

Abstract
The back door has been proposed to be an exit pathway from the myosin active site for phosphate (Pi) generated by adenosine 5′-triphosphate hydrolysis. We used molecular dynamics simulations to investigate the interaction of Pi with the back door and the plausibility of Pi release via this route. Molecular dynamics simulations were performed on the Dictyostelium motor domain with bound Mg·adenosine 5′-diphosphate (ADP) and Pi, modeled upon the Mg·ADP·BeFx and Mg·ADP·Vi structures. Simulations revealed that the relaxation of ADP and free Pi from their initial positions reduced the diameter of the back door via motions of switch 1 and switch 2 located in the upper and lower 50-kDa subdomains, respectively. In neither simulation could Pi freely diffuse out the back door. Water molecules, however, could flux through the back door in the Mg·ADP·BeFx-based simulation but not in the Mg·ADP·Vi-based simulation. In neither structure was water observed fluxing through the main (front door) entrance. These observations suggest that the ability of Pi to leave via the back door is linked tightly to conformational changes between the upper and lower 50-kDa subdomains. The simulations offer structural explanations for 18O-exchange with Pi at the active site, and Pi release being the rate-limiting step in the myosin adenosine 5′-triphosphatase.
INTRODUCTION
Myosin is the motor protein responsible for converting the chemical energy of adenosine 5′-triphosphate (ATP) into the mechanical energy of movement in muscle contraction and many other cellular processes. The release of phosphate (Pi) from the myosin·ADP·Pi complex is a crucial step in the chemomechanical transduction process, in that Pi release accompanies the transition to force generating states in actively contracting muscle (reviewed in Cooke, 1997). To understand Pi release, and its influence on force production, the interaction of Pi with the protein and the exit route of the ion must be characterized.
The most common mechanism for product release in enzymes has the products leaving the active site via the original entrance route for the substrate. However, the x-ray structures of myosin suggest the existence of a secondary entrance/exit to the active site that, drawing upon terminology developed for tryptophan synthase (Hyde et al., 1988), has been termed the back door (Yount et al., 1995). In recent years, several additional enzymes have been demonstrated or postulated to utilize back door mechanisms for product release: acetylcholinesterase (Gilson et al., 1994), Rap1A (Nassar et al., 1995), the α-subunit of G-protein (Coleman et al., 1994), actin (Wriggers and Schulten, 1997), creatine kinase (Schlattner et al., 1998), polyamine oxidase (Binda et al., 1999), formiminotransferase-cyclodeaminase (Kohls et al., 2000), and glutamine amidotransferases (Krahn et al., 1997; Larsen et al., 1999; Tesmer et al., 1996; Thoden et al., 1997).
Molecular modeling (Yount et al., 1995) of ATP into the chicken skeletal myosin subfragment 1 (S1sk) crystal structure (Rayment et al., 1993b) first suggested a possible secondary entrance/exit to the active site. The docked protein-ligand structure suggested vectorial binding of ATP within the active site. In the model, the adenine ring of ATP projected out of the protein and into the solvent via the front door, whereas the triphosphate moiety, on the opposite end of the molecule, was tightly bound in a tube-like structure, termed the phosphate tube. The phosphate tube pointed into the interior of the protein, and was formed by three structurally conserved elements, the P-loop, switch 1, and switch 2, found in all members of the G-protein superfamily (Smith and Rayment, 1996a; Vale, 1996; Kull et al., 1998) and extended into the interior of myosin, well beyond the binding location of the γ-phosphate of ATP. The rear opening of the phosphate tube, the proposed back door, was located at the apex of a large cleft that splits the 50-kDa tryptic fragment into upper and lower subdomains (the 50-kDa cleft). Thus the back door appeared to be both a likely approach route for the water molecule that attacks the γ-phosphate during hydrolysis, and a likely exit route of the cleaved γ-phosphate after hydrolysis.
The existence of the phosphate tube, and its opening into the 50-kDa cleft, was subsequently confirmed in the crystal structure of truncated Dictyostelium myosin II motor domain (MDdc) complexed with Mg·ADP·BeFx at the active site (Fisher et al., 1995). In this structure there is a clear opening of the phosphate tube into the 50-kDa cleft (Fig. 1, c and d). It is interesting to note, however, that in the MDdc structure complexed with Mg·ADP·Vi (Smith and Rayment, 1996b), the opening of the phosphate tube is blocked off by a closure of the 50-kDa cleft (Fig. 1, a and b). The crystal structure of scallop adductor muscle subfragment 1 (S1sc) complexed with Mg·ADP (Fig. 1, e and f) also shows the back door opening into the 50-kDa cleft (Houdusse et al., 1999), but it is clearly in a different state than the state observed in the MDdc·Mg·ADP·BeFx structure. Although it is clear that the phosphate tube and the opening into the 50-kDa cleft exist in at least some conformational states of myosin, evidence that Pi could potentially leave the active site via this route comes from a combination of kinetic data and molecular modeling. Biochemical data suggest that Pi can exchange in and out of the active site, whereas ADP remains bound (Webb et al., 1978; Wells and Yount, 1979; Sleep and Hutton, 1980). Modeling of the active site area based upon the myosin x-ray structures, however, indicates that Pi is unlikely to egress through the nucleotide-binding pocket via the front door without ADP dissociating first. This is because in all myosin x-ray structures, the phosphate tube very tightly encloses α- and β-phosphates via numerous side-chain-nucleotide interactions. Thus, there would be severe steric hindrance of Pi egress through the front door in the presence of bound ADP. Posthydrolysis, there would additionally be electrostatic repulsion from the charges on the α- and β-phosphates that would inhibit the cleaved Pi from moving toward the front door. Furthermore, it could actually help repel Pi down the phosphate tube toward the back door. Additionally, 18O-exchange experiments have demonstrated that the actual hydrolysis step occurs on the myosin head before product release with a readily reversible transition between M·ATP and M·ADP·Pi states (Webb et al., 1978; Sleep et al., 1980). In the presence of bound ADP, the backdoor again yields an appealing entry site for water.
FIGURE 1.

