such as Pro, Arg, or Trp, and a second group enclosing all cystine-containing peptides (Andreu and Rivas, 1998). Membrane permeabilization by amphipathic α-helical peptides can proceed via either one of the two mechanisms: 1), transmembrane pore formation via a “barrel-stave” mechanism; and 2), membrane destruction or solubilization via a “carpet-like” mechanism (Oren and Shai, 1998). Bechinger (1999) additionally mentioned the “wormhole model,” “detergent-like effects,” and the “in-plane diffusion model.”
Structural parameters, such as peptide helicity (α), hydrophobicity (H), hydrophobic moment (〈μ〉), peptide charge, and the size of the hydrophobic/hydrophilic domain generally influence the peptide activity toward bacteria but also modify the undesired membrane-disturbing activity toward normal eukaryotic cells such as erythrocytes (Dathe and Wieprecht, 1999; Dathe et al. 2002).
Dathe et al. (1996) synthesized the cationic 18-mer peptide KLAL (KLALKLALKALKAALKLA-NH2) as a model compound to form amphipathic helices, as well as its complete double d-amino acid replacement set to analyze lipid peptide interaction (Krause et al., 1995; Dathe et al., 1996). This slight structural modification does not change structural parameters such as mean hydrophobicity per residue or the total charge, but results in a position-dependent gradual reduction of the helicity with the consequence of changes in amphipathicity (Krause et al., 1995). All analogs exhibit in buffer solution a random coil structure. Addition of trifluoroethanol induces a helicity of 72% in KLAL while the helical content of 42 and 34% for KLALKLALkaLKAALKLA-NH2 (k9a10-KLAL) and KLALKLALKAlkAALKLA-NH2 (l11k12-KLAL), respectively, point to a distinctly reduced helix propensity of the two double d-substituted analogs. Biological activity investigations exhibited a high antibacterial and hemolytic activity of the KLAL (Dathe et al., 1996).
Circular dichroism studies (CD) revealed that KLAL model peptides may also form a β-structure under appropriate conditions. Thus, KLAL dissolved in 10 mM phosphoric acid became β-structured at a high peptide concentration of 5 mM and k9a10-KLAL showed β-structure when bound to negatively charged phosphatidylglycerol vesicles at a lipid/peptide ratio of 10 whereas it was helical at higher ratios (unpublished data). An α to β structural transition has been proposed as a key step in Alzheimer's disease initiating peptide aggregation and insoluble fibril formation (amyloid formation) (Janek et al., 2001).
Several studies of the secondary structure of peptides at the air/water interface have been reported. Leucine-lysine dipeptide units at the air/water interface were studied by Castano et al. (2000) using polarization-modulated infrared reflection absorption spectroscopy (IRRAS). These linear (KL)mK peptides (m = 4–7) formed intermolecular antiparallel β-sheets in pure peptide and mixed peptide/DMPC films at the air/water interface. Castano et al. (1997, 1999a,b) showed that ideally amphipathic LiKj (i = 2j) peptides folded into α-helices, whereas shorter peptides formed intermolecular antiparallel β-sheets. The peptides were oriented flat at the air/water interface. Cornut et al. (1996) showed that the amphipathic peptide L15K7 adopts a completely α-helical structure at the air/water interface with an orientation parallel to the water surface. Dieudonné et al. (1998) reported IRRAS measurements of short hydrophobic peptides to study the peptide conformational tendencies in monolayers. Short peptides of the type (K2(LA)x; x = 6, 8, 10, 12) adopt an antiparallel β-sheet structure and parallel β-sheets, the longer ones were α-helical at high surface pressures (Dieudonné et al., 1998).
In a first step, we wanted to study the secondary structure of the KLAL model peptide at the air/water interface by Fourier transform (FT) IRRAS. As mentioned above, this model peptide was primarily designed to adopt an α-helical conformation when bound to negatively charged membranes. We will show that this peptide can adopt different secondary structures when it binds to the plain air/water interface. The secondary structure changes from random coil in solution to α-helical at low concentration at the air/water interface and then in a time- and concentration-dependent fashion to an intermolecular β-sheet. We will show that FT-IRRAS is an ideal method to determine these time- and concentration-dependent changes. A critical evaluation of the hydrophobic moment calculated on different scales shows that the formation of β-sheets for these peptides is predicted when other scales instead of the consensus scale (Eisenberg, 1984) are used.
MATERIALS AND METHODS
Materials
The KLAL peptides were synthesized using the Fmoc technique and purified with RP-HPLC (Dathe et al., 1996). For the IRRAS measurements the hydrochloride salts were used to avoid additional signals due to carboxyl vibrational bands from the acetate anion. Deionized water with a resistivity of 18.2 MΩ/cm (SG Wasseraufbereitung und Regenerierstation GmbH, Barsbüttel, Germany) was used as subphase. Trishydroxymethyl ammonium chloride and sodium chloride were purchased from Sigma-Aldrich (Deisenhofen, Germany).
