

Abstract
The association between monovalent salts and neutral lipid bilayers is known to influence global bilayer structural properties such as headgroup conformational fluctuations and the dipole potential. The local influence of the ions, however, has been unknown due to limited structural resolution of experimental methods. Molecular dynamics simulations are used here to elucidate local structural rearrangements upon association of a series of monovalent Na+ salts to a palmitoyl-oleoyl-phosphatidylcholine bilayer. We observe association of all ion types in the interfacial region. Larger anions, which are meant to rationalize data regarding a Hofmeister series of anions, bind more deeply within the bilayer than either Cl− or Na+. Although the simulations are able to reproduce experimentally measured quantities, the analysis is focused on local properties currently invisible to experiments, which may be critical to biological systems. As such, for all ion types, including Cl−, we show local ion-induced perturbations to headgroup tilt, the extent and direction of which is sensitive to ion charge and size. Additionally, we report salt-induced ordering of the water well beyond the interfacial region, which may be significant in terms of hydration repulsion between stacked bilayers.
INTRODUCTION
The predominant themes in ion-channel structure research focus on how transmembrane ionic gradients influence gating, and on the interactions between ions and the channel along the permeation pathway. However, cellular regulation of membrane lipid composition may play a critical role in channel structure and function. Thus, recent studies have begun to probe the influence of lipid type on channel behavior (Lundbaek and Andersen, 1994; Moczydlowski et al., 1985; Rostovtseva et al., 1998; Elmore and Dougherty, 2003). The lipid headgroup is one biologically variable chemical feature that has been shown to dramatically impact channel conductivity (Aguilella and Bezrukov, 2001). The underlying mechanisms by which headgroups interact with both channels and ions, however, remain unknown in full molecular detail.
Biophysical studies of pure bilayer systems suggest that salt can fundamentally alter the structure and dynamics of dipolar phosphatidylcholine (PC) headgroups, and that they do so as a function of ionic size and valency (Parsegian, 1975; Brown and Seelig, 1977; Loosley-Milman et al., 1982; Tatulian, 1987; Cunningham et al., 1988; Macdonald and Seelig, 1988; Roux and Bloom, 1990; Rydall and Macdonald, 1992; Clarke and Lupfert, 1999). Upon insertion of a protein, such as an ion channel, the relevant ion-lipid interactions may involve those lipids directly interacting with the protein (boundary lipids), as opposed to those outside of the sphere of influence of the protein (bulk lipids). For example, localized spikes in ion concentration near the mouth of a gating channel may increase the likelihood of ionic-interactions with boundary lipids, but not with bulk lipids. Current experimental techniques, however, struggle to distinguish between local and global properties of bilayers. All-atom molecular dynamics (MD) simulations, on the other hand, are capable of revealing such local interactions between single molecules. In this vein, this study focuses on the localized structural changes of PC headgroups in a pure bilayer induced by a series of monovalent Na+ salts.
Based on global changes in headgroup conformations upon ion association, the headgroup dipole has been predicted to act as a charge sensor whose tilt relative to the bilayer responds as a voltmeter would to changes in electrostatic potential (Seelig et al., 1987; Akutsu and Nagamori, 1991). Monovalent anions strongly affect conformational fluctuations of the headgroups, with the magnitude of this effect following a Hofmeister series: Cl− < Br− < 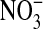 < I− < SCN− <
< I− < SCN− < 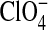 (Rydall and Macdonald, 1992). Changes in the dipole potential of dimyristoylphosphatidylcholine vesicles were correlated with the relative Gibbs solvation free energy of these anions, which suggested that the chaotropic (water structure breaking) anions (e.g., I− and
(Rydall and Macdonald, 1992). Changes in the dipole potential of dimyristoylphosphatidylcholine vesicles were correlated with the relative Gibbs solvation free energy of these anions, which suggested that the chaotropic (water structure breaking) anions (e.g., I− and 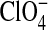 ) may penetrate more deeply into the bilayer interior than do nonchaotropic anions (e.g., Cl− and Br−) (Clarke and Lupfert, 1999). The behavior of the chaotropic anions at the relatively complex bilayer-water interface is reminiscent of their partitioning in the air-water interface (Randles, 1963).
) may penetrate more deeply into the bilayer interior than do nonchaotropic anions (e.g., Cl− and Br−) (Clarke and Lupfert, 1999). The behavior of the chaotropic anions at the relatively complex bilayer-water interface is reminiscent of their partitioning in the air-water interface (Randles, 1963).
The observations regarding the impact of large chaotropic anions on headgroup dynamics support early measurements on the electrophoretic mobility of PC lipid vesicles. The mobility of the vesicle is directly proportional to the ζ-potential, which is assumed to reflect ion-headgroup association. McLaughlin et al. (1975) showed that the effect of NaSCN and NaClO4 on the ζ-potential is more than 10-fold that of NaCl, suggesting greater surface adsorbtion in the case of the chaotropic anions. It has been generally accepted that for PC lipids, ζ ≈ 0 in NaCl (Hanai et al., 1965; Bangham, 1968; Eisenberg et al., 1979; McDaniel et al., 1984; Winiski et al., 1986). Given this result for the ζ-potential, there exist two possibilities regarding NaCl association with PC lipids. First, it could be concluded that PC bilayers bind neither Na+ nor Cl−. A second strong possibility is that the binding is approximately equal for the two ion types.
In addition to changes in headgroup structure and dynamics, one consequence of ion-headgroup association is that salt strongly influences the forces acting between stacks of bilayers in multilamellar vesicles. Divalent cations and chaotropic anions have the largest effect on these forces (Lis et al., 1981a,b; Tatulian, 1987; Macdonald and Seelig, 1988; Roux and Bloom, 1990; Rydall and Macdonald, 1992), though NaCl does affect the spacing between stacked bilayers as well (Cunningham and Lis, 1989; Korreman and Posselt, 2001). The relationship between salt-lipid interactions and interbilayer forces have been investigated through experimental studies employing osmotic pressure techniques (Homola and Robertson, 1976; LeNeveu et al., 1976; Parsegian et al., 1979; McIntosh and Simon, 1986) or the surface forces apparatus (Marra and Israelachvili, 1985; Claesson et al., 1989). The current paradigm categorizes attractive (van der Waals (vdW)) and repulsive (hydration, double layer) interbilayer forces (Cowley et al., 1978; Parsegian et al., 1979; Rand and Parsegian, 1989; Israelachvili and Wennerstrom, 1996), the balance of which shifts as the interbilayer spacing is varied. Theories related to these forces begin with descriptions of the water and lipid molecules (Israelachvili, 1992). Thus, there is a natural connection between water behavior at the bilayer interface (hydration repulsion) and salt effects. One critically debated topic is the source of the elusive hydration force (McIntosh, 2000). Two competing, though not exclusive, theories are that the hydration force is due to the molecular ordering of water, or due to steric interactions between individual protruding lipids and the overlap of undulating bilayers. Perturbations induced by salt are typically treated with a Debye screening formalism (Mahantay and Ninham, 1976) and related continuum Poisson-Boltzmann calculations (Peitzsch et al., 1995).