Comparison of solvent-accessible surface representations of the nucleotide-binding and back door region of myosin in three different conformations. This figure shows relative states of the back door in the three crystal structure observed conformations of myosin and the associated subdomain movements within the motor domain. Also shown is the position of the nucleotide/nucleotide analog. The MDdc·Mg·ADP·Vi crystal structure (a and b) (Smith and Rayment, 1996b) shows that the solvent accessible surface is discontinuous between the phosphate tube and the 50-kDa cleft indicating a closed back door state. The MDdc·Mg·ADP·BeFx crystal structure (c and d) (Fisher et al., 1995) shows the solvent-accessible surface extending all the way from the nucleotide-binding pocket and phosphate tube into the 50-kDa cleft revealing a partially open back door state. The S1sc·Mg·ADP crystal structure (e and f) (Houdusse et al., 1999) shows a 50-kDa cleft and back door region that appears even more open than the MDdc·Mg·ADP·BeFx structure (note the increased size of the accessible surface in the 50-kDa cleft relative to the rigor state). In a, c, and e, the 25-kDa tryptic fragment is colored green, the 50-kDa fragment is red, and the 20-kDa fragment is blue. The black arrows in a and e indicate the repositioning of the lower 50-kDa subdomain relative to the MDdc·Mg·ADP·BeFx structure in c.
This study examines the relative states of the back door (i.e., open versus closed) in three different myosin motor domain conformations that have been observed in x-ray crystal structures. To accomplish this, molecular modeling was used to determine the structural features of the motor domain that are involved in the opening and closing of the back door. In addition, molecular dynamics simulations of the active site/back door region of MDdc were performed with Mg·ADP·Pi bound at the active site in both the MDdc·Mg·ADP·BeFx and MDdc·Mg·ADP·Vi conformations. During the simulations, the movement of ADP, Pi, and side chains lining the route from the γ-phosphate/Pi-binding site to back door were also monitored. The ability of water to exchange between the γ-phosphate/Pi-binding site and the exterior solvent was also determined during the time course of the simulations.
METHODS
Nucleotide and protein parameters
Parameters were assigned for all protein and solvent atoms, as well as the Mg+2 ion, according to the AMBER forcefield (Weiner et al., 1986) as implemented in the Accelrys InsightII 3.0.0 software package (San Diego, CA). Parameters for ADP and Pi were assigned using a custom parameter set based on AMBER parameters and Wriggers' ab initio ADP and Pi parameters (Wriggers and Schulten, 1997). Note that the force constant used for the phosphate O–P–OH bond was double the AMBER value to maintain the ion's tetrahedral structure. The parameters are available upon request from David Lawson (dlawson@wsu.edu).
Modeling and molecular dynamics simulations
All modeling and visualization was performed on a SGI Indigo R4400 workstation (Mountain View, CA) using Accelrys InsightII 3.0.0 software. All minimizations and dynamics simulations were performed using Accelrys Discover 2.9.7 software with the AMBER forcefield (Weiner et al., 1986).
The structures used as a starting point for the molecular dynamics simulations in this study were x-ray crystal structures of the truncated Dictyostelium myosin subfragment 1 complexed with Mg·ADP·BeFx (PDB accession code: 1MMD) (Fisher et al., 1995) and MDdc complexed with Mg·ADP·Vi (PDB accession code: 1VOM) (Smith and Rayment, 1996b). To avoid problems with the large amount of unresolved structure in the C-terminal converter domain portion of the motor domain, only residues 2–693 from the MDdc·Mg·ADP·Vi structure were used. The structures of scallop adductor muscle myosin S1 complexed with Mg·ADP (PDB accession code: 1B7T) (Houdusse et al., 1999) was also used for modeling of the back door region.
Pi was modeled in the singly protonated state that dominates at physiological pH. The ADP·BeFx complex in the MDdc·Mg·ADP·BeFx structure was converted to ADP·Pi by placing the Pi with its three sp2 oxygens overlaying the fluorides of BeFx. The hydroxyl group was pointed down the phosphate tube toward the back door. The BeFx moiety was then deleted. Similarly, for the MDdc·Mg·ADP·Vi structure, ADP·Vi was converted to ADP·Pi by overlaying Pi with its three sp2 oxygens as closely as possible with the equatorial oxygens of the Vi moiety. The hydroxyl group was oriented down the phosphate tube. The Vi moiety was then subsequently deleted.
All hydrogens and sulfur lone pairs were represented explicitly and were added using the Insight interface. Ionizable groups were protonated to represent pH = 7.0. Histidines were modified as follows to be consistent with local hydrogen bonding patterns. Histidines 408, 550, and 572 in the MDdc·Mg·ADP·BeFx structure were protonated on their δ-nitrogens. Histidines 12, 297, 408, 550, and 572 in the MDdc·Mg·ADP·Vi structure were protonated on their δ-nitrogens and His-484 was doubly protonated. All other histidines in both structures were protonated on their ɛ-nitrogens. In addition, the imidizole rings of His-12 and His-408 were rotated 180° in the MDdc·Mg·ADP·Vi structure.
Unresolved residues and regions of the crystal structure were added as follows. All residues with unresolved side chains were added manually using Insight's “Change Residue” function. Unresolved loops were added by modeling a peptide based on the sequence of each loop and constraining its ends to a distance equal to the distance between resolved residues on either side of the loop. The loop was then minimized for 250 steps of steepest descent and 750 steps of conjugate gradient minimization and subsequently merged into the MDdc structure. All unresolved residues and loops were solvated with a 10-Å shell of water. This inner shell of water was surrounded by another 4-Å semifixed shell of water. The oxygens of this outer shell of water were fixed to keep the solvent from spreading along the fixed protein surface or diffusing into vacuum. Crystal waters were likewise free to move (0–10 Å), semifixed (10–14 Å) or fixed (>14 Å), based on their distance from the subset. In the subsequent minimization and dynamics, the unresolved residues were free to move freely, whereas the remainder of the protein and ligands were fixed in place. Note that none of the unresolved loops were mobile in the data production simulation described below.
The data production simulations were carried out as follows. A protein subset was created in a 14-Å radius around ADP·Pi. Residues were added and deleted from this subset to provide structural continuity within the subset. The resulting subset (the dynamic protein subset) was allowed to move freely during the simulations, whereas the remainder of the protein was fixed. The dynamic subset was solvated with a 10-Å shell of water. This inner shell of water was surrounded by a 4-Å semifixed (oxygen fixed and hydrogens free) shell of water. Crystal waters were likewise free to move (0–10 Å), semifixed (10–14 Å) or fixed (>14 Å), based on their distance from the subset.
A total of 17961 atoms were contained in MDdc·Mg·ADP·BeFx conformation simulations of which 6854 atoms were free to move. This system contains 375 crystal waters, 969 dynamic waters, 564 semifixed waters, and 761 protein residues (plus Mg·ADP·Pi). A total of 17333 atoms were contained in MDdc·Mg·ADP·Vi conformation simulations of which 7584 atoms were free to move. This system contains 705 crystal waters, 810 dynamic waters, 575 semifixed waters, and 761 protein residues (plus Mg·ADP·Pi).