Methods
Monolayer preparation
All experiments were performed with a Wilhelmy film balance (Riegler and Kirstein, Berlin, Germany), using a filter paper as Wilhelmy plate. The teflon trough consisted of a sample (300 × 60 × 3 mm3) and a reference compartment (60 × 60 × 3 mm3) linked by three small tubes to ensure equal height of the air/water interface in both troughs. The teflon barriers of the sample trough were computer controlled. Before each experiment both troughs were cleaned with a Tickopur R33 solution (Carl Roth GmbH, Karlsruhe, Germany) and rinsed thoroughly with pure water. They were filled with pure water or a buffer solution (10 mM Tris, 154 mM NaCl, pH 7.4); the temperature of the subphase was maintained at 20 ± 0.5°C. Two types of experiments were performed. First, the appropriate volume of the respective peptide stock solutions (∼1 mg/ml) were injected into the subphase and the time-dependent surface pressure increase due to the adsorption at the air/water interface was recorded. Secondly, the monolayer films of the peptides were formed by directly spreading the respective aqueous solution (∼1 mg/ml) onto the subphase. After an equilibration period of at least 15 min, the surface pressure area isotherms (π/A isotherms) were recorded with a compression speed of 0.01 or 0.02 nm2 per amino acid residue per minute. No systematic expansion measurements were performed.
IRRAS
Infrared (IR) spectra were recorded with an Equinox 55 FT-IR spectrometer (Bruker, Karlsruhe, Germany) connected to an XA 511 reflection attachment (Bruker) with the trough system and an external narrow band MCT detector. The IR beam can be directed along a system of mirrors onto the water surface at different angles of incidence. A computer-controlled rotating KRS-5 polarizer (>98% degree of polarization) is used to generate parallel and perpendicularly polarized light. The trough system is positioned on a moveable platform to be able to shuttle between the sample and the reference trough. This shuttle technique diminishes the spectral interference due to the water vapor absorption in the light beam (Flach et al., 1994; Mendelsohn et al., 1995).
In the adsorption experiments, the acquisition of the IRRA spectra started directly after the injection of the peptide into the subphase. In the experiments with spread peptide films, these were compressed to a desired area per molecule, the barriers were then stopped and the IRRA spectra were recorded. In both types of experiment the angle of incidence of the infrared beam with respect to the normal of the water surface was 40°. Perpendicular or parallel polarized radiation was used. Spectra were recorded at a spectral resolution of 8 cm−1 using Blackman-Harris four-term apodization and a zero filling factor of 2. For each spectrum 1000 or 2000 scans were co-added over a total acquisition time of ∼4.5 or 6 min, respectively. The single-beam reflectance spectrum of the reference trough surface was ratioed as background to the single-beam reflectance spectrum of the monolayer on the sample trough to calculate the reflection absorption spectrum as −log(R/R0). Spectral calculations were performed using a Visual Basic program with an implementation of the formalism published by Mendelsohn et al. (1995) and Flach et al. (1997).
RESULTS
KLAL adsorption at the air/water interface
The injection of KLAL into pure water did not result in any significant surface pressure increase after several hours. Similar results have been observed with melittin (Blaudez et al., 1996). Only when a subphase with sufficient ionic strength was used (10 mM Tris, 150 mM NaCl, pH 7.4), a pressure increase could be observed indicating the peptide surface activity. The surface pressure versus time curves are shown in Fig. 1 A. Depending on the peptide concentration in the subphase the surface pressure starts to increase immediately after the injection (500 and 250 nM) or after 25 or 33 min (100 and 25 nM, respectively). Typically the surface pressure shows a maximum after a fast steep rise (20.2, 17.5, 17.0, and 4.1 mN/m, respectively) and a slow pressure relaxation with a decrease of the surface pressure of ∼2–3.5 mN/m. Some of the pressure versus time curves show pressure oscillations of ∼0.1–1.0 mN/m due to the shuttling between reference and sample trough. These are probably instrumental artifacts and not related to film pressure changes. Fig. 1 B displays the IRRA spectra of the film with a peptide concentration of 25 nM in the subphase at the marked positions a–d of the respective pressure versus time curve in Fig. 1 A. The first spectrum at position a at almost zero surface pressure and just before the pressure increase shows a band in the amide I region at ∼1658 cm−1, indicating that the KLAL peptide has an α-helical secondary structure (Goormaghtigh et al., 1994). Additionally, a band at ∼1545 cm−1 can be seen in the amide II region, which is also an indication of an α-helical secondary structure. The intensity of the amide I band increases strongly during the increase in pressure until the maximum of the surface pressure is reached (position b). When the surface pressure decreases (position c) the amide I band at 1658 cm−1 decreases in intensity and another band at 1620 cm−1 appears, which is now the main band, the higher frequency band remaining as a shoulder. Another band appears at higher wavenumber at ∼1688 cm−1 as an additional shoulder. Together with the main band this indicates that an antiparallel β-sheet secondary structure is formed by the KLAL peptide. In the amide II region an additional band at ∼1529 cm−1 can be seen, which is also a sign for β-sheets. Finally, at position d, when the surface pressure has further decreased, the IRRA spectrum shows large bands characteristic of only the β-sheet secondary structure (at 1620 and 1688 cm−1); the bands related to α-helices (1658 cm−1) have almost completely vanished. The bands in the amide II region barely change. Especially the band at ∼1545 cm−1 can still be seen, which is normally an indication of α-helices, but the explanation of this is unknown. The IRRA spectra of the film with a peptide concentration of 100 nM in the subphase also show a transition of α-helices to β-sheets when the pressure increases steeply, whereas the spectra obtained with higher peptide concentrations (250 and 500 nM) reveal only bands of the β-sheet secondary structure at all times (data not shown). In the two experiments with the highest concentration the time to collect the respective spectra was too short to resolve the transition in the time span of the steep pressure increase.