As already mentioned, MD simulations can offer a guide to the underlying structural and dynamic rearrangement of the lipid headgroups and solvating waters upon addition of salt not directly available from experiments or continuum descriptions. Several simulations have investigated the interactions between bilayers and water in the interfacial region, showing the influence of the lipids on the ordering of water molecules relative to their aqueous phase behavior, changes in hydration level associated with the hydration force, as well as the impact of water on the bilayer dipole potential (Alper et al., 1993; Marrink et al., 1996; Perera et al., 1996; Lopez-Cascales et al., 1996; Hyvonen et al., 1997; Shinoda et al., 1998; Pasenkiewicz-Gierula et al., 1999; Jedlovszky and Mezei, 2001; Saiz and Klein, 2002).
In the past several years, simulations that address long-range salt effects have become more reliable due to the application of the Ewald summation technique for calculating electrostatic interactions (Sagui and Darden, 1999). Using MD, we have demonstrated deep penetration of the chaotropic anions into the PC bilayer interface (Sachs and Woolf, 2003). We showed that the anion binding, which occurs in between the phosphate and carbonyl groups (with a peak at 16 Å from the bilayer center), is correlated with a localized but profound change in headgroup tilt. Two recent PC/NaCl simulations under constant surface tension have obtained a clearly defined Na+ binding site within the PC headgroup region (Pandit et al., 2003; Bockmann et al., 2003). Our constant area simulations presented here show a weak and approximately equal association for Na+ and Cl−. The differences in the two ensembles have been discussed extensively (Feller and Pastor, 1999).
Here, we extend our analysis of the salt effects and investigate local molecular perturbations to lipid headgroup tilt and global water ordering induced by a series of monovalent Na+ salts. We present analysis from six molecular dynamics simulations of a palmitoyl-oleoyl-phosphatidylcholine (POPC) bilayer with varied anion type and salt concentration. Three simulations capture NaCl concentration effects (0.25 M, 0.5 M, and 1 M), and two (analyzed in part previously (Sachs and Woolf, 2003)) are designed to better understand the Hofmeister series effect on headgroup behavior described above, with minimal representations of the chaotropic anions I− and 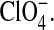 The final simulation is of the bilayer solvated by pure water, i.e., no salt, which sets the baseline for comparison. We find that all of the ion types associate transiently in the headgroup region. The association of Na+ is rather weak, and is found on the choline side of the phosphate groups. Additionally, the separation between the Na+ and Cl− association regions is <5 Å. In this regard, we reemphasize the strong and deep binding of the chaotropic anions relative to both Na+ and Cl−. The mean of the bilayer averaged headgroup tilt is relatively insensitive to salt. However, the shape of the headgroup tilt distribution is shown to change (increasing second moment) with NaCl concentration. We correlate this change with significant changes in headgroup tilt brought on by local association of both Na+ and Cl− ions. We further demonstrate that even in the case of weak NaCl association, addition of salt causes orientational ordering of waters well into the aqueous phase (>35 Å from the bilayer center), which is absent in a salt-free setting. Finally, the set of salt-induced perturbations are related to global interbilayer and local lipid-protein interactions.
The final simulation is of the bilayer solvated by pure water, i.e., no salt, which sets the baseline for comparison. We find that all of the ion types associate transiently in the headgroup region. The association of Na+ is rather weak, and is found on the choline side of the phosphate groups. Additionally, the separation between the Na+ and Cl− association regions is <5 Å. In this regard, we reemphasize the strong and deep binding of the chaotropic anions relative to both Na+ and Cl−. The mean of the bilayer averaged headgroup tilt is relatively insensitive to salt. However, the shape of the headgroup tilt distribution is shown to change (increasing second moment) with NaCl concentration. We correlate this change with significant changes in headgroup tilt brought on by local association of both Na+ and Cl− ions. We further demonstrate that even in the case of weak NaCl association, addition of salt causes orientational ordering of waters well into the aqueous phase (>35 Å from the bilayer center), which is absent in a salt-free setting. Finally, the set of salt-induced perturbations are related to global interbilayer and local lipid-protein interactions.
METHODS
Bilayer simulation details
As described previously (Sachs and Woolf, 2003), simulations were performed using CHARMM version 26, parameter set 27, with the TIP3P water model. Periodic boundary conditions were used with a constant number of atoms (N), temperature (T = 298 K), lateral area (A/lipid = 64 Å), and normal pressure (PN = 1 atm) to generate NAPNT ensembles. Electrostatics were calculated using particle mesh Ewald. Bilayers consisted of 72 lipid molecules. The number of solvating waters and salts varied according to the concentration, and are given in Table 1. We note that the actual concentration of the 0.25 M system was 0.24 M, but has been referred to as 0.25 M for simplicity. An initial salt-free POPC structure, provided by the Feller group, was run for 5 ns and provides the no-salt data. Ions were then added one at a time in randomly chosen locations, alternating between anion and cation. Each ion replaced the water molecule whose removal and replacement caused the smallest increase in total system energy. This was then followed by minimization and equilibration, and then 5 ns of dynamics for the 0.25, 0.5, and 1 M NaCl systems. The last configuration of the 1 M NaCl simulation was used as the starting point for both of the large anion simulations. In these cases, after enlargement of the anions, the systems were allowed to relax through another set of minimization and equilibration before the production dynamics.
TABLE 1.
Simulation details
| Salt | Total waters | Total ions | Salt concentration |
|---|---|---|---|
| NaCl | 3199 | 28 | 0.25 M |
| NaCl | 3167 | 60 | 0.50 M |
| NaCl | 3107 | 120 | 1 M |
| NaI | 3107 | 120 | 1 M |
| NaX | 3107 | 120 | 1 M |
| – | 3229 | 0 | 0 M |
Ion parameterization and characterization
Simulations of large, chaotropic anions are designed to isolate the role of anion size from shape and polarizability. Large anions were constructed by increasing the effective vdW radius of Cl− (Roux and Karplus, 1995). Hence, the large anions, like Cl−, are modeled as vdW spheres with a centrally located point charge. Increasing the position of the minimum in the Lennard-Jones potential, σ, while keeping the well depth, ε, unchanged simplifies the analysis and ensures that results are dependent only on ion size effects. Such an approach has been taken previously for anions such as I− (Koneshan et al., 1998). We have used thermochemical radii (Roobottom et al., 1999) to rationally estimate the radial expansion from Cl−. Although we could have parameterized anions based on solvation data alone, the effort here is to isolate the role of size and to maintain as straightforward an analysis as possible.