All minimizations and dynamics simulations were run with a 7.5-Å cutoff and a 2.0-Å switching distance. All dynamic water molecules (solvent waters and crystal waters ≤10 Å from the dynamic protein subset) were minimized for 250 steps of steepest descent and 750 steps of conjugate gradients. The dynamic protein subset and solvent was then minimized for 250 steps of steepest descent and 750 steps of conjugate gradients with the oxygens in the outer 4-Å shell of water fixed in place.
The data collection simulation was then initiated with the dynamic solvent and dynamic protein subsets free to move. The system was equilibrated by increasing the temperature from 0 K to 300 K in 30-K steps. Each step was run for 2.5 ps. Trajectory data were recorded every 500 fs during the equilibration. The production phase of the simulations then continued for another 200 ps or 500 ps, with trajectory data recorded every 100 fs. No significant difference was seen between 200-ps and 500-ps simulations suggesting that an equilibrium had been reached.
Analysis of water movement
For analysis of water movement in and around the back door area, trajectory structures were sampled every 5 ps to determine if a given water molecule had approached within 10.0 Å of the phosphorus atom in Pi. A second check was made to see if the water's position changed more than 5.0 Å during the simulation (checked every 5 ps). The trajectories of waters that met these standards were visually analyzed along with a solvent-accessible surface of the back door area.
RESULTS AND DISCUSSION
The phosphate tube structures in the x-ray structures of the myosin II motor domain can be classified into three different conformations, MDdc·Mg·ADP·Vi-like, MDdc·Mg·ADP·BeFx-like, and S1sc·Mg·ADP-like. Each conformation is defined by the relative positions of the various subdomains within the motor domain that can undergo rigid body rotation and translations relative to each other. Two of these subdomains (the upper and lower 50-kDa subdomains) act as a communications link between the actin-binding site and the nucleotide-binding site. The state of bound nucleotide can affect the position of the switch 2 loop, so called due to its homology to the G-protein nucleotide sensing switch 2 loop. This can in turn move the lower 50-kDa subdomain away from or toward the upper 50-kDa subdomain, opening or closing (respectively) the 50-kDa cleft between the two subdomains (Fig. 1, a, c, and e). The state of the 50-kDa cleft and the relationship of the upper and lower 50-kDa subdomains influences the state of the back door to various extents in each of the conformational classes. Fig. 1, b, d, and f, shows solvent-accessible surfaces of the nucleotide-binding region and the back door region in each of the three conformational classes.
Modeling the back door
The modeling of the back door region in the MDdc·Mg·ADP·BeFx conformation (Fig. 1, c and d) reveals an open 50-kDa cleft and an open back door. Immediately below the position of the BeFx γ-phosphate analog, the phosphate tube is slightly constricted by the side chains of residues Ser-181, Arg-238, Ser-456, and Glu-459, as well as by the backbone of Phe-239. There is room, however, for the phosphate to pass through this constriction as shown in Fig. 2. It should be noted that, with the exception of Ser-456, all of these residues are universally conserved within the myosin family of proteins (Cope et al., 1996). Mutation of Ser-181 to an Ala or Thr residue does not have an affect on myosin function (Li et al., 1998). This is consistent with our interpretation of the back door crystal structure because Ser-181 forms only a small part of the phosphate tube constriction. Because of this, an additional methylene (i.e., Ser→Thr) could easily be accommodated. Likewise, the mutation of Ser-456 to Ala has no affect on myosin functionality (Sasaki et al., 1998). Again, this is a consistent result since the less bulky alanine occupies this position in many myosin species. Furthermore, some species have a much larger tyrosine or phenylalanine at this position (Cope et al., 1996). It should also be noted that studies by Patterson and co-workers biochemically characterizing mutants of several residues in this region have shown that many of the residues are functionally linked to the rate of Pi release (Wu et al., 1999; Patterson, 1998).
FIGURE 2.

A comparison of the back door constriction and the size of Pi in the MDdc·Mg·ADP·BeFx structure. Three of the residues (Arg-238, Ser-456, and Glu-495) contributing to the constriction in the back door opening are shown. Pi (shown as a CPK representation with a yellow phosphorous atom) was modeled into the MDdc·Mg·ADP·BeFx crystal structure (Fisher et al., 1995) demonstrating that there is just enough room for Pi to pass through this constriction. The dot matrix represents the van der Waals surface of the displayed protein atoms.
In contrast to the MDdc·Mg·ADP·BeFx open conformation, the MDdc·Mg·ADP·Vi (Fig. 1, a and b) conformation reveals a closed back door. This results from the switch 2 loop moving closer to the γ-phosphate position, which causes Ser-456 to increase its blockage of the phosphate tube constriction. The movement of the switch 2 loop also allows the salt bridge between Arg-238 and Glu-459, which are 5.5 Å apart in the open conformation, to close down to 2.4 Å. Together, these three residues cause a complete blockage of the back door. Mutational studies of Arg-238 and Glu-459 suggest that these residues are necessary for the isomerization of myosin into a hydrolytically active conformation (Shimada et al., 1997; Sasaki et al., 1998; Li et al., 1998; Furch et al., 1999; Friedman et al., 1998). This, in turn, suggests that the Arg-238/Glu-459 salt bridge is necessary for local closure of the 50-kDa cleft and concurrent closure of the back door.
The third conformation, represented by the scallop S1 (S1sc) structure complexed with Mg·ADP (Houdusse et al., 1999) (Fig. 1, e and f) reveals a back door that is even more open than the MDdc·Mg·ADP·BeFx open conformation. The lower 50-kDa domain is rotated slightly relative to its position in the MDdc·Mg·ADP·BeFx conformation, and the switch 2 loop has a slightly altered position. The overall displacement of the lower 50-kDa subdomain in the back door area is only ∼1.5 Å and primarily occurs in a direction parallel to the 50-kDa cleft (rather than the perpendicular movement seen in the opening/closing movement discussed above). The change of Ser-456 to its smaller homolog, Ala-462 helps alleviate the phosphate tube constriction slightly, as does the movement of the lower 50-kDa domain. However, the major factor contributing to the increased openness of the back door is the flexibility of the Glu-465 (homologous to MDdc Glu-459) side chain. The rotation of the lower 50-kDa subdomain and rearrangement of switch 2 cause the side chain of Glu-465 to angle away from its salt bridge partner, Arg-242 (homologous to MDdc Arg-238) and thus destabilizes the interaction. In the S1sc·Mg·ADP structure, only the Cβ of the Glu-465 side chain is resolved. It is likely that the side-chain fluxes between a position similar to that seen in the MDdc·Mg·ADP·BeFx structure and a position closer to Arg-236 (homologous to MDdc Arg-232). Another factor contributing to the apparent openness of the S1sc·Mg·ADP structure is the repositioning of Arg-236. This repositioning, however, is not due to a subdomain rearrangement but rather to a difference in sequence. In the MDdc·Mg·ADP·BeFx structure, the side chain of Arg-232 is positioned in the gap between the switch 2 loop, the P-loop, and the lower 50-kDa subdomain and forms a ∼4.0-Å salt bridge with Glu-180. In the S1sc·Mg·ADP structure, Arg-236 moves ∼4.3 Å away from the back door and forms a bifurcated salt bridge between Glu-177 (homologous to MDdc Glu-180) and Glu-675. This is shown in Fig. 3. The reason that this conformation does not occur in the MDdc·Mg·ADP·BeFx structure is that Glu-675 is replaced by an asparagine (Asn-660) that cannot form a salt bridge with the arginine.