FIGURE 1.

(A) Surface pressure versus time course of KLAL films with different peptide concentrations starting with the injection of the appropriate volume of the stock solution into the subphase: 500 nM (▾), 250 nM (▴), 100 nM (•), and 25 nM (▪). (B) IRRA spectra of the KLAL film with the 25 nM peptide concentration at the respective positions a–d of the surface pressure time curve in Fig. 1 A. All spectra have been recorded at an angle of incidence of 40° and with s-polarized light.
The corresponding d,l-amino acid analogs of KLAL, namely k9a10-KLAL and l11k12-KLAL, show a completely different adsorption behavior. Fig. 2 A displays the respective surface pressure versus time curve of l11k12-KLAL with a peptide concentration of 500 nM in the subphase. k9a10-KLAL shows a similar behavior. The surface pressure-time curves probably indicate a two-step process of the adsorption of the peptides at the air/water interface. It is clearly seen that after a short lag time the pressure increases very fast up to ∼15 mN/m due to the adsorption of the peptides at the interface. After ∼25 min the rate of the pressure increase is reduced until after ∼150 min the equilibrium pressure of ∼21.5 mN/m is reached. This value is ∼5 mN/m higher than that found for surface accumulated KLAL in the equilibrium. In addition, the process of the total peptide adsorption is slower compared to KLAL, where the adsorption at the same peptide concentration (500 nM) is almost immediately complete before the pressure decreases slightly due to the formation of intermolecular β-sheets (see Fig. 1 A).
FIGURE 2.

(A) Surface pressure versus time course of a l11k12-KLAL film with a peptide concentration of 500 nM starting with the injection of the appropriate volume of the stock solution into the subphase. (B) IRRA spectra of the l11k12-KLAL film at the respective positions a–d of the surface pressure time curve in Fig. 2 A. All spectra have been recorded at an angle of incidence of 40° and with p-polarized light.
The IRRA spectra recorded at times a–d are shown in Fig. 2 B. At all times the spectra display a main band in the amide I region at ∼1621 cm−1 indicating a β-sheet secondary structure of the l11k12-KLAL peptide. Interestingly, the intensity of this main band in spectrum a at a surface pressure of ∼0 mN/m is almost as high as in the spectra with higher surface pressures. Therefore the surface concentration and the overall orientation of the peptide do not seem to change very much in the further course of the adsorption. All bands reveal an asymmetric band shape to higher wavenumber. The last spectrum shows a small band at 1690 cm−1, also characteristic of β-sheet secondary structures. The asymmetric band shape is not due to residual α-helical or unordered structure elements. A band shape simulation using the approach described by Flach et al. (1997) for the amide I and II bands of β-sheets lying flat on the water surface is shown in Fig. 3 (solid line). From the simulation it becomes clear that even for a purely Lorentzian band centered at 1625 cm−1 the band shape is asymmetric at an intermediate angle of incidence (40°) with a shoulder at higher frequency. In addition, all amide bands have negative intensity, because the transitional dipole moments are oriented parallel to the water surface. The IRRA spectra of k9a10-KLAL are in general very similar to those of l11k12-KLAL (not shown), but additionally show a stronger β-sheet amide I band at 1692 cm−1.
FIGURE 3.

Simulation of IRRA spectra of a β-sheet secondary structure on H2O. The calculation has been performed for p-polarized light and an angle of incidence of 40° for the amide I bands at 1625 and 1690 cm−1 and the amide II band at 1535 cm−1. The respective β-sheet is lying flat at the air/water interface (solid line), oriented perpendicular to the interface with the peptide chains parallel to the interface (dashed line), or oriented perpendicular to the interface with the direction of the peptide chains perpendicular to the interface (dotted line).