The change in σ for the three anions are given in Table 2. We have run a set of four 2 ns simulations of each ion type (one ion per simulation) in a pure water box consisting of 216 waters. These short simulations were run with periodic boundary conditions, constant temperature and pressure, and employed Ewald summation for electrostatic calculations. The position of the peaks and minima in the radial distribution functions for the four ions with water oxygens from these simulations are given in Table 2. The results for the Cl− and I−-like anions closely match those reported previously (Koneshan et al., 1998).
TABLE 2.
Ion parameters and properties
| Ion | σ (Å) | ε (kcal/mol) | nhs* | 1st peak | 1st min | 2nd peak |
|---|---|---|---|---|---|---|
| Na+ | 2.74 | −0.1 | 6.7 | 2.3 | 3.1 | 4.4 |
| Cl− | 4.29 | −0.15 | 9.9 | 3.2 | 3.8 | 4.6 |
| I− | 4.82 | −0.15 | 11.4 | 3.5 | 4.1 | 4.6 |
| X− | 5.89 | −0.15 | 30.2 | 4.1 | 5.8 | 7.1 |
Number of waters in the primary ionic hydration shell (hs) defined by the first minimum of the g(r) functions.
We will refer to the larger anion, modeled to mimic 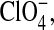 as X−. Results from the medium-sized anion (modeling I−) were generally intermediate to those of Cl− and X− and are thus, for the most part, omitted for clarity. Likewise, the results from the 0.50 M NaCl simulation were intermediate to those of the 0.25 M and 1 M simulations and are also occasionally omitted.
as X−. Results from the medium-sized anion (modeling I−) were generally intermediate to those of Cl− and X− and are thus, for the most part, omitted for clarity. Likewise, the results from the 0.50 M NaCl simulation were intermediate to those of the 0.25 M and 1 M simulations and are also occasionally omitted.
Free energy calculations
Our free energy calculations involved the interconversion of ions within a pure water box. Three sets of simulations were run in which Na+ was converted to Cl−, Cl− was converted to I−, and finally I− was converted to X−. In each simulation, the initial ion was solvated in a system of 216 waters. Simulations were run using periodic boundary conditions at constant pressure and temperature as above.
Interconversion between ion types proceeded, as for example in the case of Na+ to Cl−, by running dynamics under the following modified potential:
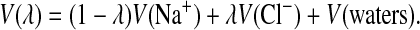 |
(1) |
For each interconversion, λ was incremented from 0 to 1 in steps of 0.1. For each λ-value, we ran 1 ns of equilibration followed by 1 ns of dynamics. The free energy perturbation method was then used to compute the free energy for each λ-step directly using the equation
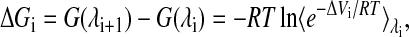 |
(2) |
where 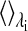 indicates an averaging over the simulation at λi, and
indicates an averaging over the simulation at λi, and
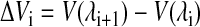 |
(3) |
is the difference between the potential energies of states λi+1 and λi. In our calculations, double-wide sampling was used such that the perturbation was to the halfway point between the λ-values. Then, the total free energy difference between two ions (e.g., Na+ and Cl−) is given by the summation over all ΔGi values from the set of λ-simulations.
Residency time calculations
To investigate the local dynamic behavior of ions, residency times for ions, τ, at a given z position were calculated from fits to the time correlation function
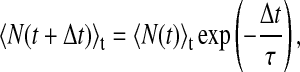 |
(4) |
where N(t) is the number of ions present at time t, and N(t + Δt) is the number of ions still present at time t + Δt. The brackets indicate averaging over all time frames within the simulation. As was done previously (Oyen and Hentschke, 2002), the z dimension was broken into 4 Å regions, and the average z for that region is given in the plots. Thus, the range of values for τ reflect the characteristic times for an ion to remain in one 4 Å bin, and therefore depend upon the choice of bin size. The acquisition and fitting of these functions is time consuming and suffers from relatively poor sampling. Thus, the plots appear somewhat noisy. Despite this, we are confident in the order of magnitude of the temporal association between the ions and headgroups.
RESULTS
Molecular distributions/density profiles
To set the stage for our main results, Fig. 1 A plots electron density profiles for molecular groups at the lipid/water interface from the 1 M simulations. Data are projected onto the dimension normal to the bilayer (z), and has been symmetrized by averaging over the two monolayer leaflets (z = 0 corresponds to the center of the bilayer). As known from previous salt-free bilayer simulations (Marrink and Berendsen, 1994), the phosphate distribution is located slightly closer to the bilayer center than the choline distribution. Water penetrates deeply into the bilayer interior, past the headgroup choline and phosphate groups. Likewise, all ions penetrate into the headgroup region, though less deeply than the water. Na+ and Cl− distributions from the 0.25 M, 0.5 M, and 1 M NaCl simulations are given in Fig. 1 B. At all three concentrations, there is a double layering of the ions: proceeding from the hydrocarbon interior toward the aqueous phase, there is first an excess of Na+ relative to Cl−, followed by a Cl− enriched region. The distributions do not show a clear peak, as would be expected from a strong and site-specific binding.
FIGURE 1.

Component distributions for various groups in the simulations as a function of distance from the center of the bilayer. (A) Electron densities for water, headgroup phosphate and choline groups, and salt from the 1 M NaCl simulation, plus the profile for X− from the NaX simulation. (B) Concentration profiles for Na+ and Cl− from the 0.25 M, 0.50 M, and 1 M simulations. (C) Concentration profiles for the three anion types in the interfacial region.
Fig. 1 C compares the distributions of the three different anion types in the interfacial region from three simulations. The larger anions penetrate deeper into the bilayer than do either the Cl− or Na+ ions (Sachs and Woolf, 2003). The Na+ ion distribution from all 1 M simulations (not shown) are nearly identical. Ionic layering is also present in the chaotropic anion simulations as well (not shown), with an additional layer of anions inside of the first Na+ layer.
Salt dynamics
To quantify the dynamic character of the ionic ordering in the bilayer interface, residency times (τ) in the z dimension have been calculated for the various ions (see Methods). Fig. 2 shows that for all ion types there is a slightly increased τ in the headgroup region as compared to the aqueous phase, though the association is relatively short-lived. The z locations for which τ is maximum are within 5 Å for Na+ and Cl−, and both are to the water side of the phosphate distributions. There is only one peak for Na+ and Cl− in Fig. 2, which is due to one broadly defined association site in the headgroup region for each ion (Sachs and Woolf, 2002). The aqueous phase value is higher by approximately twofold at 1 M as compared to 0.25 M, due to a greater number of ion-ion interactions in the more ion-dense system. For the larger anions, the residency time calculations capture an additional, stronger association site deep in the bilayer interior. This additional layer of anions stabilizes the Na+ association and thus increases τmax for the Na+ in the NaX simulation relative to that of the NaCl simulation (Fig. 2 A).