FIGURE 3.

Comparison of the back door region of the MDdc·Mg·ADP·BeFx and S1sc·Mg·ADP crystal structures. The MDdc·Mg·ADP·BeFx (Fisher et al., 1995) (purple) and S1sc·Mg·ADP crystal structures (Houdusse et al., 1999) (green) were overlaid based on the Cα atoms of the upper 50-kDa and 25-kDa subdomains. Mg·ADP·BeFx (orange) is included for reference. The difference in the degree of constriction in the back door route, as seen in the solvent accessible surfaces of Fig. 1, d and f, arises from the movement of Arg-236 (Arg-232 in MDdc) to form a new salt bridge with Glu-675 (Asn-660 in MDdc). Also, the side chain of Glu-465 beyond the Cβ is not resolved in the S1sc structure. This is likely due to the breaking of its weak salt bridge with Arg-242 that in turn is caused by the slight movement of switch 2 and the lower 50-kDa subdomain.
Active site rearrangements
During the molecular dynamics simulations of MDdc·Mg·ADP·Pi, significant rearrangements of both protein and nucleotide were observed in the active site and the back door regions (Figs. 4 and 5). The driving force of these rearrangements appears to be the Coulombic repulsion between the nucleotide diphosphate and the Pi. Because all simulations were started with ADP·Pi but based on a crystal structure with an ATP analog, some rearrangement is expected as the protein relaxes to optimize its nonbonded interactions with the new ligands.
FIGURE 4.

Protein and ligand movements in the active site and back door regions during a 200-ps simulation based on the MDdc·Mg·ADP·BeFx protein conformation. The movements of ADP·Pi, protein, side chains, and the switch 1 backbone during the simulation are represented by gold arrows on the MDdc·Mg·ADP·BeFx structure (purple). Note that the BeFx (left) and Pi (right) moieties are colored yellow for clarity. The average structure of the last 5 ps of the simulation is shown in green. As Pi moves away from ADP, it causes switch 1 to bulge into the 50-kDa cleft. Arg-238 and Lys-241 then swing into the γ-phosphate-binding site to coordinate Pi in its new position. Mg2+ is represented by a sphere in both structures.
FIGURE 5.

Protein and ligand movements in the active site and back door regions during a 200-ps simulation based on the MDdc·Mg·ADP·Vi protein conformation. The movements of ADP·Pi, protein, side chains, and the switch 1 backbone during the simulation are represented by gold arrows on the MDdc·Mg·ADP·Vi structure (purple). Note that the BeFx (left) and Pi (right) moieties are colored yellow for clarity. The average structure of the last 5 ps of the simulation is shown in green. As Pi moves away from ADP, it causes switch 1 to bulge into the 50-kDa cleft. Also, Glu-459 forms a new salt bridge with Arg-232. Mg2+ is represented by a sphere in both structures.
In the simulation performed using the MDdc·Mg·ADP·BeFx crystal structure (Fig. 4), the movement of Pi is associated with a protein rearrangement. There is an increased constriction of the phosphate tube, which leads to a closed back door state during the simulation. The average structure of the last 5 ps of the 200-ps simulation is compared with the MDdc·Mg·ADP·BeFx crystal structure in Fig. 4. During the simulation, Pi moves away from the diphosphate moiety of ADP toward the switch 1 loop. This slightly bulges out the backbone of switch 2 into the 50-kDa cleft. As this occurs, Pi maintains its electrostatic interaction with Arg-238 in switch 1 and forms a new 3.8-Å salt bridge with Lys-241. The charge-charge interactions with these two side chains appear to help stabilize Pi in its new position. Although Pi maintains its hydrogen bond with Ser-237, its hydrogen bond with Ser-236 is lost as Ser-236 moves to form a new hydrogen bond with the β-phosphate. Furthermore, with the increase in positive charge due to the movement of the Lys-241 side chain toward Pi, and the increased separation between Pi and Pβ, the Mg+2 ion moves to coordinate Pα and Pβ instead of Pβ and Pi. Unlike the many rearrangements of the phosphates and their ligands, there is relatively little movement of the ribose and the adenine ring. Both maintain all of their interactions with the protein. Finally, switch 2 moves up toward the active site ∼1.5 Å along with Glu-459 (not shown). This combination of switch 1 bulging out and switch 2 moving up causes a decrease in the cross section of the phosphate tube that effectively closes the back door.
In these first simulations of the myosin back door, we have modeled the posthydrolysis leaving group as 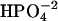 . This choice was motivated by the fact that it is the species that predominates at physiological pH. The precise phosphate species has not been experimentally determined, and an alternative could be
. This choice was motivated by the fact that it is the species that predominates at physiological pH. The precise phosphate species has not been experimentally determined, and an alternative could be 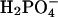 . In this case, the dissociating force between MgADP− and
. In this case, the dissociating force between MgADP− and 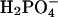 would still remain, although diminished in magnitude. Likewise, the interaction with the positively charged side chains of Lys-241 and Arg-238 would still be possible. However, the increased size of
would still remain, although diminished in magnitude. Likewise, the interaction with the positively charged side chains of Lys-241 and Arg-238 would still be possible. However, the increased size of 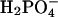 relative to
relative to 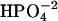 would make it even more difficult for the diprotonated species to egress through the closed back door. Thus, our fundamental conclusion (that Pi is sterically blocked from exiting via the back door in both discussed conformations) remains valid irrespective of the precise phosphate species involved.
would make it even more difficult for the diprotonated species to egress through the closed back door. Thus, our fundamental conclusion (that Pi is sterically blocked from exiting via the back door in both discussed conformations) remains valid irrespective of the precise phosphate species involved.