KLAL surface pressure area isotherms
The KLAL peptides can also be applied directly from an aqueous solution to the water surface. The peptides seem to form metastable films which can be compressed to relatively large surface pressures. The π/A isotherms of all three KLAL isomers were in close agreement (KLAL: Fig. 4 A; k9a10-KLAL: Fig. 5 A; and l11k12-KLAL: Fig. 6 A). The only deviation is found for the normal KLAL which shows at large areas a significant surface pressure of ∼0.7 mN/m at 0.66 nm2/amino acid residue, whereas the other two analogs display no measurable surface pressure at this area. During the compression of the KLAL film, the surface pressure relaxes to a lower value of 0.1 mN/m at 0.50 nm2/residue. At an area of 0.25 nm2/residue the surface pressure starts to increase steeply. k9a10-KLAL and l11k12-KLAL show similar compression behavior. The areas where the sudden pressure increase occurs are 0.35 and 0.27 nm2/residue, respectively. All three films collapse at surface pressures between 30 and 35 mN/m and areas of ∼0.16 nm2/amino acid residue. The pressure obtained by compression is 5–10 mN/m higher than the surface pressure reached after injection of the peptides into the subphase at high concentration. Assuming an antiparallel β-sheet for all peptides, model building shows that the surface area of a peptide dimer is ∼6 nm2 when most of the lysine side chains are oriented toward the water phase and the C=O···H–N bonds are parallel to the water surface. The dimer has 36 amino acids. The calculated area per amino acid residue is therefore ∼0.17 nm2. This value is indeed the area at which all surface films of the peptides start to collapse. One can conclude therefore that the surface films obtained by spreading an aqueous solution are metastable; the desorption into the subphase is slow compared to the compression speed so that a highly condensed β-sheet film can be obtained.
FIGURE 4.

(A) Surface pressure versus area per amino acid residue of a KLAL film spread from an aqueous buffer solution. (B) IRRA spectra of the KLAL film during the compression at the respective areas per amino acid residues a–f of the surface pressure area curve in Fig. 3 A. The intensity of the spectra taken at positions e and f has been reduced by a factor of 2 for clarity. All spectra have been recorded at an angle of incidence of 40° and with p-polarized light.
FIGURE 5.

(A) Surface pressure versus area per amino acid residue of a k9a10-KLAL film spread from an aqueous buffer solution. (B) IRRA spectra of the k9a10-KLAL film during the compression at the respective areas per amino acid residues a–d of the surface pressure area curve in Fig. 4 A. All spectra have been recorded at an angle of incidence of 40° and with p-polarized light.
FIGURE 6.

(A) Surface pressure versus area per amino acid residue of a l11k12-KLAL film spread from an aqueous buffer solution. (B) IRRA spectra of the l11k12-KLAL film during the compression at the respective areas per amino acid residues a–d of the surface pressure area curve in Fig. 5 A. All spectra have been recorded at an angle of incidence of 40° and with p-polarized light.
The IRRA spectra of KLAL, k9a10-KLAL, and l11k12-KLAL obtained during the compression cycle are shown in Figs. 4 B, 5 B, and 6 B, respectively. For KLAL at large areas the surface pressure is nonzero, namely ∼0.7 mN/m. At this large area an α-helical secondary structure of the peptide is observed because the amide I band is centered at ∼1660 cm−1.
With decreasing surface pressure during the compression (positions b–d) the spectra show the development of the amide I band at ∼1620 cm−1. The band at 1660 cm−1 is reduced to a shoulder of the main amide I band. The bands in the amide II region are relatively small. With decreasing area per molecule the intensity of the IRRAS bands at 1695 and 1620 cm−1 increase, especially during the steep rise of the surface pressure. The main band in the IRRA spectra of the two d,l-isomers is centered at ∼1618–1620 cm−1 at all surface pressures. The l11k12-KLAL spectra (Fig. 6 B) show a stronger intensity for the amide I band at ∼1692 cm−1 and in the amide II region at ∼1529 cm−1 compared to the two other KLAL peptides.
For both experiment types (adsorption as well as compression isotherm) the amide A band of all the KLAL analogs appears only at higher surface pressures at a frequency of ∼3280 cm−1, indicating a β-sheet secondary structure. In the spectra for the peptide in an α-helical form as indicated by the frequency of the amide I and II modes, no amide A band can be observed.
DISCUSSION
The isotherms of the different KLAL isomers are comparable with the π/A-isotherms of the β-sheets of the peptide Fmoc-(LK)3L (DeGrado and Lear, 1985) and of the peptides K2(LA)8 and K2(LA)10 as studied by Dieudonné et al. (1998). A pure α-helix shows an amide I band centered at ∼1655 cm−1. The amide I band of a β-sheet is normally expected at ∼1630 cm−1, whereas a lower value of 1620 cm−1 is usually assigned to mostly aggregated β-sheet secondary structure. Together with the band at ∼1688 cm−1 an antiparallel β-sheet secondary structure can be proposed. From the position of the amide I band and the size of the peptides, one has to assume intermolecular hydrogen bonds in β-sheet structures.