FIGURE 2.

Residency times, τ, for ions as a function of distance from the bilayer center, (a) for cations and (b) anions at 1 M (solid lines), 0.25 M (long-dashed lines) or from the NaX simulation (short-dashed lines).
Radial distribution functions for the headgroup-salt association are given in Fig. 3. Na+ is calculated relative to the headgroup phosphorus, and Cl− relative to the nitrogen. The position of the first peak for 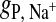 (Fig. 3 A) is at z = 5.5 Å, whereas for
(Fig. 3 A) is at z = 5.5 Å, whereas for 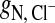 (Fig. 3 B) it is at z = 4.7 Å. Thus, Cl− approaches the choline nitrogen nearly 1 Å closer than the Na+ approaches the phosphate phosphorus, despite being the larger of the two ions. Thus, the fact that
(Fig. 3 B) it is at z = 4.7 Å. Thus, Cl− approaches the choline nitrogen nearly 1 Å closer than the Na+ approaches the phosphate phosphorus, despite being the larger of the two ions. Thus, the fact that 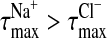 reflects a complex balance of sterics and electrostatics: it is both more difficult for a Na+ to get past the choline and deeper into the bilayer, and more difficult to leave once it has done so.
reflects a complex balance of sterics and electrostatics: it is both more difficult for a Na+ to get past the choline and deeper into the bilayer, and more difficult to leave once it has done so.
FIGURE 3.

Radial distribution functions for the lipid headgroup components with their respective counterions from the 1 M NaCl simulation (solid line) or the NaX simulation (long-dashed line). (A) 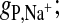 (B) gN,anion.
(B) gN,anion.
Headgroup tilt
To explore the specific local interaction between ions and headgroups, Fig. 4 A shows the effect of ionic proximity on headgroup tilt. The figure shows the average tilt angle for lipids for which there is an ion (Na+, Cl−, or X−) within a given distance of the headgroup (either the phosphorus or nitrogen, for cations or anions respectively). Headgroup tilt is defined as the angle made by the PN dipole with the bilayer normal. Headgroups closely associated with ions experience a large change in their tilt. The average headgroup tilt for those lipids that are near a Na+ ion is significantly increased relative to the bilayer average. The ion effectively pushes the headgroup down toward the bilayer plane, a result of electrostatic repulsion between the choline group and the cation. An equal and approximately opposite effect is seen with the Cl−, with the approaching anions pulling the headgroup out toward the aqueous phase. As the quantity measured in Fig. 4 A is itself an average, there are instances where the magnitude of the effect of Na+ and/or Cl− on headgroup tilt approaches that of the large anions. The impact of X− association with the choline group appears diminished because of the more deeply bound anions. Larger anions associated with the more peripheral of the two sites draw the headgroup dipole out toward the aqueous phase, as Cl− ions do. Those larger anions in the second, deeper binding site, on the other hand, are stabilized electrostatically by an opposite change in headgroup tilt. In these cases, the PN dipole swings toward the bilayer plane, as in the case of Na+, hence accommodating the negatively charged anion with the positively charged choline. Unlike for Cl−, then, the large anions have two opposing effects on the local changes in headgroup tilt.
FIGURE 4.

Effect of ionic approach to the headgroup. (A) Trajectory averaged angle (θ) between the bilayer normal and the PN dipole (0° ≡ dipole straight out into the aqueous phase; 90° ≡ dipole parallel to the bilayer plane) for lipids with a Na+ (dashed lines) within a certain distance of the headgroup P or anion (solid lines) within a certain distance of the headgroup N, from the 1 M NaCl simulation or the NaX simulation. (B) Two snapshots taken from the 1 M NaCl simulations illustrating the effect of Na+ approach on the headgroup tilt, taken 250 ps apart. (C) Dynamic changes in z position (top) and headgroup angle (bottom) for one Na+ from the 0.25 M NaCl simulation showing a correlated transition as the ion departs the headgroup region. Dashed line has been added to highlight the transition.
A snapshot of a Na+ ion approaching a headgroup is given in Fig. 4 B and illustrates the effect on tilt. To demonstrate the reversibility of these dynamics, Fig. 4 C shows a time series for one Na+ ion departing the headgroup region taken from the 0.25 M NaCl simulation. The top part of the figure gives the z position of the Na+, with a rapid transition shifting the position of the cation ∼2.5 Å toward the aqueous phase. The lower portion of the figure shows the tilt angle of the headgroup closest to this particular cation during the same time frame. As can be seen, there is a transition in the headgroup angle. Although future studies will more exhaustively investigate such correlated motions, we have visually inspected the trajectories and these types of events are easy to find. The results are presented here only to provide a qualitative picture of the salt and headgroup dynamics. Further, although these tilt calculations were done one lipid at a time, ions associated with the headgroup do affect the tilts of neighboring lipids in a consistent, though diminished, manner.
As has been reported previously, even in the no-salt case there are large fluctuations in the headgroup tilts, leading to a broad distribution ranging between 0° ≤ θ ≤ 160° (data not shown) (Hyvonen et al., 1997). The mean value (first moment) of the tilt distribution is ∼73° and is relatively insensitive (within an error of ±0.5°) to the introduction of either NaCl (Sachs and Woolf, 2002) or the larger anions. However, increasing salt concentration does change the higher order moments of the tilt distributions. Most notably, the standard deviation (second moment) of the distributions increases by ∼10% (from ≈23 (0 M) to ≈26 (1 M)) . In conjunction, these results suggest that the Na+ and Cl− ions may be affecting local headgroup tilt in approximately equal and opposite manners.