In the simulation performed using the MDdc·Mg·ADP·Vi crystal structure, the movement of Pi causes a reorganization of salt bridges near the Pi-binding site. The average structure of the last 5 ps of the 200-ps simulation is compared with the MDdc·Mg·ADP·Vi crystal structure in Fig. 5. As Pi moves away from the nucleotide diphosphate moiety, it causes the switch 2 loop to bulge ∼2.0 Å into the 50-kDa cleft. This movement and resulting concentration of negative charge (in conjunction with Glu-459) causes Arg-232 to move from its starting position between the lower 50-kDa domain and the P-loop to a new position in which it forms a salt bridge with both Pi (∼4.5 Å) and Glu-459 (∼3.7 Å). Unlike the MDdc·Mg·ADP·BeFx-based simulation, this simulation also resulted in a movement of the adenine ring. The ring angles away from its starting position by ∼35° but still remains in the purine-binding cleft and retains a majority of its protein contacts.
The Pi induced rearrangements of switch 1 observed in the simulations appear to be roughly similar to those observed in the crystal structures of mutated G-protein Giα1 subunits (Berghuis et al., 1996; Raw et al., 1997). Comparison of the crystal structures of mutant Giα1 complexed with ATPγS (an ATP analog) and ADP·Pi show that the switch 1 loop in Giα1 bulges out ∼1.8 Å upon ATP hydrolysis. This is comparable to the 1.5–2.0-Å movement of switch 1 seen in our MDdc simulations. In addition, the Pi-Pβ distance in the Giα1·ADP·Pi structure is ∼4.7 Å, which is comparable to the ∼4.6 Å seen in our simulations. It should be noted, however, that a part of the increase in the Pi-Pβ distance during the MDdc simulations is due to the movement of ADP, whereas only minimal ADP movement is observed in the Giα1 structures.
Water access to the active site
Hydrolysis of ATP is widely postulated to occur via an in-line attack by a water molecule on the γ-phosphate of ATP bound at the active site. Experiments with 18O-labeled Pi have shown multiple exchanges with water in the active site of myosin (Webb et al., 1978; Sleep and Hutton, 1980; Sleep et al., 1980), and experiments using 18O-labeled waters have shown multiple exchanges with Pi at the active site (Shaw and Yount, 2000a,b). These observations imply that the hydrolysis reaction, M·ATP↔M·ADP·Pi, is readily reversible and can involve multiple water molecules. The x-ray structures and molecular dynamics simulations indicate that there is insufficient free space at the γ-phosphate location to accommodate a large number of water molecules. This suggests that the recruitment of additional waters is necessary to explain the 18O-exchange experiments. Molecular dynamics simulations of MDdc·Mg·ADP·Pi were thus analyzed to determine the ability of water to access the Pi-binding site via the back door, with the protein in either the MDdc·Mg·ADP·BeFx or MDdc·Mg·ADP·Vi conformation. In the simulation of the MDdc·Mg·ADP·BeFx conformation, 28 waters were seen to pass within 10 Å of the Pi phosphorous atom and move at least 5 Å during the simulation. As noted previously, the presence of the free Pi in the MDdc·Mg·ADP·BeFx simulations resulted in some additional closing of the back door entrance. However, the opening was still sufficiently large to allow the passage of water molecules, and 17 of these waters actually were observed to move through the back door during a 200-ps simulation (Fig. 6). In a simulation of the MDdc·Mg·ADP·Vi conformation, only 22 waters were observed passing within 10 Å of the Pi phosphorous atom and moving at least 5 Å. However, none of these waters was seen to pass through the more constricted back door of the MDdc·Mg·ADP·Vi structure. All were confined exclusively in the exterior solvent. In addition, no waters in either simulation were observed accessing the Pi-binding site via the front door. This supports our previous suggestion that the large amount of steric hindrance created by the binding of Mg·ADP would preclude water attack on the γ-Pi position from this direction. We additionally note that 500-ps simulations were also performed (data not shown) with similar results.
FIGURE 6.

Mobility of waters through the back door during a 200-ps simulation. This figure demonstrates the ability of waters to exchange between the γ-phosphate/Pi-binding site and the exterior solvent during a simulation of MDdc·Mg·ADP·Pi in the MDdc·Mg·ADP·BeFx conformation. The simulation was performed at 300 K. The trajectories of 17 different water molecules sampled at 100-fs intervals throughout the simulation are represented by purple spheres. The backbone shown is that of the starting conformation with the 25-kDa tryptic fragment colored green, the 50-kDa fragment colored red, and the 20-kDa fragment colored blue. Mg·ADP·BeFx (orange) from the MDdc·Mg·ADP·BeFx structure is shown for reference.
Phosphate rotation
The 18O-exchange experiments likewise indicate that the γ-phosphate oxygens are equivalent during reversible hydrolysis (Sleep et al., 1980), implying an ability of the Pi moiety to rotate in the active site. During the molecular dynamics simulations, the orientation of Pi was specifically monitored to detect rotational events. Although only partial rotations were observed during the 300-K simulations (data not shown), a full rotation (i.e., in which three of the oxygens interchange their protein ligands) was observed in a simulation performed at 450 K using the MDdc·Mg·ADP·BeFx conformation for the protein starting structure. As previously observed by others (Minehardt et al., 2001), the phosphate tube remained intact during the simulation at higher temperature. The rotation was detected by monitoring the angles between each Pi oxygen, the Pi phosphorous atom, and an arbitrary fixed point during the course of the simulation. Fig. 7 shows the angle at one of the Pi oxygens as a function of time. Fig. 8 shows the rotation monitored in Fig. 7. The angular change in Fig. 7 is slightly different to that expected from a perfect tetraheydral arrangement of the oxygens. This is due to both the different bond angle for a hydroxyl oxygen and steric influences of the protein on the hydroxyl group.
FIGURE 7.

Rotation of Pi in the active site during a 200-ps simulation. During the course of the simulation, the relative rotational orientation of Pi was monitored by measuring the angles between each Pi oxygen, the Pi phosphorous atom, and an arbitrary fixed point during the course of the simulation. This plot shows the angle changes for oxygen O1G during 25 ps heating/equilibration and 200 ps production simulation run at 450 K.
FIGURE 8.

Relative orientations of Pi before and after rotation. The trajectory data from the simulation shown in Fig. 7 were averaged over 3 ps immediately before (160–163 ps) and immediately after (180–183 ps) the rotational event. The relative orientations of the Pi molecule in the two averages are shown. The orientation before rotation is on the left and that after rotation is on the right.