The pure KLAL peptide underwent a secondary structure transition from an α-helix to a β-sheet even at large areas per molecule. When a peptide solution was injected into the subphase the surface pressure began to rise steeply and the peptide adopted first an α-helical structure which immediately began to transform into β-sheets, the surface pressure decreasing concomitantly. The same process could be observed during a compression of the KLAL peptide film deposited onto the surface. At a large area per molecule the peptides adopted an α-helical structure that changed into β-sheets upon compression. Because the compression started shortly after the initial spreading, it remains unclear whether after waiting for longer times, a conversion into β-sheets would also occur. It is likely that the α-helix to β-sheet transition is concentration- as well as time-dependent. Using CD spectroscopy a concentration-dependent formation of β-sheet structures from random coil was observed in bulk solution but at much higher concentration of 5 mM (unpublished results). Sal-Man et al. (2002) found a similar result by linking together five d,l-peptide monomers to a covalently bound bundle. The respective monomers formed almost only random coil secondary structures, whereas the preassembled peptides adopted mostly β-sheet structures.
To our knowledge it has not been reported before that a decrease in surface pressure was observed when a peptide film was compressed, nor that a decrease in pressure was seen after peptide adsorption to the air/water interface. The formation of a KLAL-α-helix after spreading from the aqueous stock solution is comparable to the adsorption of KLAL from the subphase. In both cases, the peptide is in the random coil conformation when in aqueous solution but converts first to the α-helix when it reaches the air/water interface. The amphipathic α-helical secondary structure is therefore energetically more favorable than the random coil when the peptide is located at the interface. The lysine side chains are oriented toward the water and the hydrophobic amino acids toward the air. However, in a concentration- and time-dependent manner this structure converts to intermolecular β-sheets, obviously because of an energy gain. The reason for this energy gain will be discussed below. Slight changes in primary structure by introducing two d-amino acid residues modify the behavior. The two d,l-isomers showed a β-sheet structure under all experimental conditions, which is not very surprising because the d-amino acids reduce or suppress the α-helical secondary structure. Oren et al. (1999) found by using attenuated total reflectance FTIR a change from the predominant α-helical structure of a wild-type peptide to a β-sheet structure in the respective d,l-diastereomer in phospholipid membranes.
The hydrophobic moments for an ideal α-helix for the three peptides were calculated starting from KLAL using an angle ω of 100° and substituting the d,l-isomers at the appropriate position with extended side chains and without altering the angle ω. While the calculated hydrophobic moments are not much different (see Table 1), the experiments showed that the two d,l-isomers did not form any α-helices at large areas per molecule but immediately β-sheets. Also, in the adsorption experiments, no α-helical intermediate structures were observed.
TABLE 1.
Hydrophobic moment 〈μ〉 and polar angle Φ of KLAL, l11k12-KLAL and k9a10-KLAL calculated for an α-helical and a β-sheet secondary structure with different hydrophobicity scales
| Peptide | 〈μ〉 (consensus) | 〈μ〉 (OMH) | 〈μ〉 (WRH) | Φ | |
|---|---|---|---|---|---|
| KLAL | α-helix | 0.334 | 0.120 | 0.377 | 80 |
| β-sheet | 0.328 | 0.543 | 0.600 | ||
| l11k12-KLAL | α-helix | 0.267 | 0.060 | 0.254 | 160 |
| β-sheet | 0.214 | 0.419 | 0.422 | ||
| k9a10-KLAL | α-helix | 0.296 | 0.105 | 0.375 | 75 |
| β-sheet | 0.381 | 0.545 | 0.645 | ||
The angle ω between the amino acid side chains for the α-helix and the β-sheet structure was fixed at 100° and 167°, respectively. The mean hydrophobicity per residue, H, according to the different hydrophobicity scales of the peptides is −0.016 (consensus), 0.147 (OMH), and −0.344 (WRH).
When compressed to smaller areas per molecule all peptide isomers showed a similar behavior, namely they adopted a β-sheet structure. The amount of antiparallel oriented β-sheets increased, indicated by the increasing intensity of the band at ∼1690 cm−1 (Dieudonné et al., 1998). Following this scheme, the proportion of antiparallel β-sheets increased from k9a10-KLAL to KLAL and l11k12-KLAL, but the reason for this order remains unclear. Castano et al. (2000) found for (KL)mK peptides (m = 4–7) only ideally amphipathic antiparallel β-sheet structures. All these peptides showed a strict clustering of the charged K residues on one face of the β-sheet while the opposite face was constituted by only L residues. Our experiments revealed a contradictory scheme, because the peptide with the lowest hydrophobic moment showed the highest proportion of antiparallel β-sheets (l11k12-KLAL).