Water ordering and ionic hydration shell properties
In a salt-free setting, water molecules would undergo full isotropic rotational motion in the long time limit, if not for the headgroup dipoles that present both a steric and electrostatic perturbation to this intrinsic water ordering (Klose et al., 1985; Gawrisch et al., 1992; Volke et al., 1994a,b; Alper et al., 1993; Hyvonen et al., 1997; Shinoda et al., 1998; Jedlovszky and Mezei, 2001; Saiz and Klein, 2002). Fig. 5 shows the effect of salt on the average dipole orientation of waters as a function of distance from the bilayer center. In the salt-free case, there is no orientational preference in the aqueous phase, 〈cos θ〉 = 0 for z > 27 Å (≈7 Å from the average phosphate position). The presence of salt changes the water orientation in the interface and, significantly, the aqueous phase out to a distance of >35 Å from the bilayer center (≈15 Å from the average phosphate position). This corresponds to greater than three water layers extending from each monolayer surface. Fig. 5 B highlights additional ordering of the water dipoles beyond the headgroup region in all of the salt simulations. In these cases, there are two flips in orientation not seen in the salt-free simulation, the location and magnitude of which are dependent upon the ionic concentration, but not anion size. This ordering extends well into the aqueous phase (to ∼35 Å) for both concentrations. The magnitudes of these orientations are on the order of 1–3°. As can be seen, in all of the simulations the effect on water orientation also propagates deep within the bilayer. Increasing salt concentration might be expected to decrease the decay length for the water ordering due to increased screening. However, we see no such effect. Instead, we see an increase in the magnitude of the ordering in the first layers when NaCl concentration is increased from 0.25 M to 1 M. This effect is due to the increased probability of transient ion-headgroup association, and hence the number of oriented waters.
FIGURE 5.

Water dipole orientations as a function of distance from the bilayer center from the no-salt, 0.25 M and 1 M NaCl simulations, along with results from the NaX simulation. cos θ > 0 corresponds to water oxygens pointing inward, and cos θ < 0 to water oxygens pointing outward, as illustrated in the figure. (A) The full profile, (B) details of the oscillations originating in the headgroup region and propagating well into the aqueous phase.
Ion-headgroup association also impacts upon the structure of the ionic hydration shell. As has been noted previously (Pandit et al., 2003; Bockmann et al., 2003), as ions penetrate the headgroup region they lose waters from their primary hydration shell. For example, inside of 19 Å from the bilayer center, Na+ ions lose on the order of 1 water per Å. How does this loss of waters affect the ionic hydration shell structure? Fig. 6 A plots the distributions of oxygen-ion-oxygen angles (φ) within the first hydration shell of each ion in the aqueous phase. The extent of the hydration shell layers are defined by the location of the minima in the water-ion g(r) functions, which along with the number of waters in each shell is given in Table 2. There is a clear geometric arrangement of waters around the Na+ ions, with the peak at cos φ = 0 being approximately consistent with an octahedral arrangement (Spohr et al., 1996; Sachs et al., 2003a). The water structure around the Cl− ions is less well-defined, whereas the peak in the large anion distribution implies a significant perturbation to the water packing. Fig. 6 B plots the angular distribution of waters around Na+ ions within the first hydration shell of headgroup phosphorus atoms from the 1 M NaCl simulation. The data suggest that despite a shedding of some waters, the hydration shell geometry for those waters still bound is essentially unperturbed upon ion-headgroup association. Since the water arrangements around the different ion types are different, the relative solvation free energy of the ions is expected to be different. To verify this expectation, we have calculated the relative (to Na+) free energy of solvation in water (ΔG) using the free-energy perturbation method (Methods). Fig. 6 C plots ΔG for the three anions, and shows an increase in solvation free energy with anion size.
FIGURE 6.

Effect of ion size on the structure of water. (A) Hydration shell geometry, defined by the probability distribution of the angle φ formed by water oxygen-ion-water oxygen as described in the text, for ions in pure water. I− data is intermediate to Cl− and X− and is omitted for clarity. (B) Hydration shell geometry distributions for Na+ in the aqueous phase (dashed line, as in A) and for those Na+ ions within the first headgroup phosphorus hydration shell. (C) ΔG of solvation, relative to a Na+ ion, of the three anion types, as a function of the vdW radius, σ.
DISCUSSION
The effects of NaCl on globally averaged, pure lipid bilayer properties are subtle and have often been lost among the more profound effects of divalent and chaotropic salts. Our results show that both NaCl and Na+ salts with larger anions (NaX) have negligible impact on the global average of headgroup tilts. One aspect of our analysis has therefore focused on the interaction between approaching ions and PC headgroups, and how these events impact the shape of the headgroup tilt distributions. We report large local changes in headgroup tilt when Na+ and Cl− associate with the headgroup, despite a relatively unchanged mean value for the global tilt. Additionally, we show that deep binding of large chaotropic anions causes a very significant change in tilt in the same direction as that caused by the Na+, and opposite to that of the Cl−. This is due to a second binding site for the more deeply penetrant X− anions whose negative charge drives the positively charged choline to swing inward.
How can we explain this remarkable headgroup behavior? We understand that in addition to direct electrostatic interactions between the ions and the headgroups, the partitioning of ions is also subject to their relative stabilities in solution. Thus, Fig. 6 C suggests that the second, deeper binding site for the larger anions is inaccessible to Cl− because these smaller anions are less able to dehydrate than the larger ones. The more peripheral association of Cl− and Na+ also comes with a shedding of water, and is met with large changes in headgroup tilt. Although the connection between water structure and free energy is quite complex (Grossfield et al., 2003; Omta et al., 2003), these free-energy differences do help to explain why there is not a second, deeper association site for Na+ and Cl−.
It is interesting to compare the results from our simulations with those of Pandit et al. (2003) and Bockmann et al. (2003), who obtain different results as regards the extent and location of ion binding, as well as in the degree of headgroup tilt. Although these previous studies differed somewhat in the strength of the Na+ association, both agreed in principle to a binding site within the carbonyl region (peak density at ≈16 Å from the bilayer center), which is both stronger and deeper than our association region (peak τ at ≈20–25 Å, Fig. 2 A). Additionally, both reports show that Cl− binds ∼10 Å further from the bilayer center than the Na+, whereas our results show a separation in association regions of <5 Å. The two earlier reports differ from each other in the effect of NaCl of the bilayer averaged headgroup tilt, with the Bockmann results suggesting a large decrease (from 〈θ〉 = 69° to 〈θ〉 = 60.7°) and the Pandit results suggesting a negligible change (〈θ〉 ≈ 78°). Our results also show negligible change in mean tilt (〈θ〉 ≈ 73°). The differences are due to force fields, ensemble choice, and simulation time (united-atom GROMOS under constant surface tension versus all-atom CHARMM force field under constant surface area). It should be noted that both GROMACS simulations report significant reduction in the area per lipid upon ion binding, whereas we have imposed a constant area restraint.
The localized tilt effects described in this report are not easily seen in the bilayer average. However, by looking at the second moment of the tilt distributions, the NaCl concentration dependence arises. Increasing salt concentration leads to an increase in the likelihood of ionic approach (Fig. 1 B), and hence in the associated changes in tilt (Fig. 4 A). This leads to greater spread in the tilt distribution, and the observed increase in the second moment. With an approximate PN length of 4–5 Å (depending upon the extension of the group), such changes in tilt could significantly alter the distance between single lipid molecules in two neighboring bilayers in a multilamellar vesicle. Based on the water ordering that we see, and the changes of headgroup tilt, it is reasonable to expect that hydration forces are modified by salt. Our results also suggest that features from both protrusion models and water-ordering models should be considered in a theory meant to include salt effects.