The role of the back door in the contractile cycle
Our analyses allow us to relate structural conformations of the back door to the myosin ATPase cycle. The MDdc·Mg·ADP·BeFx crystal structure of myosin shows the opening of the back door to be sufficiently dilated to allow free Pi to pass through the opening (Fig. 2) and out into the 50-kDa cleft. Our molecular dynamics simulations now permit us to investigate the subsequent, posthydrolysis interaction of the free Pi with the adjacent protein in the back door region. The simulations imply that this Pi-protein interaction results in the back door now narrowing sufficiently to prevent the unimpeded passage of Pi in and out of the phosphate tube due to motions of switch 1 and switch 2. However, the back door remains sufficiently open to allow the passage of water. Via homology with other ATPases, hydrolysis is widely postulated to occur via an in-line water attack on the γ-phosphate. In the MDdc·Mg·ADP·Vi structure, which represents this hydrolysis transition-state intermediate, the back door completely shuts off to preclude additional water exchange.
The interaction of myosin with actin is obligatory to convert the free energy of ATP hydrolysis into biologically useful force and motion. Thus myosin has evolved to have an extremely low ATPase rate in the absence of actin (0.01 s−1; reviewed in Bagshaw, 1982), resulting in a highly efficient relaxed state in living muscle. At physiological temperatures, the rate-limiting step in the myosin ATPase is the release of Pi from the M·ADP·Pi complex (reviewed in Taylor, 1979). Thus our molecular dynamics simulations of static x-ray structures now provide a structural explanation for both 18O-exchange with water and the low basal ATPase rate via a slow rate of Pi release after ATP hydrolysis on the myosin head.
Our original working hypothesis was that the back door would also provide an egress route for Pi after hydrolysis. The simulations still do not preclude this possibility. The dissociation rate for Pi has been measured in the range from 0.01 s−1 to 0.06 s−1 (reviewed in Woledge et al., 1985). Thermal fluctuations could potentially result in a dilation of the back door sufficient for Pi release on this timescale. This, of course, would be an extremely rare, and thus unseen, event on the 200-ps timescale of our molecular dynamics simulations.
It is clear that the back door as represented by the MDdc·Mg·ADP·BeFx and MDdc·Mg·ADP·Vi structures is not able to release Pi at the dramatically enhanced, actin-activated rate of 35 s−1 (Seow et al., 2001). Thus, the conformation of myosin in the strongly bound actomyosin complex must be different from that observed in either of these structures. As suggested by a number of investigators, a possible model of the strongly bound state is that the prepowerstroke state corresponds to the MDdc·Mg·ADP·Vi structure, and the strongly bound rigor state corresponds to the MDdc·Mg·ADP·BeFx structure or some slight variation thereof (Holmes, 1996; Dominguez et al., 1998; Houdusse et al., 1999). Comparison of the MDdc·Mg·ADP·BeFx and S1sc·Mg·ADP structures demonstrates that small changes in the position of the lower 50-kDa subdomain relative to the upper 50-kDa subdomain can lead to significant changes in the state of the back door. Such changes could be induced by actin binding, for which ΔG is ∼−8 kcal/mol, based on a Kd = 10−6 M for the actoS1 complex (Ritchie et al., 1993). Similarly, crystal-packing forces have been reported as high as 3 kcal/mol (Takahashi, 1997). Such forces could influence the conformation observed in the crystal structure.
Other studies have suggested that the 50-kDa cleft is closed during strong binding and an alternate route for Pi release opens. The closing of the 50-kDa cleft was first suggested by Rayment and co-workers (Rayment et al., 1993a) comparing the chicken skeletal myosin subfragment 1 x-ray crystal structure (Rayment et al., 1993b) to the actomyosin complex from cryoelectron microscopy data. Fluorescence studies have also been interpreted to imply that the 50-kDa cleft closes in the strongly bound state (Yengo et al., 2002; Conibear et al., 2003). Higher resolution cryoelectron microscopy studies supported the original hypothesis of Rayment and co-workers, with the additional conclusion that the closing of the 50-kDa cleft was accompanied by an opening of the phosphate tube via a displacement of switch 1 (Holmes et al., 2003). Biochemical, cross-linking, spectroscopic, and biomechanical studies first demonstrated that the phosphate tube opened, with the suggestion that hydrolysis products release could be modulated via a “trap door” mechanism involving a displacement of switch 1 (Pate et al., 1997). The trap door has received additional support from an x-ray structure of myosin with a displaced switch 1 (Reubold et al., 2003). However, the physiological relationship between this structure and all previous myosin structures showing a closed phosphate tube remains unresolved.
It should also be noted that recent evidence suggests that an alternative route for Pi release may modulate force production in myosin. Shaw and co-workers (Shaw and Yount, 2002a,b) used 18O-exchange between the solvent and the cleaved Pi to study the kinetics of several nucleotide and nanalog (Wang et al., 1993) nonnucleoside triphosphate substrates. The species had varying abilities to promote force transduction in myosin. Modeling of the 18O-exchange data suggested two separate kinetic pathways occurring in myosin. It was concluded that the Pi release route plays an integral role in the production of force. Substrates with exchange data similar to ATP supported force transduction and used a back door mechanism for Pi release. Substrates that uncoupled hydrolysis from mechanics appeared to use a diphosphate-first release pathway via the front door.
In summary, to our knowledge, our studies provide the first structural explanation for 18O-exchange in myosin. We also provide the first structural explanation for the rate-limiting step of myosin ATPase activity in the absence of actin. The precise relationship between Pi release and force production remains unresolved. Indeed, one cannot rule out back door release being the preferred pathway in the absence of actin and an alternative structural pathway resulting in an acceleration of Pi release in the presence of actin.
Acknowledgments
The authors thank Willy Wriggers for the use of his ADP and Pi parameters in the early phases of this work. The authors also thank Toshiko Ichiye, Brian Beck, Paul Swartz, and Rob Yelle for their many insightful discussions.
This work was supported by National Institutes of Health grants DK05195 (R.G.Y.), AR35186 (I.R.), and AR39643 (E.P.) and by a National Institutes of Health Biotechnology Training grant GM08336 (J.D.L.).
J. David Lawson's present address is Concurrent Pharmaceuticals, 502 W. Office Center Dr., Fort Washington, PA 19034.
References
- Bagshaw, C. R. 1982. Muscle Contraction. Chapman and Hall, New York.