The orientation of the peptides at the air/water interface can be derived from the analysis of the amide I and II bands (Cornut et al., 1996; Castano et al., 1999b, 2000). The transition dipole moment of the amide I band at ∼1620 cm−1 of the β-sheets is oriented perpendicular to the peptide chain, whereas for the amide I band at ∼1690 cm−1 and the amide II band the transition dipole moments are oriented along the peptide chains. The simulation of the IRRA spectra of the different orientations of a β-sheet secondary structure are shown in Fig. 3, where i), a β-sheet lying flat on the water interface gives a weak negative amide I band at ∼1690 cm−1, a strong negative amide I band at ∼1620 cm−1, and a negative amide II band (solid line); ii), when the β-sheet is oriented perpendicular to the interface with the peptide chains parallel to the interface, a negative amide I band at 1690 cm−1, a strong positive amide I band at 1620 cm−1, and a negative amide II band is observed (dashed line); and iii), when the β-sheet is oriented perpendicular to the interface and the direction of the peptide chains is also perpendicular to the interface, a positive amide I band at ∼1690 cm−1, a strong negative amide I band at ∼1620 cm−1, and a positive amide II band is seen (dotted line). The simulation and the measurements were performed for H2O as subphase; therefore the asymmetric band shape of the main amide I band and especially the baseline in case 2 is caused by the underlying H2O deformation band. The simulations of the IRRA spectra clearly demonstrate that the β-sheets as well as the α-helices (simulations not shown) of the different KLAL isomers were aligned almost parallel to the water surface.
The KLAL peptides were designed for optimal surface activity with large hydrophobic moments 〈μ〉 and optimal polar angles Φ to facilitate the adsorption to a membrane interface. The hydrophobic moment 〈μ〉 and the polar angle Φ of all the three investigated peptides are shown in Table 1. As mentioned above, the hydrophobic moments were calculated for the ideal α-helical structure with an angle ω between the side chains of 100°. For the β-sheet structures an angle ω of 167° was taken for the twisted extended conformation which was the optimal angle with the highest hydrophobicity for KLAL, a similar value found for other β-sheet structures (Eisenberg et al., 1984). The mean hydrophobicity per residue, H, according to the different hydrophobicity scales of the peptides is −0.016 (consensus scale; Eisenberg, 1984), 0.147 (optimal matching hydrophobicity (OMH) scale; Eisenberg, 1984), and −0.344 (whole residue hydrophobicity (WRH) scale: octanol minus polar interface; White and Wimley, 1999). According to the consensus scale the α-helix and the β-sheet structure have almost the same hydrophobic moment 〈μ〉. The l11k12-KLAL peptide has a higher hydrophobic moment for the α-helix compared to the β-sheet structure, whereas the k9a10-KLAL has a higher hydrophobic moment for the β-sheet structure. The OMH and the WRH scales both favor the β-sheet structure for the three different sequences as the one with the higher hydrophobic moment 〈μ〉.
A decrease of the polar angle Φ, the angle subtended by the polar face of a helix, increases the apparent effectiveness for membrane permeabilization (Dathe et al., 1997, 2002; Uematsu and Matsuzaki, 2000). The polar angle Φ for k9a10-KLAL and l11k12-KLAL was estimated from the molecular representations after substitution with the d,l-isomers without changing the angle ω. KLAL and k9a10-KLAL have almost the same values for the polar angle for the α-helix whereas l11k12-KLAL has a much larger polar angle. One could speculate that this (a large polar helix surface) would lead to a better stabilization of an α-helix at the air/water interface compared to the other two isomers. However, no α-helix was observed for pure l11k12-KLAL films.
Based on the results of the calculations a model of the twisted helical ribbon β-sheet structure of KLAL at the air/water interface with an angle ω of 167° between the amino acid side chains has been set up, shown in Fig. 7 for a single strand. In the single strand of KLAL all lysine side chains are indeed pointing to more or less one side. Any angle closer to 180° reduces the hydrophobic moment. For an angle of 180° between the amino acids the side chains would alternate between up and down position. Probably a compromise between the optimal angle ω and the hydrophobic moment is found by the peptide. The IRRA spectra also showed that the high frequency component at 1690 cm−1 has different intensities for the KLAL analogs. Possibly, parallel β-sheets can also be formed at the interface. In all cases, the molecular area at total compression of the peptide films indicated that the surface was completely covered with β-sheet structures.
FIGURE 7.