As was mentioned earlier, the impact of salt on the strength of headgroup-water binding has been postulated to affect the hydration force. Upon ion association, there is a local increase in the water density in the interfacial region due to the incomplete shedding of ion hydration shell waters. In addition, our results suggest that the largest impact of monovalent ions on water structure occurs beyond the headgroup region. We showed in Fig. 5 that in salt solution waters have a dipolar orientation stretching >35 Å from the bilayer center. The double layer of ions induces this order. With interbilayer water spacings typically <35 Å, the extent of this ordering is very likely relevant to interbilayer forces.
A discussion of simulation scale is now warranted. Large-scale simulations are now capable of capturing global properties of lipid bilayers, including undulations (Lindahl and Edholm, 2000), and salt-induced changes in lipid chain order (Pandit et al., 2003; Bockmann et al., 2003) and lipid mobility (Bockmann et al., 2003). Smaller-scale simulations, like those presented here, are able to capture less demanding features of the bilayer, such as the electron density profile (Sachs et al., 2003b), and are well-suited for studying the local structuring of lipid headgroups and salt solutions. The perspective of this study addresses the relationship between, and the relative importance of, individual molecular interactions (e.g., between headgroups and salt ions) and global bilayer properties. Do local changes in headgroup tilt and hydration impact upon global properties measured by current techniques such as x-ray and NMR? In MD we see strong localized effects in tilt that are essentially lost in global averaging. However, these changes may in fact underlie experimentally measurable changes in hydration forces, for example. We have recently shown that measurements on the order of 1–2 Å are less than the uncertainty inherent in the Fourier analysis of x-ray scattering intensities (Sachs et al., 2003b). Is the impact of single molecule structural transitions lost in the experimental smoothing? Might it be the case that these local interactions are the more important ones for biologically relevant phenomena, such as ion channel physiology? For example, using MD we have shown (Petrache et al., 2002) that even at a high protein/lipid ratio, averaging lipid properties (such as bilayer thickness) over the entire bilayer misses key adaptations of boundary lipids.
Many simulations have addressed the interaction of water with lipid bilayers, and have offered valuable insight into the molecular underpinnings of critical interbilayer interactions. Here, we have extended these analyses to probe lipid interactions with salt. We have shown that monovalent ions perturb both local headgroup tilt and global water ordering, both of which may impact on ion channels and other transmembrane proteins. The relative strengths and locations of ion association are critical to comparing the simulation results to experimental observations such as electrophoretic mobility. Discrepancies between the simulation results presented here and elsewhere, particularly in regard to the strength and location of NaCl association, can be attributed to force-field parametrization and ensemble choice. However, our main results regarding the effect of local ionic association on headgroup tilt should not be particularly sensitive to these factors.
All of the salt effects presented here and elsewhere may be enhanced in the case of charged lipid headgroups and divalent cations, as well as polyatomic ions. Additionally, we note that although the ion, water, and lipid models used in current simulations are adequate in certain regards, inclusion of atomic polarizability may significantly influence the degree of molecular ordering in the system. For example, recent MD simulations of the nitrate anion at the air-water interface have shown the importance of polarizability in determining solvation (Salvador et al., 2003). Thus, improvements to MD force fields will ultimately be critical in explaining all of the phenomena associated with the hydration force and interbilayer interactions.
Acknowledgments
The authors thank Dr. Alan Grossfield, Dr. Hyunbum Jang, and Naveen Michaud-Agrawal for helpful discussions, and Dr. Scott E. Feller for providing the POPC starting structure. J.N.S. thanks the Whitaker Foundation for Biomedical Engineering for graduate fellowship support, and the National Computational Science Alliance and Pittsburgh Supercomputer Center for a generous computational grant and supercomputer resources.
Partial support of the research was from the American Cancer Society under grant RSG-01-048-01-GM.
Jonathan N. Sachs' present address is Dept. of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520.
References
- Aguilella, V. M., and S. M. Bezrukov. 2001. Alamethicin channel conductance modified by lipid charge. Eur. Biophys. J. 30:233–241. [DOI] [PubMed] [Google Scholar]
- Akutsu, H., and T. Nagamori. 1991. Conformational-analysis of the polar head group in phosphatidylcholine bilayers: a structural-change induced by cations. Biochemistry. 30:4510–4516. [DOI] [PubMed] [Google Scholar]
- Alper, H. E., D. Bassolino-Klimas, and T. R. Stouch. 1993. The limiting behavior of water hydrating a phospholipid monolayer: a computer-simulation study. J. Chem. Phys. 99:5547–5559. [Google Scholar]
- Bangham, A. D. 1968. Membrane models with phospholipids. Prog. Biophys. Mol. Biol. 18:29–95. [DOI] [PubMed] [Google Scholar]
- Bockmann, R. A., A. Hac, T. Heimburg, and H. Grubmuller. 2003. Effect of sodium chloride on a lipid bilayer. Biophys. J. 85:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M. F., and J. Seelig. 1977. Ion-induced changes in head group conformation of lecithin bilayers. Nature. 269:721–723.16072355 [Google Scholar]
- Claesson, P., A. M. Camona-Ribeiro, and K. Kurihara. 1989. Dihexadecyl phosphate monolayers: intralayer and interlayer interactions. J. Phys. Chem. 93:917–922. [Google Scholar]
- Clarke, R. J., and C. Lupfert. 1999. Influence of anions and cations on the dipole potential of phosphatidylcholine vesicles: a basis for the Hofmeister effect. Biophys. J. 76:2614–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley, A. C., N. L. Fuller, R. P. Rand, and V. A. Parsegian. 1978. Measurement of repulsive forces between charged phospholipid bilayers. Biochemistry. 17:3163–3168. [DOI] [PubMed] [Google Scholar]
- Cunningham, B. A., E. Gelerinter, and L. J. Lis. 1988. Mono-valent ion-phosphatidylcholine interactions: an electron-paramagnetic resonance study. Chem. Phys. Lipids. 46:205–211. [DOI] [PubMed] [Google Scholar]
- Cunningham, B. A., and L. J. Lis. 1989. Interactive forces between phosphatidylcholine bilayers in mono-valent salt-solutions. J. Colloid Interface Sci. 128:15–25. [Google Scholar]
- Eisenberg, M., T. Gresalfi, T. Riccio, and S. McLaughlin. 1979. Adsorption of monovalent cations to bilayer membranes containing negative phospholipids. Biochemistry. 18:5213–5223. [DOI] [PubMed] [Google Scholar]
- Elmore, D., and D. Dougherty. 2003. Investigating lipid composition effects on the mechanosensitive channel of large conductance (MscL) using molecular dynamics simulations. Biophys. J. 85:1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller, S. E., and R. W. Pastor. 1999. Constant surface tension simulations of lipid bilayers: the sensitivity of surface areas and compressibilities. J. Chem. Phys. 111:1281–1287. [Google Scholar]
- Gawrisch, K., D. Rustond, J. Zimmerberg, V. A. Parsegian, R. P. Rand, and N. Fuller. 1992. Membrane dipole potentials, hydration forces, and the ordering of water at membrane surfaces. Biophys. J. 61:1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossfield, A., P. Y. Ren, and J. W. Ponder. 2003. Ion solvation thermodynamics from simulation with a polarizable force field. J. Am. Chem. Soc. 125:15671–15682. [DOI] [PubMed] [Google Scholar]
- Hanai, T., D. A. Haydon, and J. Taylor. 1965. Polar group orientation and the electrical properties of lecithin bimolecular leaflets. J. Theor. Biol. 9:278–296. [DOI] [PubMed] [Google Scholar]
- Homola, A., and A. A. Robertson. 1976. Compression method for measuring forces between colloidal particles. J. Colloid Interface Sci. 54:286–297. [Google Scholar]
- Hyvonen, M. T., T. T. Rantala, and M. Ala-Korpela. 1997. Structure and dynamic properties of diunsaturated 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphatidylcholine lipid bilayer from molecular dynamics simulation. Biophys. J. 73:2907–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelachvili, J. N. 1992. Intermolecular and Surface Forces. Academic Press, London.