- Berghuis, A. M., E. Lee, A. S. Raw, A. G. Gilman, and S. R. Sprang. 1996. Structure of the GDP-Pi complex of Gly203→Ala Giα1: a mimic of the ternary product complex of Gα-catalyzed GTP hydrolysis. Structure. 4:1277–1290. [DOI] [PubMed] [Google Scholar]
- Binda, C., A. Coda, R. Angelini, R. Federico, P. Ascenzi, and A. Mattevi. 1999. A 30-angstrom-long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure. 7:265–276. [DOI] [PubMed] [Google Scholar]
- Coleman, D. E., A. M. Berghuis, E. Lee, M. E. Linder, A. G. Gilman, and S. R. Sprang. 1994. Structures of active conformations of Giα1 and the mechanism of GTP hydrolysis. Science. 265:1405–1412. [DOI] [PubMed] [Google Scholar]
- Conibear, P. B., C. R. Bagshaw, P. Fajer, M. Kovacs, and A. Malnasi-Csizmadia. 2003. Myosin cleft movement and its coupling to actomyosin dissociation. Nat. Struct. Biol. 10:831–835. [DOI] [PubMed] [Google Scholar]
- Cooke, R. 1997. Actomyosin interaction in striated muscle. Physiol. Rev. 77:671–697. [DOI] [PubMed] [Google Scholar]
- Cope, M. J. T., J. Whisstock, I. Rayment, and J. Kendrick-Jones. 1996. Conservation within the myosin motor domain: implications for structure and function. Structure. 4:969–987. [DOI] [PubMed] [Google Scholar]
- Dominguez, R., Y. Freyzon, K. M. Trybus, and C. Cohen. 1998. Crystal structure of a vertebrate smooth muscle myosin motor domain and its complex with the essential light chain: visualization of the pre-power stroke state. Cell. 94:559–571. [DOI] [PubMed] [Google Scholar]
-
Fisher, A. J., C. A. Smith, J. B. Thoden, R. Smith, K. Sutoh, H. M. Holden, and I. Rayment. 1995. X-ray structures of the myosin motor domain of Dictyostelium discoideum complexed with MgADP·BeFx and
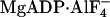 . Biochemistry. 34:8960–8972. [DOI] [PubMed] [Google Scholar]
. Biochemistry. 34:8960–8972. [DOI] [PubMed] [Google Scholar] - Friedman, A. L., M. A. Geeves, D. J. Manstein, and J. A. Spudich. 1998. Kinetic characterization of myosin head fragments with long-lived myosin.ATP states. Biochemistry. 37:9679–9687. [DOI] [PubMed] [Google Scholar]
- Furch, M., S. Fujita-Becker, M. A. Geeves, K. C. Holmes, and D. J. Manstein. 1999. Role of the salt-bridge between switch-1 and switch-2 of Dictyostelium myosin. J. Mol. Biol. 290:797–809. [DOI] [PubMed] [Google Scholar]
- Gilson, M. K., T. P. Straatsma, J. A. McCammon, D. R. Ripoll, C. H. Faerman, P. H. Axelsen, I. Silman, and J. L. Sussman. 1994. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science. 263:1276–1278. [DOI] [PubMed] [Google Scholar]
- Holmes, K. C. 1996. Muscle proteins–their actions and interactions. Curr. Opin. Struct. Biol. 6:781–789. [DOI] [PubMed] [Google Scholar]
- Holmes, K. C., I. Angert, F. J. Kull, W. Jahn, and R. R. Schroder. 2003. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 425:423–427. [DOI] [PubMed] [Google Scholar]
- Houdusse, A., V. N. Kalabokis, D. Himmel, A. G. Szent-Gyorgyi, and C. Cohen. 1999. Atomic structure of scallop myosin subfragment S1 complexed with MgADP: a novel conformation of the myosin head. Cell. 97:459–470. [DOI] [PubMed] [Google Scholar]
- Hyde, C. C., S. A. Ahmed, E. A. Padlan, E. W. Miles, and D. R. Davies. 1988. Three-dimensional structure of the tryptophan synthase α2β2 multienzyme complex from Salmonella typhimurium. J. Biol. Chem. 263:17857–17871. [PubMed] [Google Scholar]
- Kohls, D., T. Sulea, E. O. Purisima, R. E. MacKenzie, and A. Vrielink. 2000. The crystal structure of the formiminotransferase domain of formiminotransferase-cyclodeaminase: implications for substrate channeling in a bifunctional enzyme. Struct. Fold. Des. 8:35–46. [DOI] [PubMed] [Google Scholar]
- Krahn, J. M., J. H. Kim, M. R. Burns, R. J. Parry, H. Zalkin, and J. L. Smith. 1997. Coupled formation of an amidotransferase interdomain ammonia channel and a phosphoribosyltransferase active site. Biochemistry. 36:11061–11068. [DOI] [PubMed] [Google Scholar]
- Kull, F. J., R. D. Vale, and R. J. Fletterick. 1998. The case for a common ancestor: kinesin and myosin motor proteins and G proteins. J. Muscle Res. Cell Motil. 19:877–886. [DOI] [PubMed] [Google Scholar]
- Larsen, T. M., S. K. Boehlein, S. M. Schuster, N. G. Richards, J. B. Thoden, H. M. Holden, and I. Rayment. 1999. Three-dimensional structure of Escherichia coli asparagine synthetase B: a short journey from substrate to product. Biochemistry. 38:16146–16157. [DOI] [PubMed] [Google Scholar]
- Li, X. D., T. E. Rhodes, R. Ikebe, T. Kambara, H. D. White, and M. Ikebe. 1998. Effects of mutations in the γ-phosphate binding site of myosin on its motor function. J. Biol. Chem. 273:27404–27411. [DOI] [PubMed] [Google Scholar]
- Minehardt, T. J., R. Cooke, E. Pate, and P. A. Kollman. 2001. Molecular dynamics study of the energetic, mechanistic, and structural implications of a closed phosphate tube in ncd. Biophys. J. 80:1151–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar, N., G. Horn, C. Herrmann, A. Scherer, F. McCormick, and A. Wittinghofer. 1995. The 2.2 Å crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature. 375:554–560. [DOI] [PubMed] [Google Scholar]
- Pate, E., N. Naber, M. Matuska, K. Franks-Skiba, and R. Cooke. 1997. Opening of the myosin nucleotide triphosphate binding domain during the ATPase cycle. Biochemistry. 36:12155–12166. [DOI] [PubMed] [Google Scholar]
- Patterson, B. 1998. Intragenic suppressors of Dictyostelium myosin G680 mutants demarcate discrete structural elements. Implications for conformational states of the motor. Genetics. 149:1799–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raw, A. S., D. E. Coleman, A. G. Gilman, and S. R. Sprang. 1997. Structural and biochemical characterization of the GTPγS- GDP·Pi-, and GDP-bound forms of a GTPase-deficient Gly42 → Val mutant of Giα1. Biochemistry. 36:15660–15669. [DOI] [PubMed] [Google Scholar]