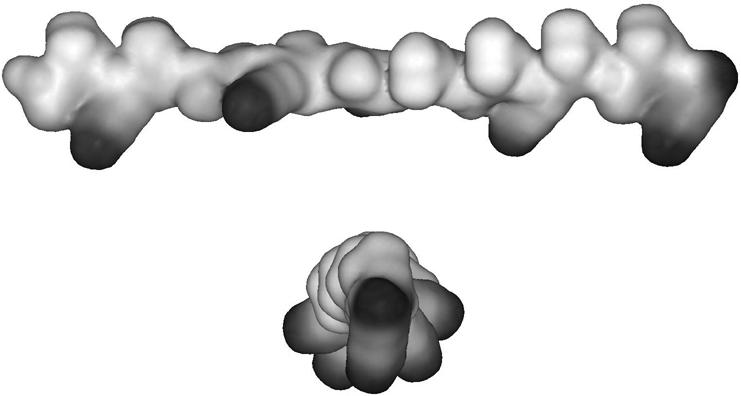
CPK model representation of KLAL in the extended twisted β-sheet conformation (single chain). Top: side view of the molecule to visualize the orientation of the lysine side chains (black). Bottom: view onto the end of the molecule with the lysine side chains pointing to the bottom.
SUMMARY AND CONCLUSIONS
The secondary structure of model peptides for antimicrobial peptides adsorbed to the air/water interface was characterized using the FT-IRRAS method. The lysine-bearing peptides had several positively charged lysine residues and were designed for maximum efficiency in binding to and permeabilizing bacterial membranes when they adopt an α-helical secondary structure in the bound form. In water their structure is random coil. At the air/water interface, however, all three analogs form intermolecular β-sheet structures, though the air/water interface is similar to a liquid interface between polar and nonpolar liquids. Only for KLAL at low surface concentration at the air/water interface could the formation of α-helices be detected, which convert to β-sheet structures in a time- and concentration-dependent manner. This peptide shows a peculiar sequence of secondary structure changes, namely from random coil to α-helix when it first binds to the air/water surface and then a conversion to β-sheets when the surface concentration is high enough. The reason for this behavior is probably the fact that the hydrophobic moments of α-helix and β-sheet are very similar. For the double-d substituted KLAL analogs this behavior is not found. These peptides form β-sheets at all observed surface concentrations. The data show that FT-IRRAS is a powerful method to detect secondary structure changes at the air/water interface. The results of further investigations of the secondary structure of these peptides when bound to lipid monolayers will be reported in the future.
Acknowledgments
The invaluable help by Dr. A. Gericke, Kent State University, in establishing the IRRAS technique in our laboratory is gratefully acknowledged.
This work was supported by grants from Max-Planck-Gesellschaft, the Deutsche Forschungsgemeinschaft (SFB 198), and the Fonds der Chemischen Industrie.
Andreas Erbe's present address is Max-Planck-Institut für Kolloid- und Grenzflächenforschung, 14424 Potsdam, Germany.
References
- Andreu, D., and L. Rivas. 1998. Animal antimicrobial peptides: an overview. Biopolymers. 47:415–433. [DOI] [PubMed] [Google Scholar]
- Bechinger, B. 1999. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta. 1462:157–183. [DOI] [PubMed] [Google Scholar]
- Blaudez, D., J.-M. Turlet, J. Dufourcq, D. Bard, T. Buffeteau, and B. Desbat. 1996. Investigations at the air/water interface using polarization modulation IR spectroscopy. J. Chem. Soc. Faraday Trans. 92:525–530. [Google Scholar]
- Castano, S., I. Cornut, K. Büttner, J. L. Dasseux, and J. Dufourcq. 1999a. The amphipathic helix concept: length effects on ideally amphipathic LiKj(i = 2j) peptides to acquire optimal hemolytic activity. Biochim. Biophys. Acta. 1416:161–175. [DOI] [PubMed] [Google Scholar]
- Castano, S., B. Desbat, I. Cornut, P. Meleard, and J. Dufourcq. 1997. α-Helix to β-sheet transition within the LeuiLysj (i = 2j) series of lytic amphipathic peptides by decreasing their size. Lett. Peptide Sci. 4:195–200. [Google Scholar]
- Castano, S., B. Desbat, and J. Dufourcq. 2000. Ideally amphipathic β-sheeted peptides at interfaces: structure, orientation, affinities for lipids and hemolytic activity of (KL)mK peptides. Biochim. Biophys. Acta. 1463:65–80. [DOI] [PubMed] [Google Scholar]
- Castano, S., B. Desbat, M. Laguerre, and J. Dufourcq. 1999b. Structure, orientation and affinity for interfaces and lipids of ideally amphipathic lytic LiKj(i = 2j) peptides. Biochim. Biophys. Acta. 1416:176–194. [DOI] [PubMed] [Google Scholar]
- Cornut, I., B. Desbat, J. M. Turlet, and J. Dufourcq. 1996. In situ study by polarization modulated Fourier transform infrared spectroscopy of the structure and orientation of lipids and amphipathic peptides at the air-water interface. Biophys. J. 70:305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dathe, M., J. Meyer, M. Beyermann, B. Maul, C. Hoischen, and M. Bienert. 2002. General aspects of peptide selectivity towards lipid bilayers and cell membranes studied by variation of the structural parameters of amphipathic helical model peptides. Biochim. Biophys. Acta. 1558:171–186. [DOI] [PubMed] [Google Scholar]