- Israelachvili, J. N., and H. Wennerstrom. 1996. Role of hydration and water structure in biological and colloidal interactions. Nature. 379:219–225. [DOI] [PubMed] [Google Scholar]
- Jedlovszky, P., and M. Mezei. 2001. Orientational order of the water molecules across a fully hydrated DMPC bilayer: a Monte Carlo simulation study. J. Phys. Chem. B. 105:3614–3626. [Google Scholar]
- Klose, G., K. Arnold, G. Peinel, H. Binder, and K. Gawrisch. 1985. The structure and dynamics of water near membrane surfaces. Colloid. Surface. 14:21–30. [Google Scholar]
- Koneshan, S., J. C. Rasaiah, R. M. Lynden-Bell, and S. H. Lee. 1998. Solvent structure, dynamics, and ion mobility in aqueous solution at 25°C. J. Phys. Chem. B. 102:4193–4204. [Google Scholar]
- Korreman, S. S., and D. Posselt. 2001. Modification of anomalous swelling in multilamellar vesicles induced by alkali halide salts. Eur. Biophys. J. 30:121–128. [DOI] [PubMed] [Google Scholar]
- LeNeveu, D. M., R. P. Rand, and V. A. Parsegian. 1976. Measurement of forces between lecithin bilayers. Nature. 259:601–603. [DOI] [PubMed] [Google Scholar]
- Lindahl, E., and O. Edholm. 2000. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 79:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis, L. J., V. A. Parsegian, and R. P. Rand. 1981a. Binding of divalent-cations to dipalmitoylphosphatidylcholine bilayers and its effect on bilayer interaction. Biochemistry. 20:1761–1770. [DOI] [PubMed] [Google Scholar]
- Lis, L. J., W. T. Lis, V. A. Parsegian, and R. P. Rand. 1981b. Adsorption of divalent-cations to a variety of phosphatidylcholine bilayers. Biochemistry. 20:1771–1777. [DOI] [PubMed] [Google Scholar]
- Loosley-Millman, M. E., R. P. Rand, and V. A. Parsegian. 1982. Effects of monovalent ion binding and screening on measured electrostatic forces between charged phospholipid bilayers. Biophys. J. 40:221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Cascales, J. J., J. Garcia de la Torre, S. J. Marrink, and H. J. C. Berendsen. 1996. Molecular dynamics simulation of a charged biological membrane. J. Chem. Phys. 104:2713–2720. [Google Scholar]
- Lundbaek, J. A., and O. Andersen. 1994. Lysophospholipids modulate channel function by altering the mechanical-properties of lipid bilayers. J. Gen. Phys. 104:645–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, P. M., and J. Seelig. 1988. Anion binding to neutral and positively charged lipid-membranes. Biochemistry. 27:6769–6775. [DOI] [PubMed] [Google Scholar]
- Mahantay, J., and B. Ninham. 1976. Dispersion Forces. Academic Press, New York. 1976.
- Marra, J., and J. N. Israelachvili. 1985. Direct measurements of forces between phosphatidylcholine and phosphatidylethanolamine bilayers in aqueous-electrolyte solutions. Biochemistry. 24:4608–4618. [DOI] [PubMed] [Google Scholar]
- Marrink, S. J., and H. J. C. Berendsen. 1994. Simulation of water transport through a lipid membrane. J. Phys. Chem. 98:4155–4168. [Google Scholar]
- Marrink, S. J., D. P. Tieleman, A. R. vanBuuren, and H. J. C. Berendsen. 1996. Membranes and water: an interesting relationship. Faraday Discuss. 103:191–201. [Google Scholar]
- McDaniel, R. V., A. McLaughlin, A. P. Winiski, M. Eisenberg, and S. McLaughlin. 1984. Bilayer-membranes containing the ganglioside-GM1: models for electrostatic potentials adjacent to biological-membranes. Biochemistry. 23:4618–4624. [DOI] [PubMed] [Google Scholar]
- McIntosh, T. J. 2000. Short-range interactions between lipid bilayers measured by X-ray diffraction. Curr. Opin. Struct. Biol. 10:481–485. [DOI] [PubMed] [Google Scholar]
- McIntosh, T. J., and S. A. Simon. 1986. Hydration force and bilayer deformation: a reevaluation. Biochemistry. 25:4058–4066. [DOI] [PubMed] [Google Scholar]
- McLaughlin, S., A. Bruder, S. Chen, and C. Moser. 1975. Chaotropic anions and the surface potential of bilayer membranes. Biochim. Biophys. Acta. 394:304–313. [DOI] [PubMed] [Google Scholar]
- Moczydlowski, E., O. Alvarez, C. Vergara, and R. Latorre. 1985. Effect of phospholipid surface-charge on the conductance and gating of a Ca2+-activated K+ channel in planar lipid bilayers. J. Membr. Biol. 83:273–282. [DOI] [PubMed] [Google Scholar]
- Omta, A. W., M. F. Kropman, S. Woutersen, and H. J. Bakker. 2003. Negligible effect of ions on the hydrogen-bond structure in liquid water. Science. 301:347–349. [DOI] [PubMed] [Google Scholar]
- Oyen, E., and R. Hentschke. 2002. Molecular dynamics simulation of aqueous sodium chloride solution at the NaCl(001) interface with a polarizable water model. Langmuir. 18:547–556. [Google Scholar]
- Pandit, S. A., D. Bostick, and M. L. Berkowitz. 2003. Molecular dynamics simulation of a dipalmitoylphosphatidylcholine bilayer with NaCl. Biophys. J. 84:3743–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsegian, V. A. 1975. Ion-membrane interactions as structural forces. Ann. N. Y. Acad. Sci. 264:161–171. [DOI] [PubMed] [Google Scholar]