- Rayment, I., H. M. Holden, M. Whittaker, C. B. Yohn, M. Lorenz, K. C. Holmes, and R. A. Milligan. 1993a. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 261:58–65. [DOI] [PubMed] [Google Scholar]
- Rayment, I., W. R. Rypniewski, K. Schmidt-Base, R. Smith, D. R. Tomchick, M. M. Benning, D. A. Winkelmann, G. Wesenberg, and H. M. Holden. 1993b. Three-dimensional structure of myosin subfragment-1: a molecular motor. Science. 261:50–58. [DOI] [PubMed] [Google Scholar]
- Reubold, T. F., S. Eschenburg, A. Becker, F. J. Kull, and D. J. Manstein. 2003. A structural model for actin-induced nucleotide release in myosin. Nat. Struct. Biol. 10:826–830. [DOI] [PubMed] [Google Scholar]
- Ritchie, M. D., M. A. Geeves, S. K. Woodward, and D. J. Manstein. 1993. Kinetic characterization of a cytoplasmic myosin motor domain expressed in Dictyostelium discoideum. Proc. Natl. Acad. Sci. USA. 90:8619–8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, N., T. Shimada, and K. Sutoh. 1998. Mutational analysis of the switch II loop of Dictyostelium myosin II. J. Biol. Chem. 273:20334–20340. [DOI] [PubMed] [Google Scholar]
- Schlattner, U., M. Forstner, M. Eder, O. Stachowiak, K. Fritz-Wolf, and T. Wallimann. 1998. Functional aspects of the X-ray structure of mitochondrial creatine kinase: a molecular physiology approach. Mol. Cell. Biochem. 184:125–140. [PubMed] [Google Scholar]
- Seow, C. Y., H. D. White, and L. E. Ford. 2001. Effects of substituting uridine triphosphate for ATP on the crossbridge cycle of rabbit muscle. J. Physiol. 537:907–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, M. A., and R. G. Yount. 2000a. Kinetic and 18O exchange studies of the hydrolysis of nucleoside triphosphates by myosin S1 and their implications in force production. Biophys. J. 78:272a. (Abstr.) [Google Scholar]
- Shaw, M. A., and R. G. Yount. 2000b. Measurement of intermediate 18O exchange during hydrolysis of NANTP and related analogs by myosin S1. Biophys. J. 78:273a. (Abstr.) [Google Scholar]
- Shimada, T., N. Sasaki, R. Ohkura, and K. Sutoh. 1997. Alanine scanning mutagenesis of the switch I region in the ATPase site of Dictyostelium discoideum myosin II. Biochemistry. 36:14037–14043. [DOI] [PubMed] [Google Scholar]
- Sleep, J. A., D. D. Hackney, and P. D. Boyer. 1980. The equivalence of phosphate oxygens for exchange and the hydrolysis characteristics revealed by the distribution of [18O]Pi species formed by myosin and actomyosin ATPase. J. Biol. Chem. 255:4094–4099. [PubMed] [Google Scholar]
- Sleep, J. A., and R. L. Hutton. 1980. Exchange between inorganic phosphate and adenosine 5′-triphosphate in the medium by actomyosin subfragment 1. Biochemistry. 19:1276–1283. [DOI] [PubMed] [Google Scholar]
- Smith, C. A., and I. Rayment. 1996a. Active site comparisons highlight structural similarities between myosin and other P-loop proteins. Biophys. J. 70:1590–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, C. A., and I. Rayment. 1996b. X-ray structure of the magnesium(II) ·ADP·vanadate complex of the Dictyostelium discoideum myosin motor domain to 1.9 Å resolution. Biochemistry. 35:5404–5417. [DOI] [PubMed] [Google Scholar]
- Takahashi, T. 1997. Significant role of electrostatic interactions for stabilization of protein assemblies. Adv. Biophys. 34:41–54. [DOI] [PubMed] [Google Scholar]
- Taylor, E. W. 1979. Mechanism of actomyosin ATPase and the problem of muscle contraction. CRC Crit. Rev. Biochem. 6:103–164. [DOI] [PubMed] [Google Scholar]
- Tesmer, J. J., T. J. Klem, M. L. Deras, V. J. Davisson, and J. L. Smith. 1996. The crystal structure of GMP synthetase reveals a novel catalytic triad and is a structural paradigm for two enzyme families. Nat. Struct. Biol. 3:74–86. [DOI] [PubMed] [Google Scholar]
- Thoden, J. B., H. M. Holden, G. Wesenberg, F. M. Raushel, and I. Rayment. 1997. Structure of carbamoyl phosphate synthetase: a journey of 96 Å from substrate to product. Biochemistry. 36:6305–6316. [DOI] [PubMed] [Google Scholar]
- Vale, R. D. 1996. Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J. Cell Biol. 135:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D., E. Pate, R. Cooke, and R. G. Yount. 1993. Synthesis of non-nucleotide ATP analogues and characterization of their interaction with muscle fibres. J. Muscle Res. Cell Motil. 14:484–497. [DOI] [PubMed] [Google Scholar]
- Webb, M. R., G. G. McDonald, and D. R. Trentham. 1978. Kinetics of oxygen-18 exchange between inorganic phosphate and water catalyzed by myosin subfragment 1, using the 18O shift in 31P NMR. J. Biol. Chem. 253:2908–2911. [PubMed] [Google Scholar]
- Weiner, S. J., P. A. Kollman, D. T. Nguyen, and D. A. Case. 1986. An all atom forcefield for simulations of proteins and nucleic acid. J. Comput. Chem. 7:230–252. [DOI] [PubMed] [Google Scholar]
- Wells, J. A., and R. G. Yount. 1979. Active site trapping of nucleotides by crosslinking two sulfhydryls in myosin subfragment 1. Proc. Natl. Acad. Sci. USA. 76:4966–4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woledge, R. C., N. A. Curtin, and E. Homsher. 1985. Energetic Aspects of Muscle Contraction. Academic Press, London, UK. [PubMed]
- Wriggers, W., and K. Schulten. 1997. Stability and dynamics of G-actin: back-door water diffusion and behavior of a subdomain 3/4 loop. Biophys. J. 73:624–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Y., M. Nejad, and B. Patterson. 1999. Dictyostelium myosin II G680V suppressors exhibit overlapping spectra of biochemical phenotypes including facilitated phosphate release. Genetics. 153:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yengo, C. M., E. M. De La Cruz, L. R. Chrin, D. P. Gaffney 2nd, and C. L. Berger. 2002. Actin-induced closure of the actin-binding cleft of smooth muscle myosin. J. Biol. Chem. 277:24114–24119. [DOI] [PubMed] [Google Scholar]
- Yount, R. G., D. Lawson, and I. Rayment. 1995. Is myosin a “back door” enzyme? Biophys. J. 68:44S–47S; discussion 47S–49S. [PMC free article] [PubMed] [Google Scholar]