- Dathe, M., M. Schürmann, T. Wieprecht, A. Winkler, M. Beyermann, E. Krause, K. Matsuzaki, O. Murase, and M. Bienert. 1996. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry. 35:12612–12622. [DOI] [PubMed] [Google Scholar]
- Dathe, M., and T. Wieprecht. 1999. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta. 1462:71–87. [DOI] [PubMed] [Google Scholar]
- Dathe, M., T. Wieprecht, H. Nikolenko, L. Handel, W. L. Maloy, D. L. MacDonald, M. Beyermann, and M. Bienert. 1997. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 403:208–212. [DOI] [PubMed] [Google Scholar]
- DeGrado, W. F., and J. D. Lear. 1985. Induction of peptide conformation at apolar/polar interfaces. 1. A study with model peptides of defined hydrophobic periodicity. J. Am. Chem. Soc. 107:7684–7689. [Google Scholar]
- Dieudonné, D., A. Gericke, C. R. Flach, X. Jiang, R. S. Farid, and R. Mendelsohn. 1998. Propensity for helix formation in the hydrophobic peptides K2(LA)x (x = 6, 8, 10, 12) in monolayer, bulk, and lipid-containing phases. Infrared and circular dichroism studies. J. Am. Chem. Soc. 120:792–799. [Google Scholar]
- Eisenberg, D. 1984. Three-dimensional structure of membrane and surface proteins. Annu. Rev. Biochem. 53:595–623. [DOI] [PubMed] [Google Scholar]
- Eisenberg, D., R. M. Weiss, and T. C. Terwilliger. 1984. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA. 81:140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand, R. M., and H. J. Vogel. 1999. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta. 1462:11–28. [DOI] [PubMed] [Google Scholar]
- Flach, C. R., J. W. Brauner, J. W. Taylor, R. C. Baldwin, and R. Mendelsohn. 1994. External reflection FTIR of peptide monolayer films at the air/water interface: experimental design, spectra-structure correlations, and effects of hydrogen-deuterium exchange. Biophys. J. 67:402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flach, C. R., A. Gericke, and R. Mendelsohn. 1997. Quantitative determination of molecular chain tilt angles in monolayer films at the air/water interface: infrared reflection/absorption spectroscopy of behenic acid methyl ester. J. Phys. Chem. B. 101:58–65. [Google Scholar]
- Goormaghtigh, E., V. Cabiaux, and J.-M. Ruysschaert. 1994. Determination of soluble and membrane protein structure by Fourier transform infrared spectroscopy. III. Secondary structures. Physicochemical methods in the study of biomembranes. In Subcellular Biochemistry, Vol. 23. H. J. Hilderson and G. B. Ralston, editors. Plenum Press, New York. 405–450. [DOI] [PubMed]
- Janek, K., S. Rothemund, K. Gast, M. Beyermann, J. Zipper, H. Fabian, M. Bienert, and E. Krause. 2001. Study of the conformational transition of A β(1–42) using d-amino acid replacement analogues. Biochemistry. 40:5457–5463. [DOI] [PubMed] [Google Scholar]
- Krause, E., M. Beyermann, M. Dathe, S. Rothemund, and M. Bienert. 1995. Location of an amphipathic α-helix in peptides using reversed-phase HPLC retention behavior of d-amino acid analogs. Anal. Chem. 67:252–258. [DOI] [PubMed] [Google Scholar]
- Mendelsohn, R., J. W. Brauner, and A. Gericke. 1995. External infrared reflection absorption spectrometry of monolayer films at the air-water interface. Annu. Rev. Phys. Chem. 46:305–334. [DOI] [PubMed] [Google Scholar]
- Oren, Z., J. Hong, and Y. Shai. 1999. A comparative study on the structure and function of a cytolytic α-helical peptide and its antimicrobial β-sheet diastereomer. Eur. J. Biochem. 259:360–369. [DOI] [PubMed] [Google Scholar]
- Oren, Z., and Y. Shai. 1998. Mode of action of linear amphipathic α-helical antimicrobial peptides. Biopolymers. 47:451–463. [DOI] [PubMed] [Google Scholar]
- Sal-Man, N., Z. Oren, and Y. Shai. 2002. Preassembly of membrane-active peptides is an important factor in their selectivity toward target cells. Biochemistry. 41:11921–11930. [DOI] [PubMed] [Google Scholar]
- Uematsu, N., and K. Matsuzaki. 2000. Polar angle as a determinant of amphipathic α-helix-lipid interactions: a model peptide study. Biophys. J. 79:2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]