- Parsegian, V. A., N. Fuller, and R. P. Rand. 1979. Measured work of deformation and repulsion of lecithin bilayers. Proc. Natl. Acad. Sci. USA. 76:2750–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasenkiewicz-Gierula, M., Y. Takaoka, H. Miyagawa, K. Kitamura, and A. Kusumi. 1999. Charge pairing of headgroups in phosphatidylcholine membranes: a molecular dynamics simulation study. Biophys. J. 76:1228–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peitzsch, R. M., M. Eisenberg, K. A. Sharp, and S. McLaughlin. 1995. Calculations of the electrostatic potential adjacent to model phospholipid-bilayers. Biophys. J. 68:729–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera, L., U. Essmann, and M. L. Berkowitz. 1996. Role of water in the hydration force acting between lipid bilayers. Langmuir. 12:2625–2629. [Google Scholar]
- Petrache, H. I., D. M. Zuckerman, J. N. Sachs, J. A. Killian, R. E. Koeppe II, and T. B. Woolf. 2002. Helix tilt and bilayer deformations induced by hydrophobic mismatch: molecular dynamics calculations. Langmuir. 18:1340–1351. [Google Scholar]
- Rand, R. P., and V. A. Parsegian. 1989. Hydration forces between phospholipid bilayers. Biochim. Biophys. Acta. 988:351–376. [Google Scholar]
- Randles, J. E. B. 1963. The interface between aqueous electrolyte solutions and the gas phase. Adv. Electroch. El. Eng. 3:1–30. [Google Scholar]
- Roobottom, H. K., D. B. Jenkins, J. Passmore, and L. Glasser. 1999. Thermochemical radii of complex ions. J. Chem. Educ. 76:1570–1573. [Google Scholar]
- Rostovtseva, T. K., V. M. Aguilella, I. Vodyanoy, S. M. Bezrukoz, and V. A. Parsegian. 1998. Membrane surface-charge titration probed by gramicidin A channel conductance. Biophys. J. 75:1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux, M., and M. Bloom. 1990. Ca2+, Mg2+, Li+, Na+, and K+ distributions in the headgroup region of binary membranes of phosphatidylcholine and phosphatidylserine as seen by deuterium NMR. Biochemistry. 29:7077–7089. [DOI] [PubMed] [Google Scholar]
- Roux, B., and M. Karplus. 1995. Potential energy function for cation-peptide interactions: an ab initio study. J. Comput. Chem. 16:690–704. [Google Scholar]
- Rydall, J. R., and P. M. Macdonald. 1992. Investigation of anion binding to neutral lipid membranes using 2H NMR. Biochemistry. 31:1092–1099. [DOI] [PubMed] [Google Scholar]
- Sachs, J. N., H. I. Petrache, D. M. Zuckerman, and T. B. Woolf. 2003a. Molecular dynamics simulations of ionic concentration gradients across model bilayers. J. Chem. Phys. 118:1957–1969. [Google Scholar]
- Sachs, J. N., H. I. Petrache, and T. B. Woolf. 2003b. Interpretation of small angle X-ray measurements guided by molecular dynamics simulations of lipid bilayers. Chem. Phys. Lipids. 126:211–223. [DOI] [PubMed] [Google Scholar]
- Sachs, J. N., and T. Woolf. 2002. Toward understanding of salt behavior near bilayer surfaces: molecular dynamics simulations. Biophys. J. 82:761. (Abstr.) [Google Scholar]
- Sachs, J. N., and T. B. Woolf. 2003. Understanding the Hofmeister effect in interactions between chaotropic anions and lipid bilayers: molecular dynamics simulations. J. Am. Chem. Soc. 125:8742–8743. [DOI] [PubMed] [Google Scholar]
- Sagui, C., and T. A. Darden. 1999. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 28:155–179. [DOI] [PubMed] [Google Scholar]
- Saiz, L., and M. L. Klein. 2002. Electrostatic interactions in a neutral model phospholipid bilayer by molecular dynamics simulations. J. Chem. Phys. 116:3052–3057. [Google Scholar]
- Salvador, P., J. E. Curtis, D. J. Tobias, and P. Jungwirth. 2003. Polarizability of the nitrate anion and its solvation at the air/water interface. Phys. Chem. Chem. Phys. 5:3752–3757. [Google Scholar]
- Seelig, J., P. M. Macdonald, and P. G. Scherer. 1987. Phospholipid head groups as sensors of electric charge in membranes. Biochemistry. 26:7535–7541. [DOI] [PubMed] [Google Scholar]
- Shinoda, W., M. Shimizu, and S. Okazaki. 1998. Molecular dynamics study on electrostatic properties of a lipid bilayer: polarization, electrostatic potential, and the effects on structure and dynamics of water near the interface. J. Phys. Chem. B. 102:6647–6654. [Google Scholar]
- Spohr, E., G. Toth, and K. Heinzinger. 1996. Structure and dynamics of water and hydrated ions near platinum and mercury surfaces as studied by MD simulations. Electrochim. Acta. 41:2131–2144. [Google Scholar]
- Tatulian, S. A. 1987. Binding of alkaline-earth metal-cations and some anions to phosphatidylcholine liposomes. Eur. J. Biochem. 170:413–420. [DOI] [PubMed] [Google Scholar]
- Volke, F., S. Eisenblater, J. Galle, and G. Klose. 1994a. Dynamic properties of water at phosphatidylcholine lipid-bilayer surfaces as seen by deuterium and pulsed field gradient proton NMR. Chem. Phys. Lipids. 70:121–131. [DOI] [PubMed] [Google Scholar]
- Volke, F., S. Eisenblater, and G. Klose. 1994b. Hydration force parameters of phosphatidylcholine lipid bilayers as determined from 2H-NMR studies of deuterated water. Biophys. J. 67:1882–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winiski, A. P., A. C. McLaughlin, R. V. McDaniel, M. Eisenberg, and S. McLaughlin. 1986. An experimental test of the discreteness-of-charge effect in positive and negative lipid bilayers. Biochemistry. 25:8206–8214. [DOI] [PubMed] [Google Scholar]